

Abstract
Hyper-beta-alaninemia is a rare metabolic condition that results in elevated plasma and urinary β-alanine levels and is characterized by neurotoxicity, hypotonia, and respiratory distress. It has been proposed that at least some of the symptoms are caused by oxidative stress; however, only limited information is available on the mechanism of reactive oxygen species generation. The present study examines the hypothesis that β-alanine reduces cellular levels of taurine, which are required for normal respiratory chain function; cellular taurine depletion is known to reduce respiratory function and elevate mitochondrial superoxide generation. To test the taurine hypothesis, isolated neonatal rat cardiomyocytes and mouse embryonic fibroblasts were incubated with medium lacking or containing β-alanine. β-alanine treatment led to mitochondrial superoxide accumulation in conjunction with a decrease in oxygen consumption. The defect in β-alanine-mediated respiratory function was detected in permeabilized cells exposed to glutamate/malate but not in cells utilizing succinate, suggesting that β-alanine leads to impaired complex I activity. Taurine treatment limited mitochondrial superoxide generation, supporting a role for taurine in maintaining complex I activity. Also affected by taurine is mitochondrial morphology, as β-alanine-treated fibroblasts undergo fragmentation, a sign of unhealthy mitochondria that is reversed by taurine treatment. If left unaltered, β-alanine-treated fibroblasts also undergo mitochondrial apoptosis, as evidenced by activation of caspases 3 and 9 and the initiation of the mitochondrial permeability transition. Together, these data show that β-alanine mediates changes that reduce ATP generation and enhance oxidative stress, factors that contribute to heart failure.
Keywords: Electron transport chain, Oxidative stress, Respiration, Taurine, Mitochondrial fragmentation, Apoptosis
Introduction
β-alanine is a non-essential, β-amino acid that exists in several different forms in vivo. In the brain, it is present as a free amino acid, where it is thought to function as either a neurotransmitter or a neuromodulator [1, 2]. In energy demanding tissues, such as the heart, it is a component of coenzyme A, which is required for the metabolism of fatty acids and the function of the citric acid cycle [3]. In skeletal muscle, it is a component of the dipeptide, carnosine, which has been implicated in multiple physiological actions, the most important being its ergogenic potential arising from intramyocellular buffering [4]. In the liver, β-alanine is degraded to malonate semialdehyde, a reaction catalyzed by either β-alanine-α-ketoglutarate transaminase or β-alanine-pyruvate transaminase. Malonate semialdehyde is further converted to acetyl CoA, which is utilized by the citric acid cycle [5].
In humans, excessive accumulation of β-alanine leads to a rare metabolic condition known as hyper-beta-alaninemia, which is characterized by neurotoxicity (seizures, lethargy, malaise or sleep disorder, encephalopathy), hypotonia, and respiratory distress [6]. In addition to elevations in plasma and urinary levels of β-alanine, the condition is also associated with elevated urinary levels of GABA and beta-aminoisobutyric acid. Because animal models of hyper-beta-alaninemia can be generated by either excessive β-alanine administration, injection of the aminotransferase inhibitor, (aminooxy)acetate, or treatment with an inhibitor (6-azauridine triacetate) of β-alanine degrading enzymes, it has been suggested that the clinical condition is caused by impaired β-alanine transaminase activity [7, 8].
However, the mechanisms responsible for the symptoms of the disease are largely unknown. Gemelli et al. [9] have suggested that the neurotoxicity might be caused by oxidative stress. In β-alanine-treated rats, they observed an increase in catalase activity of the cerebral cortex and cerebellum and a reduction in the activity of superoxide dismutase, which together are capable of elevating H2O2 and superoxide anion. In the presence of iron, H2O2 and superoxide anion react to form hydroxyl radical via the Haber–Weiss reaction. Hydroxyl radical is extremely reactive and can cause severe oxidative damage. However, the targets of oxidative damage examined by the authors were cytosolic enzymes, with the response being different in the cerebral cortex than in the cerebellum, raising questions regarding the validity of their hypothesis.
We recently found that rats fed β-alanine exhibit impaired mitochondrial energy metabolism and cardiac weakness [10], effects consistent with the reports of β-alanine-mediated hypotonia. In the present study, we examine the effect of β-alanine exposure on isolated cardiomyocytes and fibroblasts, which are the two major cell types of the heart. Because β-alanine exposure was found to adversely affect mitochondrial energy production, the present study tests the hypothesis that β-alanine excess acts at the level of the mitochondria to reduce respiratory chain function and cause oxidative damage, which in turn initiates apoptosis. The data support the idea that β-alanine excess mediates oxidative stress through reductions in cellular taurine content.
Materials and methods
All reagents used were purchased from Sigma-Aldrich, St. Louis, MO unless otherwise specified. Dulbecco's modified Eagle's medium (DMEM) was obtained from Gibco, while the source of fetal bovine serum (FBS) was from Atlanta Biologicals. The following antibodies were used: caspase-3 (Cell signaling, #9962), Bcl-2 (Cell signaling, #2876), and Bax (Cell signaling, #2772). All dyes were purchased from Life Technologies and dissolved in DMSO for use.
Cell culture
All imaging experiments were performed using mouse embryonic fibroblasts. For storage, cells were maintained in medium (45 % DMEM, 45 % FBS, and 10 % DMSO) and stored at −80 °C. The fibroblasts were seeded in 35 mm glass bottom culture dishes (MatTek) and cultured in DMEM supplemented with 10 % FBS and a 1 % penicillin/streptomycin solution. To produce the hyper-beta-alanine model, the fibroblasts were incubated with DMEM medium containing 5 mM β-alanine; controls were incubated with medium lacking β-alanine. In cells incubated in medium lacking taurine, changes in mitochondrial size could be detected as low as 5 μM; however, the effect was small (20 %), was slow in developing, and was not observed when taurine was present in the medium. Therefore, we subsequently used 5 mM β-alanine to clearly evaluate the effects of β-alanine when taurine was present in the medium. Trypsinization was used to harvest the cells for subculture. However, in some experiments, cells were exposed to cold PBS and then scraped from the culture dishes.
Determination of mitochondrial respiratory function utilized neonatal rat cardiomyocytes. The cells were prepared according to the method described previously [11]. The cells were incubated with serum-free medium supplemented with either 0 or 5 mM β-alanine for 48 h. The cells were then suspended in buffer supplemented with either 4 mM glutamate and 2 mM malate or 5 mM succinate and placed in a 2 ml sealed chamber fitted with a Clark electrode to measure oxygen consumption. The cell membrane was permeabilized by addition of 20 μl of saponin (5 mg/ml). After obtaining a stable rate of state 2 respiration, 20 μl of 50 mM ADP was added to generate state 3 respiration. Following complete conversion of ADP to ATP via oxidative phosphorylation, a new slower rate of oxygen consumption was achieved, referred to as state 4 respiration. The respiratory control ratio (RCR) represents the ratio of state 3/state 4. The P/O ratio was determined from the amount of ATP formed (ADP added) and the oxygen consumed during state 3.
Oxygen consumption
Fibroblasts were incubated for 48 h in medium containing either 0 mM (Control) or 5 mM β-alanine. Approximately, (1 × 10−7) cells were scraped from the culture dishes and then concentrated by centrifugation. The cell pellet was suspended in growth medium (DMEM, 10 % FBS, and 1 % penicillin/streptomycin solution). For measuring oxygen consumption, cell suspensions were analyzed in a gas-tight vessel fitted with a calibrated Clark-type oxygen electrode connected to a chart recorder and maintained at 37 °C.
Measurement of intracellular taurine levels
The fibroblasts were scraped from the dish and homogenized in ice-cold 1 M perchloric acid and 2 mM EDTA. Homogenates were centrifuged at 10,000×g for 10 min at °C. The supernatant was neutralized to a pH between 6.0 and 7.0 using 2 M KOH and centrifuged again at 10,000×g for 10 min at 4 °C. The resulting supernatant was passed through a Dowex-50 × 8 (200–400 mesh, 9 × 40–55 mm) column and then eluted with 2 ml of deionized water. To assay taurine content, an aliquot (0.4 ml) of the eluant was added to 0.1 ml of dinitrofluorobenzene (DNFB), 0.1 ml of 1 M NaOH, and 0.5 ml DMSO. The reaction was stopped by the addition of 0.1 ml HCl to lower the pH to 1.5–2. After increasing the final volume to 5 ml by addition of water, 20 ml of acetyl acetate was added. After thoroughly mixing the reaction solution for 10 min, the upper acetyl acetate layer was removed by aspiration. The taurine concentration in the lower layer was measured at 355 nm. A taurine standard curve was developed using 0.312–5 μg taurine/5 ml for each assay.
Mitochondrial superoxide detection
Fibroblasts were incubated with either 0 mM (Control) or 5 mM β-alanine. After the desired time of β-alanine exposure, the growth medium was aspirated, and the cells were loaded with 10 μM MitoSox Red (mitochondrial superoxide indicator) and 200 nM of MitoTracker Green to stain mitochondria for 15 min at 37 °C in Krebs buffer (supplemented with 250 μM d-Glucose and 15 μM Dextran). After loading, the cells were washed twice with Krebs buffer before being incubated in 2 ml of Krebs buffer. Digital images were obtained by a Nikon confocal microscope system (A1) at 1024 × 1024 pixels. Images were captured using 60× water immersion objective lens. MitoSox was excited by laser at 514 nm and its emission was detected at 595 nm, whereas MitoTracker Green was excited by laser at 488 nm and the emission was detected at 525 nm.
Cytoplasmic superoxide detection
Fibroblasts were seeded in 35-mm glass bottom culture dish (MatTek) and then incubated for 48 h in medium supplemented with either 0 mM (Control) or 5 mM β-alanine. The growth medium was aspirated, and the cells were loaded with 10 μM dihydroethidium (DHE), a cytoplasmic superoxide indicator. After 15-min incubation at 37 °C in Krebs buffer (supplemented with 250 μM d-Glucose and 15 μM Dextran), the cells were washed twice with dihydroethidium-free buffer and then maintained in 2 ml of dihydroethidium-free Krebs buffer. Digital images were obtained as described above. Dihydroethidium was excited by laser at 514 nm and the emission was detected at 595 nm.
Mitochondrial morphology
After seeding fibroblasts in 35-mm glass bottom culture dishes (MatTek), they were incubated for 48 h in medium containing either 0 mM (Control) or 5 mM β-alanine. The growth medium was aspirated, and the cells were loaded with 150 nM MitoTracker Deep Red for 30 min in Krebs buffer (supplemented with 250 μM d-Glucose and 15 μM Dextran) at 37 °C. The cells were washed twice with MitoTracker-free medium before being incubated in 2 ml of MitoTracker-free Krebs buffer. Digital images of mitochondrial length were obtained as described in the text. MitoTracker Deep Red was excited by laser at 633 nm and the emission was detected at 650 nm.
Western Blot analyses
The relative content of specific proteins was determined by Western Blot analysis. Fibroblasts were incubated in medium containing either 0 mM (Control) or 5 mM β-alanine for 48 h. The medium was then aspirated, the cells were washed with cold PBS, and then scraped from the culture dish with cold PBS. The pellet was obtained by centrifugation at 500 g for 5 min. The pellet was then lysed in RadioImmuno Precipitation Assay (RIPA) lysis buffer (50 mM Tris-base (pH 8.0) containing 150 mM NaCl, 0.5 % deoxycholic acid (DOC), 1 % NP-40, 0.1 % sodium dodecyl sulfate), 1 mM sodium fluoride (NaF), 1 mM sodium orthovanadate (Na3VO4), 1 % phenylmethylsulfonyl fluoride (PMSF), and protease inhibitors (a 1:100 dilution of protease inhibitor cocktail; Catalog No. 78410, Thermo Scientific; Rockford, IL). The cell lysate was maintained on ice for 15 min and then centrifuged at 10,000×g for 10 min. Some supernatant was reserved for the protein assay, in which the protein concentration was determined spectrophotometrically using the bicinchoninic acid assay (Pierce® BCA Protein Assay Kit from Thermo Scientific), with bovine serum albumin (BSA) serving as a standard. Equal concentrations of protein samples were mixed with 5× electrophoresis sample buffer (1.25 mM tris(hydroxymethyl)aminomethane hydrochloride (Tris–HCl), pH 6.8, containing 1 % SDS, 10 % glycerol [vol/vol], and 5 % mercaptoethanol) at a 5: 1 (v/v) ratio and boiled for 5 min. The boiled protein samples were subjected to Western Blot analysis after one-dimensional electrophoresis using 10–15 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were then transferred to a nitrocellulose membrane and the membrane was blocked in blocking buffer (5 % milk in Tris buffer saline with Tween 20). The membrane was incubated with appropriate antibody overnight at 4 °C; the membrane was washed and incubated with a secondary antibody. The Western Blot was developed and visualized by enhanced chemiluminescence. Succinate dehydrogenase and glyceraldehyde-3-phosphate dehydrogenase were used as loading controls to normalize protein content of isolated mitochondria and total cell homogenates, respectively.
Cell volume using calcein AM
After seeding, fibroblasts were incubated for 48 h in medium containing either 0 mM (Control) or 5 mM β-alanine. The growth medium was aspirated and the cells were then loaded for 15 min at 37 °C with Krebs buffer (supplemented with 250 μM d-Glucose and 15 μM Dextran) containing 250 μM Calcein AM. The cells were then washed twice with Calcein-free medium and then maintained in 2 ml Calcein-free Krebs buffer. Digital images were obtained as mentioned above. Calcein AM was excited by laser at 495 nm and the emission was detected at 515 nm.
Effects of taurine on β-alanine-treated fibroblasts
Fibroblasts were incubated with 5 mM β-alanine for 48 h. Some of the β-alanine-treated cells were harvested by trypsinization. The remainder of the cells were incubated for 48 h with medium containing either 0 mM or 10 mM taurine. The incubation medium was then aspirated and the cells were loaded for 15 min at 37 °C with Krebs buffer containing both 10 μM MitoSox Red (mitochondrial superoxide indicator) and MitoTracker Green (200 nM). The cells were washed twice with MitoSox and Mito-Tracker-free medium before being placed in 2 ml of MitoSox and MitoTracker-free Krebs buffer. Digital images of superoxide content and mitochondrial size were obtained as described in the text.
Stimulation of mitochondrial permeability transition by β-alanine
Four different conditions were used to evaluate the mitochondrial permeability transition. Isolated neonatal cardiomyocytes were incubated for 48 h with either [1] normal medium, [2] medium containing 5 mM β-alanine, [3] cyclosporin A (200 nM) supplemented medium, or [4] medium containing both 5 mM β-alanine and 200 nM cyclosporin A. The number of apoptotic cells was determined by the TACS Annexin V-FITC kit method. The percent of apoptotic cells was determined in five separate fields of the microscope. Cyclosporin A was used as an inhibitor of the mitochondrial permeability transition. Cyclosporin A was effective in preventing initiation of the mitochondrial permeability transition, as seen with β-alanine treatment. However, cyclosporin A has no effect on the control cells, as the mitochondrial permeability transition is not initiated.
Statistical analysis
All results are reported as mean ± S.E.M. The statistical significance of the data was determined using the Student's t test for comparison within 2 groups or analysis of variance followed by Newman Keuls test for comparisons within more than two groups. Values of p < 0.05 were considered statistically significant.
Results
β-alanine reduces oxygen consumption
Gemelli et al. [9] proposed that oxidative stress might contribute to the symptoms of hyper-beta-alaninemia. A major source of superoxide production in the cytosol of most cells is NADPH oxidase. However, we have previously shown that β-alanine has no influence on NADPH oxidase activity. Moreover, exposure of fibroblasts to β-alanine for 48 h had no influence on dihydroethidium (DHE) fluorescence, which is commonly used to detect cytosolic superoxide content (data not shown). Nonetheless, β-alanine treatment mediated a 25 % decrease in oxygen consumption (Fig. 1), raising the possibility that the diminished rate of respiratory chain function might cause diversion of electrons away from the respiratory chain to the acceptor oxygen, forming superoxide anion. As seen in Figs. 2a, b, fibroblasts treated with β-alanine containing medium for 12, 24, or 48 h exhibited a time-dependent increase in mitochondrial superoxide content, as evidenced by a nearly 3-fold increase in MitoSox fluorescence over the time period of the 12–48 h incubation.
Fig. 1.
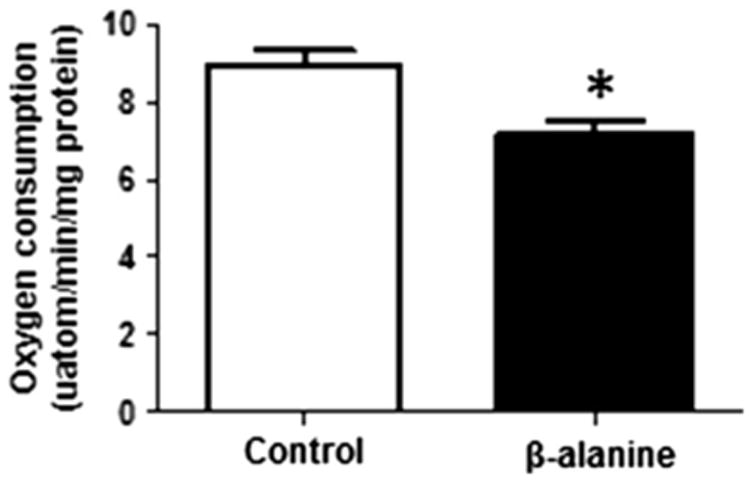
Reduction in oxygen consumption of β-alanine-treated fibroblasts. Cultured fibroblasts were exposed to either control or β-alanine containing medium for 48 h. The rate of oxygen consumption for both groups of cells was determined using a Clark electrode. β-alanine-treated cells consumed less oxygen than the control cells. Data shown represent mean ± S.E.M. of 6 different preparations. The asterisk denotes a significant difference between control and β-alanine-treated cells (p < 0.05)
Fig. 2.
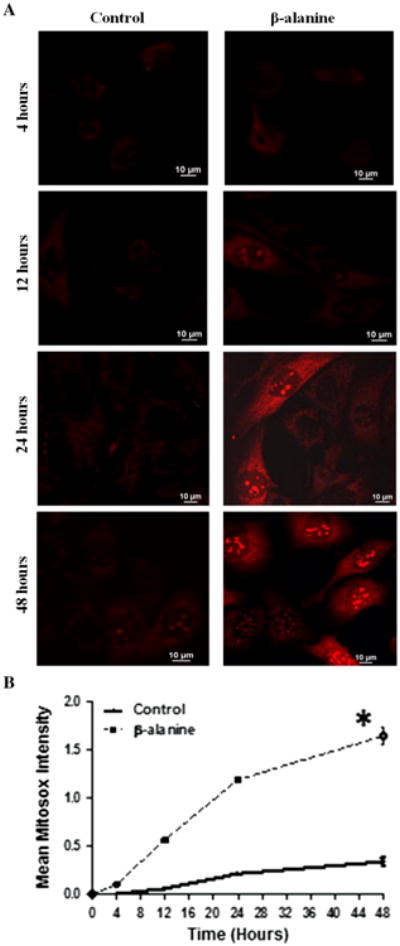
Time course of mitochondrial superoxide production by β-alanine-treated fibroblasts. a Representative images of MitoSox fluorescence (superoxide generation) in control and β-alanine-treated cells. While control cells exhibited a slow rate of superoxide generation, a significant elevation in mitochondrial superoxide levels was noted as early as 12 h of β-alanine exposure, an effect that became magnified with prolonged incubation. b The time course of superoxide generation in control and β-alanine-treated cells. The data represent means of 2 different preparations for 4, 12, and 24 h incubation periods, but mean ± S.E.M of 4 different preparations for the 48 h incubation period. The asterisk denotes a significant difference between β-alanine-treated and control cells (p < 0.05)
The two major sites of superoxide generation in the mitochondria are complexes I and III [12, 13]. To provide insight into the source of superoxide generation in the mitochondria, the substrate dependence of the β-alanine-mediated respiratory chain defect was examined. Control and β-alanine-treated cardiomyocytes were first exposed to saponin, which permeabilizes the cell membrane. The permeabilized cells were then incubated with medium containing one of two substrates, a complex I-specific substrate combination, malate/glutamate, or a complex II-specific substrate, succinate. Respiratory function of permeabilized cells (both control and β-alanine-treated cells) incubated with medium containing 5 mM succinate was virtually identical, indicating that the function of the respiratory chain complexes II–VI was unaffected by β-alanine treatment (Table 1). On the other hand, state 3 and the respiratory quotient (RQ) were significantly less in permeabilized β-alanine-treated cells than in permeabilized control cells exposed to glutamate/malate, suggesting that complex I activity is specifically reduced by β-alanine treatment. Because the P/O ratio was also lower in the β-alanine-treated cells exposed to glutamate/malate, the defect in complex I also decreases the efficiency of oxidative phosphorylation.
Table 1. Mitochondrial respiration of β-alanine treated and untreated permeabilized cardiomyocytes.
| Condition | Substrate | State 3 rate | State 4 rate | RCR | P/O ratio |
|---|---|---|---|---|---|
| Control | Glu-Mal | 1.0 (43.61) | 1.0 (11.03) | 1.0 (3.7) | 2.65 ± 0.2 |
| β-alanine | Glu-Mal | 0.78 ± 0.08* | 0.9 ± 0.09 | 0.8 ± 0.1* | 2.24 ± 0.17* |
| Control | Succinate | 1.0 (65.4) | 1.0 (15.8) | 1.0 (3.3) | 1.47 ± 0.12 |
| β-alanine | Succinate | 0.98 ± 0.09 | 1.2 ± 0.1 | 0.95 ± 0.07 | 1.31 ± 0.08 |
All of the control groups were normalized to 1.0, with the actual values shown in parentheses. The control and β-alanine-treated cardiomyocytes were incubated in medium containing either 4 mM glutamate + 2 mM malate or 5 mM succinate. After a steady-state rate of state 2 respiration was observed, 1 μmol of ADP was added and the rate of state 3 respiration was determined. When ADP levels were depleted, state 4 respiration was determined
RCR respiratory control ratio. Values shown represent means ± S.E.M. of 3–4 different experiments. Asterisks denote a significant difference between the control and β-alanine-treated groups (p < 0.05). Because the experiments were performed with different preparations, all data were normalized relative to the control cells
β-alanine causes mitochondrial fragmentation
Ahmad et al. [14] have shown that changes in mitochondrial morphology are often associated with increases in ROS production. To elucidate if β-alanine-mediated oxidative stress is also accompanied by changes in mitochondrial morphology, control and β-alanine-treated cells loaded with the mitochondrial imaging agent, MitoTracker Deep Red, were monitored using confocal laser scanning microscopy. Images of MitoTracker Deep Red shown in Fig. 3a reveal that β-alanine promotes mitochondrial fragmentation, with the mitochondria acquiring small round fragmented morphology, whereas mitochondria from control fibroblasts assumed a tubular shape. Fibroblasts in culture exhibit various mitochondrial lengths, depending on the status of the cells relative to division, age, and viability. The histograms for mitochondrial length (Fig. 3b) indicate that the mitochondrial length of control fibroblasts follows a normal distribution pattern with an average length of 5.07 ± 0.106 μm. By comparison, mitochondrial length of β-alanine-treated cells was shifted to the left, resulting in an average length equal to 1.98 ± 0.048 μm (Fig. 3c).
Fig. 3.
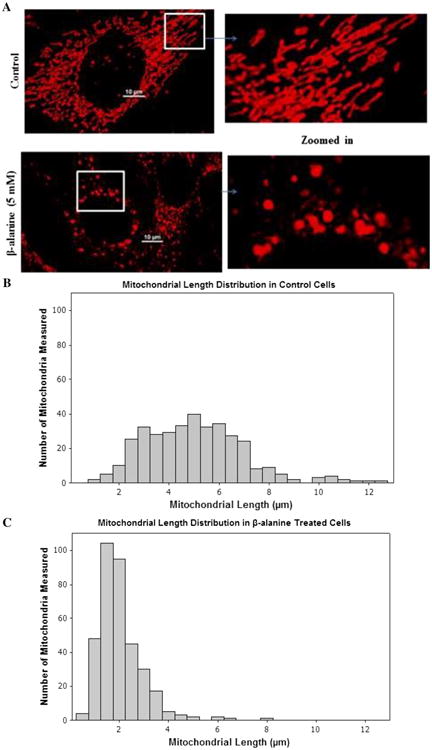
β-alanine mediates mitochondrial fragmentation. Control and β-alanine-treated cells were incubated for 48 h. a Shown are representative MitoTracker Deep Red (150 nM) loaded control fibroblasts (upper panel), which contain tubular mitochondria, and β-alanine-treated fibroblasts (lower panel), which contain fragmented mitochondria. b Average distribution of mitochondrial length of control fibroblasts in culture. c Average distribution of mitochondrial length of β-alanine-treated fibroblasts. The distribution pattern of mitochondrial length was shifted to the left by β-alanine treatment
It has been documented that the mechanism of mitochondrial fragmentation could be mediated through an increase in Drp1 activity, a decrease in Opa1 activity, or both [15]. In the case of β-alanine-treated fibroblasts, Drp1 appeared to mediate mitochondrial fragmentation (Fig. 4); no difference in Opa1 protein levels was observed between control and β-alanine-treated cells (data not shown).
Fig. 4.
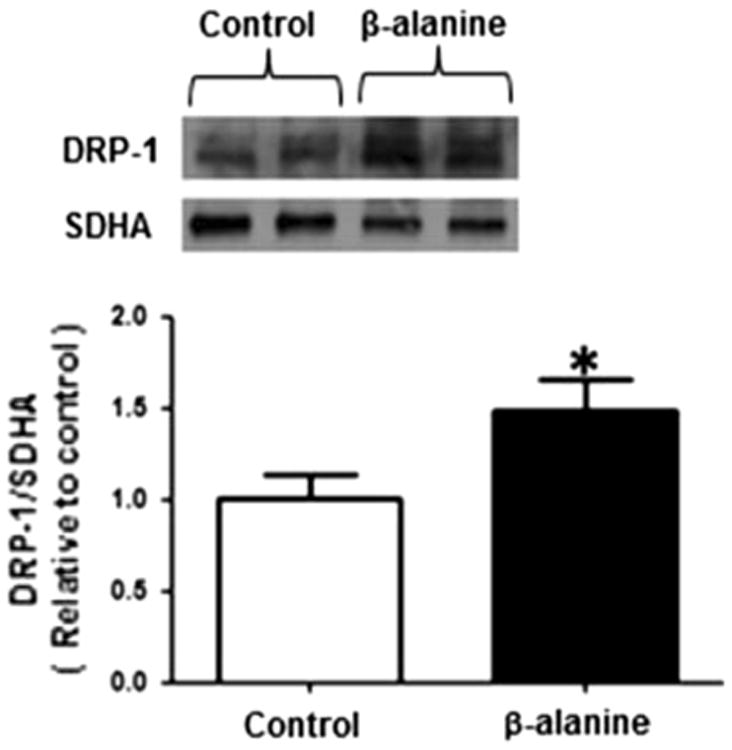
β-alanine treatment is associated with an increase in mitochondrial Drp1 content. Mitochondria were isolated from control and β-alanine-treated fibroblasts and then subjected to Western blot analysis of Drp1. The upper panel depicts a representative Western blot for mitochondrial Drp1 content of control and β-alanine-treated cells, with succinate dehydrogenase (SDHA) serving as a loading control. Data shown in the lower panel represent mean ± S.E.M. of eight different preparations. The asterisk denotes a significant difference in mitochondrial Drp1 content of control and β-alanine-treated cells (p < 0.05)
β-alanine induces apoptosis
Mitochondrial oxidative stress and fragmentation are considered signs of unhealthy mitochondria that are capable of initiating cellular apoptosis. To determine if β-alanine treatment triggers mitochondria-linked apoptosis, the levels of the cleaved, active form of the effector caspase, caspase-3, and of the inactive, procaspase form of the initiator caspase, caspase-9, were determined via Western blot analysis. After 48 h of β-alanine treatment, cellular content of the active, cleaved form of caspase-3 was elevated (Fig. 5a), while the levels of the uncleaved, inactive pro-caspase 9 form were reduced (Fig. 5b), suggesting that initiation of the apoptosome led to an activation of both caspases 3 and 9. Moreover, approximately 20 % of the β-alanine-treated fibroblasts underwent a change in morphology, characterized by a reduction in cell size and the appearance of cell membrane blebs (Fig. 5c). These changes were prevented by the mitochondrial permeability transition pore inhibitor, cyclosporin A, suggesting that β-alanine initiates apoptosis after release of cytochrome c from permeabilized mitochondria (Fig. 5d).
Fig. 5.
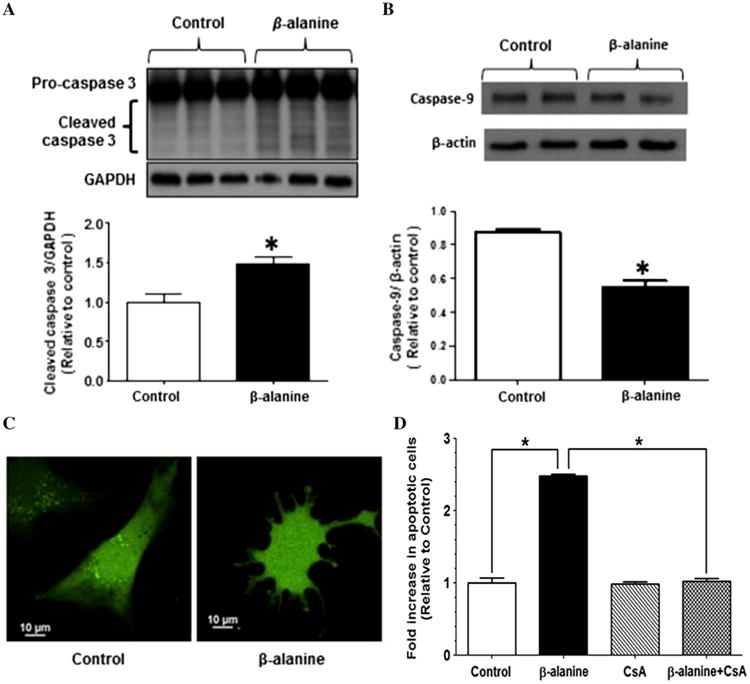
Initiation of apoptosis by β-alanine treatment. a Western blot analysis of procaspase 3 and cleaved caspase-3 content of whole homogenates of control and β-alanine-treated fibroblasts. The upper panel depicts a representative Western blot for procaspase-3 and cleaved caspase-3 content, with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) serving as a loading control. Data in the lower panel represent mean ± S.E.M of four different preparations. The asterisk denotes a significant difference between control and β-alanine-treated fibroblasts (p < 0.05). b Western blot analysis of procaspase-9 content of whole homogenates of control and β-alanine-treated fibroblasts, with β-actin serving as a loading control. The upper panel depicts a representative gel of procaspase-9 content. Data in the lower panel represent mean ± S.E.M of four different preparations. The asterisk denotes a significant difference between the control and β-alanine-treated fibroblasts (p < 0.05). c. Control and β-alanine-treated fibroblasts were loaded with calcein AM (250 μM) Representative image of control and apoptotic, β-alanine-treated fibroblasts. Control fibroblasts appear elongated with a smooth cell surface, while the apoptotic fibroblasts treated for 48 h with β-alanine appear rounded with budding of apoptotic bodies. d Effect of cyclosporin A (200 nM) on percent of β-alanine-treated cardiomyocytes undergoing apoptosis
Role of taurine in β-alanine induced mitochondrial superoxide production and mitochondrial fragmentation
Both β-alanine and taurine share the same β-amino acid transporter [16]; therefore, we investigated whether addition of β-alanine to the incubation medium might reduce taurine content of the fibroblasts in culture. As seen in Fig. 6, exposure of the fibroblasts to β-alanine for 48 h resulted in a 50 % decrease in intracellular taurine content (from 0.40 ± 0.03 μmol/μg protein to 0.21 ± 0.02/μg protein).
Fig. 6.
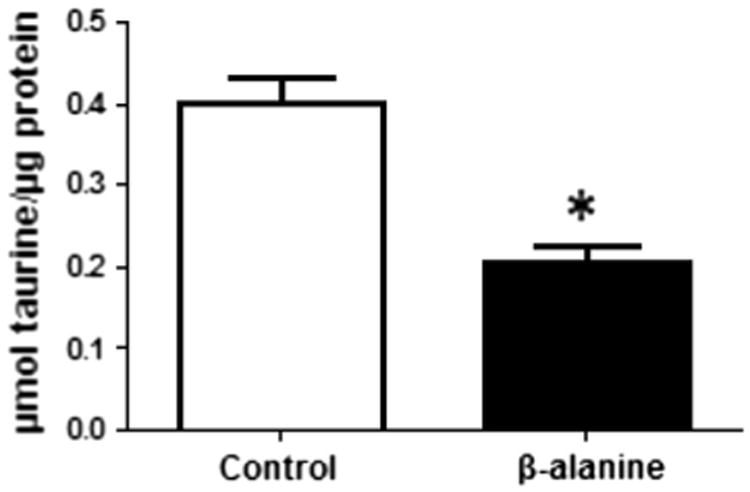
Decrease in the taurine level in the β-alanine-treated fibroblasts. Intracellular taurine levels of control and β-alanine-treated fibroblasts were determined as described in methods. β-alanine treatment reduced taurine content 50 % relative to that of the control. Data represent mean ± S.E.M of four different preparations. The asterisk denotes a significant difference between the control and β-alanine-treated fibroblasts (p < 0.05)
Taurine is a β-amino acid, which serves several physiological functions, one of which is the maintenance of respiratory chain activity [17]. Therefore, we proposed that normal taurine content might favor normal mitochondrial morphology and redox balance. To test our hypothesis, mitochondrial morphology and superoxide generation were measured in β-alanine-treated cells whose taurine content was restored by exposure to medium containing 10 mM taurine. Fibroblasts, which were co-loaded with MitoSox Red and MitoTracker Green, were incubated for 48 h in medium containing β-alanine, which turned the mitochondria yellow (mixture of red from MitoSox Red and green from MitoTracker Green). The yellow cells were then incubated for an additional 48 h with medium containing either 0 or 10 mM taurine. The β-alanine-treated cells that were incubated for 48 h with medium lacking taurine retained their characteristic yellow fluorescence; however, the yellow fluorescence disappeared in the β-alanine-treated cells incubated with medium containing taurine, being replaced by green fluorescence, as taurine treatment appeared to significantly reduce MitoSox fluorescence, allowing the fluorescence of MitoTracker Green to dominate (Fig. 7a, lower panel).
Fig. 7.
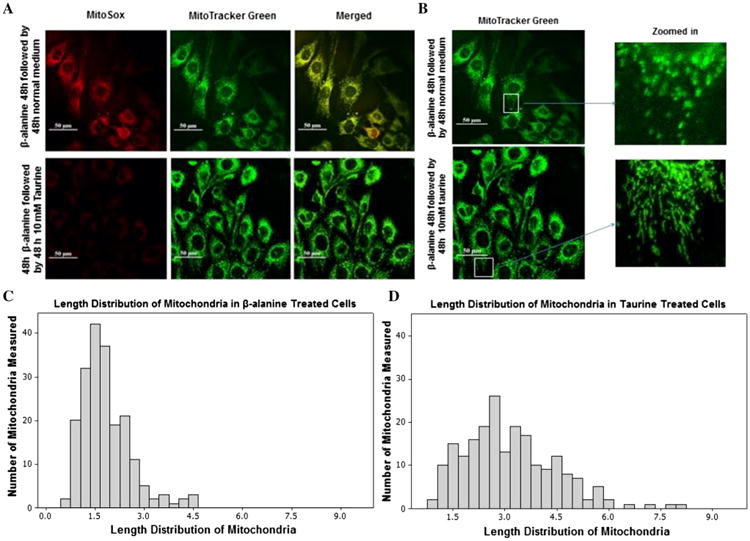
Reversal of β-alanine-mediated mitochondrial toxicity by taurine treatment. a The upper panel shows representative confocal microscopic images of fibroblasts treated initially for 48 h with β-alanine followed by a 48 h incubation with normal β-alanine-free medium. In the lower panel, fibroblasts were treated initially for 48 h with β-alanine and then placed in medium containing taurine for 48 h. All cells were loaded with MitoSox (10 μM) and MitoTracker Green (200 nM). In the upper left hand panel is shown MitoSox fluorescence (red) following the exposure of β-alanine-treated fibroblasts to normal medium for 48 h. In the lower left hand panel is shown MitoSox fluorescence (red) of β-alanine-treated fibroblasts exposed for 48 h to taurine containing medium. In the upper middle and lower middle panels is shown MitoTracker green fluorescence of mitochondria from β-alanine-treated fibroblasts exposed for 48 h to normal medium or to taurine containing medium, respectively. In the upper left hand and lower left hand panels is shown a merged image of MitoSox and MitoTracker green fluorescence of mitochondria from β-alanine-treated fibroblasts exposed for 48 h to normal medium or to taurine containing medium, respectively. Taurine supplementation partially reversed the increase in MitoSox fluorescence mediated by β-alanine treatment. b Magnified, MitoTracker green fluorescent β-alanine-treated fibroblasts were exposed for 48 h to medium containing or lacking taurine. Taurine supplementation partially reversed mitochondrial fragmentation mediated by β-alanine treatment. c The distribution of mitochondrial length in fibroblasts treated initially for 48 h with β-alanine followed by a further 48 h incubation with normal medium. The distribution pattern is shifted to the left, indicating an increase in the number of mitochondria having reduced diameter. The mean length of mitochondria of β-alanine-treated fibroblasts exposed for 48 h to normal medium or to taurine containing medium were 1.81 ± 0.053 and 3.12 ± 0.095 μm, respectively
To determine whether taurine treatment was also capable of reversing β-alanine-mediated mitochondrial fragmentation, fibroblasts were loaded with MitoTracker Green and then incubated with medium containing β-alanine, which caused fragmentation of most of the mitochondria. Once fragmented, the removal of β-alanine from the medium did not restore normal mitochondrial size. However, a significant decrease in mitochondrial fragmentation was detected in β-alanine-treated fibroblasts that were transferred to medium containing taurine (Fig. 7b). Analysis of the distribution of mitochondrial length after 48 h revealed that taurine-treated fibroblasts contained mitochondria ranging in length from 0.75 to 8.06 μm, with the mean mitochondrial length being 3.12 ± 0.09. By comparison, the length of mitochondria in β-alanine-treated cells that had been transferred to normal medium lacking both β-alanine and taurine varied from 0.56 μm to 4.6 μm, with mean mitochondrial length being 1.81 ± 0.05 μm (Figs. 7c, d).
Discussion
The most important finding of the present study is that incubation of fibroblasts with medium containing β-alanine caused mitochondrial fragmentation and oxidative stress, effects that were reversed by transferring the cells to taurine containing medium. Because β-alanine treatment reduces cellular taurine content while taurine treatment restores it, decreases in taurine content appear to play an important role in the toxicity of β-alanine excess.
It has been suggested that taurine-mediated alterations in citric acid cycle flux, ATP production and mitochondrial ROS generation relate to the posttranslational conversion of tRNALeu(UUR) to 5-taurinomethyluridine-tRNALeu(UUR) [10, 18, 19]. The uridine base modified by taurine is located in the wobble position of the anticodon (AAU) of tRNALeu(UUR). The taurine conjugation reaction improves mitochondrial function by strengthening the interaction between the anticodon (AAU) of tRNALeu(UUR) and the UUG codon, thereby facilitating UUG decoding. Although the levels of 5-taurinomethyluridine-tRNALeu(UUR) were not measured in the present study, the levels of 5-taurinomethyluridine-tRNALeu(UUR) have been previously shown to regulate UUG decoding [18–20]. Because UUG decoding defects alter the expression of key complex I subunits, taurine deficiency leads to diminished complex 1 activity [18]. The present study shows that respiratory chain function (state 3 and respiratory quotient) of β-alanine-treated cells utilizing the complex II substrate, succinate, is identical to that of control cells utilizing succinate. However, β-alanine treatment specifically reduces respiratory chain function of cells incubated in medium containing the complex I substrate combination, malate/glutamate. Together these data support the view that β-alanine specifically reduces complex I activity.
The increase in superoxide production by β-alanine-treated fibroblasts appears to be directly related to the reduction in complex I activity. Interestingly, β-alanine treatment resembles the effects of the complex I inhibitor, rotenone, as both agents decrease respiration, specifically inhibit NADH dehydrogenase activity and divert electrons from the respiratory chain to oxygen forming superoxide anion [21]. The present study shows that exposing the mitochondria to succinate bypasses the β-alanine-mediated defect in complex I by preventing the decrease in respiration and improving the efficiency of oxidative phosphorylation. It is likely that β-alanine treatment reduces the rate of oxidative phosphorylation (reduced P/O ratio) by diverting oxygen from the respiratory chain to form superoxide anion rather than utilizing oxygen efficiently to produce ATP.
It is widely recognized that severe mitochondrial oxidative stress can induce cellular apoptosis [22]. The initiation of mitochondrial apoptosis by ROS has been attributed to several mechanisms but most involve permeabilization of mitochondria and release of cytochrome c (and other proapoptotic factors) from the mitochondria [23, 24]. In the cytosol, cytochrome c interacts with Apaf 1 (apoptotic protease-activating factor 1) and ATP to form an apoptosome, which recruits and activates the caspase 9, which in turn activates caspase 3 to mediate cell death. In the present study, β-alanine treatment was shown to activate both caspase 9 and caspase 3.
The permeabilization of the outer mitochondrial membrane is mediated by proapoptotic members of the Bcl-2 family, such as Bax and or Bak, which oligomerize to form a pore in the mitochondrial membrane [23]. According to D'Alessio et al. [24], oxidative modification of Bax causes its dimerization and translocation to the mitochondria. ROS also oxidize the unique mitochondria-associated phospholipid, cardiolipin, which upon oxidation translocates from the inner mitochondrial membrane to the outer mitochondrial membrane, where it facilitates the oligomerization of Bax and the release of cytochrome c from the mitochondria [22]. However, the antiapoptotic Bcl-2 family members, such as Bcl-2, can prevent the oligomerization of Bax and Bak. In the present study, β-alanine treatment diminished the Bcl-2/Bax ratio, but the effect was small and unlikely to significantly influence mitochondrial permeabilization.
Oxidative stress also initiates protein kinase-containing signaling pathways that terminate in programmed responses, such as differentiation, inflammation, and apoptosis. Among the protein kinases that are activated by stress signaling, such as ROS, are the mitogen-activated protein (MAP) kinases, which include p38 MAPK, JNK, and ERK [25], JNK has attracted considerable attention because of its well-known role in activating apoptotic pathways. Worthy of consideration is the work of Leboucher et al. [26], who described a novel JNK-dependent, redox-signaling pathway that leads to both mitochondrial fragmentation and apoptosis. Although we did not attempt to directly associate mitochondrial fragmentation and apoptosis in the present study, the ROS dependency of β-alanine-mediated apoptosis does not appear to involve activation of JNK, as we have previously shown that the phosphorylation and activation of JNK are not mediated solely by β-alanine treatment [27].
Normally, the inner mitochondrial membrane is impermeable to small solutes and molecules. However, elevations in mitochondrial Ca2+ and ROS, as well as other proapoptotic factors, cause the formation of a mitochondrial permeability transition (MPT) pore that allows sufficient mitochondrial swelling to rupture the outer mitochondrial membrane and release cytochrome c and other proapoptotic factors. Although the composition of the MPT pore and the mechanism underlying the effect of ROS on MPT pore formation have been controversial, it is generally accepted that cyclosporin A specifically inhibits MPT pore formation. Figure 5d shows that β-alanine-mediated apoptosis is completely prevented by cyclosporin A, suggesting that activation of the MPT pore is an important contributor to β-alanine-mediated apoptosis. It is interesting that β-alanine exposure reduces the relaxation phase of the Ca2+ transient, largely by decreasing sarcoplasmic reticular Ca2+ ATPase activity secondary to diminished phospholamban phosphorylation [28]. These changes in [Ca2+]i may act synergistically with oxidative stress to initiate the mitochondrial permeability transition.
We have recently shown that hearts of β-alanine-treated rats exhibit reduced contractile function, an effect related in part to impaired mitochondrial energy metabolism [10]. In the present study, we found that the major site of β-alanine action is complex I of the respiratory chain. By reducing flux through complex I, β-alanine treatment decreases both oxygen consumption and ATP generation of the β-alanine-treated heart [10]. Moreover, the similarity between the actions of rotenone and those of β-alanine [21] implies that β-alanine-mediated reductions in complex I activity also cause a diversion of electrons from complex I to the acceptor oxygen, forming in the process superoxide. Thus, β-alanine treatment is associated with diminished ATP generation and energy metabolism, as well as elevated oxidative stress, factors that are capable of altering the contractile state of the failing heart [28, 29]. These results support the suggestion that β-alanine-mediated oxidative stress contributes to the development of tissue dysfunction, including diminished muscle function and hypotonia.
Acknowledgments
We appreciate the financial support of Taisho Pharmaceutical Co. and the intellectual input of our deceased colleague, Dr. Junichi Azuma, Osaka, Japan.
References
- 1.Wu FS, Gibbs TT, Farb DH. Dual activation of GABAA and glycine receptors by beta-alanine inverse modulation by progesterone and 5 alpha-pregnan-3 alpha-ol-20-one. Eur J Pharmacol. 1993;246:239–246. doi: 10.1016/0922-4106(93)90037-a. [DOI] [PubMed] [Google Scholar]
- 2.Tiedje KE, Stevens K, Barnes S, Weaver DF. β-alanine as a small molecule neurotransmitter. Neurochem Int. 2010;57:177–188. doi: 10.1016/j.neuint.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 3.Hayaishi O, Nishizuka Y, Tatibana M, Takeshita M, Kuno S. Enzymatic studies on the metabolism of β-alanine. J Biol Chem. 1961;236:781–790. [PubMed] [Google Scholar]
- 4.Derave W, Everaert I, Beeckman S, Baguet A. Muscle carnosine metabolism and β-alanine supplementation in relation to exercise and training. Sports Med. 2010;40:247–263. doi: 10.2165/11530310-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 5.Yamada EW, Jakoby WB. Aldehyde oxidation. V. Direct conversion of malonic semialdehyde to acetyl-coenzyme A. J Biol Chem. 1960;235:589–594. [PubMed] [Google Scholar]
- 6.Gibson MK, Jakobs C. Disorder of β- and γ-amino acids in free and peptide-linked forms. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. McGraw Hill; New York: 2001. pp. 2079–2105. [Google Scholar]
- 7.Slavik M, Blanc O, Smith KJ, Slavik J. 6-azauridine triacetate induced hyper beta-alaninemia and its decrease by administration of pyridoxine. J Nutr Sci Vitaminol (Tokyo) 1983;29:631–635. doi: 10.3177/jnsv.29.631. [DOI] [PubMed] [Google Scholar]
- 8.Kurozumi Y, Abe T, Yao WB, Ubuka T. Experimental beta-alaninuria induced by (aminooxy)acetate. Acta Med Okayama. 1999;53:13–18. doi: 10.18926/AMO/31647. [DOI] [PubMed] [Google Scholar]
- 9.Gemelli T, de Andrade RB, Rojas DB, Bonorino NF, Mazzola PN, Tortorelli LS, Filho CSD, Wannmacher CMD. Mol Cell Biochem. 2013;380:161–170. doi: 10.1007/s11010-013-1669-8. [DOI] [PubMed] [Google Scholar]
- 10.Schaffer SW, Shimada-Takaura K, Jong CJ, Ito T, Takahashi K. Impaired energy metabolism of the taurine-deficient heart. Amino Acids. 2016;7:1–10. doi: 10.1007/s00726-015-2110-2. [DOI] [PubMed] [Google Scholar]
- 11.Grishko V, Pastukh V, Solodushko V, Gillespie M, Azuma J, Schaffer S. Apoptotic cascade initiated by angiotensin II in neonatal cardiomyocytes: role of DNA damage. Am J Physiol. 2003;285:H2364–H2372. doi: 10.1152/ajpheart.00408.2003. [DOI] [PubMed] [Google Scholar]
- 12.Kussmaul L, Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci USA. 2006;103(20):7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muller FL, Liu Y, van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279(47):49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 14.Ahmad T, Aggarwal K, Pattnaik B, Mukherjee S, Sethi T, Tiwari BK, Kumar M, Micheal A, Mabalirajan U, Ghosh B, Roy SS, Aggarwal A. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis. 2013;4:1–10. doi: 10.1038/cddis.2012.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators fis1, drp1 and opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caruso J, Charles J, Unruh K, Giebel R, Learmonth L, Potter W. Ergogenic effects of β-alanine and carnosine: proposed future research to quantify their efficacy. Nutrients. 2012;4:586–601. doi: 10.3390/nu4070585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jong CJ, Ito T, Mozaffari M, Azuma J, Schaffer S. Effects of β-alanine treatment on mitochondrial taurine level and 5-taurinomethyluridine content. J Biomed Sci. 2010;17(S1):525–532. doi: 10.1186/1423-0127-17-S1-S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jong CJ, Azuma J, Schaffer S. Mechanism underlying the antioxidant activity of taurine: prevention of mitochondrial oxidant production. Amino Acids. 2012;42(6):2223–2232. doi: 10.1007/s00726-011-0962-7. [DOI] [PubMed] [Google Scholar]
- 19.Schaffer SW, Jong CJ, Ito T, Azuma J. Role of taurine in the pathologies of MELAS and MERRF. Amino Acids. 2014;46(1):47–56. doi: 10.1007/s00726-012-1414-8. [DOI] [PubMed] [Google Scholar]
- 20.Kirino Y, Goto Y, Campos Y, Arenas J, Suzuki T. Specific correlation between the wobble modification deficiency in mutant tRNAs and the clinical features of a human mitochondrial disease. Proc Natl Acad Sci USA. 2005;102:7127–7132. doi: 10.1073/pnas.0500563102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turrens J, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu CC, Bratton SB. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid Redox Signal. 2013;19(6):546–558. doi: 10.1089/ars.2012.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11(9):621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 24.D'Alessio M, De Nicola M, Coppola S, Gualandi G, Pugliese L, Cerella C, Cristofanon S, Civtareale P, Ciriolo MR, Bergamaschi A, Magrini A, Ghibelli L. Oxidative Bax dimerization promotes its translocation to mitochondria independently of apoptosis. FASEB J. 2005;19:1504–1506. doi: 10.1096/fj.04-3329fje. [DOI] [PubMed] [Google Scholar]
- 25.Matsuzawa A, Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim Biophys Acta. 2008;1780(11):1325–1336. doi: 10.1016/j.bbagen.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 26.Leboucher GP, Tsai YC, Yang M, Shaw KC, Zhou M, Veenstra TD, Glickman MH, Weissman AM. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptoSsis. Mol Cell. 2012;47(4):547–557. doi: 10.1016/j.molcel.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schaffer S, Solodushko V, Pastukh V, Ricci C, Azuma J. Possible cause of taurine deficient cardiomyopathy: potentiation of angiotensin II action. J Cardiovasc Pharmacol. 2003;41(5):751–759. doi: 10.1097/00005344-200305000-00012. [DOI] [PubMed] [Google Scholar]
- 28.Ramila KC, Jong CJ, Pastukh V, Ito T, Azuma J, Schaffer SW. Role of protein phosphorylation in excitation-contraction coupling in taurine deficient hearts. Am J Physiol. 2015;308(3):H232–H239. doi: 10.1152/ajpheart.00497.2014. [DOI] [PubMed] [Google Scholar]
- 29.Doenst T, Nguyen TD, Abel ED. Heart failure compendium: cardiac metabolism and heart failure: implications beyond ATP production. Circ Res. 2013;113:709–724. doi: 10.1161/CIRCRESAHA.113.300376. [DOI] [PMC free article] [PubMed] [Google Scholar]