

Abstract
Innovative host-directed drug therapies are urgently required to treat sepsis. We tested the effect of a small-volume 0.9% NaCl adenosine, lidocaine, and Mg2+ (ALM) bolus and a 4-h intravenous infusion on survivability in the rat model of polymicrobial sepsis over 6 days. ALM treatment led to a significant increase in survivability (88%) compared to that of controls (25%). Four controls died on day 2 to 3, and two died on day 5. Early death was associated with elevated plasma and lung inflammatory markers (interleukin-6 [IL-6], IL-1β, C-reactive protein), reduced white blood cell (WBC) count, hypoxemia, hypercapnia, acidosis, hyperkalemia, and elevated lactate, whereas late death was associated with a massive cytokine storm, a neutrophil-dominated WBC rebound/overshoot, increased lung oxidant injury, edema, and persistent ischemia. On day 6, seven of eight ALM survivors had inflammatory and immunological profiles not significantly different from those of sham-treated animals. We conclude in the rat model of experimental sepsis that small-volume ALM treatment led to higher survivability at 6 days (88%) than that of controls (25%). Early death in controls (day 2 to 3) was associated with significantly elevated plasma levels of IL-1β, IL-6, and C-reactive protein, severe plasma lymphocyte deficiency, reduced neutrophils, and acute lung injury. Late death (day 5) was associated with a massive neutrophil inflammatory storm, increased lung injury, and persistent ischemia. Possible mechanisms of ALM protection are discussed.
INTRODUCTION
Globally, 1,000 people die from sepsis every hour, claiming more lives than trauma, heart attack, or cancer (1–3). Sepsis develops from the host's response to an infection involving an overexpression of systemic inflammation, coagulopathy, immune dysfunction, and eventually multiple-organ failure (4, 5). Over the past 2 decades, despite new antimicrobial/antifungal agents and advanced life support systems, the case fatality rate for patients with sepsis has remained at 20% to 30% and up to 50% in low-income countries (3, 4, 6). An ongoing concern is why do some patients who receive state-of-the-art treatment die and others live? The reasons are complex but appear to be related to the type and unpredictable nature of pathogen progression (2, 5, 7), the degree of autonomic and cardiac dysfunction that develops (8), and the lack of a suitable frontline drug therapy to protect against the inability of the immune system to discriminate self from non-self, causing widespread tissue injury and eventually death.
Previously, we have shown in the rat model of polymicrobial sepsis, induced by cecal ligation and puncture (CLP), that small-volume adenosine, lidocaine, and Mg2+ (ALM) induced a stable, hypotensive state over 5 h with no arrhythmias, significantly less pulmonary edema, and corrected coagulopathy (9). The ALM concept has also translated to the pig model of lipopolysaccharide endotoxemia (10). In comparison to controls, ALM infusion led to a mild hibernation-like state that decreased mean arterial pressure (MAP) to 47 mm Hg over 5 h and provided improved left ventricular-arterial coupling, cardiac output, and tissue O2 delivery, significantly lower plasma tumor necrosis factor alpha (TNF-α), reduced pulmonary arterial pressures and resistances, higher PaO2/FiO2 ratios, and less edema (10). Single-bolus ALM has also been shown to resuscitate the heart and rapidly correct coagulopathy after severe hemorrhagic shock (11) and cardiac arrest (12) and exhibits potent antiarrhythmic (13), anti-ischemic (14) and anti-inflammatory properties (15, 16). The aim of the present study was to investigate the effect of small-volume ALM therapy in the conscious rat model of CLP over 6 days. Our hypothesis was that ALM infusion administered over the first 4 h will bolster the host's stress response to the pathogen load (i.e., prevent an overexpression of the host's immune and inflammatory systems) and improve survivability in this model.
MATERIALS AND METHODS
Animals and reagents.
Adult male Sprague-Dawley rats (360 to 420 g) had free access to food and water. Nonheparinized animals were administered buprenorphine (subcutaneously [s.c.]) and isoflurane anesthetic (5% via inhalation and then 1.5% throughout surgery). The study conformed to the Guide for the Care and Use of Laboratory Animals (17) and was approved by the JCU Animal Ethics Committee (approval no. A2029). Adenosine (catalog no. A9251), lidocaine-HCl (catalog no. L5647), and MgSO4 (catalog no. M7506) were purchased from Sigma-Aldrich (Castle Hill, New South Wales, Australia).
Surgical protocol.
The left femoral vein and artery were catheterized aseptically for infusions and blood sampling. Dual-access vascular ports were fitted within a customized jacket. In CLP rats, a 3-cm midline laparotomy was performed and the cecum was ligated at half the distance between the distal pole and the ileocecal valve (9, 18). The ligated portion was punctured once through-and-through with a 21-gauge needle, and a small droplet of stool was milked through each puncture. Sham-treated animals were subjected to the same procedures but with no CLP (Fig. 1A). Animals were allowed to recover on a warming pad (37°C), administered buprenorphine s.c., and placed in isolation cages for treatment and monitoring.
FIG 1.

(A) Schematic representation of the in vivo rat protocol of polymicrobial sepsis. Following isoflurane anesthesia, the femoral artery and vein were cannulated and a laparotomy, cecum isolation, cecal ligation, and puncture were performed in control and ALM groups. Sham-treated animals received the same procedures but no cecal ligation or puncture, i.e., cannulation of femoral artery and vein, laparotomy, and manipulation of the organs and 4-h i.v. saline injections. (B) Kaplan-Meier survival relationship. Symbols: ■, 0.9% NaCl group (control); ▲, ALM group; ●, sham group over the 6-day period. P < 0.05, controls compared to sham- or ALM-treated animals.
Experimental design.
Rats were randomly assigned to different groups in the acute and chronic arms of the study (Fig. 1A). The groups comprised baseline (n = 8) (group 1), 0.9% NaCl sham (n = 8) (group 2), 0.9% NaCl control (n = 8) (group 3), and 0.9% NaCl ALM (n = 8) (group 4) groups. The acute-arm animals were used for 2 h and day 1 data sampling and then sacrificed (n = 8 each group), and the chronic-arm animals were used for blood sampling on days 2, 4, and 6 (n = 8 each group). After day 1, groups 2 to 4 (chronic arm) were reanesthetized, a laparotomy was performed again, and the gangrenous cecum was removed to reduce the pathogen load (cecum was surgically isolated but not resected in sham-treated animals). Preliminary studies showed that if the cecum was not removed, death was imminent (n = 10).
Infusion protocol.
After 15 min, control and sham group animals received a 0.3-ml intravenous (i.v.) bolus of 0.9% NaCl, followed by a 4-h infusion (1 ml/kg of body weight/h) (Fig. 1). ALM animals received a 0.3-ml bolus of 1 mM adenosine, 3 mM lidocaine, and 2.5 mM MgSO4 in 0.9% NaCl (9), followed by an ALM infusion developed from rat and pig studies (adenosine at 6 mg/kg/h, lidocaine at 12 mg/kg/h, and MgSO4 at 6.72 mg/kg/h in 0.9% NaCl) (9) (10). After 4 h, the rats were detached from the infusion tether and had free access to food and water.
Moribund score and survival.
Each animal was assessed twice daily using the humane endpoints described by Nemzek and colleagues (19) and Toth (20). Scoring was done for the following: alertness (grades: 0, normal; 1, low attention; 2, very low attention), mobility (grades: 0, normal; 1, low motion; 2, motionless), piloerection (grades: 0, none; 1, mild; 2, moderate; 3, severe), diarrhea (grades: 0, none; 1, moderate; 2, severe), encrusted eyes, dirty nose and tail (grades: 0, none; 1, one place; 2, two places; 3, three places), and food intake (grades: 0, normal; 1, low; 2, very low; 3, none). If any animal scored ≥12, it was categorized as being in a moribund state that preceded imminent death (19, 20). Euthanasia was performed under 5% isoflurane inhalation, and blood and tissues were rapidly removed.
Whole-blood cell count, gases, and electrolytes.
Blood cell counts were measured on a VetScan HM5 hematology system (REM Systems, Australia). Arterial blood gases, oxygen saturation (sO2), pH, electrolytes, and lactate were measured on an ABL 8000 blood gas analyzer (Radiometer, Pacific).
Cytokine assays and CRP.
Plasma and lung tissue supernatants (pg/ml) were used to measure cytokines with Magpix technology and MilliplexMap rat cytokine/chemokine magnetic bead panel kits (Abacus ALS, Queensland, Australia). Plasma C-reactive protein (CRP) (ng/ml) was measured using Cusabio rat CRP enzyme-linked immunosorbent assay (ELISA) kits (Resolving Images).
Lung injury and edema.
Lung neutrophil infiltration and pro-oxidant activity were estimated from myeloperoxidase (MPO) activity (21), and and malondialdehyde ELISA kits were used to quantify lipid peroxidation (Sigma-Aldrich, Australia). Lung water content was calculated as a percentage based on the formula [(wet weight − dry weight)/wet weight] × 100 (9).
Statistical analysis.
SPSS Statistics 22.0 was used for all data analysis (IBM, Armonk, NY). A priori power analysis was conducted using G*Power3 to determine sample size to minimize type 1 errors (survival; n = 8). Survival was assessed using a Kaplan-Meier test, expressed as a percentage, with a log rank test used to determine differences. Animals were grouped within their treatment group depending on whether they died within the early phase (1 to 3 days) or late phase (4 to 6 days) or if they survived (22, 23). The data were evaluated between groups when there were sufficient sample sizes (n ≥ 2 or 3) by using a one-way analysis of variance (ANOVA) in conjunction with Levene's test of homogeneity to ensure that the assumption of equal variance was met. This was followed by multiple comparisons (Fisher's least significant difference). Within-group differences were evaluated using a repeated-measure ANOVA. Nonparametric data were analyzed with a Kruskal-Wallis test, followed by a Mann-Whitney U test to analyze the difference between two groups. All values are expressed as means ± standard errors of the mean (SEM) with significance set at a P of <0.05.
The data sets supporting the conclusions of this article are available by contacting the corresponding author.
RESULTS
Mortality and blood physiological status.
Mortalities were 0% (sham-treated rats), 75% (saline-treated controls), and 12% (ALM-treated rats) (Fig. 1B); 50% of the controls died on day 2 to 3, and two died on day 5 (as did one ALM rat). Early death was associated with a PaO2 of 73 mm Hg, 60% saturation, PaCO2 of 59 mm Hg, blood pH 7.31, hyperkalemia (5.7 mM), and blood lactate (2.1 mM) (Table 1). Late death was associated with no hypoxemia, no hypercapnia, normal pH, normokalemia, and 2.0 mM lactate. ALM-treated rats with late death had normal PaO2, 97% saturation, hypercapnia, hypokalemia, 159 mM Na+, 1.82 mM Ca2+, and baseline lactate (Table 1).
TABLE 1.
Blood gases, plasma ions, and lactate at baseline and in sham-treated rats, saline controls, and ALM-treated rats over 6 daysa
| Rat group (n)b and time point | pH | pCO2 (mm Hg) | pO2 (mm Hg) | sO2 (%) | K+ (mmol/liter) | Na+ (mmol/liter) | Ca2+ (mmol/liter) | Cl− (mmol/liter) | Lactate (mmol/liter) |
|---|---|---|---|---|---|---|---|---|---|
| Baseline (8) | 7.43 ± 0.01 | 38 ± 1 | 109 ± 1 | 100 ± 0.2 | 3.9 ± 0.2 | 138 ± 2.4 | 1.28 ± 0.03 | 102 ± 1.2 | 0.9 ± 0.1 |
| 2 h | |||||||||
| Sham (8) | 7.44 ± 0.01 | 44 ± 1.5* | 86 ± 2* | 97 ± 0.5* | 4.3 ± 0.1 | 141 ± 1.5 | 1.30 ± 0.04 | 104 ± 0.6 | 0.9 ± 0.1 |
| Control (8) | 7.44 ± 0.01 | 41 ± 0.6 | 86 ± 3* | 96 ± 1.2* | 4.7 ± 0.2* | 139 ± 1.5 | 1.27 ± 0.03 | 104 ± 0.8 | 0.9 ± 0.1 |
| ALM (8) | 7.43 ± 0.02 | 41 ± 3 | 91 ± 5* | 97 ± 1* | 4.7 ± 0.1* | 139 ± 0.9 | 1.22 ± 0.01 | 105 ± 1.2 | 0.9 ± 0.1 |
| Day 1 | |||||||||
| Sham (8) | 7.42 ± 0.01 | 48 ± 2* | 88 ± 3* | 96 ± 0.7 | 3.6 ± 0.0 | 141 ± 1.8 | 1.33 ± 0.03 | 100 ± 0.7 | 1.4 ± 0.1 |
| Control (8) | 7.42 ± 0.03 | 50 ± 5* | 73 ± 10* | 82 ± 11* | 3.9 ± 0.2 | 141 ± 1.0 | 1.31 ± 0.02 | 101 ± 0.5 | 1.9 ± 0.5* |
| ALM (8) | 7.41 ± 0.01 | 47 ± 1.8 | 88 ± 3* | 95 ± 0.6 | 3.8 ± 0.2 | 142 ± 1.5 | 1.34 ± 0.03 | 101 ± 0.8 | 1.6 ± 0.2 |
| Day 2 | |||||||||
| Sham (8) | 7.49 ± 0.01* | 36 ± 1.4 | 92 ± 3* | 98 ± 0.8* | 4.5 ± 0* | 141 ± 0.5 | 1.33 ± 0.01 | 103 ± 0.4 | 1.3 ± 0.1 |
| Control (2) | 7.53 ± 0.01* | 36 ± 0.8 | 100 ± 0.1 | 101 ± 0.5 | 4.4 ± 0 | 141 ± 0.5 | 1.33 ± 0.05 | 104 ± 1.5 | 0.7 ± 0.3 |
| ALM (7) | 7.52 ± 0.02* | 34 ± 1.6* | 101 ± 6 | 99 ± 0.7 | 4.6 ± 0.1* | 141 ± 0.9 | 1.34 ± 0.02* | 104 ± 0.7 | 1.2 ± 0.3 |
| Day 4 | |||||||||
| Sham (8) | 7.51 ± 0.01* | 35 ± 1.4 | 91 ± 2* | 98 ± 0.6 | 4.1 ± 0.0 | 139 ± 1.4 | 1.32 ± 0.03 | 103 ± 0.2 | 1.3 ± 0.2 |
| Control (2) | 7.53 ± 0.04* | 33 ± 4.0 | 113 ± 9* | 100 ± 0 | 4.4 ± 0.1 | 140 ± 0.5 | 1.34 ± 0.03 | 104 ± 0.0 | 1.3 ± 0.4 |
| ALM (7) | 7.53 ± 0.01* | 33 ± 1.4* | 102 ± 7 | 98 ± 0.7 | 4.2 ± 0.1 | 142 ± 0.7 | 1.36 ± 0.01* | 105 ± 1.1 | 1.1 ± 0.1 |
| Day 6 | |||||||||
| Sham (8) | 7.45 ± 0.04 | 42 ± 5.7 | 84 ± 9* | 97 ± 1.6 | 3.9 ± 0.2 | 140 ± 1.4 | 1.34 ± 0.02 | 103 ± 0.6 | 1.1 ± 0.1 |
| Control (2) | 7.45 ± 0.00 | 42 ± 2.3 | 85 ± 5 | 97 ± 2.8 | 3.0 ± 0.0 | 142 ± 2 | 1.36 ± 0.02 | 102 ± 0.0 | 1.5 ± 0.6 |
| ALM (7) | 7.47 ± 0.02 | 39 ± 1.0 | 93 ± 3 | 97 ± 1.1 | 3.4 ± 0.1 | 147 ± 3* | 1.40 ± 0.04* | 105 ± 1.7 | 1.2 ± 0.2 |
| Death | |||||||||
| CED (4) | 7.31 ± 0.08 | 59 ± 12* | 73 ± 34 | 64 ± 22* | 5.7 ± 0.7* | 141 ± 1.3 | 1.37 ± 0.02 | 103 ± 2 | 2.1 ± 0.7* |
| CLD (2) | 7.45 ± 0.02 | 39 ± 5.1 | 85 ± 21 | 90 ± 9 | 3.6 ± 0.3 | 145 ± 0.5 | 1.39 ± 0.01 | 107 ± 1 | 2.0 ± 0.5* |
| ALM LD (1) | 7.30 | 59 | 108 | 97 | 2.9 | 159 | 1.81 | 107 | 1.20 |
Values are for survivors through day 6, followed by animals with early and late death. *, P < 0.05, relative to baseline. CED, control early death; CLD, control late death; LD, late death.
n = number of animals.
Plasma cytokines (6-day survivors).
Interleukin-6 (IL-6) was 2-fold higher in controls than in sham- or ALM-treated rats after 2 h (P < 0.05) and peaked on day 1 (not detected in the sham or ALM group) (Fig. 2). TNF-α was significantly higher than baseline at 2 h in all groups, as was IL-1β and RANTES, which decreased day 1. IL-10 was significantly increased in all groups at 2 h over baseline; IL-10 was 2-fold higher in controls and the ALM group than in sham-treated animals (P < 0.05). On day 1, IL-10 in sham-treated animals and the ALM group decreased toward baseline, whereas that in controls remained significantly higher. C-reactive protein was significantly higher in controls than in all other groups on day 1 (Fig. 2). No significant changes in IL-1α, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-2, gamma interferon (IFN-γ), and IL-4 occurred at 2 h or day 1. On day 2, no significant differences were found in the cytokines except for IFN-γ, with that in controls being 2-fold higher than in the ALM group. Thus, the redone laparotomy and surgical removal of the necrotic cecum did not appear to have a significant effect on inflammatory or immune responses, because the values for sham-treated animals on day 2 were not significantly different from those on day 1 (Fig. 2; Table 2). On day 4, RANTES was 2-fold higher in controls than in sham-treated animals or the ALM group. On day 6, with the exception of RANTES and C-reactive protein, all other cytokines were significantly elevated in controls, and ALM cytokine levels were similar to those in sham-treated animals (not IL-10) (Fig. 2).
FIG 2.

Plasma inflammatory cytokines in survivors. ■, baseline (n = 8);  , sham (n = 8);
, sham (n = 8);  , control survivors (n = 8, 2 h, day 1; n = 2, day 2, 4, 6);
, control survivors (n = 8, 2 h, day 1; n = 2, day 2, 4, 6);  , ALM (n = 8, 2 h, day 1; n = 7, day 2, 4, 6). Data are presented as means ± SEM. *, P < 0.05 versus baseline group; ^, P < 0.05 versus all groups; ∼, P < 0.05 versus sham group.
, ALM (n = 8, 2 h, day 1; n = 7, day 2, 4, 6). Data are presented as means ± SEM. *, P < 0.05 versus baseline group; ^, P < 0.05 versus all groups; ∼, P < 0.05 versus sham group.
TABLE 2.
Total WBC, neutrophil, lymphocyte, and monocyte counts and neutrophil/lymphocyte ratios at baseline and for sham-treated rats, controls, and ALM-treated rats over 6 daysa
| Rat group (n) and time pointb | Total WBC count (109/liter) | Neutrophil count (109/liter) | Lymphocyte count (109/liter) | Monocyte count (109/liter) | Neutrophil/lymphocyte ratio |
|---|---|---|---|---|---|
| Baseline (8) | 9.07 ± 0.71 | 1.60 ± 0.19 | 7.21 ± 0.62 | 0.26 ± 0.05 | 0.2 ± 0.03 |
| 2 h | |||||
| Sham (8) | 7.41 ± 0.75 | 4.75 ± 0.51* | 2.2 ± 0.36* | 0.47 ± 0.09 | 2.4 ± 0.4* |
| Control (8) | 8.56 ± 1.21 | 4.92 ± 1.09* | 3.05 ± 0.51* | 0.59 ± 0.14* | 1.8 ± 0.4* |
| ALM (8) | 7.8 ± 0.83 | 4.79 ± 0.59* | 2.37 ± 0.25* | 0.64 ± 0.12* | 2.1 ± 0.2* |
| Day 1 | |||||
| Sham (8) | 7.85 ± 0.5 | 4.82 ± 0.46* | 2.6 ± 0.13* | 0.43 ± 0.1 | 1.9 ± 0.2 |
| Control (8) | 4.82 ± 0.66^ | 3.11 ± 0.49^ | 1.5 ± 0.23+ | 0.21 ± 0.03∼ | 2.1 ± 0.2* |
| ALM (8) | 6.96 ± 0.59* | 4.8 ± 0.42* | 1.8 ± 0.2* | 0.37 ± 0.08 | 2.8 ± 0.3* |
| Day 2 | |||||
| Sham (8) | 8.11 ± 0.92 | 4.41 ± 0.77* | 3.35 ± 0.38* | 0.35 ± 0.04 | 1.4 ± 0.4* |
| Control (2) | 6.37 ± 0.04 | 3.40 ± 0.02 | 2.69 ± 0.07* | 0.29 ± 0.02 | 1.3 ± 0.04* |
| ALM (7) | 7.74 ± 0.61 | 4.27 ± 0.34* | 3.13 ± 0.31* | 0.33 ± 0.03 | 1.4 ± 0.1* |
| Day 4 | |||||
| Sham (8) | 11.68 ± 0.98 | 6.87 ± 0.93* | 4.24 ± 0.45* | 0.57 ± 0.11 | 1.7 ± 0.3 |
| Control (2) | 12.22 ± 6.33 | 8.95 ± 5.34* | 2.61 ± 0.72* | 0.67 ± 0.28 | 3.1 ± 1.2* |
| ALM (7) | 13.43 ± 1.23* | 8.5 ± 1.12* | 3.93 ± 0.64* | 1.0 ± 0.16+ | 3.0 ± 1.0* |
| Day 6 | |||||
| Sham (8) | 9.9 ± 1.54 | 5.95 ± 0.95* | 3.23 ± 0.85* | 0.72 ± 0.19* | 2.8 ± 0.9* |
| Control (2) | 9.88 ± 0.02 | 6.68 ± 0.12* | 2.54 ± 0.2* | 0.72 ± 0.02 | 2.7 ± 0.3 |
| ALM (7) | 9.14 ± 0.65 | 5.87 ± 0.5* | 2.6 ± 0.65* | 0.67 ± 0.13* | 3.0 ± 0.6* |
| Death | |||||
| CED (4) | 2.91 ± 1.02* | 1.69 ± 0.55 | 0.95 ± 0.41* | 0.26 ± 0.1 | 2.2 ± 0.3* |
| CLD (2) | 12.79 ± 8.42 | 9.15 ± 5.94* | 2.92 ± 2.1* | 0.68 ± 0.34* | 3.5 ± 0.5* |
| ALM LD (1) | 3.17 | 2.11 | 0.78 | 0.29 | 2.7 |
Values are for survivors through day 6, followed by animals with early and late death. *, P < 0.05 versus value at baseline; ^, P < 0.05 versus values for all groups; ∼, P < 0.05 versus value for sham; +, P < 0.05 versus values for sham and baseline. CED, control early death; CLD, control late death; LD, late death.
n = number of animals.
Plasma cytokines (nonsurvivors).
In control animals with early death, IL-6, IL-1β, and C-reactive protein were all higher than in any other group (P < 0.05) (Fig. 3). In contrast, control animals with late death showed elevations in all cytokines. The ALM-treated animals with late death had increases only in IL-1β, C-reactive protein, and IL-10 (Fig. 3).
FIG 3.
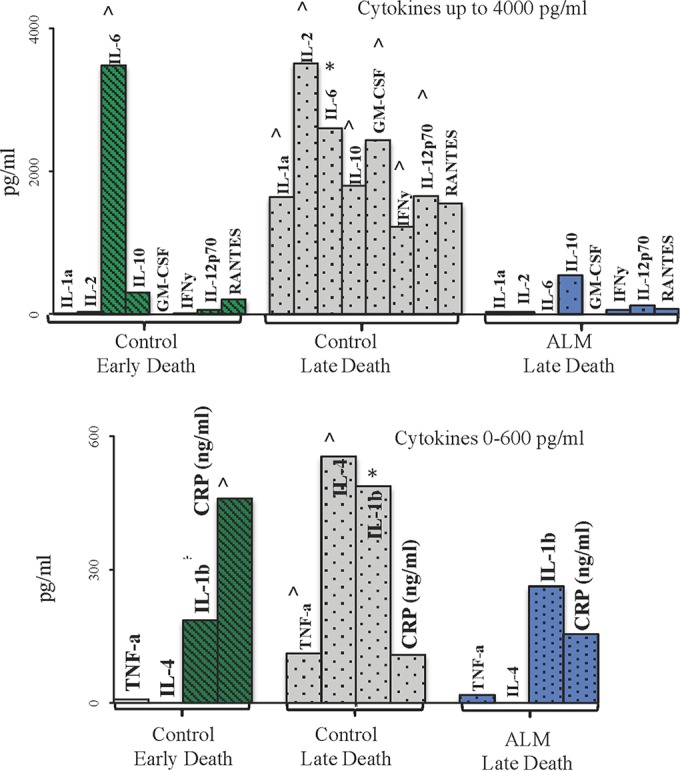
Plasma inflammatory cytokines in those animals that died. Data are presented as means ± SEM. *, P < 0.05 versus baseline group; ^, P < 0.05 versus all groups. Cytokines were grouped into high and lower activities for the three groups: controls with early death (n = 4) ( ); controls with late death (n = 2) (
); controls with late death (n = 2) ( ); ALM-treated animals with late death (n = 1) (
); ALM-treated animals with late death (n = 1) ( ).
).
Blood cell counts.
The baseline white blood cell (WBC) count was 9.07 × 109/liter and fell 6% to 18% after 2 h in all groups. After day 1, the WBC count in controls fell nearly 50% and was significantly lower than that in sham- or ALM-treated rats (Table 2). At day 2, there were no significant differences in WBC counts between groups (Table 2). On day 4, WBC counts in control survivors and in ALM- and sham-treated rats were 1.28 to 1.44 times the baseline (ALM group, P < 0.05), and on day 6 the WBC counts decreased back toward baseline. Early death in controls was associated with a 68% fall in WBC count, but controls with late death had counts 1.41 times that of the baseline. The WBC counts in ALM animals with late death fell 65% (similar to that of controls with early death) (Table 2).
The percentages of neutrophils, lymphocytes, and monocytes relative to the baseline were 18%, 79%, and 3%, respectively, for a neutrophil/lymphocyte (N/L) ratio of 0.22 (Table 2). After 2 h, neutrophils increased 3-fold and lymphocytes decreased by 66% in all groups, resulting in 10-fold increases in the N/L ratio, and monocytes increased around 2-fold. A similar trend occurred at days 1 and 2; however, the N/L ratio fell by 35% to 50% to ∼1.4 in all groups, largely from a rise in lymphocyte count. At day 4, the N/L ratio increased in control survivors and ALM rats to around 3.0 (1.8 times that of sham-treated rats) (Table 2). On day 6, the N/L ratio was 2.8 to 3.0 for all survivors (15 times the baseline). Controls with early death were WBC depleted, with a 1.7-times higher N/L ratio (2.2) than control survivors (1.3) the day before. As mentioned, control animals with late death had very high WBC counts, but these values were similar to those of control survivors the day before (day 4), indicating that the total WBC count (or high N/L ratio) was not responsible for death (Table 2).
Lung inflammation, injury markers, and water content.
Lung IL-6 changed little in survivors on day 1 or 6 but was dramatically increased in controls with early or late death (Fig. 4). TNF-α increased slightly at day 1 in all groups and significantly peaked in controls at day 6 compared to that in sham- or ALM-treated animals with late death. Similar lung TNF-α levels were found in controls with early or late death, and levels were 60% lower in ALM animals with late death. Changes in IL-1β levels were similar to those of TNF-α, and IL-1α peaked in animals with late death but showed minimal changes in animals with early death or in ALM animals with late sdeath. Lung GM-CSF increased in controls at day 1, and small changes occurred in all other groups over the 6 days. IFN-γ increased only in controls and ALM rats on day 1. IL-12p70 was higher in controls with early death and lowest in ALM rats with late death. IL-2 changed little over the 6 days. Similarly, RANTES changed little but decreased during death in all groups. Lung IL-10 and IL-4 levels were high in controls with late death but not in any other group (Fig. 4).
FIG 4.

Lung inflammatory cytokines and injury markers. ■, baseline (n = 8); , sham (n = 8); , control survivors (n = 8, day 1; n = 2, day 6); , ALM survivors (n = 8, day 1; n = 7, day 6); , controls with early death (n = 4); , controls with late death (n = 2); , ALM-treated animals with late death (n = 1). Data are presented as means ± SEM. *, P < 0.05 versus baseline group; ^, P < 0.05 versus all groups; +, P < 0.05 versus sham and baseline groups; ∼, P < 0.05 versus sham group.
Lung water content was 75 to 77% in all groups and increased to 81% in controls with late death, with no change in animals with early death (77%). Lung MPO significantly increased during early death in controls and increased a further 3 times at late death (P < 0.05). Lung MDA significantly increased in controls at day 1 and during early and late death (2.5 times that during early death). MDA levels in ALM-treated rats were 50% less (P < 0.05) than those in controls with early death (Fig. 4).
DISCUSSION
Despite significant improvements in medical care, breakthrough drug therapies are urgently required to treat sepsis (2, 3, 6, 24). We report in the rat model of experimental sepsis that small-volume ALM treatment led to higher survivability at 6 days compared to that of controls. Early death in controls occurred on day 2 to 3 and was associated with inflammatory/immune dysfunction, hypoxemia, hypercapnia, blood acidosis, high lactate, and acute lung injury. In contrast, late death (day 5) had a very different inflammatory/immune and lung injury profile, with less systemic hypoxemia, no hypercapnia, normal blood pH, and persistent lactatemia. The one ALM rat that died at day 5 had a reduced white cell count, higher plasma IL-1β, C-reactive protein, and IL-10 levels, hypercapnia, low blood pH, normal arterial pO2, low lactate, and no apparent lung injury. ALM group survivors at day 6 had an inflammatory/immune profile not significantly different from that of sham-treated animals.
Drug volume and timing.
Our study showed that ALM therapy resulted in one death at day 5 compared to 6 deaths in controls (P < 0.05) (Fig. 1B), which is remarkable given that (i) the total drug volume administered was ∼1.6 ml per animal over 4 h (∼6% of blood volume), (ii) the treatment time comprised 2.8% of the total 6-day experimental sepsis, and (iii) there were no antibiotics. We will now discuss these differences in survivability and possible reasons for ALM's ability to improve outcomes.
Early death in controls.
Early death at day 2 to 3 in controls was associated with significantly elevated IL-1β, IL-6, and C-reactive protein, a severe lymphocyte deficiency, and 50% fewer neutrophils than that in survivors (day 2 to 3) (Fig. 3; Table 2). Of the 12 inflammatory cytokines/proteins measured, only three appeared to be activated at early death, indicating a selective upregulation of the acute-phase response. The acute-phase response is one of two interrelated arms of the immune response; it is an innate, host-directed mechanism to protect against early infection (or injury) and may further limit invasion by trapping microbes within local blood clots (25). The innate response is followed by a second, temporally delayed, adaptive immune response, which generates a different recognition repertoire of seemingly limitless clonally expressed receptor combinations on immune cells to fight off infection (26). Despite the differences, both response strategies in the whole organism can overcorrect and change the system from a pro-survival to an “injurious” phenotype resulting in widespread collateral endothelial/tissue damage and, eventually, death (25–27). Our data are, in part, consistent with this scenario.
In our study, upregulation of IL-1β, IL-6, and C-reactive protein is interesting because IL-1β is one of the major activators of IL-6 expression (28) and both IL-1β and IL-6 are potent inducers of C-reactive protein synthesis in the liver (25, 27, 29). All three are also believed to be central mediators at the interface between the innate and adaptive immune responses. Curiously, plasma TNF-α, an early “master” amplifier of the acute-phase response (29), did not increase at early death (Fig. 3) or in survivors on day 2 to 4 (Fig. 2). However, TNF-α did increase in lung tissue at early death (see below) and after 2 h in response to surgery (Fig. 2). Although we did not study the mechanism of regulation, IL-1β may have been activated and upregulated via the caspase-1 inflammasome pathway (30). However, why IL-1α did not increase (Fig. 4) is unclear, because this cytokine (and IL-18) is part of the IL-1 family that is also activated by the innate caspase-1 inflammasome complex (31). The reason may be that IL-1β activation and IL-1α activation require different pattern recognition “trigger”' signals or compartmentation strategies to feed their specific precursors into the inflammasome complex (32) or may involve other independent activation pathways (33), such as platelets which produce IL-1β (25).
Although plasma IL-6 was significantly higher in controls at early death than in survivors (Fig. 2 and 3), it is unclear whether IL-6 was a marker of death or disease severity or a mediator of tissue injury. The literature appears divided on this question because several studies have shown that antibody inhibition of IL-6 improved survival in a bacterium-derived sepsis model, while others have not (34). IL-6 also has many other inflammatory functions, including enhancing the production of fibrinogen, and activates the hypothalamic-pituitary-adrenal (HPA) axis as part of the stress response to infection and trauma (35, 36). In our study, we also found higher IL-6, IL-1β, and TNF-α levels in lung tissue at early death, and this together with increased MPO activity (2.8-fold at day 1) and higher MDA (3.8-fold baseline, P < 0.05) suggests acute lung injury from neutrophil infiltration, inflammation, and lipid peroxidation damage (Fig. 4). Nonsurvivors were also hypoxic (PaO2, 73 mm Hg, 64% saturation), hypercapnic, acidotic, and hyperkalemic with a blood lactate of 2 mM (Table 1). The absence of pulmonary edema in our study (Fig. 4) may be due to the small volumes of fluids used during ALM treatment compared to what is currently recommended clinically (37).
The dramatic fall in WBC and lymphocyte numbers in our study (Table 2) is consistent with the study of Muenzer and colleagues, which reported low WBC numbers and immunosuppression in the two-hit mouse model of CLP plus pneumonia (38), and the decreased T-cell number increased apoptosis in septic humans (39). Boomer and colleagues further showed that immunosuppression was present not only in peripheral blood cells but also locally in organs in patients that died of sepsis (40). Unfortunately, we did not identify the relative contributions of B lymphocytes (B cells), T lymphocytes (T cells), or natural killer cells (NK cells) to the total WBC count. We therefore conclude that early death in controls was associated with a systemic overexpression of plasma IL-1β, IL-6, and C-reactive protein, a severe lymphocyte deficiency, and an immune-mediated acute lung injury.
Late death was a different inflammatory and immune state.
Late death in controls (day 5) was very different and was associated with a massive amplification of inflammatory and anti-inflammatory cytokines compared to the levels at day 4 (6-day survivors) and a higher neutrophil-dominated WBC count (Table 2). Compared to survivors on day 4, plasma IL-1α increased >1,600-fold, IL-1β >20-fold, IL-2 >45-fold, IL-4 >500-fold, IL-6 >2,500-fold, TNF-α 100-fold, IL-10 >16-fold, GM-CSF >2,400-fold, IFN-γ >1,200-fold, IL-12 >1,600-fold, and RANTES >7-fold. The lower plasma level of C-reactive protein at late death than at early death (Fig. 3) may indicate liver injury secondary to infection, although liver injury markers were not measured. Despite lymphocyte counts remaining relatively low (23% of total), as well as monocytes (5% of total), the cytokine storm at day 5 was associated with a significant 4.4-fold-higher WBC count than that at early death (Table 2). In contrast to findings at early death, neutrophil numbers were dominant (72% of WBC) and their activation appeared to be the potential source of the different cytokine profile (Fig. 3), 5-fold-higher lung oxidant MPO activity (Fig. 4), increased lung MDA (lipid peroxidation), and increased pulmonary edema (Fig. 4). Curiously, the arterial PaO2 of 85 mm Hg was not indicative of hypoxia (90% saturation) and there was no hypercapnia, no blood acidosis, and no hyperkalemia (Table 1). Late death in controls appears to be a “systems failure” from a neutrophil-dominated cytokine storm and persistent elevation of blood lactate (2 mM). Lactate may remain elevated from the liver's reduced ability to clear lactate (via gluconeogenesis) and/or increased peripheral mitochondrial dysfunction with a greater anaerobic contribution to sustain the body's ATP-energy requirements during ongoing sepsis.
ALM-treated animals had similar profiles as sham-treated animals.
At the termination of the experiment on day 6, the ALM-treated survivor group had inflammatory, immune cell counts and blood gas, electrolyte, and lactate levels similar to those of sham-treated animals (Fig. 2; Tables 1 and 2). ALM animals also appeared to limit acute lung injury (Fig. 4), which is a major complication of sepsis (4, 5). The one ALM animal that died on day 5 had a WBC count and inflammatory profile similar to those of controls at early death but without elevated IL-6 in plasma (Fig. 3; Table 2). The survival advantage of ALM therapy appeared to be the bolstering of host defense and the minimization of collateral damage or self-harm. Our working hypothesis is that ALM therapy controls a centrally driven frontline regulatory response that (i) reduces the body's “stress” circuits from activating with improved heart rate variability via the medulla's nucleus tractus solitarius “task matching” capability, (ii) helps to regulate the innate control of adaptive immunity, and (iii) reduces aberrant immune cell receptor-sensing mechanisms and uncontrolled gene expression programs that would otherwise lead to injury (16). In the present study, we assume that the early pathogen-associated molecular patterns (PAMPS) and pattern recognition receptors (PRRs) in ALM-treated animals were the same as those in controls, but the deleterious overexpression of inflammatory and immune responses was minimized or prevented. The ALM therapy may also increase host fitness by increasing peripheral tolerance to an ongoing pathogen challenge.
Damaging immune overexpression in response to sepsis may arise from the body's inability to distinguish self from non-self (26) and may be leveled at multiple checkpoints, including central nervous system (CNS) autonomic regulatory imbalance (41), macrophage effector dysfunction (42), “autoreactive” lymphocytes (26, 42), and aberrant neutrophil attack (43). Collectively, this system of promiscuous immune malfunction may lead to deteriorating endothelial dysfunction, cardiac compromise, multiple-organ failure, and eventually death.
At present, we do not know the relative contributions of adenosine, lidocaine, or magnesium to ALM protection in our rat model of polymicrobial sepsis (9, 16). Individually, adenosine and lidocaine have a long history in sepsis research, but neither have realized therapeutic potential in the clinical setting, and hypomagnesemia is a frequent complication of septic and critically ill patients but magnesium therapy for improving outcomes is controversial (44). In preclinical studies using knockout mice, and other animal models, blocking adenosine A1, A2b, or A3 receptors before experimental sepsis has been shown to be associated with multiple-organ dysfunction and increased mortality (45). Csóka and colleagues further showed that activating A2b adenosine receptors protected against sepsis-induced mortality by dampening excessive inflammation (46). Similarly, Wang and colleagues showed that lidocaine was protective against sepsis in rats and is linked to inhibition of serum high-mobility-group box 1 (HMGB1) expression and NF-κB in liver, kidneys, lungs, and ileum (47). Liu and colleagues also showed that lidocaine protected against renal and hepatic dysfunction in septic rats from the downregulation of Toll-like receptor 4 (48). It is noteworthy that in all of our previous work, from myocardial ischemia to hemorrhagic shock, the ALM combination is key for whole-body protection, as the individual adenosine or lidocaine groups provided suboptimal outcomes (16). Further studies are under way to understand the underlying protective mechanisms of ALM during sepsis.
Potential clinical implications.
The ALM therapy has a number of potential advantages for clinical adoption, as follows: (i) the drug combination is used at higher concentrations as a “polarizing” cardioplegia in cardiac surgery, and a recent prospective randomized trials showed its superiority over the standard of care (49); (ii) early drug infusion time for infection control is short (4 h); (iii) the small volume avoids the untoward pulmonary and cardiac problems of colloidal or larger-volume saline-based fluid therapies (37); and (iv) the therapy may help to bolster the host's early response to fight other “stressors,” i.e., viral or parasitic infections (24), including buying biological time for victims of Ebola and other viral hemorrhagic diseases.
Limitations of the study.
The rat CLP model has many clinically relevant features; however, translation of new drugs to human use has proven to be an enormous challenge (50). In the present study, we did not characterize the bacterial burden or clearance over the 6-day period, the mechanisms of innate sensing and pattern recognition, lymphocyte subtypes, regulation of adaptive immunity, endothelial function, or platelet function. Nor did we investigate cardiac, liver, kidney, or gut function over 6 days or how long any animal could survive beyond 6 days. Further studies are required to examine ALM therapy before human trials with a full cardiovascular, tissue O2 delivery, neurological, immune, and coagulation assessment can be made.
Conclusions.
We conclude in the rat model of experimental sepsis that small-volume ALM treatment led to higher survivability at 6 days (88%) than in controls (25%). Early death in controls (day 2 to 3) was associated with significantly elevated plasma levels of IL-1β, IL-6, and C-reactive protein, severe plasma lymphocyte deficiency, reduced neutrophils, and acute lung injury. Late death (day 5) was associated with a massive neutrophil inflammatory storm, increased lung injury, and persistent ischemia.
ACKNOWLEDGMENTS
G.P.D. is the sole inventor of the ALM concept for cardioplegia, surgery, infection, and trauma. Maddison J. Griffin and Hayley L. Letson have no conflicts to declare.
The experimental work and data collection were carried out by Maddison Griffin. All authors contributed to the literature search, figures, tables, study design, data analysis, data interpretation, and writing of the manuscript.
This work was supported by the College of Medicine and Dentistry and the Australian Institute of Health and Tropical Medicine, James Cook University. The funding body had no involvement in study design, in the collection, analysis, and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication.
REFERENCES
- 1.Reinhart K, Daniels R, O'Brien J, Machado FR, Jimenez E. 2013. The burden of sepsis—a call to action in support of World Sepsis Day 2013. J Crit Care 28:526–528. doi: 10.1016/j.jcrc.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Hotchkiss RS, Monneret G, Payen D. 2013. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 13:862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen J, Vincent JL, Adhikari NK, Machado FR, Angus DC, Calandra T, Jaton K, Giulieri S, Delaloye J, Opal SM, Tracey K, van der Poll T, Pelfrene E. 2015. Sepsis: a roadmap for future research. Lancet Infect Dis 15:581–614. doi: 10.1016/S1473-3099(15)70112-X. [DOI] [PubMed] [Google Scholar]
- 4.Hotchkiss RS, Monneret G, Payen D. 2013. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis 13:260–268. doi: 10.1016/S1473-3099(13)70001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angus DC, van der Poll T. 2013. Severe sepsis and septic shock. N Engl J Med 369:840–851. doi: 10.1056/NEJMra1208623. [DOI] [PubMed] [Google Scholar]
- 6.Angus DC, Opal S. 12 April 2016. Immunosuppression and secondary infection in sepsis: part, not all, of the story. JAMA. Epub ahead of print. doi: 10.1001/jama.2016.2762. [DOI] [PubMed] [Google Scholar]
- 7.Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, Sevransky JE, Sprung CL, Douglas IS, Jaeschke R, Osborn TM, Nunnally ME, Townsend SR, Reinhart K, Kleinpell RM, Angus DC, Deutschman CS, Machado FR, Rubenfeld GD, Webb SA, Beale RJ, Vincent JL, Moreno R, Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup . 2013. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 41:580–637. doi: 10.1097/CCM.0b013e31827e83af. [DOI] [PubMed] [Google Scholar]
- 8.Bollen Pinto B, Ritter C, Michels M, Gambarotta N, Ferrario M, Dal-Pizzol F, Singer M. 26 May 2016. Characterization of brain-heart interactions in a rodent model of sepsis. Mol Neurobiol. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffin MJ, Letson HL, Dobson GP. 2014. Adenosine, lidocaine and Mg2+ (ALM) induces a reversible hypotensive state, reduces lung edema and prevents coagulopathy in the rat model of polymicrobial sepsis. J Trauma Acute Care Surg 77:471–478. doi: 10.1097/TA.0000000000000361. [DOI] [PubMed] [Google Scholar]
- 10.Granfeldt A, Letson HL, Dobson GP, Shi W, Vinten-Johansen J, Tønnesen E. 2014. Adenosine, lidocaine and Mg2+ improves cardiac and pulmonary function, induces reversible hypotension and exerts anti-inflammatory effects in an endotoxemic porcine model. Crit Care 18:682. doi: 10.1186/s13054-014-0682-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Letson HL, Dobson GP. 2015. Correction of acute traumatic coagulopathy with small-volume 7.5% NaCl adenosine, lidocaine and Mg2+ (ALM) occurs within 5 min: a ROTEM analysis. J Trauma Acute Care Surg 78:773–783. doi: 10.1097/TA.0000000000000587. [DOI] [PubMed] [Google Scholar]
- 12.Djabir Y, Letson HL, Dobson GP. 2013. Adenosine, lidocaine and Mg2+ (ALM) increases survival and corrects coagulopathy after 8 min asphyxial cardiac arrest in the rat. Shock 40:222–232. doi: 10.1097/SHK.0b013e3182a03566. [DOI] [PubMed] [Google Scholar]
- 13.Canyon SJ, Dobson GP. 2004. Protection against ventricular arrhythmias and cardiac death using adenosine and lidocaine during regional ischemia in the in vivo rat. Am J Physiol Heart Circ Physiol 287:H1286–H1295. doi: 10.1152/ajpheart.00273.2004. [DOI] [PubMed] [Google Scholar]
- 14.Canyon SJ, Dobson GP. 2006. The effect of adenosine and lidocaine infusion on myocardial high energy phosphates and pH during regional ischemia in the rat model in vivo. Can J Physiol Pharmacol 84:903–912. doi: 10.1139/Y06-035. [DOI] [PubMed] [Google Scholar]
- 15.Shi W, Jiang R, Dobson GP, Granfeldt A, Vinten-Johansen J. 2012. The non-depolarizing, normokalemic cardioplegia formulation adenosine-lidocaine (adenocaine) exerts anti-neutrophil effects by synergistic actions of its components. J Thorac Cardiovasc Surg 143:1167–1175. doi: 10.1016/j.jtcvs.2011.06.045. [DOI] [PubMed] [Google Scholar]
- 16.Dobson GP, Letson HL. 2016. Adenosine, lidocaine and Mg2+ (ALM): from cardiac surgery to combat casualty care: teaching old drugs new tricks. J Trauma Acute Care Surg 80:135–145. doi: 10.1097/TA.0000000000000881. [DOI] [PubMed] [Google Scholar]
- 17.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
- 18.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. 2009. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc 41:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nemzek JA, Xiao HY, Minard AE, Bolgos GL, Remick DG. 2004. Humane endpoints in shock research. Shock 21:17–25. doi: 10.1097/01.shk.0000101667.49265.fd. [DOI] [PubMed] [Google Scholar]
- 20.Toth LA. 2000. Defining the moribund condition as an experimental endpoint for animal research. ILAR J 41:72–79. doi: 10.1093/ilar.41.2.72. [DOI] [PubMed] [Google Scholar]
- 21.Bradley PP, Christensen RD, Rothstein G. 1982. Cellular and extracellular myeloperoxidase in pyogenic inflammation. Blood 60:618–622. [PubMed] [Google Scholar]
- 22.Malmezat T, Breuillé D, Capitan P, Mirand PP, Obled C. 2000. Glutathione turnover is increased during the acute phase of sepsis in rats. J Nutr 130:1239–1246. [DOI] [PubMed] [Google Scholar]
- 23.Xiao HY, Siddiqui J, Remick DG. 2006. Mechanisms of mortality in early and late sepsis. Infect Immun 74:5227–5235. doi: 10.1128/IAI.01220-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zumla A, Rao M, Wallis RS, Kaufmann SH, Rustomjee R, Mwaba P, Vilaplana C, Yeboah-Manu D, Chakaya J, Ippolito G, Azhar E, Hoelscher M, Maeurer M, H-DTN consortium . 2016 Host-directed therapies for infectious diseases: current status, recent progress, and future prospects. Lancet Infect Dis 16:e47–63. doi: 10.1016/S1473-3099(16)00078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. 2014. Emerging roles for platelets as immune and inflammatory cells. Blood 123:2759–2767. doi: 10.1182/blood-2013-11-462432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palm NW, Medzhitov R. 2009. Pattern recognition receptors and control of adaptive immunity. Immunol Rev 227:221–233. doi: 10.1111/j.1600-065X.2008.00731.x. [DOI] [PubMed] [Google Scholar]
- 27.Vince JE, Silke J. 2016. The intersection of cell death and inflammasome activation. Cell Mol Life Sci 73:2349–2367. doi: 10.1007/s00018-016-2205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hunter CA, Jones CA. 2015. IL-6 as a keystone cytokine in health and disease. Nat Immunol 16:448–457. doi: 10.1038/ni.3153. [DOI] [PubMed] [Google Scholar]
- 29.Schulte W, Bernhagen J, Bucala R. 2013. Cytokines in sepsis: potent immunoregulators and potential therapeutic targets—an updated view. Mediators Inflamm 2013:165974. doi: 10.1155/2013/165974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raupach B, Peuschel S-K, Monack DM, Zychlinsky A. 2006. Caspase-1-mediated activation of interleukin-1β (IL-1β) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect Immun 74:4922–4926. doi: 10.1128/IAI.00417-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sollberger G, Strittmatter GE, Garstkiewicz M, Sand J, Beer H-D. 2014. Caspase-1: the inflammasome and beyond. Innate Immun 20:115–125. doi: 10.1177/1753425913484374. [DOI] [PubMed] [Google Scholar]
- 32.Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, Quadroni M, Drexler SK, Tschopp J. 2012. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 36:388–400. doi: 10.1016/j.immuni.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 33.Netea MG, Simon A, van de Veerdonk F, Kullberg BJ, Van der Meer JW, Joosten LA. 2010. IL-1beta processing in host defense: beyond the inflammasomes. PLoS Pathog 6:e1000661. doi: 10.1371/journal.ppat.1000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Remick DJ, Bolgos G, Copeland S, Siddiqui J. 2005. Role of interleukin-6 in mortality from and physiologic response to sepsis. Infect Immun 73:2751–2757. doi: 10.1128/IAI.73.5.2751-2757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burger D, Dayer JM. 2002. Cytokines, acute-phase proteins, and hormones: IL-1 and TNF-alpha production in contact-mediated activation of monocytes by T lymphocytes. Ann N Y Acad Sci 966:464–473. doi: 10.1111/j.1749-6632.2002.tb04248.x. [DOI] [PubMed] [Google Scholar]
- 36.Dobson GP. 2015. Addressing the global burden of trauma in major surgery. Front Surg 2:43. doi: 10.3389/fsurg.2015.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Acheampong A, Vincent J-L. 2015. A positive fluid balance is an independent prognostic factor in patients with sepsis. Crit Care 19:251. doi: 10.1186/s13054-015-0970-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muenzer JT, Davis CG, Chang KC, Schmidt RE, Dunne WM, Coopersmith CM, Hotchkiss RS. 2010. Characterization and modulation of the immunosuppressive phase of sepsis. Infect Immun 78:1582–1592. doi: 10.1128/IAI.01213-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Venet F, Davin F, Guignant C, Larue A, Cazalis MA, Darbon R, Allombert C, Mougin B, Malcus C, Poitevin-Later F, Lepape A, Monneret G. 2010. Early assessment of leukocyte alterations at diagnosis of septic shock. Shock 34:358–363. doi: 10.1097/SHK.0b013e3181dc0977. [DOI] [PubMed] [Google Scholar]
- 40.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD, Krupnick AS, Srivastava A, Swanson PE, Green JM, Hotchkiss R. 2011. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 306:2594–2605. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sonneville R, Verdonk F, Rauturier C, Klein IF, Wolff M, Annane D, Chretien F, Sharshar T. 2013. Understanding brain dysfunction in sepsis. Ann Intensive Care 3:15. doi: 10.1186/2110-5820-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okabe Y, Medzhitov R. 2016. Tissue biology perspective on macrophages. Nat Immunol 17:9–17. doi: 10.1038/ni.3320. [DOI] [PubMed] [Google Scholar]
- 43.Juss J, Herre J, Begg M, Bradley G, Lennon M, Amour A, House D, Hessel EM, Summers C, Condliffe AM, Chilvers ER. 2015. Genome-wide transcription profiling in neutrophils in acute respiratory distress syndrome. Lancet 385(Suppl 1):S55. doi: 10.1016/S0140-6736(15)60370-1. [DOI] [PubMed] [Google Scholar]
- 44.Velissaris D, Karamouzos V, Pierrakos C, Aretha D, Karanikolas M. 2015. Hypomagnesemia in critically ill sepsis patients. J Clin Med Res 7:911–918. doi: 10.14740/jocmr2351w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Law WR. 2006. Adenosine receptors in response to sepsis: what do receptor-specific knockouts tell us? Am J Physiol Regul Integr Comp Physiol 291:R957–R958. doi: 10.1152/ajpregu.00412.2006. [DOI] [PubMed] [Google Scholar]
- 46.Csóka B, Németh ZH, Rosenberger P, Eltzschig HK, Spolarics Z, Pacher P, Selmeczy Z, Koscsó B, Himer L, Vizi ES, Blackburn MR, Deitch EA, Haskó G. 2010. A2B adenosine receptors protect against sepsis-induced mortality by dampening excessive inflammation. J Immunol 185:542–550. doi: 10.4049/jimmunol.0901295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang HL, Xing YQ, Xu YX, Rong F, Lei WF, Zhang WH. 2013. The protective effect of lidocaine on septic rats via the inhibition of high mobility group box 1 expression and NF-κB activation. Mediators Inflamm 2013:570370. doi: 10.1155/2013/570370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu J, Zhang H, Qi Z, Zheng X. 2014. Lidocaine protects against renal and hepatic dysfunction in septic rats via downregulation of Toll-like receptor 4. Mol Med Rep 9:118–124. doi: 10.3892/mmr.2013.1799. [DOI] [PubMed] [Google Scholar]
- 49.Onorati F, San Biagio L, Abbasciano R, Fanti D, Dobson GP, Covajes C, Menon T, Gottin L, Biancari F, Mazzucco A, Faggian G. 12 April 2016. Superior myocardial protection using “polarizing” adenosine, lidocaine, and Mg2+ (ALM) cardioplegia in humans. J Am Coll Cardiol. Epub ahead of print. doi: 10.1016/j.jacc.2015.12.071. [DOI] [PubMed] [Google Scholar]
- 50.Dobson GP. 2014. Addressing the global burden of sepsis: importance of a systems-based approach. Crit Care Med 42:e797–8. doi: 10.1097/CCM.0000000000000595. [DOI] [PubMed] [Google Scholar]