

To the Editor
Ataxia-telangiectasia (AT) is an autosomal recessive disease caused by biallelic mutations in ATM encoded on chromosome 11q22.3 that result in DNA repair defects affecting nucleated cells throughout the body (1). AT is suspected in young children with progressive ataxia followed by ocular and cutaneous telangiectasia and is associated with variable forms of immune deficiency (2) (3, 4). Serum alpha-fetoprotein (AFP) is typically elevated in AT, and its origin is likely hepatic even in the absence of obvious liver disease(5) (6).
Nodular regenerative hyperplasia (NRH) is an uncommon liver disease characterized by regenerative nodules diffusely distributed throughout the liver without the fibrosis that usually accompanies chronic liver diseases (7). It is thought to result from microvascular endothelial injury or from alterations in the balance of portal venous and hepatic arterial blood flow on a microscopic level. NRH is characterized clinically by non-cirrhotic portal hypertension, splenomegaly, cytopenias, and liver enzyme abnormalities (8) (7) (9).
NRH is associated with several primary immunodeficiency diseases affecting B cells (e.g., X-linked agammaglubilinemia [XLA] and common variable immunodeficiency [CVID]), T cells (e.g., hyper IgM syndrome due to CD40 ligand deficiency) and phagocytic cells (e.g., chronic granulomatous disease [CGD]) (10) (11) (12) (13). To our knowledge, NRH has not been reported in patients with AT or other DNA repair defects.
A 6-year-old Iraqi Chaldean girl with bilateral ocular and facial telangiectasia, recurrent sino-respiratory infections since infancy, bronchiectasis, and immunodeficiency presenting as hyper-IgM syndrome (HIGM) was diagnosed with AT at the Primary Immunodeficiency Clinic, National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH). Her previous immune evaluations showed her to be slightly lymphopenic with all lymphocyte subsets within (or close to) normal ranges (Lymphocytes, 1070/uL; CD3+ 73.7% (744/uL), CD3+CD4+ 57% (576/uL), CD3+CD8+ 12.3% (124/uL), CD16+56+ 16.4% (166/uL) and CD20 10.4% (105/uL); representative data). Lymphocyte proliferation assays against mitogens and antigens was tested only once, but results were difficult to interpret, as cell viability was suboptimal. Immunoglobulin levels showed a hyper IgM phenotype (IgG 25mg/dL, IgA 36 mg/dL, IgM 1918 mg/dL and IgE <2IU/mL), and antibody responses to protein and polysaccharide antigens were impaired despite full childhood vaccination (non protective titers to tetanus toxoid, diphteria toxoid, H. influenza B, Rubella, pneumococcal antigens and negative isohemagglutinins). Sequencing showed a homozygous mutation in ATM (ATM NM_000051 c.2250G>A). Both of her parents and an older male sibling carried one mutated allele. Following confirmation of AT diagnosis, in depth neurological testing showed subtle anomalies as mild dysarthria and axial ataxia without appendicular ataxia that were unapparent on multiple previous clinical evaluations.
Additionally, the patient had failure to thrive, cytopenias, hepatosplenomegaly and lymphadenopathy without evidence of hematologic malignancy. Her liver disease was characterized by elevated AST and ALT (10 times the upper normal limit), as well as γGT (4 times the upper normal limit). Serologies done prior to her evaluation at NIH (and before IgG replacement therapy was started) were negative for hepatitis A, B, C; CMV and EBV. PCR studies for EBV, CMV, Parvovirus, Hepatitis B virus and Hepatitis C virus in peripheral blood were negative. Bacterial, fungal and mycobacterial cultures from the liver biopsy were negative too; PCR studies for the above-mentioned viruses (and Enteroviruses) were also negative when the liver tissue was tested. MRCP excluded primary sclerosing cholangitis. She had normal levels of alpha-1 antitrypsin and ceruloplasmin, and alpha-fetoprotein levels were elevated to 40.3 ng/ml (reference range 0.6–6.6 ng/ml). Transjugular hepatic biopsy with venous pressure gradient showed severe portal hypertension (16–17 mm Hg corrected hepatic wedge pressure) and typical features of NRH (Figure 1). Regenerative nodules were demonstrated on reticulin stain by 2-cell thick hepatocytes plates of enlarged hepatocytes bounded by narrowed 1-cell thick plates of small, atrophic appearing hepatocytes. The hepatocytes were generally mature and did not show characteristics of fetal hepatocytes. Mild, predominantly lobular inflammation was also present. No Cryptosporidium was seen on the sampled tissue. Portal hypertension was prospectively monitored by ultrasonography at her primary care institution. The patient’s clinical condition has recently shown a rapid and progressive deterioration compatible with portopulmonary syndrome that is under current evaluation.
Figure 1.
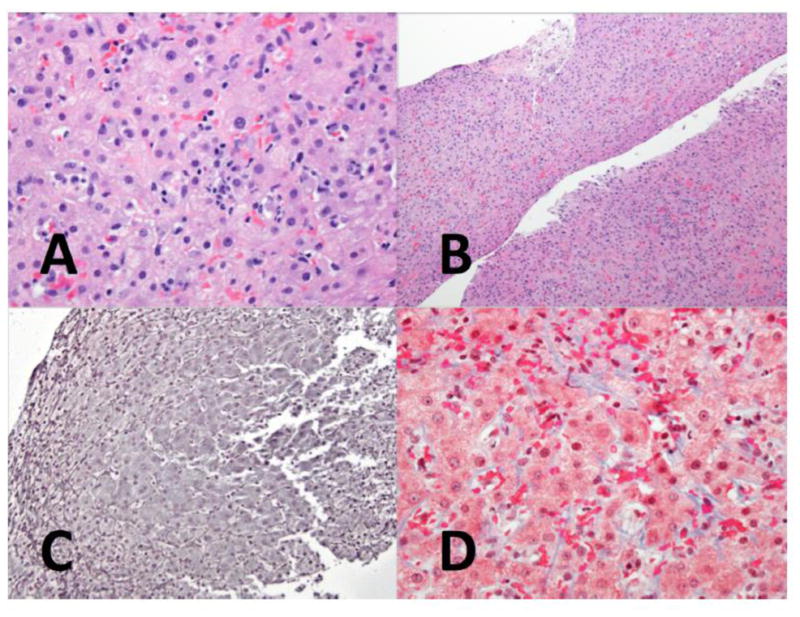
A. Mild lobular inflammation is seen in the parenchyma (H&E, 400x)
B. At medium magnification, the hepatic parenchyma shows few abnormalities. (H&E, 100x)
C. Reticulin staining shows the characteristic changes of NRH, with widened, 2-cell thick liver plates bounded by compressed atrophic liver cell plates. (Reticulin, 200x)
D. A trichrome stain for collagen shows only mild perisinusoidal fibrosis (Masson trichrome, 400x)
Discussion
AT is a rare, degenerative, multi-organ disease with median survival of 20–25 years and incidence of 1 to 2.5 per 100,000 live births (14) (15). The etiology of high AFP levels is suspected to be of liver origin based on its particular glycosylation pattern, despite the typical absence of clinical liver disease (6).
NRH is likely the consequence of vascular injury associated with drug toxicities and a variety of diseases, including immune deficiency syndromes (7) (10) (9). The diagnosis of NRH is hindered by subtle histopathological findings, particularly in needle liver biopsies (9). Even rare, it is estimated to affect 2.1%–2.6% of the general population based on post-mortem findings (16, 17). The prognosis of NRH depends on the underlying etiology and potential complications of portal hypertension (18).
NRH has been reported in 5–12% cases of CVID (10) (11) and severity of portal hypertension varied (11). NRH in CVID is felt to be autoimmune in nature, and is a static or slowly progressive condition (10). Prior authors suggest that insufficient duration of follow up underestimated reporting of NRH in CVID and subsequently led to an under appreciation of NRH related complications. When comparing CVID patients with NRH to those without, those with NRH are more likely to exhibit abnormalities including lymphoproliferation, hepatomegaly, deranged liver enzymes, granuloma, enteropathy, and cytopenias (11). Patients with NRH in the setting of CVID, HIGM, or XLA had portal and/or intra-sinusoidal T-cell infiltrates, disruption of the sinusoid lining, and portal vein endotheliitis and granulomas, which may further impair hepatic perfusion (13).
Data collected on a large NIH cohort of CGD patients suggests that NRH is predictive of CGD mortality (19). Portal venopathy with splenomegaly was present in 80% of the 194 person CGD cohort, and NRH was diagnosed in 9, including 6 of 12 autopsy specimens (12) (19).
Very limited systematic information is available regarding AFP values in NRH. Alpha-fetoprotein was reported to be normal in 8 adult cases with focal nodular hyperplasia (extracted from a larger series), but elevated in a single patient case report (20) (21). In the later, the elevated AFP was associated with the presence of hepatocytes having features of progenitor cells inside the NRH lesion. Among more than 60 NRH patients followed at NIH, including individuals with CVID, XLA and CGD, AFP values were consistently normal (personal communications, Dr. Ivan Fuss and Dr. Theo Heller). Allowing for differences in the amount of background inflammation, the histologic appearance of the NRH in our case was similar to that seen in other adults and children cases, and no increased immature hepatocytes were detected in our patient either.
Treatment of NRH is aimed at the underlying etiology and control of portal hypertension (7). Empiric therapy of NRH with corticosteroids or other immunosuppressive agents has been attempted to preserve liver function; however, the risk versus benefit of prolonged immunosuppressant therapy deserves careful consideration (10).
A recent retrospective study associated AT with abnormal liver enzymes, fatty liver and dyslipidemia: out of 53 mostly pediatric AT patients studied, 23 had elevated liver enzymes, 9 had fatty liver disease by ultrasound, and 10 had dyslipidemia. Liver biopsies in 2 patients found non-alcoholic fatty-liver disease, and no signs of NRH were described (22).
We report a novel association of NRH and AT, a disease of defective DNA repair. Although we cannot formally exclude a casual association between this two conditions in our patient, NRH belongs in the differential diagnosis of AT associated with elevated liver enzymes, cytopenias and splenomegaly. Further evaluation for portal hypertension and liver biopsy with reticulin staining is necessary for accurate diagnosis. The mechanism underlying NRH in AT remains to be determined.
Acknowledgments
We want to thank the patient and her family for participating in our research study. This research was supported in part by the Intramural Research Program of the NIH Clinical Center, NIAID, NIDDK, and NCI.
Footnotes
Conflict of Interest
The authors have no conflict of interest to disclose
References
- 1.Gatti RA, Berkel I, Boder E, Braedt G, Charmley P, Concannon P, et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22-23. Nature. 1988;336(6199):577–80. doi: 10.1038/336577a0. [DOI] [PubMed] [Google Scholar]
- 2.Cabana MD, Crawford TO, Winkelstein JA, Christensen JR, Lederman HM. Consequences of the delayed diagnosis of ataxia-telangiectasia. Pediatrics. 1998;102(1 Pt 1):98–100. doi: 10.1542/peds.102.1.98. [DOI] [PubMed] [Google Scholar]
- 3.Kraus M, Lev A, Simon AJ, Levran I, Nissenkorn A, Levi YB, et al. Disturbed B and T cell homeostasis and neogenesis in patients with ataxia telangiectasia. J Clin Immunol. 2014;34(5):561–72. doi: 10.1007/s10875-014-0044-1. [DOI] [PubMed] [Google Scholar]
- 4.Noordzij JG, Wulffraat NM, Haraldsson A, Meyts I, van’t Veer LJ, Hogervorst FB, et al. Ataxia-telangiectasia patients presenting with hyper-IgM syndrome. Arch Dis Child. 2009;94(6):448–9. doi: 10.1136/adc.2008.149351. [DOI] [PubMed] [Google Scholar]
- 5.Waldmann TA, McIntire KR. Serum-alpha-fetoprotein levels in patients with ataxia-telangiectasia. Lancet. 1972;2(7787):1112–5. doi: 10.1016/s0140-6736(72)92717-1. [DOI] [PubMed] [Google Scholar]
- 6.Ishiguro T, Taketa K, Gatti RA. Tissue of origin of elevated alpha-fetoprotein in ataxia-telangiectasia. Dis Markers. 1986;4(4):293–7. [PubMed] [Google Scholar]
- 7.Reshamwala PA, Kleiner DE, Heller T. Nodular regenerative hyperplasia: not all nodules are created equal. Hepatology. 2006;44(1):7–14. doi: 10.1002/hep.21258. [DOI] [PubMed] [Google Scholar]
- 8.Schouten JN, Garcia-Pagan JC, Valla DC, Janssen HL. Idiopathic noncirrhotic portal hypertension. Hepatology. 2011;54(3):1071–81. doi: 10.1002/hep.24422. [DOI] [PubMed] [Google Scholar]
- 9.Jharap B, van Asseldonk DP, de Boer NK, Bedossa P, Diebold J, Jonker AM, et al. Diagnosing Nodular Regenerative Hyperplasia of the Liver Is Thwarted by Low Interobserver Agreement. PLoS One. 2015;10(6):e0120299. doi: 10.1371/journal.pone.0120299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuss IJ, Friend J, Yang Z, He JP, Hooda L, Boyer J, et al. Nodular regenerative hyperplasia in common variable immunodeficiency. J Clin Immunol. 2013;33(4):748–58. doi: 10.1007/s10875-013-9873-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ward C, Lucas M, Piris J, Collier J, Chapel H. Abnormal liver function in common variable immunodeficiency disorders due to nodular regenerative hyperplasia. Clin Exp Immunol. 2008;153(3):331–7. doi: 10.1111/j.1365-2249.2008.03711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hussain N, Feld JJ, Kleiner DE, Hoofnagle JH, Garcia-Eulate R, Ahlawat S, et al. Hepatic abnormalities in patients with chronic granulomatous disease. Hepatology. 2007;45(3):675–83. doi: 10.1002/hep.21524. [DOI] [PubMed] [Google Scholar]
- 13.Malamut G, Ziol M, Suarez F, Beaugrand M, Viallard JF, Lascaux AS, et al. Nodular regenerative hyperplasia: the main liver disease in patients with primary hypogammaglobulinemia and hepatic abnormalities. J Hepatol. 2008;48(1):74–82. doi: 10.1016/j.jhep.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 14.Crawford TO, Skolasky RL, Fernandez R, Rosquist KJ, Lederman HM. Survival probability in ataxia telangiectasia. Arch Dis Child. 2006;91(7):610–1. doi: 10.1136/adc.2006.094268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swift M, Morrell D, Cromartie E, Chamberlin AR, Skolnick MH, Bishop DT. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am J Hum Genet. 1986;39(5):573–83. [PMC free article] [PubMed] [Google Scholar]
- 16.Nakanuma Y. Nodular regenerative hyperplasia of the liver: retrospective survey in autopsy series. J Clin Gastroenterol. 1990;12(4):460–5. doi: 10.1097/00004836-199008000-00023. [DOI] [PubMed] [Google Scholar]
- 17.Wanless IR. Micronodular transformation (nodular regenerative hyperplasia) of the liver: a report of 64 cases among 2,500 autopsies and a new classification of benign hepatocellular nodules. Hepatology. 1990;11(5):787–97. doi: 10.1002/hep.1840110512. [DOI] [PubMed] [Google Scholar]
- 18.Al-Mukhaizeem KA, Rosenberg A, Sherker AH. Nodular regenerative hyperplasia of the liver: an under-recognized cause of portal hypertension in hematological disorders. Am J Hematol. 2004;75(4):225–30. doi: 10.1002/ajh.20024. [DOI] [PubMed] [Google Scholar]
- 19.Feld JJ, Hussain N, Wright EC, Kleiner DE, Hoofnagle JH, Ahlawat S, et al. Hepatic involvement and portal hypertension predict mortality in chronic granulomatous disease. Gastroenterology. 2008;134(7):1917–26. doi: 10.1053/j.gastro.2008.02.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brumm C, Schulze C, Charels K, Morohoshi T, Klöppel G. The significance of alpha-fetoprotein and other tumour markers in differential immunocytochemistry of primary liver tumours. Histopathology. 1989 May;14(5):503–13. doi: 10.1111/j.1365-2559.1989.tb02186.x. [DOI] [PubMed] [Google Scholar]
- 21.Mneimneh W1, Farges O, Bedossa P, Belghiti J, Paradis V. High serum level of alpha-fetoprotein in focal nodular hyperplasia of the liver. Pathol Int. 2011 Aug;61(8):491–4. doi: 10.1111/j.1440-1827.2011.02691.x. [DOI] [PubMed] [Google Scholar]
- 22.Weiss B, Krauthammer A, Soudack M, Lahad A, Sarouk I, Somech R, et al. Liver Disease in Pediatric Patients With Ataxia Telangiectasia: A Novel Report. J Pediatr Gastroenterol Nutr. 2016;62(4):550–5. doi: 10.1097/MPG.0000000000001036. [DOI] [PubMed] [Google Scholar]