

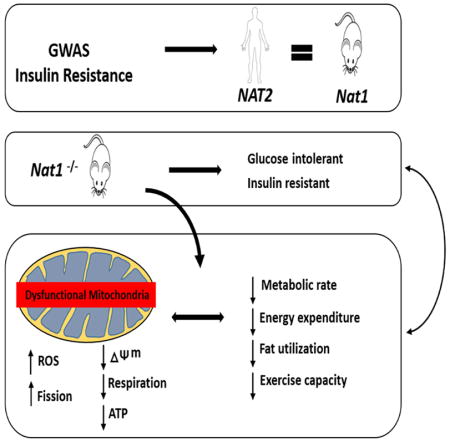
Summary
We recently identified human N-acetyltransferase 2 (NAT2) as an insulin resistance (IR) gene. Here we examine the cellular mechanism linking NAT2 to IR and find that Nat1 (mouse ortholog of NAT2) is co-regulated with key regulators of mitochondria. RNA-interference mediated silencing of Nat1 led to mitochondrial dysfunction characterized by increased intracellular reactive oxygen species and mitochondrial fragmentation as well as decreased mitochondrial membrane potential, biogenesis, mass, cellular respiration and ATP generation. These effects were consistent in 3T3-L1 adipocytes, C2C12 myoblasts and in tissues from Nat1 deficient mice including white adipose tissue, heart and skeletal muscle. Nat1 deficient mice had changes in plasma metabolites and lipids consistent with a decreased ability to utilize fats for energy and a decrease in basal metabolic rate and exercise capacity without altered thermogenesis. Collectively, our results suggest that Nat1 deficiency results in mitochondrial dysfunction, which may constitute a mechanistic link between this gene and IR.
Keywords: NAT2, Nat1, Insulin resistance, Mitochondrial dysfunction, Adipose tissue
Graphical Abstract
Introduction
The worldwide prevalence of insulin resistance and its metabolic complications have increased substantially in recent decades (Facchini et al., 2001; Frayn, 2001; Reaven, 1988; Yip et al., 1998). It is estimated that 25% to 33% of the US population is sufficiently insulin resistant to be at risk for adverse clinical consequences. At a tissue level, while decreased insulin-mediated glucose uptake in skeletal muscle is a cardinal feature of IR, it is also apparent that IR and dysfunctional adipose tissue are inextricably intertwined and co-occur in type 2 diabetes mellitus (T2D) and cardiovascular disease (Kim et al., 2007; Wang et al., 2005; Weyer et al., 2000). Hallmarks of IR in adipose tissue include decreased insulin mediated glucose uptake, inability of insulin to suppress lipolysis, dysregulation of adipocyte growth and differentiation, mitochondrial dysfunction, disrupted adipokine signaling and, in some instances, increased inflammation (De Pauw et al., 2009). Nevertheless, the genetic and cellular mechanisms underlying IR in adipose tissue remain incompletely understood.
We recently identified an association of non-synonymous coding SNPs in the human N-acetyltransferase 2 (NAT2) gene with insulin resistance (IR) using a genome wide association study (GWAS) approach, and validated these findings in vitro and in vivo through perturbations of the murine ortholog Nat1. In the murine 3T3-L1 adipocyte cell line, silencing Nat1 decreased insulin-mediated glucose uptake and increased basal and isoproterenol-stimulated lipolysis. Nat1-deficient mice had elevated fasting blood glucose, insulin, and triglyceride levels and decreased insulin sensitivity, as measured by glucose and insulin tolerance tests (Knowles et al., 2015). The exact mechanism linking the association of Nat1 with insulin resistance remains unclear and identification of the ‘missing link’ between Nat1, insulin action and cellular metabolism in adipose tissue is an important next step towards our understanding of the actions of this gene.
Here we show that Nat1 is not only strongly co-regulated with key mitochondrial genes but also that Nat1 deficiency causes significant decreases in multiple indices of mitochondrial function in vitro as well as global lipid metabolism alterations in vivo. Furthermore, Nat1 deficient mice have a marked decrease in basal metabolic rate and exercise capacity. Taken together, these data suggest that mitochondrial dysfunction may be a cellular mechanistic link between Nat1, insulin sensitivity and whole body energy expenditure.
Results
Characterization of Nat1 deficient mice on regular chow
A significant increase in mean body weight was observed in the Nat1 knockout (Nat1 ko) mice compared to their wild-type (wt) littermate controls after 15 weeks on a regular chow diet (Figure 1A), despite no difference in food or water intake (data not shown). The weights of epididymal fat and liver were significantly increased in Nat1 ko mice compared to those of the control mice (Figure 1, B and C), and hematoxylin and eosin (H&E) staining of epididymal fat tissue revealed that Nat1 deficient mice exhibit larger adipocytes compared to the wild-type controls (Figure 1D). The increased adiposity in Nat1 ko mice was accompanied by a decrease in the adipose expression of adiponectin (Adipoq) mirrored by a 17% decrease in plasma adiponectin levels (Figure 1E) while there was a non-significant increase in plasma leptin levels (Figure 1F). Livers of Nat1 deficient mice exhibit increased intrahepatic triglyceride content on regular chow (Supplemental Figure 1, A and B) compared with control mice. No differences were observed in liver enzymes (Supplemental Figure 1, C and D).
Figure 1.

Metabolic characterization of Nat1 deficient mice (A) Body weight (B) Epididymal fat and (C) Liver weights of wild-type (Wt) and Nat1 deficient mice (Ko) at 16 weeks fed a chow diet (n=10–15 mice per group). Values are mean ± SEM. (D) Haematoxylin and eosin staining (H&E) of epididymal WAT of wild type and Nat1 deficient mice. Scale bar, 100×Pi (10X magnification). (E) Plasma adiponectin (F) Leptin (G) FFA concentrations were determined in mice fasted overnight (n=10–13 per genotype). Values represent means ± SEM. (***p ≤ 0.001, **p ≤ 0.01, *p < 0.05). Gene expression profiling in white adipose tissue in wild type and Nat1 Ko mice (n = 5–6 mice per group) using real-time quantitative PCR for expression of genes involved in lipid metabolism (H) and for inflammatory markers (I). The expression values were normalized to cyclophilin. Results represent mean ± SEM (**p ≤ 0.01, *p < 0.05).
Free fatty acid (FFA) concentrations were comparable between Nat1 ko and wild-type mice (Figure 1G) despite elevated plasma TG levels in the Nat1 ko mice (9). mRNA levels of important genes involved in lipogenesis and FFA transport (e.g., Srebp1, Fas, Cd36) were not significantly elevated in Nat1 deficient mice. As expected given the decreased insulin sensitivity observed in Nat1 ko mice, the expression of Glut4 was also significantly decreased in Nat1 ko mice (Figure 1H).
In addition, we also found increased expression of the pro-inflammatory markers Tnfa, Il-1b and Mcp1 (Ccl2) in adipose tissue of Nat1 ko mice compared to wild-type littermates (Figure 1I), whereas there was no difference in the Il-10 expression levels between the groups.
Effects of Nat1 deficiency on plasma metabolome and lipidome
We performed a comprehensive metabolite and lipid profiling of fasting plasma from wild-type and Nat1 ko mice using a broad spectrum liquid chromatography coupled with mass spectrometry platform (Contrepois et al., 2015b). Comparative analysis of the metabolites and lipids revealed that lipid metabolism was profoundly perturbed in Nat1 ko mice compared to the wild-type littermate controls. In particular, the plasma level of 51 phospholipids, 30 lysophophospholipids, 20 glycerolipids and 11 acylcarnitines were decreased in Nat1 ko mice compared to wild-type littermate mice (Figure 2A). Similar changes were observed in non-fasting plasma despite more inter-individual variability due to small differences in postprandial timing after which the blood was drawn (Supplemental Figure 2). Short-chain (C2, C4OH, C6, C6DC), medium-chain (C10:1, C10:3) and long-chain (C14, C14:2, C16:0, C18:1, C18:2) acylcarnitines, intermediates of fatty acid oxidation, were diminished in the plasma of Nat1 ko mice suggesting remodeling of the oxidative phosphorylation (OXPHOS) process and changes in lipid oxidation (Figure 2B). Interestingly, most of the significantly deregulated lipids contained essential linoleic acid (18:2) such as lysophosphatidylcholines LPC(18:2), lysophosphatidylethanolamine LPE(18:2), phosphatidylcholine PC(14:0/18:2), PC(16:0p/18:2), triacylglycerol TG(18:2/20:4/22:6) and TG(18:3/18:2/22:6) (Figure 2, C and D), (Supplemental Table 1). In addition to lipid metabolism, 19 amino acids and derivatives were also significantly reduced in Nat1 deficient mice. Pathway analysis revealed a significant alteration of arginine and proline metabolism (FDR 0.0035) among which L-proline, ornithine, L-glutamic acid, and L-arginine are key metabolites (Figure 2E). Glycine, serine and threonine metabolism was also significantly altered (FDR 0.0301). Interestingly, derivatives of L-arginine (homoarginine, N,N-dimethylarginine) along with L-lysine and N6-acetyl-L-lysine were also significantly decreased. An exhaustive list of the metabolites and lipids significantly altered upon Nat1 knockdown can be found in Supplemental Table 1.
Figure 2.

Effect of Nat1 deficiency on fasting plasma metabolome and lipidome. (A) Hierarchical clustering of significantly different metabolites and lipids in fasting plasma from non-targeted metabolomics and lipidomics experiments (FDR < 0.1). Signal intensities were clustered in two dimensions (horizontal: metabolites and lipids; vertical: samples) on the basis of Euclidean distance. Colors indicate metabolite/lipid abundance as high (red), median (white), or low (blue). (B, C, D, E) Values are normalized to the median and expressed as box-and-whisker plots; n = 9–11; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. AC: acylcarnitine, DG: diacylglycerol, PC: phosphatidylcholine, PE: phosphatidylethanolamine, PI: phosphatidylinositol, SM: sphingomyelin, TG: triacylglycerol. The list of significantly changing metabolites and lipids are shown in Supplemental Table 1. Data obtained from non-fasting plasma samples are shown in Supplemental Figure 2.
Nat1 expression in adipose is co-regulated with mitochondrial genes
To provide insight into the metabolic pathways interacting with Nat1, we examined co-expression gene networks in adipose tissue collected from a large mouse cohort that segregated for obesity-induced diabetes (Tian et al., 2015; Tu et al., 2012). Gene ontology (GO) and KEGG pathway analysis was performed to identify the molecular functions and biological processes of genes that correlated with Nat1 expression. Interestingly, genes that positively correlated with Nat1 expression were significantly enriched for biological processes associated with mitochondrial biology, glucose metabolism and energy balance (Supplemental Figure 3, A and B). Genes that negatively correlated with Nat1 expression in adipose significantly enriched for growth factor binding and extracellular matrix (Supplemental Figure 3C). The list of mitochondrial genes most positively correlated with Nat1 expression in adipose tissue is shown in Supplemental Table 2.
Loss of Nat1 decreases mitochondrial function in 3T3-L1 adipocytes
Based on data indicating that Nat1 is strongly co-regulated with mitochondrial pathway genes we sought to comprehensively evaluate mitochondrial function resulting from perturbations of Nat1. Increased reactive oxygen species (ROS) production is often observed in mitochondrial dysfunction and insulin resistance (Sivitz and Yorek, 2010). ROS was measured using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA). Knockdown of Nat1 in 3T3-L1 adipocytes markedly induced ROS production (~1.3 fold) and strongly elevated the production of mitochondrial superoxide (O2−), a major source of mitochondrial ROS when evaluated with Mitosox ™ Red (Figure 3, A and B). Because the electrochemical gradient of the mitochondrial membrane is essential for ATP synthesis, we monitored mitochondrial membrane potential by fluorescence microscopy. Cells were stained with TMRM (Tetramethylrhodamine, methyl ester), imaged with a fluorescent microscope and the intensity of TMRM fluorescence was determined (Figure 3C). We found that Nat1 silencing resulted in a decrease of mitochondrial membrane potential of ~30% suggesting that loss of Nat1 markedly induced mitochondrial membrane depolarization in adipocytes from 3T3-L1 cells. Loss of membrane potential was further verified using a fluorescent plate reader and showed a decreased membrane potential of ~30% (Figure 3D).
Figure 3.

Effect of Nat1 knockdown on mitochondrial functions. (A) Intracellular ROS measured using H2DCFDA in 3T3-L1 adipocytes transfected with scrambled siRNA (scr siRNA) or with siRNA against Nat1 (si Nat1). (B) Mitochondrial superoxides were measured by fluorescence microscopy in 3T3-L1 adipocytes transduced with lenti viral controls and sh Nat1 stained with MitoSOX™ Red (10 μM) (4X magnification). Mitochondrial membrane potential (ΔΨm) was monitored using TMRM staining (50 nM) in 3T3-L1 adipocytes. (C) Fluorescent microscopic analysis (20X magnification) and intensity of TMRM was quantified. (D) Membrane potential was further analyzed using fluorescent plate reader. Data are presented as mean ± SEM of 3 separate experiments, with 8 wells per condition. Mitochondrial mass was measured in 3T3-L1 adipocytes transduced with lenti virus controls and sh Nat1 using mitotracker deep red (100 nM). (E) Fluorescence was analyzed by flow cytometry. Values are mean ± SEM of triplicates of 3 different experiments (** p ≤ 0.01). (F) Confocal microscopy in 3T3-L1 adipocytes. (G) Quantitative PCR analysis of Pgc1a expression in 3T3-L1 adipocytes. Data represents mean ± SEM. (H) Western blot analysis of Pgc1a protein levels in differentiating mouse 3T3-L1 adipocytes. (I) Quantitative PCR analysis of mitochondrial genes in 3T3-L1 adipocytes normalized to cyclophilin. Representative results are shown of n = 3 experiments. Data are presented as mean ± SEM. *p < 0.05, **p ≤ 0.01, ***p < 0.001.
Nat1 deficiency decreases mitochondrial mass and biogenesis in adipocytes
To explore whether loss of Nat1 affects mitochondrial number and biogenesis, we measured mitochondrial mass using a fluorescent probe, mitotracker deep red. Intensity of mitotracker fluorescence was analyzed by flow cytometry and was further verified by confocal imaging (Figure 3, E and F). Loss of Nat1 significantly decreased mitochondrial mass by 22%. We then measured the expression of Pgc1a, a transcriptional co-activator implicated in mitochondrial biogenesis and adaptive thermogenesis. Pgc1a was significantly down regulated at both mRNA and protein level in knockdown group compared to controls (Figure 3, G and H). To further explore the changes in mitochondrial biogenesis and oxidative phosphorylation, we also assessed the levels of key regulators of mitochondria by real time qPCR. Nrf1, Tfam, Cycs, Pgc1b were significantly down regulated in the Nat1 knockdown group compared to controls which is consistent with the in vivo results (Figure 3I). Our findings suggest that loss of Nat1 decreases mitochondrial mass and biogenesis in 3T3-L1 adipocytes.
Given these findings we also assessed the expression of genes involved in mitochondrial biogenesis and oxidative phosphorylation in white adipose tissue (WAT) of wild-type and Nat1 deficient mice. The deletion of Nat1 significantly decreased the expression of Pgc1a and Cycs (Cytochrome C) (Supplemental Figure 3D).
Nat1 knockdown promotes Drp1-dependent mitochondrial fragmentation
We evaluated the effect of Nat1 knockdown on the regulation of mitochondrial dynamics by performing confocal microscopy on 3T3-L1 cells and assessing morphology with MitoTracker Red CMXRos. Mitochondrial fragmentation is a morphological phenotype normally associated with dysfunctional mitochondria such as with ischemic reperfusion injury (Disatnik et al., 2013). We observed a marked increase in mitochondrial fragmentation in Nat1 ko cells compared to the controls (Figure 4A). Mitochondrial fragmentation can occur by a number of mechanisms including increased mitochondrial fission, decreased mitochondrial fusion or decreased mitophagy (Dorn, 2015). We first examined Drp1 (Dynamin 1- like) expression in 3T3-L1 adipocytes by Western blot, since Drp1 is the key regulator of mitochondrial fission. Knocking down Nat1 in 3T3-L1 adipocytes led to a significant increase of Drp1 in the mitochondrial fraction by 1.6 fold (Figure 4B). We then measured the levels of the mitochondrial fusion proteins Mfn1, Mfn2 and mitophagy proteins Lc3a/b and Pink1 and found no difference between groups (Figure 4, C and D).
Figure 4.

Effect of Nat1 on mitochondrial dynamics in 3T3-L1 adipocytes. Nat1 knockdown increased mitochondrial fission/fragmentation. (A) Confocal imaging of mitochondrial fragmentation in 3T3-L1 adipocytes transduced with lenti virus for controls and sh Nat1 stained with MitoTracker Red CMXRos (100 nM) and analyzed for mitochondrial area, perimeter, circularity and interconnectivity. Western blot analysis of mitochondrial proteins for (B) Mitochondrial fission quantified by imageJ, values are mean ± SEM of 2 different experiments (*p ≤ 0.05). (C) Fusion (D) Mitophagy in isolated mitochondrial and cytosolic fractions from 3T3-L1 adipocytes transduced with lenti virus for controls and sh Nat1. (E) Mitochondrial markers in isolated mitochondrial fractions from WAT of wild type and Nat1 deficient mice (n = 4).
We also measured mitochondrial markers in isolated mitochondrial fractions from WAT of wild type and Nat1 deficient mice. Nat1 deficiency increased Drp1 expression (Figure 4E). Electron microscopy of WAT showed smaller and more circular mitochondria in Nat1 ko mice compared with wild type mice (Supplemental Figure 4).
Taken together, these data indicate that the deficiency of Nat1 is associated with mitochondrial fragmentation mediated by Drp1-dependent mechanisms of mitochondrial fission and results in significant changes to the morphology and number of mitochondria, which are also observed in vivo.
Nat1 silencing decreases cellular respiration and ATP generation in adipocytes
As impaired mitochondrial respiration has been linked to the development of insulin resistance, we measured in situ respiratory capacity using the XF24 Seahorse extracellular flux analyzer with cultured adipocytes. Respiration rates were calculated as a function of changes in the oxygen consumption rate in response to oligomycin, FCCP and antimycin/rotenone. Nat1-silenced cells had a reduced oxygen consumption rate (OCR) relative to control adipocytes (Figure 5A). The results of the respiration studies indicated that Nat1 knockdown adipocytes had decreased basal and maximal respiration by 18% and 26% respectively compared to the control adipocytes (Figure 5B). Extracellular acidification rate (ECAR) was also measured as an index of lactate production. We found no difference in the basal acidification rate, however the maximal acidification rate was decreased in Nat1 knockdown adipocytes compared to the controls (Supplemental Figure 5, A and B).
Figure 5.

Effect of Nat1 ablation on cellular respiration and ATP production in differentiated 3T3-L1 adipocytes. (A) Oxygen consumption rate (OCR) measured using the XF24 Extracellular Fluid Analyzer in 3T3-L1 adipocytes transduced with control pLV and sh Nat1. (B) Basal respiration and maximal respiration rates determined from A. (n = 9–10; ** p ≤ 0.01, ***p < 0.001). (C) Oxygen consumption rate (OCR) (D) Mitochondrial DNA content (mtDNA/nDNA) measured in WAT of wild type and Nat1 deficient mice (n=5) (* p ≤ 0.05). (E) Cellular ATP levels were measured in 3T3-L1 adipocytes. Data are presented as mean ± SEM of 3 separate experiments, with 5 wells per experiment. *p ≤ 0.05.
Decreased cellular respiration was further verified in WAT of wild type and Nat1 deficient mice using an Oroboros Oxygraph-2k respirometer. Nat1 deficiency significantly decreased basal respiration (Figure 5C). Decreased OCR was accompanied by a significant decrease in mtDNA content in WAT of Nat1 deficient mice compared to wild-type controls thus indicating a decreased number of mitochondria in the WAT of Nat1 ko mice, which is consistent with decreased mitochondrial mass and biogenesis in 3T3-L1 adipocytes lacking Nat1 expression (Figure 5D).
These data suggest that inhibition of Nat1 is associated with impaired cellular respiration. Since respiration was significantly affected by Nat1 deficiency, we next assessed the cellular ATP concentrations and found that Nat1 knockdown in 3T3-L1 adipocytes resulted in a decrease of total cellular ATP levels by 24%, suggesting that loss of Nat1 markedly decreased overall mitochondrial function and ATP generation (Figure 5E).
Nat1 knockout mice display decreased metabolic rate and energy expenditure
Given the effect of Nat1 on mitochondrial function, and the observed effect on plasma metabolites and lipids, we hypothesized that Nat1 deficiency would be associated with changes in basal metabolic rate. To better understand the role of Nat1 in whole body energy expenditure under basal conditions, we subjected 4 month old wild-type and Nat1 deficient mice fed a normal chow diet to indirect calorimetry analysis in metabolic cages for 24 h. Nat1 ko mice exhibit increased respiratory exchange ratio (RER) (Figure 6A) and decreased whole body oxygen consumption rate (VO2) (Figure 6B), suggesting an inability to switch from carbohydrate to fat metabolism. Overall resting energy expenditure was significantly decreased Nat1 ko mice compared to wild-type mice (Figure 6C).
Figure 6.

Decreased metabolic rate in Nat1 Ko mice. (A) RER (B) VO2 (C) Energy expenditure was measured in 4 month-old wild-type and Nat1 deficient mice given a normal chow diet for 24 h (n = 4 mice per group) (***p ≤ 0.001). (D and E) Heat production was measured for 24 hours in 4 month-old wild-type and Nat1 deficient mice given a normal diet for 24 h (n = 4). (F) H&E staining of brown adipose tissue of wild type and Nat1 deficient mice (n = 4). (G) Thermogenic gene expression in BAT of wild type and Nat1 deficient mice. (n = 3), normalized to cyclophilin. Results represent mean ± SEM. (H) Core body temperature was measured in 4 month-old wild-type and Nat1 deficient male mice fed a chow diet (n=5–8 mice). (I) Rectal temperature was measured every 30 min for 4 h after exposure to a 4°C environment (n=5–8 mice). (J) Blood glucose (K) Non-esterified fatty acids in plasma measured in wild-type and Nat1 deficient mice (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
Nat1 deficient mice are cold tolerant
As Nat1 deficiency was associated with changes in basal metabolic rate, a reasonable hypothesis is that Nat1 deficiency would also have a primary effect on thermogenesis. Therefore, we assessed fuel metabolism by indirect calorimetry for 24 h. Heat production was measured in 4-month-old wild-type and Nat1 deficient mice fed a normal chow diet for 24 h. Over the course of 24 h, the percentage of energy generated from fatty acid oxidation was less than carbohydrate metabolism in Nat1 ko mice (Figure 6, D and E).
Brown adipose tissue (BAT) is involved in the generation of heat by burning lipids. BAT is activated during cold exposure to maintain core body temperature. Histological analysis of brown adipose tissue from wild-type and Nat1 ko mice did not show differences in lipid accumulation under basal conditions (Figure 6F). However, ablation of Nat1 resulted in reduced expression of the major thermogenic genes including Pgc1a, Ucp1, Cidea (Peroxisome proliferator-activated receptor gamma coactivator-1-alpha, Uncoupling protein 1, Cell death-inducing DFFA-like effector A, respectively) (Figure 6G). We also assessed their basal core body temperature and found no difference between the wild-type and Nat1 deficient mice (Figure 6H).
To investigate a possible role for Nat1 in BAT function, we exposed Nat1 ko and wild-type mice to a cold (4°C) environment for 4 h in order to activate BAT. Rectal temperature was measured every 30 min. The Nat1 deficient mice had no difference in core body temperature following cold exposure compared to wild-type mice (Figure 6I). Circulating glucose and free fatty acids were also measured before and after cold challenge. Nat1 deficient mice exhibited a marked increase in blood glucose levels but no change in the plasma free fatty acids after cold exposure (Figure 6, J and K).
Together, these results suggest that Nat1 deficiency is associated with a preferential use of carbohydrates over fats as fuel both under basal and cold-challenge conditions and potentially a reduction in FFA release.
Nat1 deletion impairs exercise capacity and cardiac mitochondrial respiration in Nat1 ko mice
As mitochondrial function is a key determinant of exercise capacity we sought to evaluate whether Nat1 deficiency would be accompanied by decreased exercise tolerance. We subjected 12–14 week-old Nat1 ko and wild-type littermate mice on a normal chow diet to a treadmill exercise challenge. Nat1 deficient animals exhibited a 19% decrease in exercise capacity, a 23% decrease in time to exhaustion, a lower anaerobic threshold and a 14% decrease in VO2 max compared to their wild-type controls (Figures 7, A–D).
Figure 7.

Nat1 deficiency on energy expenditure and cardiac phenotype in vivo. (A) Mean RER (B) Exercise capacity (C) Time to exhaust (D) Baseline and maximal VO2 were determined during exercise by treadmill protocol. Values are the means ± SEM of 8 mice per group (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001). (E) Representative electron micrographs of the hearts of 4 month-old mice; scale bar, 2μm. (F and G) Mitochondrial number and area were quantified blindly from 8–10 images from different fields (n = 4) (***p ≤ 0.001). (H) Oxygen consumption rate of heart isolated mitochondria was assessed using an Oroboros Oxygraph-2k respirometer (n= 5–7 mice per group, **p ≤ 0.01). (I) Relative mitochondrial DNA (mtDNA) content measured in cardiac muscle of wild type and Nat1 deficient mice (n = 3). (J) Mitochondrial markers in isolated mitochondrial fractions from hearts of wild type and Nat1 deficient mice (n = 4).
To examine the possibility that the decreased exercise capacity in Nat1 deficient mice can be attributed to effects on cardiac mitochondria, we performed electron microscopic analyses of heart tissue, which showed smaller mitochondria in Nat1 ko mice compared with wild type mice (Figure 7, E–G). Lipid droplets were present among mitochondria in knockout mice (data not shown). Oxygen consumption was significantly decreased in cardiac mitochondria from Nat1 ko mice versus control mice (Figure 7H). However, no changes were observed in the mitochondrial DNA content (Figure 7I). We also measured mitochondrial markers in isolated mitochondrial fractions from hearts. Nat1 ko mice exhibit increased expression of mitochondrial fission marker Drp1 (Figure 7J).
We also examined cardiac function in Nat1 ko and wild-type mice in the resting state with echocardiography. There was no change in left ventricular internal diameter (LVID) or percent fractional shortening (FS%) in the Nat1 ko mice compared with control animals (Supplemental Figure 6). All echocardiography measurements are shown in Supplemental Table 3.
As skeletal muscle is also a major determinant of energy expenditure, we assessed whether global deficiency of Nat1 is associated with functional defects in skeletal muscle mitochondrial function. C2C12 myoblasts with Nat1 knockdown had significantly decreased cellular respiration (Supplemental Figure 7A). Furthermore, Nat1 ko mice exhibit smaller and increased numbers of mitochondria per tibialis muscle fiber (Supplemental Figure 7, B–D) and increased expression of mitochondrial fission marker Drp1 compared to the wild-type mice (Supplemental Figure 7E).
Discussion
Our findings link deficiency of the insulin resistance gene Nat1 to profound mitochondrial dysfunction characterized by an enhanced accumulation of reactive oxygen species (ROS) and super oxides (O2−), an increase in mitochondrial membrane depolarization, decreased basal and maximal cellular respiration and decreased ATP production. Though our most detailed characterization was performed in 3T3-L1 adipocytes and WAT, the major observations of mitochondrial dysfunction were extended to skeletal muscle cells (C2C12 myoblats) and tissue as well as cardiac cells. In addition, our in vivo observations suggest that mitochondrial dysfunction seen in Nat1 ko mice is accompanied by a decrease in basal respiration and exercise capacity. Finally, multiple lines of evidence suggest a dysregulation in the ability to utilize fats for energy.
Thus, these studies provide a specific genetic mechanism (Nat1 deficiency) which may help explain prior observations that tied mitochondrial dysfunction and impaired biogenesis with insulin resistance and abnormal glucose homeostasis (Handschin et al., 2007; Johnson and Olefsky, 2013; Kelley et al., 2002; Kim et al., 2000; Kleiner et al., 2012; Morino et al., 2006; Petersen et al., 2004; Schrauwen and Hesselink, 2004; Vianna et al., 2006). Furthermore, our studies also suggest a means to partly explain the co-occurrence of mitochondrial dysfunction in adipose tissue in animal models of T2D as well as in human obesity, T2D and the metabolic syndrome (Bogacka et al., 2005a; Bogacka et al., 2005b; Choo et al., 2006; Patti and Corvera, 2010).
This mitochondrial dysfunction could be due to decreased mitochondrial number/mass or decreased mitochondrial efficiency. Our results suggest that both of these processes are affected by Nat1 deficiency. Mitochondrial mass was markedly reduced as were master regulators of mitochondrial biogenesis and function such as peroxisome proliferator-activated receptor gamma coactivator-1-alpha and beta (Pgc1a and Pgc1b), nuclear respiratory factor 1 (Nrf1) and mitochondrial transcription factor A (Tfam). Mitochondria constantly fuse and divide, processes known as fusion and fission and excessive mitochondrial fragmentation induces mitochondrial dysfunction. In our study, we observed a significant increase in mitochondrial fragmentation and Drp1 (dynamin 1-like protein) expression in Nat1 ko cells compared to the controls indicating that the deficiency of Nat1 triggers mitochondrial fragmentation by increasing Drp1. In addition, selective thermogenic genes in brown adipose tissue were significantly decreased in Nat1 deficient mice compared with wild-type controls, which may also reflect effects on Pgc1a. Finally, key parts of the electron transport chain and ATP generation, e.g. Cycs were significantly downregulated in Nat1 deficient cells and white adipose tissue.
Nat1-associated mitochondrial dysfunction may be primary or secondary (for instance related to mitochondrial substrate availability). While the former is likely, as outlined above, it is difficult to exclude the later. As an example, we found that fasted Nat1 ko mice have markedly reduced levels of multiple plasma acylcarnitines that has previously been shown to contribute to mitochondrial dysfunction (Noland et al., 2009). Acylcarnitines result from the addition of a carnitine molecule to an activated free fatty acid and allow transportation into the mitochondria and subsequent β-oxidation. Together with the fact that Nat1 ko mice have an inability to utilize fatty acids, it suggests that Nat1 deficiency is associated with an impaired fatty acid transportation and/or β-oxidation. The resulting diminution in acylcarnitines is a common feature of insulin resistance (Noland et al., 2009). However, it is not yet clear whether acylcarnitines are “reflecting or inflicting” insulin resistance (Schooneman et al., 2013).
Nat1 deficiency is also associated a reduction of numerous lysophospholipids, phospholipids and glycerolipids alterations in lipid metabolism and amino acids levels in plasma that are consistent with an insulin resistance phenotype (Roberts et al., 2014). Interestingly deregulated lipids often contain linoleic acid 18:2. LPC(18:2) is a well-known negative correlate of insulin resistance, dysglycemia and diabetes (Ferrannini et al., 2013; Floegel et al., 2013; Gall et al., 2010; Wang-Sattler et al., 2012) and LPCs have been shown to be reduced in obesity and T2D (Kim et al., 2011; Kim et al., 2013; Wang-Sattler et al., 2012). The global decrease of phospholipids may be due to a reduced biosynthesis because phospholipid precursors choline, phosphorylcholine and sn-glycero-3-phosphoethanolamine were also found in lower quantities in the plasma of Nat1 ko mice.
Amino acid metabolism was also markedly altered in Nat1 ko mice. Arginine and proline metabolism (L-aspartic acid, L-arginine, creatine, ornithine, L-proline, L-glutamic acid) pathways were the most significantly deregulated upon Nat1 depletion. Most of these metabolites have been associated with obesity and diabetes (Giesbertz et al., 2015). We also found a reduction in the quantity of glycine in Nat1 deficiency that is a well-established negative correlate to diabetes and obesity (Ferrannini et al., 2013; Giesbertz et al., 2015; Newgard et al., 2009; Wang-Sattler et al., 2012). Altogether these results suggest that Nat1 deficiency destabilizes energy homeostasis.
This altered homeostasis is also manifest in other ways. Nat1 deficient mice accumulate more fat in the form of WAT depots and liver TG without a change in daily food intake or changes in FFA levels or expression levels of genes involved in lipogenesis, which include fatty acid synthase (Fas), and sterol regulatory element-binding protein 1 (Srebp1). The increased adiposity could reflect a decrease in energy expenditure. In this regard, we observed that Nat1 deficiency significantly decreased the expression of Ucp1 and related thermogenic genes (Cidea, Pgc1a) in brown adipose tissue (BAT), suggesting decreased thermogenesis (Enerback et al., 1997; Matthias et al., 2000; Ricquier and Bouillaud, 2000; Wolf, 2009), and Ucp1 ablation which induces obesity (Feldmann et al., 2009). Nat1 ko mice also seem to derive less heat from fatty acid oxidation. Assessment of the animals’ responses to acute cold exposure showed that the Nat1 ko mice have a diminished FFA response but a more profound elevation in glucose compared to wild-type mice. However, these experiments did not reveal any gross thermogenic impairment in the Nat1 ko mice, as they maintained their core body temperature equal to wild-type mice. Such an acute challenge of mice previously housed at room temperature triggers a response in which all available heat-generating tissues are maximally activated. In such animals this consists of a combination of shivering (muscle dependent) and previously adapted uncoupled thermogenesis (BAT dependent) (Cannon and Nedergaard, 2011). While our cold challenge data do not distinguish between the relative contribution of muscle and BAT to the maintenance of heat, they strongly suggest that a primary defect in the thermogenic capacity or functionality of BAT is not causative of the change in energy balance observed in the Nat1 ko mice. Such severe impediments to BAT thermogenesis usually result in a more marked drop in core body temperature following this type of challenge (Lee et al., 2015). Subtler impairments to the efficiency of BAT or compensatory increases in muscle shivering capacity, caused by the previously highlighted changes to substrate availability and with the potential to impact energy balance, cannot be ruled out. However, longer-term chronic cold exposure and perhaps tissue-specific KO models would likely be required to dissect such adaptive changes in the Nat1 ko mouse.
A key finding in the current study is that wild-type mice run longer than Nat1 deficient mice. The decreased endurance in Nat1 deficient mice is accompanied by decreased VO2. Decreased exercise endurance in Nat1 deficient mice suggests reduced oxygen-dependent respiration during exercise. This is supported by the fact that Nat1 deficient mice reach anaerobic threshold sooner than the control mice (Figure 7C).
Impaired exercise capacity is a predictor of insulin resistance, T2D, cardiovascular diseases and overall mortality (Church et al., 2004; Fang et al., 2005) and mitochondria play a key role in the response to exercise challenge. The mitochondrial dysfunction seen in Nat1 deficiency is likely contributing to the observed decreased exercise capacity in Nat1 ko mice (Figure 7). Further, Nat1 ko mice also had a higher resting RER and decreased basal energy expenditure versus wild type mice (Figure 6), suggesting that Nat1 deficiency may involve preferential use of inefficient carbohydrate over fats as fuel during resting and active states. Our observations linking Nat1 deficiency to insulin sensitivity, mitochondrial function and exercise capacity demonstrate the interconnectedness of these phenotypes.
In conclusion, our data reveal a metabolic function of Nat1 that triggers mitochondrial dysfunction and is accompanied by changes in cellular respiration that are correlated at a whole body level with decreased basal energy expenditure and exercise tolerance. These findings shed light on the cellular mechanistic link between Nat1, IR and mitochondrial function. Additional research may identify new treatment approaches targeting mitochondria. The link between insulin sensitivity and mitochondria may be a fruitful area of discovery as new IR loci are being identified.
Materials and Methods
Animals
Nat1 targeted mice (Nat1 ko) were produced as described previously (Knowles et al., 2015). Mice were housed in a pathogen-free barrier facility with a 12-hour light/dark cycle and fed standard rodent chow diet and water ad libitum. Male mice between 3 and 4-month old were used in all the experiments.
Systemic metabolic parameters
Overnight fasting blood samples were collected by retro-orbital bleeding from anesthetized mice and EDTA plasma was prepared immediately. Plasma concentrations of adiponectin (Crystal Chem, Inc.), leptin (Crystal Chem, Inc.), Insulin (Crystal Chem, Inc.) and FFA (WAKO diagnostics) were determined enzymatically according to the manufacturer’s protocols. Overnight fasting blood glucose levels from the tail vein were measured with a glucometer (TRUEbalance, Nipro Diagnostics, Inc.).
Study approval for animal work
All animal protocols were approved by the Administrative Panel on Laboratory Animal Care at Stanford University and were performed in accordance with the guidelines of the American Association for the Accreditation of Laboratory Animal Care.
Histology
Histological analysis was performed according to standard protocols for paraffin embedded samples.
Cell culture
The murine 3T3-L1 and C2C12 myoblasts were obtained from American Type Culture Collection (Manassas, VA), cultured and differentiated according to the manufacturer’s instructions. Described details in Supplemental Methods.
Nat1 knockdown using siRNA in 3T3-L1 adipocytes
For knockdown experiments, differentiated 3T3-L1 adipocytes were transfected with 50 nM synthetic predesigned siRNA targeting Nat1 or non-silencing siRNA (scr siRNA) (Origene) using lipofectamine 2000 transfection reagent (Life Technologies) following the manufacturer’s recommended protocol. After 48 h of transfection, the media was changed.
Nat1 knockdown using lentiviral constructs
Cells were transduced with a combination of lentiviral particles for two knockdown constructs at a MOI of 100 each. After overnight incubation, medium containing viral particles was removed and replaced with fresh medium. The transduction efficiency was assessed by quantifying the percentage of GFP-positive cells by flow cytometry on a BD FACS Calibur. Nat1 knockdown efficiency was confirmed by qPCR.
RNA extraction, reverse transcription and real-time PCR
RNA extraction, reverse transcription and real time PCR was performed as described in Supplemental Methods. Primer sequences are given in the Supplemental Table 3. The gene expression values were normalized to cyclophilin A as a housekeeping gene. The data were analyzed by the public domain program Relative Expression Software Tool – REST. Values are presented as mean ± SEM (Chennamsetty et al., 2011).
Protein extraction and immunoblotting
Immunoblotting was performed as described in Supplemental Methods. The primary antibodies used were as follows: LC3A/B (Cell signaling), Porin/anti VDAC1 (Abcam), Drp1 (BD transduction laboratories), Pgc1a (H 300), PINK1, β-actin (Santa Cruz Biotechnology Inc.), Mfn1 (Abcam), Mfn2 (Abnova).
Intracellular Reactive Oxygen Species (ROS)
Intracellular ROS generation was determined using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA; 10uM) (Sigma) as per the manufacturers protocol. In brief, adipocytes were washed and incubated with H2DCFDA (Sigma, St. Louis, MO, USA) for 20 min. Cells were then washed twice gently and fluorescence was measured using a microplate reader (Tecan, Salzburg, Austria) with an excitation/emission wavelength of 485/515 nm.
Mitochondrial superoxide measurement
MitoSOX™ Red (Molecular Probes) was used to measure mitochondrial specific superoxide. Cells were stained, washed twice with PBS and live cell images were taken using fluorescence microscope.
Mitochondrial membrane potential (ΔΨm)
Mitochondrial membrane potential was measured using tetramethylrhodamine ethylester (TMRM) kit (Abcam, Cambridge, MA). The 3T3-L1 cells in 96-well black-walled culture plates were incubated with 100 nM TMRE for 20 min at 37 °C in PBS containing 0.2% BSA. After the cells were washed, the TMRM fluorescence was monitored using fluorescent microscope and imaged the cells. Membrane potential was further verified using a fluorescent plate reader (Tecan, Salzburg, Austria). The excitation and emission wavelengths were set at 445 nm and 580 nm, respectively.
ATP Production Assay
The ATP content of the adipocytes was measured using a Luminescent ATP Detection Assay Kit (Perkin Elmer) according to the manufacturer’s protocol. Cells were mixed with the detection reagent and luminescence was measured using a 10-second integration time with a microplate luminometer and SOFTmax PRO software (Molecular Devices), and was normalized to protein concentration.
Mitochondrial DNA content
Genomic DNA was isolated from tissues of wild-type and Nat1 ko mice (n = 5 per group) using the DNeasy Blood & Tissue Kit (Qiagen) following manufacturer’s instructions. Their mtDNA content were assessed by Quantitative real-time PCR. The relative ratio between mitochondrial DNA (Cox1) compared with nuclear DNA (18s rRNA).
Mitochondrial Mass Measurement
3T3-L1 adipocytes were incubated with 50 nM MitoTracker Deep-Red FM (Invitrogen) in serum free DMEM for 30 min at 37 °C. Cells were the n washed thoroughly with PBS, trypsinized, centrifuged and resuspended in PBS. Fluorescent intensity was analyzed using a FACS Calibur™ (BD Pharmingen).
Confocal laser microscopy for visualizing mitochondria
3T3-L1 adipocytes transduced with lentivirus for controls and sh Nat1 stained with mitochondria specific fluorescent dye, MitoTracker Red CMXRos (100 nM), fixed in 4% paraformaldehyde, and mitochondria visualized by confocal microscopy and analyzed for area, perimeter, circularity and interconnectivity by Image J.
XF24 Oxygen Consumption Assay and Bioenergetics Profile
Oxygen consumption was measured using the XF24 extracellular flux analyzer from Seahorse Bioscience according to their protocol. Cells were seeded in a 24-well XF24 cell culture microplate (Seahorse Bioscience) at a density of 10,000 cells per well in 200 μL DMEM supplemented with 10% FBS. Cells were differentiated. The rate of oxygen consumed by the cells as an indicator of mitochondrial respiration was measured using XF cell mito stress test kit (Seahorse) as per manufacturer’s instructions. Three basal oxygen consumption rate (OCR) measurements were performed using the Seahorse analyzer, and measurements were repeated following injection of oligomycin, FCCP and rotenone/antimycin A. Basal extracellular acidification rate (ECAR) was determined from data collected at basal measurement points.
Measurement of endogenous respiration using Oxygraph 2K
Isolated mitochondria and tissues were used for oxygraphic measurements (Oxygraph 2K; Oroboros Instruments) according to the manufacturer’s protocol. The rate of oxygen consumed was recorded. Respiration rates were normalized to total protein content.
Mitochondrial isolation
Mitochondria were isolated from 3T3-L1 adipocytes and tissues using Mitochondria Isolation Kit according to the manufacturer’s protocol (Abcam).
Metabolic expenditure measurements
For basal metabolic expenditure measurements, mice were placed in an Oxymax metabolic Chamber (Columbus Instruments, Columbus, OH) and oxygen consumption (VO2) and carbon dioxide expiration (VCO2) were measured using an open circuit volumetric method of gas analysis (Desai et al., 1997). Reparatory exchange ratio (RER) was calculated as VCO2/VO2. The rate of basal metabolic expenditure (BME) was calculated using the subjects VO2 * calorific value (CV), which is the relationship between heat and the volume of consumed oxygen. CVs are derived from historical empirical data. BME were then normalized to whole body weight (kg). Analysis of the oxidation of carbohydrates and fats were calculated as previously published.
Body temperature and cold exposure
Body temperatures were assessed in 4-month-old mice using a RET-3 rectal probe for mice (Physitemp). For the cold exposure experiment, mice were housed individually and transferred to a cold room with an ambient temperature of 4 °C. Temperature was measured every 30 min for 4 h, or until mouse core body temperature dropped below 25° C.
Triglyceride quantification
Frozen liver tissue (50 mg) was homogenized, total lipids were extracted by Folch method. Lipids were dried and reconstituted by brief sonication in 0.1% Triton-X 100. TG content was quantified by using enzymatic assays (Wako Chemicals).
Animal exercise protocol
Mice were exercised on a treadmill equipped for simultaneous measurement of O2 consumption (VO2) and CO2 production (VCO2) (Columbus Instruments), and distance run, total work and respiratory exchange ratio (RER) determined using established protocols (Desai et al., 1997). Exercise capacity was assessed using a protocol that incrementally increased the degree of elevation of the treadmill by 2° and the speed by 2 m/min every 3 minutes. Mice were exercised until exhaustion while monitoring VO2/CO2. Mice were not forced to exercise after exhaustion.
Echocardiographic assessment of cardiac function
Resting In vivo heart function was evaluated by echocardiography at 14–15 weeks. Mice were anesthetized with isoflurane (2% inhalation). 2D clips and M-mode images were obtained in the short axis view from the mid-LV at the tips of the papillary muscles with with a Sonos 5500 echocardiograph (Philips, Eindhoven, The Netherlands) (Details in Supplemental Methods).
Non-targeted Metabolomic and Lipidomic by LC-MS
Metabolites were extracted from mouse plasma as described previously (Contrepois et al., 2015a) and lipids were extracted using the Folch method with some modifications. Pathway analysis was performed using the metabolites with a HMDB accession number in the web tool Metaboanalyst (Xia et al., 2015). (Described details in Supplemental Methods).
Generation of B6×BTBR cross F2 Mice and genotyping and gene expression data
Construction of the diabetes-segregating F2 cohort has been previously described (Tian et al., 2015; Tu et al., 2012). Described details in Supplemental Methods.
Analysis of Nat1 correlated gene sets
Gene ontology (GO) and KEGG pathway enrichment analysis was performed using the DAVID bioinformatic database at the NIH (https://david.ncifcrf.gov/). The 500 most correlated genes to Nat1 (both positive and negative correlates) were identified from ~500 adipose samples from the F2 mice described above. The correlated gene sets were uploaded to DAVID, resulting in the GO and KEGG enrichments presented in Supplemental Table 1. Mitochondrial genes were identified from the mouse MitoCarta database at the Broad (http://www.broadinstitute.org/files/shared/metabolism/mitocarta/mouse.mitocarta.2.0.html). The mitochondrial genes showing the strongest positive correlation to the adipose expression of Nat1 are illustrated in Supplemental Table 1.
Statistical analyses
Statistical analyses of the experiments were performed with GraphPad Prism 5.0. 2-tailed unpaired Student’s t test and ANOVA were applied to determine statistical significance. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Supplementary Material
Acknowledgments
Joshua W. Knowles was supported by an American Heart Association Fellow-to-Faculty Transition Award (10FTF3360005) and by NIH grant U01HL107388. Thomas Quertermous was supported by NIH grants (U01HL107388, HL109512, R21HL120757) and a grant from the LeDucq Foundation. Michael Coronado was supported by NIH K99 award HL12947401A1. Kévin Contrepois was supported by NIH grant 3U54DK10255603S1. Ivan Carcamo-Orive was supported by NIH grant U01HL107388. Daniel Bernstein was supported by NIH grant HL061535. The authors thank Erik Ingelsson and Mark Walker for helpful comments.
Footnotes
Author contributions
Indumathi Chennamsetty contributed to basic science experiments in vitro and in vivo and writing the manuscript
Michael Coronado contributed to the mitochondrial experiments, helped write sections of the manuscript and provided critical comments
Kévin Contrepois contributed to the generation and analysis of metabolomics and lipidomics data, helped write sections of the manuscript and provided critical comments
Mark P. Keller contributed to co-expression gene networks analysis, helped write sections of the manuscript and provided critical comments
Ivan Carcamo-Orive contributed to basic science experiments
John Sandin contributed to basic science experiments in vivo
Giovanni Fajardo contributed to basic science experiments
Andrew J, Whittle contributed to basic science experiments
Mohsen Fathzadeh contributed to basic science experiments
Michael Snyder contributed to study design, discussion, manuscript preparation
Gerald Reaven contributed to study design, discussion, manuscript preparation
Alan D. Attie contributed to co-expression gene networks analysis and provided helpful comments and critical review of manuscript
Daniel Bernstein contributed to the mitochondrial experiments in vitro and in vivo and also provided critical comments on the manuscript
Thomas Quertermous contributed to study design, discussion, manuscript preparation
Joshua W. Knowles conceived the work, contributed to the overall design and writing the manuscript
Conflict of Interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bogacka I, Ukropcova B, McNeil M, Gimble JM, Smith SR. Structural and functional consequences of mitochondrial biogenesis in human adipocytes in vitro. J Clin Endocrinol Metab. 2005a;90:6650–6656. doi: 10.1210/jc.2005-1024. [DOI] [PubMed] [Google Scholar]
- Bogacka I, Xie H, Bray GA, Smith SR. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes. 2005b;54:1392–1399. doi: 10.2337/diabetes.54.5.1392. [DOI] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. Nonshivering thermogenesis and its adequate measurement in metabolic studies. Journal of Experimental Biology. 2011;214:242–253. doi: 10.1242/jeb.050989. [DOI] [PubMed] [Google Scholar]
- Chennamsetty I, Claudel T, Kostner KM, Baghdasaryan A, Kratky D, Levak-Frank S, Frank S, Gonzalez FJ, Trauner M, Kostner GM. Farnesoid X receptor represses hepatic human APOA gene expression. J Clin Invest. 2011;121:3724–3734. doi: 10.1172/JCI45277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo HJ, Kim JH, Kwon OB, Lee CS, Mun JY, Han SS, Yoon YS, Yoon G, Choi KM, Ko YG. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia. 2006;49:784–791. doi: 10.1007/s00125-006-0170-2. [DOI] [PubMed] [Google Scholar]
- Church TS, Cheng YJ, Earnest CP, Barlow CE, Gibbons LW, Priest EL, Blair SN. Exercise Capacity and Body Composition as Predictors of Mortality Among Men With Diabetes. Diabetes Care. 2004;27:83–88. doi: 10.2337/diacare.27.1.83. [DOI] [PubMed] [Google Scholar]
- Contrepois K, Jiang L, Snyder M. Optimized Analytical Procedures for the Untargeted Metabolomic Profiling of Human Urine and Plasma by Combining Hydrophilic Interaction (HILIC) and Reverse-Phase Liquid Chromatography (RPLC)-Mass Spectrometry. Mol Cell Proteomics. 2015a;14:1684–1695. doi: 10.1074/mcp.M114.046508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contrepois K, Jiang L, Snyder M. Optimized Analytical Procedures for the Untargeted Metabolomic Profiling of Human Urine and Plasma by Combining Hydrophilic Interaction (HILIC) and Reverse-Phase Liquid Chromatography (RPLC)–Mass Spectrometry. Molecular & Cellular Proteomics: MCP. 2015b;14:1684–1695. doi: 10.1074/mcp.M114.046508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pauw A, Tejerina S, Raes M, Keijer J, Arnould T. Mitochondrial (Dys)function in Adipocyte (De)differentiation and Systemic Metabolic Alterations. The American Journal of Pathology. 2009;175:927–939. doi: 10.2353/ajpath.2009.081155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai KH, Sato R, Schauble E, Barsh GS, Kobilka BK, Bernstein D. Cardiovascular indexes in the mouse at rest and with exercise: new tools to study models of cardiac disease. American Journal of Physiology - Heart and Circulatory Physiology. 1997;272:H1053–H1061. doi: 10.1152/ajpheart.1997.272.2.H1053. [DOI] [PubMed] [Google Scholar]
- Disatnik MH, Ferreira JCB, Campos JC, Gomes KS, Dourado PMM, Qi X, Mochly-Rosen D. Acute Inhibition of Excessive Mitochondrial Fission After Myocardial Infarction Prevents Long-term Cardiac Dysfunction. Journal of the American Heart Association: Cardiovascular and Cerebrovascular Disease. 2013;2:e000461. doi: 10.1161/JAHA.113.000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW. Mitochondrial dynamism and heart disease: changing shape and shaping change. EMBO Molecular Medicine. 2015;7:865–877. doi: 10.15252/emmm.201404575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enerback S, Jacobsson A, Simpson EM, Guerra C, Yamashita H, Harper ME, Kozak LP. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature. 1997;387:90–94. doi: 10.1038/387090a0. [DOI] [PubMed] [Google Scholar]
- Facchini FS, Hua N, Abbasi F, Reaven GM. Insulin resistance as a predictor of age-related diseases. J Clin Endocrinol Metab. 2001;86:3574–3578. doi: 10.1210/jcem.86.8.7763. [DOI] [PubMed] [Google Scholar]
- Fang ZY, Sharman J, Prins JB, Marwick TH. Determinants of Exercise Capacity in Patients With Type 2 Diabetes. Diabetes Care. 2005;28:1643–1648. doi: 10.2337/diacare.28.7.1643. [DOI] [PubMed] [Google Scholar]
- Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. UCP1 Ablation Induces Obesity and Abolishes Diet-Induced Thermogenesis in Mice Exempt from Thermal Stress by Living at Thermoneutrality. Cell Metabolism. 2009;9:203–209. doi: 10.1016/j.cmet.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Ferrannini E, Natali A, Camastra S, Nannipieri M, Mari A, Adam KP, Milburn MV, Kastenmüller G, Adamski J, Tuomi T, et al. Early Metabolic Markers of the Development of Dysglycemia and Type 2 Diabetes and Their Physiological Significance. Diabetes. 2013;62:1730–1737. doi: 10.2337/db12-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floegel A, Stefan N, Yu Z, Mühlenbruch K, Drogan D, Joost HG, Fritsche A, Häring HU, Hrabĕ de Angelis M, Peters A, et al. Identification of Serum Metabolites Associated With Risk of Type 2 Diabetes Using a Targeted Metabolomic Approach. Diabetes. 2013;62:639–648. doi: 10.2337/db12-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frayn KN. Adipose tissue and the insulin resistance syndrome. Proceedings of the Nutrition Society. 2001;60:375–380. doi: 10.1079/pns200195. [DOI] [PubMed] [Google Scholar]
- Gall WE, Beebe K, Lawton KA, Adam KP, Mitchell MW, Nakhle PJ, Ryals JA, Milburn MV, Nannipieri M, Camastra S, et al. α-Hydroxybutyrate Is an Early Biomarker of Insulin Resistance and Glucose Intolerance in a Nondiabetic Population. PLoS ONE. 2010;5:e10883. doi: 10.1371/journal.pone.0010883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesbertz P, Padberg I, Rein D, Ecker J, Höfle AS, Spanier B, Daniel H. Metabolite profiling in plasma and tissues of ob/ob and db/db mice identifies novel markers of obesity and type 2 diabetes. Diabetologia. 2015;58:2133–2143. doi: 10.1007/s00125-015-3656-y. [DOI] [PubMed] [Google Scholar]
- Handschin C, Choi CS, Chin S, Kim S, Kawamori D, Kurpad AJ, Neubauer N, Hu J, Mootha VK, Kim YB, et al. Abnormal glucose homeostasis in skeletal muscle–specific PGC-1α knockout mice reveals skeletal muscle–pancreatic β cell crosstalk. The Journal of clinical investigation. 2007;117:3463–3474. doi: 10.1172/JCI31785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson Andrew MF, Olefsky Jerrold M. The Origins and Drivers of Insulin Resistance. Cell. 2013;152:673–684. doi: 10.1016/j.cell.2013.01.041. [DOI] [PubMed] [Google Scholar]
- Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of Mitochondria in Human Skeletal Muscle in Type 2 Diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Kim JH, Noh S, Hur HJ, Sung MJ, Hwang JT, Park JH, Yang HJ, Kim MS, Kwon DY, et al. Metabolomic Analysis of Livers and Serum from High-Fat Diet Induced Obese Mice. Journal of Proteome Research. 2011;10:722–731. doi: 10.1021/pr100892r. [DOI] [PubMed] [Google Scholar]
- Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA. Lipid oxidation is reduced in obese human skeletal muscle. American Journal of Physiology - Endocrinology and Metabolism. 2000;279:E1039–E1044. doi: 10.1152/ajpendo.2000.279.5.E1039. [DOI] [PubMed] [Google Scholar]
- Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. The Journal of clinical investigation. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Yang HJ, Kim JH, Ahn CW, Lee JH, Kim KS, Kwon DY. Obesity-Related Metabolomic Analysis of Human Subjects in Black Soybean Peptide Intervention Study by Ultraperformance Liquid Chromatography and Quadrupole-Time-of-Flight Mass Spectrometry. Journal of Obesity. 2013;2013:874981. doi: 10.1155/2013/874981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner S, Mepani RJ, Laznik D, Ye L, Jurczak MJ, Jornayvaz FR, Estall JL, Chatterjee Bhowmick D, Shulman GI, Spiegelman BM. Development of insulin resistance in mice lacking PGC-1α in adipose tissues. Proceedings of the National Academy of Sciences. 2012;109:9635–9640. doi: 10.1073/pnas.1207287109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles JW, Xie W, Zhang Z, Chennemsetty I, Assimes TL, Paananen J, Hansson O, Pankow J, Goodarzi MO, Carcamo-Orive I, et al. Identification and validation of N-acetyltransferase 2 as an insulin sensitivity gene. The Journal of clinical investigation. 2015;125:1739–1751. doi: 10.1172/JCI74692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Ellis Jessica M, Wolfgang Michael J. Adipose Fatty Acid Oxidation Is Required for Thermogenesis and Potentiates Oxidative Stress-Induced Inflammation. Cell Reports. 2015;10:266–279. doi: 10.1016/j.celrep.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthias A, Ohlson KBE, Fredriksson JM, Jacobsson A, Nedergaard J, Cannon B. Thermogenic Responses in Brown Fat Cells Are Fully UCP1-dependent: UCP2 OR UCP3 DO NOT SUBSTITUTE FOR UCP1 IN ADRENERGICALLY OR FATTY ACID-INDUCED THERMOGENESIS. Journal of Biological Chemistry. 2000;275:25073–25081. doi: 10.1074/jbc.M000547200. [DOI] [PubMed] [Google Scholar]
- Morino K, Petersen KF, Shulman GI. Molecular Mechanisms of Insulin Resistance in Humans and Their Potential Links With Mitochondrial Dysfunction. Diabetes. 2006;55:S9–S15. doi: 10.2337/db06-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, et al. A Branched-Chain Amino Acid-Related Metabolic Signature that Differentiates Obese and Lean Humans and Contributes to Insulin Resistance. Cell Metabolism. 2009;9:311–326. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noland RC, Koves TR, Seiler SE, Lum H, Lust RM, Ilkayeva O, Stevens RD, Hegardt FG, Muoio DM. Carnitine Insufficiency Caused by Aging and Overnutrition Compromises Mitochondrial Performance and Metabolic Control. Journal of Biological Chemistry. 2009;284:22840–22852. doi: 10.1074/jbc.M109.032888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti ME, Corvera S. The Role of Mitochondria in the Pathogenesis of Type 2 Diabetes. Endocrine Reviews. 2010;31:364–395. doi: 10.1210/er.2009-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaven GM. Role of Insulin Resistance in Human Disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- Ricquier D, Bouillaud F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochemical Journal. 2000;345:161–179. [PMC free article] [PubMed] [Google Scholar]
- Roberts LD, Koulman A, Griffin JL. Towards metabolic biomarkers of insulin resistance and type 2 diabetes: progress from the metabolome. The Lancet Diabetes & Endocrinology. 2014;2:65–75. doi: 10.1016/S2213-8587(13)70143-8. [DOI] [PubMed] [Google Scholar]
- Schooneman MG, Vaz FM, Houten SM, Soeters MR. Acylcarnitines: reflecting or inflicting insulin resistance? Diabetes. 2013;62:1–8. doi: 10.2337/db12-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrauwen P, Hesselink MKC. Oxidative Capacity, Lipotoxicity, and Mitochondrial Damage in Type 2 Diabetes. Diabetes. 2004;53:1412–1417. doi: 10.2337/diabetes.53.6.1412. [DOI] [PubMed] [Google Scholar]
- Sivitz WI, Yorek MA. Mitochondrial Dysfunction in Diabetes: From Molecular Mechanisms to Functional Significance and Therapeutic Opportunities. Antioxidants & Redox Signaling. 2010;12:537–577. doi: 10.1089/ars.2009.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Keller MP, Oler AT, Rabaglia ME, Schueler KL, Stapleton DS, Broman AT, Zhao W, Kendziorski C, Yandell BS, et al. Identification of the Bile Acid Transporter Slco1a6 as a Candidate Gene That Broadly Affects Gene Expression in Mouse Pancreatic Islets. Genetics. 2015;201:1253–1262. doi: 10.1534/genetics.115.179432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Z, Keller MP, Zhang C, Rabaglia ME, Greenawalt DM, Yang X, Wang IM, Dai H, Bruss MD, Lum PY, et al. Integrative Analysis of a Cross-Loci Regulation Network Identifies App as a Gene Regulating Insulin Secretion from Pancreatic Islets. PLoS Genetics. 2012;8:e1003107. doi: 10.1371/journal.pgen.1003107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vianna CR, Huntgeburth M, Coppari R, Choi CS, Lin J, Krauss S, Barbatelli G, Tzameli I, Kim YB, Cinti S, et al. Hypomorphic Mutation in PGC1β causes mitochondrial dysfunction and liver insulin resistance. Cell metabolism. 2006;4:453–464. doi: 10.1016/j.cmet.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Rimm EB, Stampfer MJ, Willett WC, Hu FB. Comparison of abdominal adiposity and overall obesity in predicting risk of type 2 diabetes among men. The American Journal of Clinical Nutrition. 2005;81:555–563. doi: 10.1093/ajcn/81.3.555. [DOI] [PubMed] [Google Scholar]
- Wang-Sattler R, Yu Z, Herder C, Messias AC, Floegel A, He Y, Heim K, Campillos M, Holzapfel C, Thorand B, et al. Novel biomarkers for pre-diabetes identified by metabolomics. Molecular Systems Biology. 2012;8:615–615. doi: 10.1038/msb.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang-Sattler R, Yu Z, Herder C, Messias AC, Floegel A, He Y, Heim K, Campillos M, Holzapfel C, Thorand B, et al. Novel biomarkers for pre- diabetes identified by metabolomics. Molecular Systems Biology. 2012;8 doi: 10.1038/msb.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer C, Foley JE, Bogardus C, Tataranni PA, Pratley RE. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts Type II diabetes independent of insulin resistance. Diabetologia. 2000;43:1498–1506. doi: 10.1007/s001250051560. [DOI] [PubMed] [Google Scholar]
- Wolf G. Brown adipose tissue: the molecular mechanism of its formation. Nutrition Reviews. 2009;67:167–171. doi: 10.1111/j.1753-4887.2009.00184.x. [DOI] [PubMed] [Google Scholar]
- Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0--making metabolomics more meaningful. Nucleic Acids Res. 2015;43:W251–257. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip J, Facchini FS, Reaven GM. Resistance to insulin-mediated glucose disposal as a predictor of cardiovascular disease. J Clin Endocrinol Metab. 1998;83:2773–2776. doi: 10.1210/jcem.83.8.5005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.