

Abstract
To characterize the structures of N-glycans derived from human serum, we report a strategy that combines microchip electrophoresis, standard addition, enzymatic digestion, and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS). We compared: (i) electrophoretic mobilities of known N-glycans from well-characterized (standard) glycoproteins through standard addition, (ii) the electrophoretic mobilities of N-glycans with their molecular weights determined by MALDI-MS, and (iii) electrophoretic profiles of N-glycans enzymatically treated with fucosidase. The key step to identify the sialylated N-glycans was to quantitatively neutralize the negative charge on both α2,3- and α2,6-linked sialic acids by covalent derivatization with methylamine. Both neutralized and nonsialylated N-glycans from these samples were then reacted with 8-aminopyrene-1,3,6-trisulfonic acid (APTS) to provide a fluorescent label and a triple-negative charge, separated by microchip electrophoresis, and detected by laser-induced fluorescence. The methylamidation step leads to a 24% increase in the peak capacity of the separation and direct correlation of electrophoretic and MALDI-MS results. In total, 37 unique N-glycan structures were assigned to 52 different peaks recorded in the electropherograms of the serum samples. This strategy ensures the needed separation efficiency and detectability, easily resolves linkage and positional glycan isomers, and is highly reproducible.
Keywords: microfluidics, microchip electrophoresis, sialic acid neutralization, methylamidation, human blood serum, N-glycan structural identification, glycomics, cancer biomarkers
TOC image
Introduction
Nearly two decades ago, the first link between changes in glycosylation and the onset and development of cancer was established.1–4 These altered patterns in glycosylation can be expressed on serum proteins as an immunological response, on acute-phase proteins synthesized by the liver and secreted into the blood, and on membrane-bound proteins of tumor cells which may be shed into the blood stream. Consequently, identification and reliable quantitation of glycans derived from serum samples may lead to non-invasive analytical methods for cancer diagnostic and prognostic measurements. An important step towards developing accurate screening methods is to determine which glycans are impacted most by the disease and how the glycan profile changes during early stages and progression of the disease. Consequently, we need to accurately quantify changes in the glycosylation profile and to demonstrate if what is measured actually correlates to a disease state.
Mass spectrometry (MS) methods effectively track alterations in glycan profiles associated with various cancers, including breast cancer,5–9 ovarian cancer,10–12 prostate cancer,13–14 hepatocellular carcinoma,15 colon cancer,16 pancreatic cancer,17–18 and esophageal adenocarcinoma.19 These MS-based methods are well-suited for structural characterization of unknown glycans and high-throughput analysis of known glycan structures, but do not readily distinguish among the structural isomers of various glycans. On the other hand, capillary-20–27 and microchip-based28–32 electrophoresis provide sufficient separation efficiency to easily resolve glycans and their positional and linkage isomers.25–29,31 Moreover, electrophoretic-based methods have been used to characterize N-glycans from therapeutic and immunological proteins,24–26,33 to diagnose liver disease noninvasively,30,34 and to compare N-glycan profiles from control groups and patients with esophageal adenocarcinoma32 and ovarian cancer.35
However, reliable methods are still needed to identify N-glycans resolved by capillary electrophoresis (CE). One method to identify N-glycan structures in CE separations is to compare the electrophoretic mobilities of unknown samples and purified standards.21,36–37 Although this strategy is straightforward, availability of well-characterized glycan standards is limited. Full enzymatic sequencing allows comprehensive structural determination of N-glycan mixtures,25–26,34,38–39 but is both time and labor intensive, especially when sample complexity is high. Another method is glucose unit (GU) analysis40–43 where an oligoglucose ladder is used as a migration standard. A GU value is calculated from the migration time of the N-glycan of interest relative to the migration times of the leading and trailing oligoglucose peaks. GU values of known and unknown structures are then compared to identify the unknown structure. However, as discussed below, fluctuations between migration times of standards and unknowns can lead to misidentification. More recently, some N-glycan structures have been identified by direct hyphenation of CE with MS through electrospray ionization (ESI).44–47 With CE-MS, we are not yet able to resolve structural isomers with sufficient sensitivity to identify the same serum N-glycans reported here. This shortfall in separation efficiency is due, in part, to a mismatch in optimum buffer compositions for CE and MS and band-broadening attributed to the ESI interface.
To identify N-glycans derived from serum samples, we correlate here various sets of data from microchip electrophoresis and MALDI-MS. The first step is to ensure the N-glycans separated by microchip electrophoresis vary systematically with their hydrodynamic volumes. Because terminal sialic acid (N-acetylneuraminic acid) residues carry a negative charge and are present in many glycan structures, we want to neutralize this additional charge prior to fluorescent labeling of the samples with 8-aminopyrene-1,3,6-trisulfonic acid (APTS).48 Often, sialic residues can be neutralized by methylamidation49 and other chemical modifications, e.g., permethylation,50–51 to prevent their dissociation from glycan structures and improve detection sensitivity during MS analysis. Here, we adapt a similar strategy to neutralize the charge of both α2,3- and α2,6-linked sialic acids by covalent derivatization with methylamine. Consequently, the methylamidated N-glycans carry solely the triple-negative charge from the APTS label, so their migration order is based purely on their hydrodynamic volume (or size).
With N-glycans from several well-characterized (standard) glycoproteins, we confirmed that the electrophoretic mobility varied systematically (mostly linearly) with the molecular weight determined by MALDI-MS of derivatized N-glycans. N-Glycan structures from other common glycoproteins were assigned by correlating electrophoretic mobilities with molecular weights and relevant information from previous reports for fibrinogen,52 immunoglobulin G,26,53–55 ribonuclease B,56 haptoglobin,57 lactoferrin,58 and α-1-acid glycoprotein.49,59 With this suite of 36 N-glycan standards, we used standard addition to identify a majority of the structures in the electropherograms of the serum samples. For peaks without a corresponding standard N-glycan, the MALDI-MS data provided insight as to whether a particular N-glycan could be present in the sample. Additionally, unique isomers were assigned to individual peaks in the electropherogram by comparing our results with structures and intensities reported in the literature and the migration order of known sialic acid isomers. To resolve or confirm several ambiguous assignments, we treated the samples with fucosidase to differentiate between fucosylated and non-fucosylated structures. In total, we assigned 37 distinct N-glycan structures to 52 different detectable peaks from the serum samples, which include some positional and linkage isomers.
Experimental Section
Materials
We purchased the following materials: high-purity 8-aminopyrene-1,3,6-trisulfonic acid (APTS) from Beckman Coulter, Inc.; 2,3-sialyllactose and 2,6-sialyllactose from Carbosynth Ltd.; peptide-N-glycosidase F (PNGase F) of Chryseobacterium menigosepticum (EC 3.2.2.18) from Northstar BioProducts; α(1–2,3,4,6) fucosidase (bovine kidney; GKX-5006 kit) from ProZyme, Inc.; haptoglobin from Abcam; human α-1-acid glycoprotein (AGP), human fibrinogen, human immunoglobulin G (IgG), bovine lactoferrin, and bovine ribonuclease B (RNase B) from Sigma-Aldrich Co.; and activated carbon and amino (NH2) micro-spin columns from Harvard Apparatus. All other chemicals were purchased from Sigma-Aldrich Co. Human serum samples were purchased from BioreclemationIVT and also obtained from an experimental drug trial from patients with late-stage recurrent ovarian cancer.60 The sample collection was approved through institutional review board-approved clinical protocols (HOG-Breast120 and HOG-Gyn062).
Preparation of N-Glycan Samples
N-Glycans were enzymatically cleaved from standard and serum glycoproteins with PNGase F and purified.35 For methylamidation of sialic acids on N-glycans,49 we modified the protocol to minimize sample loss and maximize purity. Dried samples of sialyllactose (0.5 μg) and N-glycans derived from serum and standard glycoproteins were dissolved in 5 μL of dimethyl sulfoxide (DMSO) containing 2 M methylamine hydrochloride and 1 M 4-methylmorpholine. Subsequently, 5 μL of 100 mM (7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyAOP) in DMSO was added to these samples. The reaction proceeded for 60 min under ambient conditions and was quenched with 240 μL of an 85:15 acetonitrile (ACN)/H2O solution, and the N-glycans were purified with amino (NH2) micro-spin columns. Briefly, micro-spin columns were conditioned with three 300-μL aliquots of a solution composed of H2O/ACN/TFA (95:5:0.1) and equilibrated with three 300-μL aliquots of the 85:15 ACN/H2O solution. The samples were loaded onto the columns, the columns were centrifuged, the sample solution was collected in microcentrifuge tubes and reloaded onto the columns, and the entire process was repeated. The samples were washed with two 200-μL aliquots of the 85:15 ACN/H2O solution and finally eluted with 300-μL of a 20:80 ACN/H2O solution. Samples requiring no further treatment or derivatization were labeled with APTS48 by established procedures29 to impart a negative charge for electrophoresis and a fluorescent tag for detection.
N-Glycan Treatment with a Non-Specific Fucosidase
Several samples of methylamidated N-glycans derived from serum, IgG, and haptoglobin were enzymatically treated with a fucosidase prior to electrophoretic analysis. These samples were first dissolved in 2 μL of a 5× reaction buffer from the GKX-5006 kit (sodium citrate phosphate, pH 6.0) and then incubated with 8 μL of α(1–6) fucosidase enzyme at 37 °C for 48 h. After incubation, the samples were purified by solid-phase extraction on activated carbon columns, vacuum dried, and then labeled with APTS.
Fabrication and Operation of Microfluidic Devices
The microfluidic devices were fabricated in glass substrates29,35 and had a serpentine channel with a 22-cm separation length and asymmetrically tapered, 180° turns (Figure 1b).61 To minimize electroosmotic flow and prevent sample adsorption, the microchannels were coated with linear poly(acrylamide). Samples were introduced into the analysis channel by standard and modified pinched injections29,62 and detected by laser-induced fluorescence with excitation from the 465-nm line of an argon ion laser (Melles Griot, Inc.). Peak areas and migration times were extracted from the electropherograms by fitting the peaks with a Gaussian function in OriginPro 2015.
Figure 1.
N-Glycans are attached to protein backbones through N-acetylglucosamine (GlcNAc; blue square) and often contain terminal N-acetylneuraminic acids (NeuAc or sialic acids; purple diamond). (a) After N-glycans are extracted from blood serum samples with PNGase F (cleaved), the sialic acids are neutralized by covalent derivatization with methylamine, and the reducing end of GlcNAc is labeled with 8-aminopyrene-1,3,6-trisulfonic acid (APTS; derivatized). Other symbols represent mannose (green circle), galactose (yellow circle), and fucose (red triangle). These samples are analyzed on (b) a microfluidic device that has a serpentine channel with a separation length of 22 cm and two asymmetrically tapered 180° turns with taper ratio 3.
MALDI-MS
For MALDI-MS analysis,12 the samples were interrogated in positive-ion mode by an Applied Biosystems 4800 MALDI-TOF/TOF mass spectrometer in the m/z range from 1500 to 5000. Spectra were acquired from a total of 1000 laser shots for each sample, spotted in triplicate. Spectra were baseline corrected, filtered, and converted to text files in Data Explorer 4.0. The data were normalized by expressing the intensity of each glycan ion as a percentage of the total intensity for all glycans in the spectra. N-Glycan structures and expected masses were drawn and calculated, respectively, with GlycoWorkBench software.
Results and Discussion
Neutralization of the Charge on α2,3- and α2,6-linked Sialic Acids by Methylamidation
N-Glycans often contain terminal sialic acids, N-acetylneuraminic (NeuAc) or N-glycolylneuraminic acid (NeuGc). After enzymatic release and purification of N-glycans, the charge on both α2,3- and α2,6-linked sialic acids was neutralized by covalent derivatization with methylamine (Figure 1a). Following this derivatization step, the electrophoretic migration order of the glycans is based on their hydrodynamic volume. Without derivation, the electrophoretic mobilities of similarly sized N-glycans can vary dramatically if one or more sialic acids with their associated charges are present. To evaluate the methylamidation reaction, 2,3-sialyllactose and 2,6-sialyllactose were used as model compounds and were analyzed by positive-ion mode MALDI-MS (Figure S1a–b in Supporting Information) and microchip electrophoresis (Figure S1c in Supporting Information). The results from this reaction are indicative of quantitative derivatization of both α2,3- and α2,6-linked sialic acids. The reaction is also carried out under mild reaction conditions, i.e., pH 8 and ambient temperatures, which further minimizes undesired byproducts.
Microchip Electrophoresis of Methylamidated N-Glycans
For electrophoretic analysis and fluorescence detection, the methylamidated N-glycan structures must be compatible with subsequent labeling of the reducing end GlcNAc with 8-aminopyrene-1,3,6-trisulfonic acid (APTS).48 Chemical derivatization to neutralize the charge of sialic acids44 and fluorescent tagging with APTS are both quantitative and, as shown here, can be used in combination. All samples in this study were analyzed in 50 mM HEPES buffer (pH 6.8) with the microfluidic device shown in Figure 1b with a 22-cm long separation channel that was operated at 1250 V/cm.
Our derivatization strategy is illustrated through electrophoretic analysis of 2,3-sialyllactose and 2,6-sialyllactose and N-glycan samples derived from RNase B, fibrinogen, and human serum. After the samples were treated with methylamine and labeled with APTS, the electrophoretic separations provided excellent resolution between linkage isomers, e.g., 2,3-sialyllactose and 2,6-sialyllactose (Figure S1c), and positional isomers, e.g., mannose 7 and 8 isomers (Figure S2c in Supporting Information). In the separation of N-glycans from fibrinogen (Figure S2a–b in Supporting Information), the shift to longer migration times and switch in migration order are dramatic for N-glycans with one and two sialic acids.
Note that the high-mannose structures do not have sialic acids, but were subjected to the same amidation reaction as a control. Because the methylamidation reaction did not impact analysis of the N-glycans from RNase B, these glycans were spiked into all samples prior to analysis and used as a migration reference to manually align electropherograms to minimize migration time variation from small fluctuations in temperature, buffer composition, and separation conditions. Initially, we tested an oligoglucose ladder as an internal standard and intended to calculate glucose units for the N-glycans derived from serum. However, for reasons unknown to us, addition of the oligoglucose ladder to the serum N-glycan samples resulted in irreproducible, random shifts in migration times in the electropherograms that could not be corrected with internal standards. Because accurate glucose units could not be calculated, we were not able to compare our results to other reported data.
Human serum contains a variety of N-glycan structures, both sialylated and non-sialylated (neutral). After being labeled with APTS, each N-glycan gains a -3 charge, in addition to any native charge present from sialic acids. For non-methylamidated N-glycans with sialic acids, this variation in the number of charges on each N-glycan adds complexity to electropherogram (Figure 2a), in which there are a large number of peaks corresponding to structures with one to four or more sialic acids, all migrating in the window from 67 to 72 s. Without extensive enzymatic digestion and analysis of the native serum N-glycans, deconvolution and assignment of structures in the electrophogram are difficult. After neutralization of the native charge by methylamidation, structures with one or more sialic acids shift to longer migration times, effectively inverting the migration order (Figure 2b) of these sialylated structures. Structures not containing sialic acids (neutral structures) are not affected by the methylamidation reaction, which results in consistent migration times before and after this derivatization step, as indicated in Figure 2. In addition to a migration order that correlates directly with hydrodynamic volume, the shift of sialylated structures to longer migration times also expands the migration window without a loss in separation efficiency. This larger window contributes to an increase in peak capacity from 85 peaks to 105 peaks, an increase of 24%.
Figure 2.
Electropherograms of (a) native and (b) methylamidated N-glycans derived from serum samples. The methylamidated N-glycans with neutralized sialic acids shift to longer migration times (lower electrophoretic mobilities) compared to the native N-glycans. A region of peaks that overlaps between the two separations from 80 and 96 s indicates many of these peaks correspond to non-sialylated N-glycans because their migration times are unaffected by the neutralization reaction. The electric field strength was 1250 V/cm, the separation length was 22 cm, and the samples were labeled with APTS. The y-axis is in arbitrary units of fluorescence.
Correlation of Hydrodynamic Volume (Size) and Molecular Weight
After covalent derivatization of N-glycan samples with methylamine, the negative charge on the sialic acids is neutralized, and after fluorescent labeling with APTS, all N-glycan structures have the same effective charge (i.e., -3 from APTS at pH 6.8). Consequently, the migration order of the N-glycans is based solely on their hydrodynamic volume (size). Additionally, the change in mass of the glycan structures after derivatization with methylamine, i.e., 13 Da for each sialic acid, does not significantly influence the electrophoretic mobility. Figure 3 shows the correlation of average electrophoretic mobility and molecular weight of methylamidated N-glycans that were derived from immunoglobulin G and human serum that are either neutral or contain ≥ 1 sialic acids. For N-glycans with more than one structural isomer, the average electrophoretic mobility of these isomers is plotted. To spot-check proper alignment of the data in Figure 3, we compared the electrophoretic mobilities of the peaks with the largest areas in the electropherograms with the molecular weights of the most intense peaks in the MALDI spectra. Table S1 in the Supporting Information lists the N-glycan structures under consideration and their relative intensities and peak areas. To calculate the estimated electrophoretic mobilities, the data in Figure 3 were fitted with linear regressions, and the fit parameters are reported in Table S2 in the Supporting Information.
Figure 3.
Variation of average electrophoretic mobility (μep) with molecular weight for methylamidated N-glycans derived from IgG and serum. Electrophoretic mobilities from the separations can be compared to the molecular weight of N-glycan structures that have been neutralized by methylamidation or do not contain sialic acids. Bisected structures have higher mobilities than non-bisected structures of similar molecular weight, whereas fucosylated and nonfucosylated structures of similar molecular weight have similar mobilities. Underivatized N-glycans that contain ≥ 1 sialic acids do not exhibit the same correlation between electrophoretic mobility and molecular weight. Error bars are ±σ for n = 3 replicate injections.
Similar, but separate, linear trends are observed for bisected and non-bisected N-glycans. The bisected structures have a smaller hydrodynamic volume and, consequently, a higher electrophoretic mobility than a non-bisected structure of similar molecular weight. This unique correlation between electrophoretic mobility and molecular weight can help identify whether an unknown structure is bisected. Underivatized N-glycans with one or more sialic acids do not exhibit similar correlations due to considerable variation in the charge on these structures. Because electrophoretic separations easily resolve small differences in the hydrodynamic volumes of N-glycans, structural isomers of both bisected and non-bisected structures can be readily separated, which is difficult with MS-based analysis. Also, as seen in Figure 3, fucosylated and nonfucosylated N-glycans with similar molecular weights have comparable electrophoretic mobilities. Several derivatization strategies54,57,63–65 have been employed for the separation of sialic acid isomers with MS analyses, but isomers with other monosaccharides remain beyond the scope of current non-MSn-based techniques.
Structural Identification of Serum N-Glycans
We identified the structures shown in Figure 4 through a multi-step process. We compared the electrophoretic mobilities of N-glycans from standard glycoproteins through standard addition. These standard glycoproteins included fibrinogen, lactoferrin, AGP, IgG, haptoglobin, and RNase B. We also analyzed the same serum samples by MALDI-MS to determine what structures were present and their relative intensities. The electrophoretic mobilities of the methylamidated N-glycans were then correlated with molecular weights of N-glycans determined by MALDI-MS. To help resolve ambiguous assignments, IgG, haptoglobin, and serum samples were enzymatically treated with a fucosidase, and electropherograms prior to and after enzymatic treatment were compared.26,52,55–56
Figure 4.
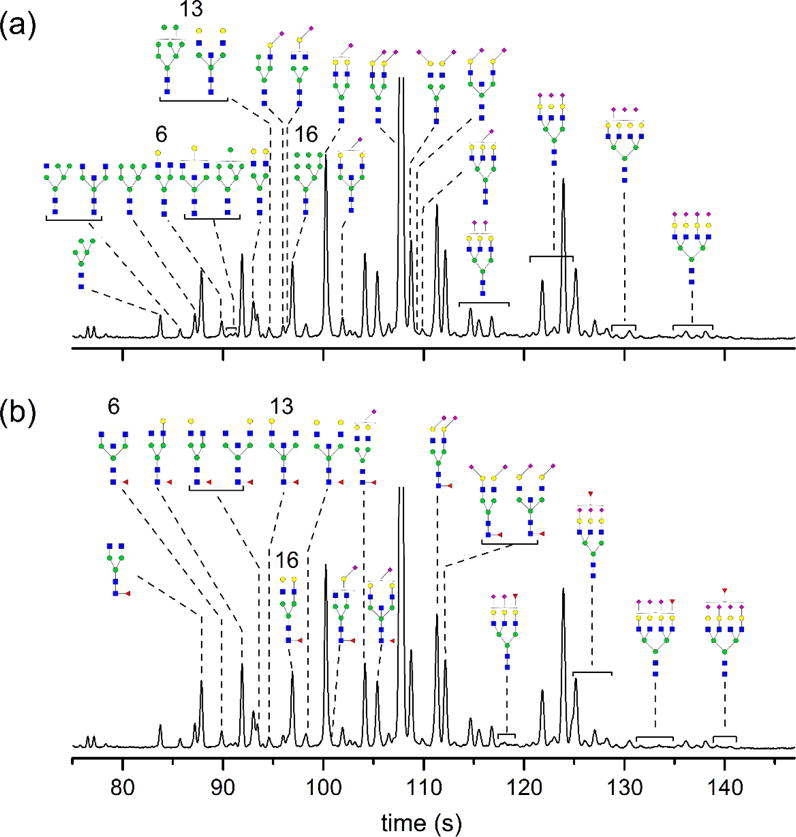
Electropherogram labeled with (a) non-fucosylated and (b) fucosylated N-glycans derived from serum. N-glycan structures were identified by matching their electrophoretic mobilities with known N-glycans and correlating their electrophoretic mobilities with molecular weights determined by MALDI-MS. Known N-glycans were derived from fetuin, haptoglobin, α-1 acid glycoprotein, lactoferrin, immunoglobulin G, and ribonuclease B. Peaks 6, 13, and 16 are identified as containing both fucosylated and non-fucosylated structures. The electric field strength was 1250 V/cm, the separation length was 22 cm, and the samples were labeled with APTS. The y-axis is in arbitrary fluorescence units. See Figure 1 for definitions of monosaccharide symbols.
Fibrinogen,52 IgG,26,53–55 and ribonuclease B56 have been previously characterized in substantial detail which allowed for a relatively simple assignment of structures reported in the literature to peaks in our electropherograms. Haptoglobin,57 lactoferrin,58 and AGP49,59 proved to be more complicated sources of N-glycan structures to identify in serum due to less rigorous enzymatic studies and large, multiply sialylated structures. Electropherograms from these glycoproteins were analyzed by comparing reported structures and intensities, if available, for each glycoprotein to electropherograms and MALDI-MS spectra for the same samples. Haptoglobin and AGP also contained several structures with multiple possible isomers. Because each glycan is labeled with a single fluorophore, the relative peak areas in the electropherograms can be compared quite reliably. However, the relative intensities of MALDI-MS data are not quantitative and could not be directly compared to the peak areas of the electropherograms (see Table S1 in the Supporting Information for a comparison of the relative intensities). After a library of structures from each glycoprotein was generated, the electrophoretic mobilities of N-glycans from these glycoproteins were matched to peaks from the serum sample.
Further structural assignments were refined through the use of fucosidase-treated IgG. Due to the presence of several sets of peaks that were identified as containing both fucosylated and nonfucosylated N-glycans, the enzyme fucosidase was used to further separate these structures and definitively assign structures to each peak. IgG was used as a simple analog of serum to optimize reaction conditions and confirm expected peak shifts. For serum samples, peaks corresponding to fucosylated structures and their de-fucosylated forms were monitored for their relative peak area reductions and increases, respectively. If the peak behavior was in agreement with behavior of a fucosylated N-glycan, the structural assignment remained unchanged. Conversely, if the peak area did not decrease after enzymatic treatment, neighboring peaks were monitored, and in some cases, the structural assignment was revised.
Figure 4 shows the culmination of these structural assignments with an annotated electropherogram, which has been separated into non-fucosylated structures (Figure 4a) and fucosylated structures (Figure 4b) for clarity. The numbered peaks correspond to peaks that have been identified as being comprised of both fucosylated and non-fucosylated structures with mobilities similar enough that we could not distinguish them. Enzymatic treatment with fucosidase resulted in a decrease in the peak area, but a portion of the peak remained after treatment. Also, in several instances, structures are shown with core fucosylation. These structures may be fucosylated on the antennae; however, the standards, e.g., IgG, used to determine peak position have only core fucosylation. In total, 37 unique structures were assigned to 52 different peaks, which include positional and linkage isomers of the 37 unique structures.
Conclusion
In conclusion, we demonstrated that the charge on α2,3- and α2,6-linked sialic acid containing N-glycans can be effectively neutralized by covalent derivatization with methylamine prior to fluorescent labeling with APTS. These modifications allowed the structures of serum N-glycans to be identified by comparing the electrophoretic mobilities from microchip electrophoresis and molecular weights of structures identified in the same samples by MALDI-MS analysis. Because both methylamidation and fluorescent labeling reactions are quantitative, electrophoretic analysis of serum samples can be used to determine quantitative differences among specific N-glycans structures from patients with cancer and disease-free individuals. More importantly, structural identification of N-glycans that change as a function of disease-state is necessary for biomarker discovery and validation.
Supplementary Material
Acknowledgments
This work was supported by NIH R01 GM106084 and NIH R01 GM024349. The authors thank Dr. Daniela E. Matei and Nancy Menning from the Indiana University School of Medicine (Indianapolis, IN) for providing the samples and the Indiana University Nanoscale Characterization Facility for use of its instruments.
Footnotes
Supporting Information Available. MALDI-MS spectra of 2,3-sialyllactose and 2,6-sialyllactose; electropherogram of 2,3-sialyllactose and 2,6-sialyllactose; electropherograms of native and methylamidated N-glycans derived from fibrinogen; electropherogram of N-glycans derived from ribonuclease B; table of N-glycan structures, their masses, relative intensities, and relative peak areas; and table of linear regression parameters. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hakomori S. Cancer Res. 1996;56:5309–5318. [PubMed] [Google Scholar]
- 2.Kobata A. Glycoconjugate J. 1998;15:323–331. doi: 10.1023/a:1006961532182. [DOI] [PubMed] [Google Scholar]
- 3.Dennis JW, Granovsky M, Warren CE. Bioessays. 1999;21:412–421. doi: 10.1002/(SICI)1521-1878(199905)21:5<412::AID-BIES8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 4.Dube DH, Bertozzi CR. Nat Rev Drug Discov. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 5.Kirmiz C, Li BS, An HJ, Clowers BH, Chew HK, Lam KS, Ferrige A, Alecio R, Borowsky AD, Sulaimon S, Lebrilla CB, Miyamoto S. Mol Cell Proteomics. 2007;6:43–55. doi: 10.1074/mcp.M600171-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.Kyselova Z, Mechref Y, Kang P, Goetz JA, Dobrolecki LE, Sledge GW, Schnaper L, Hickey RJ, Malkas LH, Novotny MV. Clin Chem. 2008;54:1166–1175. doi: 10.1373/clinchem.2007.087148. [DOI] [PubMed] [Google Scholar]
- 7.Abd Hamid UM, Royle L, Saldova R, Radcliffe CM, Harvey DJ, Storr SJ, Pardo M, Antrobus R, Chapman CJ, Zitzmann N, Robertson JF, Dwek RA, Rudd PM. Glycobiology. 2008;18:1105–1118. doi: 10.1093/glycob/cwn095. [DOI] [PubMed] [Google Scholar]
- 8.Goetz JA, Mechref Y, Kang P, Jeng MH, Novotny MV. Glycoconjugate J. 2009;26:117–131. doi: 10.1007/s10719-008-9170-4. [DOI] [PubMed] [Google Scholar]
- 9.Pierce A, Saldova R, Abd Hamid UM, Abrahams JL, McDermott EW, Evoy D, Duffy MJ, Rudd PM. Glycobiology. 2010;20:1283–1288. doi: 10.1093/glycob/cwq090. [DOI] [PubMed] [Google Scholar]
- 10.An HJ, Miyamoto S, Lancaster KS, Kirmiz C, Li BS, Lam KS, Leiserowitz GS, Lebrilla CB. J Proteome Res. 2006;5:1626–1635. doi: 10.1021/pr060010k. [DOI] [PubMed] [Google Scholar]
- 11.Saldova R, Royle L, Radcliffe CM, Hamid UMA, Evans R, Arnold JN, Banks RE, Hutson R, Harvey DJ, Antrobus R, Petrescu SM, Dwek RA, Rudd PM. Glycobiology. 2007;17:1344–1356. doi: 10.1093/glycob/cwm100. [DOI] [PubMed] [Google Scholar]
- 12.Alley WR, Vasseur JA, Goetz JA, Svoboda M, Mann BF, Matei DE, Menning N, Hussein A, Mechref Y, Novotny MV. J Proteome Res. 2012;11:2282–2300. doi: 10.1021/pr201070k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabares G, Radcliffe CM, Barrabes S, Ramirez M, Aleixandre RN, Hoesel W, Dwek RA, Rudd PM, Peracaula R, de Llorens R. Glycobiology. 2006;16:132–145. doi: 10.1093/glycob/cwj042. [DOI] [PubMed] [Google Scholar]
- 14.Kyselova Z, Mechref Y, Al Bataineh MM, Dobrolecki LE, Hickey RJ, Vinson J, Sweeney CJ, Novotny MV. J Proteome Res. 2007;6:1822–1832. doi: 10.1021/pr060664t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldman R, Ressom HW, Varghese RS, Goldman L, Bascug G, Loffredo CA, Abdel-Hamid M, Gouda I, Ezzat S, Kyselova Z, Mechref Y, Novotny MV. Clin Cancer Res. 2009;15:1808–1813. doi: 10.1158/1078-0432.CCR-07-5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vercoutter-Edouart AS, Slomianny MC, Dekeyzer-Beseme O, Haeuw JF, Michalski JC. Proteomics. 2008;8:3236–3256. doi: 10.1002/pmic.200800151. [DOI] [PubMed] [Google Scholar]
- 17.Zhao J, Simeone DM, Heidt D, Anderson MA, Lubman DM. J Proteome Res. 2006;5:1792–1802. doi: 10.1021/pr060034r. [DOI] [PubMed] [Google Scholar]
- 18.Zhao J, Qiu WL, Simeone DM, Lubman DM. J Proteome Res. 2007;6:1126–1138. doi: 10.1021/pr0604458. [DOI] [PubMed] [Google Scholar]
- 19.Mechref Y, Hussein A, Bekesova S, Pungpapong V, Zhang M, Dobrolecki LE, Hickey RJ, Hammoud ZT, Novotny MV. J Proteome Res. 2009;8:2656–2666. doi: 10.1021/pr8008385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu JP, Shirota O, Wiesler D, Novotny M. Proc Natl Acad Sci U S A. 1991;88:2302–2306. doi: 10.1073/pnas.88.6.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nashabeh W, El Rassi Z. J Chromatogr A. 1992;600:279–287. doi: 10.1016/0021-9673(92)85559-c. [DOI] [PubMed] [Google Scholar]
- 22.Kakehi K, Susami A, Taga A, Suzuki S, Honda S. J Chromatogr A. 1994;680:209–215. doi: 10.1016/0021-9673(94)80069-3. [DOI] [PubMed] [Google Scholar]
- 23.Callewaert N, Contreras R, Mitnik-Gankin L, Carey L, Matsudaira P, Ehrlich D. Electrophoresis. 2004;25:3128–3131. doi: 10.1002/elps.200406020. [DOI] [PubMed] [Google Scholar]
- 24.Luo RJ, Archer-Hartmann SA, Holland LA. Anal Chem. 2010;82:1228–1233. doi: 10.1021/ac902052m. [DOI] [PubMed] [Google Scholar]
- 25.Archer-Hartmann SA, Sargent LM, Lowry DT, Holland LA. Anal Chem. 2011;83:2740–2747. doi: 10.1021/ac103362r. [DOI] [PubMed] [Google Scholar]
- 26.Mittermayr S, Bones J, Doherty M, Guttman A, Rudd PM. J Proteome Res. 2011;10:3820–3829. doi: 10.1021/pr200371s. [DOI] [PubMed] [Google Scholar]
- 27.Mittermayr S, Guttman A. Electrophoresis. 2012;33:1000–1007. doi: 10.1002/elps.201100681. [DOI] [PubMed] [Google Scholar]
- 28.Dang FQ, Zhang LH, Jabasini M, Kaji N, Baba Y. Anal Chem. 2003;75:2433–2439. doi: 10.1021/ac034110a. [DOI] [PubMed] [Google Scholar]
- 29.Zhuang Z, Starkey JA, Mechref Y, Novotny MV, Jacobson SC. Anal Chem. 2007;79:7170–7175. doi: 10.1021/ac071261v. [DOI] [PubMed] [Google Scholar]
- 30.Vanderschaeghe D, Szekrenyes A, Wenz C, Gassmann M, Naik N, Bynum M, Yin HF, Delanghe J, Guttman A, Callewaert N. Anal Chem. 2010;82:7408–7415. doi: 10.1021/ac101560a. [DOI] [PubMed] [Google Scholar]
- 31.Zhuang Z, Mitra I, Hussein A, Novotny MV, Mechref Y, Jacobson SC. Electrophoresis. 2011;32:246–253. doi: 10.1002/elps.201000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitra I, Zhuang Z, Zhang Y, Yu C-Y, Hammoud ZT, Tang H, Mechref Y, Jacobson SC. Anal Chem. 2012;84:3621–3627. doi: 10.1021/ac203431s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gahoual R, Biacchi M, Chicher J, Kuhn L, Hammann P, Beck A, Leize-Wagner E, Francois YN. mAbs. 2014;6:1464–1473. doi: 10.4161/mabs.36305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Callewaert N, Van Vlierberghe H, Van Hecke A, Laroy W, Delanghe J, Contreras R. Nat Med. 2004;10:429–434. doi: 10.1038/nm1006. [DOI] [PubMed] [Google Scholar]
- 35.Mitra I, Alley WR, Goetz JA, Vasseur JA, Novotny MV, Jacobson SC. J Proteome Res. 2013;12:4490–4496. doi: 10.1021/pr400549e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hermentin P, Doenges R, Witzel R, Hokke CH, Vliegenthart JFG, Kamerling JP, Conradt HS, Nimtz M, Brazel D. Anal Biochem. 1994;221:29–41. doi: 10.1006/abio.1994.1374. [DOI] [PubMed] [Google Scholar]
- 37.Guttman A, Pritchett T. Electrophoresis. 1995;16:1906–1911. doi: 10.1002/elps.11501601314. [DOI] [PubMed] [Google Scholar]
- 38.Guttman A. Electrophoresis. 1997;18:1136–1141. doi: 10.1002/elps.1150180719. [DOI] [PubMed] [Google Scholar]
- 39.Ma S, Nashabeh W. Anal Chem. 1999;71:5185–5192. doi: 10.1021/ac990376z. [DOI] [PubMed] [Google Scholar]
- 40.Yamashita K, Mizuochi T, Kobata A. Methods Enzymol. 1982;83:105–126. doi: 10.1016/0076-6879(82)83008-5. [DOI] [PubMed] [Google Scholar]
- 41.Guttman A, Herrick S. Anal Biochem. 1996;235:236–239. doi: 10.1006/abio.1996.0118. [DOI] [PubMed] [Google Scholar]
- 42.Guttman A, Ulfelder KW. J Chromatogr A. 1997;781:547–554. doi: 10.1016/s0021-9673(97)00724-3. [DOI] [PubMed] [Google Scholar]
- 43.Szabo Z, Guttman A, Rejtar T, Karger BL. Electrophoresis. 2010;31:1389–1395. doi: 10.1002/elps.201000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gennaro LA, Salas-Solano O, Ma S. Anal Biochem. 2006;355:249–258. doi: 10.1016/j.ab.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 45.Jayo RG, Li J, Chen DDY. Anal Chem. 2012;84:8756–8762. doi: 10.1021/ac301889k. [DOI] [PubMed] [Google Scholar]
- 46.Bunz S-C, Rapp E, Neusuess C. Anal Chem. 2013;85:10218–10224. doi: 10.1021/ac401930j. [DOI] [PubMed] [Google Scholar]
- 47.Bunz S-C, Cutillo F, Neusuess C. Anal Bioanal Chem. 2013;405:8277–8284. doi: 10.1007/s00216-013-7231-4. [DOI] [PubMed] [Google Scholar]
- 48.Evangelista RA, Chen FTA, Guttman A. J Chromatogr A. 1996;745:273–280. [Google Scholar]
- 49.Liu X, Qiu H, Lee RK, Chen W, Li J. Anal Chem. 2010;82:8300–8306. doi: 10.1021/ac101831t. [DOI] [PubMed] [Google Scholar]
- 50.Kang P, Mechref Y, Novotny MV. Rapid Comm Mass Spectrom. 2008;22:721–734. doi: 10.1002/rcm.3395. [DOI] [PubMed] [Google Scholar]
- 51.Alley WR, Jr, Madera M, Mechref Y, Novotny MV. Anal Chem. 2010;82:5095–5106. doi: 10.1021/ac100131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adamczyk B, Struwe WB, Ercan A, Nigrovic PA, Rudd PM. J Proteome Res. 2013;12:444–454. doi: 10.1021/pr300813h. [DOI] [PubMed] [Google Scholar]
- 53.Schwedler C, Kaup M, Petzold D, Hoppe B, Braicu EI, Sehouli J, Ehlers M, Berger M, Tauber R, Blanchard V. Electrophoresis. 2014;35:1025–1031. doi: 10.1002/elps.201300414. [DOI] [PubMed] [Google Scholar]
- 54.Bondt A, Rombouts Y, Selman MHJ, Hensbergen PJ, Reiding KR, Hazes JMW, Dolhain RJEM, Wuhrer M. Mol Cell Proteomics. 2014;13:3029–3039. doi: 10.1074/mcp.M114.039537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adamczyk B, Tharmalingam-Jaikaran T, Schomberg M, Szekrenyes A, Kelly RM, Karlsson NG, Guttman A, Rudd PM. Carbohydr Res. 2014;389:174–185. doi: 10.1016/j.carres.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 56.Schwedler C, Kaup M, Weiz S, Hoppe M, Braicu EI, Sehouli J, Hoppe B, Tauber R, Berger M, Blanchard V. Anal Bioanal Chem. 2014;406:7185–7193. doi: 10.1007/s00216-014-8168-y. [DOI] [PubMed] [Google Scholar]
- 57.Tousi F, Bones J, Hancock WS, Hincapie M. Anal Chem. 2013;85:8421–8428. doi: 10.1021/ac4018007. [DOI] [PubMed] [Google Scholar]
- 58.van Leeuwen SS, Schoemaker RJW, Timmer CJAM, Kamerling JP, Dijkhuizen L. Biochim Biophys Acta, Gen Subj. 2012;1820:1444–1455. doi: 10.1016/j.bbagen.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 59.Nakano M, Kakehi K, Tsai MH, Lee YC. Glycobiology. 2004;14:431–441. doi: 10.1093/glycob/cwh034. [DOI] [PubMed] [Google Scholar]
- 60.Matei D, Emerson RE, Schilder J, Menning N, Baldridge LA, Johnson CS, Breen T, McClean J, Stephens D, Whalen C, Sutton G. Cancer. 2008;113:723–732. doi: 10.1002/cncr.23605. [DOI] [PubMed] [Google Scholar]
- 61.Ramsey JD, Jacobson SC, Culbertson CT, Ramsey JM. Anal Chem. 2003;75:3758–3764. doi: 10.1021/ac0264574. [DOI] [PubMed] [Google Scholar]
- 62.Jacobson SC, Hergenröder R, Koutny LB, Warmack RJ, Ramsey JM. Anal Chem. 1994;66:1107–1113. [Google Scholar]
- 63.Alley WR, Jr, Novotny MV. J Proteome Res. 2010;9:3062–3072. doi: 10.1021/pr901210r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou H, Warren PG, Froehlich JW, Lee RS. Anal Chem. 2014;86:6277–6284. doi: 10.1021/ac500298a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reiding KR, Blank D, Kuijper DM, Deelder AM, Wuhrer M. Anal Chem. 2014;86:5784–5793. doi: 10.1021/ac500335t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.