

Abstract
N-terminal acetylation (Nt-acetylation) by N-terminal acetyltransferases (NATs) is one of the most common protein modifications in eukaryotes. The NatC complex represents one of three major NATs of which the substrate profile remains largely unexplored. Here, we defined the in vivo human NatC Nt-acetylome on a proteome-wide scale by combining knockdown of its catalytic subunit Naa30 with positional proteomics. We identified 46 human NatC substrates, expanding our current knowledge on the substrate repertoire of NatC which now includes proteins harboring Met-Leu, Met-Ile, Met-Phe, Met-Trp, Met-Val, Met-Met, Met-His and Met-Lys N termini. Upon Naa30 depletion the expression levels of several organellar proteins were found reduced, in particular mitochondrial proteins, some of which were found to be NatC substrates. Interestingly, knockdown of Naa30 induced the loss of mitochondrial membrane potential and fragmentation of mitochondria. In conclusion, NatC Nt-acetylates a large variety of proteins and is essential for mitochondrial integrity and function.
Protein N-terminal (Nt)1 acetylation is the transfer of an acetyl moiety from acetyl coenzyme A (Ac-CoA) to the N-terminal alpha-amino group of proteins. Nt-acetylation is one of the most common protein modifications in eukaryotes (1–4) and has a wide range of functional implications and impacts protein-protein interactions (5), protein subcellular targeting (6–10), protein folding (11), and ubiquitin-dependent degradation of specific proteins thereby controlling protein stoichiometry and quality (12, 13).
Nt-acetylation is catalyzed by evolutionarily conserved Nt-acetyltransferases (NATs). NatA-NatF differ both with respect to substrate specificity and subunit composition, where the catalytic subunits are members of the GNAT acetyltransferase superfamily. NATs can bind to ribosomes where they perform Nt-acetylation in a co-translational manner (14). However, post-translational Nt-acetylation of proteins has also been reported (15–17). The NatC complex was originally identified in yeast (18, 19) and we previously described the human NatC complex, which is conserved from yeast both with respect to subunit composition, ribosome binding, and in vitro oligopeptide substrate specificity (19, 20). NatC is composed of the catalytic subunit Naa30 (Mak3) and the auxiliary subunits Naa35 (Mak10) and Naa38 (Mak31), all of which cosediment with ribosomes (20). Naa30 plays a role in stress resistance in Caenorhabditis elegans (21). Previously it was found that NatC Nt-acetylates Met-Leu-, Met-Ile-, Met-Phe- and Met-Trp- N termini which are relatively rare as compared with NatA and NatB type N termini (22–24). Of note is that NatC does not acetylate all proteins matching these N-terminal sequences and typically NatC substrates are less Nt-acetylated as compared with NatA and NatB substrates (1, 25). As such, the full basis for substrate recognition by NatC is not fully understood.
Several studies have indirectly linked NatC to organelle traffic, as several Arf(-like) GTPases (i.e., hARFRP1, hArl8b) have been put forward as NatC substrates. In the case of yArl3 and yGrh1, loss of Nt-acetylation in yNatC deletion strains resulted in disruption of their localization to the Golgi apparatus (6–8). Furthermore, hArl8b depends on Naa30-mediated acetylation for correct lysosomal localization (20, 26).
Mitochondrial morphology is highly dynamic. Fission and fusion reactions regulate the number, size, and movement of mitochondria (27). Dysfunctional mitochondria are often associated with fragmentation, by which damaged mitochondria are isolated to prevent their reentry into the mitochondrial network. General defects in mitochondrial dynamics are found in a number of neurodegenerative diseases including Alzheimer's, Huntington's, and Parkinson's disease (27).
Although fragmentation of mitochondria represents a feature of apoptosis (28), mitochondrial damage can also occur and can be exacerbated by reactive oxygen species (ROS). Under oxidative stress, damaged mitochondria are targeted by mitophagy, a selective form of autophagy where membranes encompass damaged mitochondria, giving rise to membranous particles delivered to lysosomes for degradation (27).
Currently, no studies have looked at human NatC mediated Nt-acetylation at a proteome-wide scale. In this study we aimed to define the in vivo substrate profile of hNatC by positional proteomics (29), thereby aiding in elucidating the functional role of NatC. Previously, only a handful of endogenous yeast NatC substrates have been identified. Our proteome analysis revealed 46 human NatC substrates, thereby greatly expanding our knowledge on the in vivo substrate repertoire of NatC and including a wide variety of substrates including the previously identified substrate hArl8b. Interestingly and in line with the reduced levels of various mitochondrial (matrix) proteins observed, depletion of NatC's catalytic subunit Naa30 led to mitochondrial fragmentation and loss of membrane potential. On the other hand, Naa30 depletion did not affect the morphology and distribution patterns of the endoplasmic reticulum (ER), endosome, and peroxisome, nor the architecture of microtubules and the actin cytoskeleton, suggesting that the observed mitochondrial fragmentation is not the result of general disruption of organelles or microtubules. Overall, our results place Naa30 in mitochondrial function and Nt-acetylation of a wide range of iMet-starting proteins.
EXPERIMENTAL PROCEDURES
Plasmids, Antibodies, and Vital Dyes
The following antibodies were used for immunoblotting and/or immunofluorescence: anti-UCRI (Santa Cruz Biotechnology, Santa Cruz, CA; sc-13561), anti-EEA1 (Santa Cruz Biotechnology, sc-33585), anti-PDI (Thermo Scientific, MA3–018), anti-PMP70 (Sigma-Aldrich, SAB4200181), anti-COX IV-rabbit (Cell Signaling, 4850), anti-COX IV-mouse (Cell Signaling, Danvers, MA; 11967), anti-α-actin (AbCam ab6276), anti-β-tubulin (Sigma-Aldrich, St. Louis, MO; T5293) and anti-Naa35 (Sigma-Aldrich, HPA021547). Anti-Naa30 (Biogenes, Berlin, Germany) was generated by immunizing rabbits with purified full-length recombinant human Naa30 protein produced in E. coli, followed by IgG isolation from the resulting sera (20). Horseradish peroxidase-coupled anti-mouse and anti-rabbit from Amersham Biosciences Biosciensce were used as secondary antibodies for immunoblot detection.
Cell culture and transfection
Human A-431 cells (epidermoid carcinoma cell line) (ATCC, CRL-1555) were cultured in Glutamax-containing DMEM medium supplemented with 10% dialyzed fetal bovine serum (Invitrogen, Carlsbad, CA), 100 units/ml penicillin (Invitrogen) and 100 μg/ml streptomycin (Invitrogen). Cells were grown in media containing either natural (12C6) or 13C615N4 l-arginine (Cambridge Isotope Labs, Andover, MA) at a concentration of 80 μm (i.e. 16.8 mg/l or 20% of the suggested concentration present in DMEM at which arginine to proline conversion was not detectable for A-431 cells). Cells were cultured for at least six population doublings to ensure complete incorporation of the labeled arginine. Cells were cultured at 37 °C in a humidified atmosphere of 5% CO2 in air. HeLa cells (epithelial cervix adenocarcinoma; ATCC, CRL-1573) and CAL-62 cells (anaplastic carcinoma, 8305C, DSMZ, ACC 448) were cultured and transfected as described previously (30). Cells were harvested 72 h post siRNA transfection. siRNA mediated knockdown was performed using Dharmafect 1 transfection reagent (Dharmacon) or HiPerFect (Qiagen) according to the vendors' instructions. Gene-specific siRNAs were purchased from Dharmacon and used at the final concentration indicated and ranging between 20–100 nm to silence Naa30. For all siRNA validation experiments two different siRNAs targeting Naa30 were used to ensure that phenotypes were specific for Naa30 depletion: sihNAA30–1 (Dharmacon, LAF, Colorado; d-009961–01), and sihNAA30–2 (Dharmacon, d-009961–05). Knockdown efficiency of Naa30 was inspected by immunoblotting. Non-targeting siRNA (Dharmacon, d-001819–10) was used as a control. For siRNA transfections in A-431 cells, 10 μm Z-VAD-fmk pan-caspase inhibitor (R&D Systems Europe Ltd.) was added throughout the experiment to avoid cell death, whereas 5 μm Z-VAD-fmk was used for siRNA transfections in HeLa cells.
N-terminal COFRADIC: Experimental Design and Statistical Rationale
siRNA transfection was performed as described above and an experimental workflow is given in Fig. 1. For transfections maintained for more than 72 h, cells were retransfected using the same conditions 48 h after the first transfection. Cells were harvested 96 h post-siRNA transfection, lysed in 50 mm sodium phosphate buffer pH 7.5 and 50 mm NaCl supplemented with a complete protease inhibitor mixture tablet (Roche Diagnostics GmbH, Mannheim, Germany) using multiple cycles of freeze-thawing (3×) and total lysates subjected to N-terminal COFRADIC analysis as described previously (1). The organelle fraction was isolated as described below and subjected to a modified sample preparation protocol optimized for fractions containing less soluble proteins. In brief, all modifications were performed using similar conditions as in (31), however in between all modification steps, the insoluble organelle fraction was pelleted, washed, and resuspended in the respective buffer supplemented with the modifying agent(s), and sonicated with a probe sonicator for 5 min using 3 s pulses in an ice/water bath. Following these protein modification steps, the insoluble pellet was dissolved in 10 mm ammonium bicarbonate buffer (pH 8.0) by sonication and digested overnight with sequencing-grade modified trypsin (1/100 (w/w trypsin/substrate)) at 37 °C. Subsequent steps of the N-terminal COFRADIC procedures were performed as in (31).
Fig. 1.
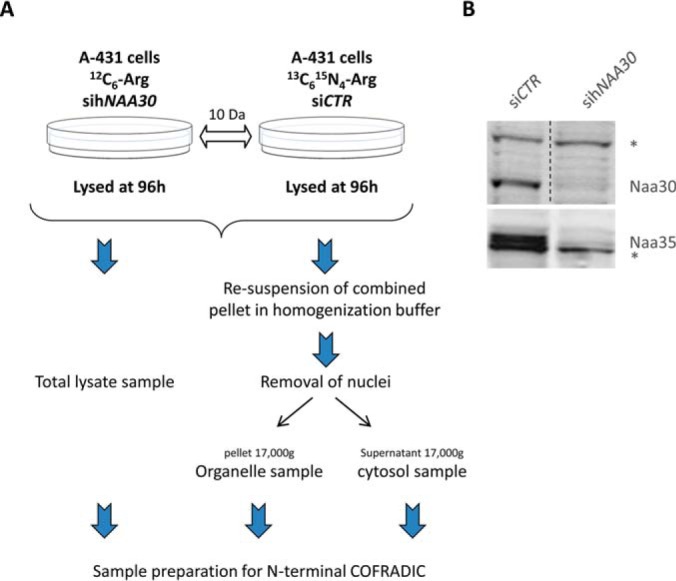
Experimental workflow and confirmation of the efficiency of hNAA30-knockdown. A, Workflow of sample preparations for N-terminal COFRADIC. B, Immunoblots of A-431 cell lysates treated with non-targeting siRNA (siCTR) or sihNAA30. Cells were harvested 96 h post siRNA transfection. Blots were probed with anti-Naa30 and anti-Naa35 to assess levels of endogenous Naa30 and Naa35. The asterisk indicates nonspecific bands that serve as loading control.
The analysis of the fractionated samples served to increase the total proteome coverage and to uncover possible differences in the degree of Nt-acetylation of proteins found in both fractions, if any. In both setups analyzed, making use of an N-hydroxysuccinimide ester of (stable isotopic encoded) acetate at the protein level (i.e. NHS esters of 13C2D3 acetate), in vitro 13C2D3-acetylation was used in combination with differential l-Arg SILAC (stable isotope-labeling by amino acids in cell culture) labeling, which allows for the calculation of the extent of Nt-acetylation as well as the relative quantification of N-terminal peptides (2, 32). Further, given the known and expected substrate specificities of the different NATs, we enriched for NatB, NatC, NatE, and NatF substrates, i.e. Met-starting N-terminal peptides, during the COFRADIC sorting step (32). For this, a methionine oxidation step was introduced between the primary and secondary RP-HPLC separations, thereby shifting all methionine-containing N-terminal peptides to earlier elution times, allowing an enrichment of these (32).
All quantifications (12C6 l-arginine versus 13C615N4 l-arginine) were carried out using the Mascot Distiller Quantitation Tool (version 2.2.1). The quantification method details were as follows: constrain search: yes; protein ratio type: average; report detail: yes; minimum peptides: 1; protocol: precursor; allow mass time match: yes; allow elution shift: no; all charge states: yes; fixed modifications: mass values. Mascot Distiller tries to fit an ideal isotopic distribution on the experimental data based on the peptide average amino acid composition. This fitting is followed by extraction of the XIC signal of all light (12C6 l-arg) and heavy (13C615N4 l-arg) peptide components from the raw data. Subsequently, peptide ratios are calculated by comparing the XIC peak areas of all matched light versus heavy peptides (integration method “trapezium,” integration source “survey”). To calculate this ratio value, a least squares fit to the component intensities from the different scans in the XIC peak is created. MS scans used for this ratio calculation are situated in the elution peak of the precursor determined by the Distiller software (XIC threshold 0.3, XIC smooth 1, Max XIC width 250). To validate the calculated ratio, the standard error on the least square fit has to be below 0.16 and correlation coefficient of the isotopic envelope should be above 0.97. The calculated ratios originally reported as FALSE were all verified by visual inspection of the highest scoring MS spectra. All protein N termini were corrected according to the deviation of a 1:1 ratio of protein-N termini in both analyses.
A distribution of all individual peptide ratios was determined followed robust statistics. Peptides displaying a ratio that was significantly up- or downregulated (95% confidence interval) were considered affected by the hNAA30 knockdown. Quantification of the degree of Nt-acetylation was performed as described in (33). When comparing the degrees of Nt-acetylation from two independent control experiments (with the degrees of Nt-acetylation of more than 1000 unique N termini calculated) and taking into account a [x – 10%, x + 10%] interval around the calculated x-value (the x-value being the degree (%) of Nt-acetylation for the calculated data point in one data set), the p value was calculated to be p ≤ 0.01, indicating that upon setting these limits, less than 1% of all measured Nt-Ac values differed more than 10%. Therefore, a significant variation in the degree of Nt-acetylation was set to 10% or more.
LC-MS/MS Analysis and Data Storage
The obtained peptide mixtures were introduced into an LC-MS/MS system, the Ultimate 3000 (Dionex, Amsterdam, The Netherlands) in-line connected to an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) and LC-MS/MS analysis was performed as described previously (1, 2). The generated MS/MS peak lists were searched with Mascot using the Mascot Daemon interface (version 2.2.0, Matrix Science). Searches were performed in the Swiss-Prot database with taxonomy set to human (UniProtKB/Swiss-Prot database version 2014_02, containing 20,256 human entries). 13C2D3-acetylation of lysine side-chains, carbamidomethylation of cysteine and methionine oxidation to methionine-sulfoxide were set as fixed modifications for the N-terminal COFRADIC analyses. Variable modifications were 13C2D3-acetylation and acetylation of protein N termini. Pyroglutamate formation of N-terminal glutamine was additionally set as a variable modification. Mass tolerance on precursor ions was set to 10 ppm (with Mascot's C13 option set to 1) and on fragment ions to 0.5 Da. Endoproteinase semi-Arg-C/P (Arg-C specificity with arginine-proline cleavage allowed) was set as enzyme allowing no missed cleavages. The peptide charge was set to 1+, 2+, 3+, and instrument setting was put to ESI-TRAP. Only peptides that were ranked one, have a minimum amino acid length of seven and scored above the threshold score, set at 99% confidence, were withheld. The estimated false discovery rate by searching decoy databases (a shuffled version of the yeast Swiss-Prot database made by the DBToolkit algorithm (34)) was below 1.5% on the spectrum level and below 2% at the peptide level. All data management was done in ms_lims (35). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (36) with the data set identifier PXD000930 and DOI 10.6019/PXD000930.
Cell Lysis and Organelle Sedimentation
The fractionated samples used for COFRADIC analysis were harvested, resuspended in cell homogenization buffer (0.25 m sucrose, 1 mm EDTA, 20 mm HEPES-NaOH, pH 7.4), and homogenized using six strokes through a cell crusher with 4 mm gap. Approximately 5 × 107 cells (A-431) were used per sample. Nuclei were removed through sedimentation at 3000 × g for 5 min. To separate organelles from cytosol, the homogenate was further centrifuged at 17,000 × g for 20 min. The resulting pellet contains organelles and plasma membrane, whereas the supernatant contains the cytosol devoid of membranous and organelle compartments. The supernatant and the pellet were further prepared for analysis by N-terminal COFRADIC, as described above. In parallel, cells were analyzed for the efficiency of knockdown, harvested by scraping and pelleted by centrifugation at 2000 × g for 5 min. Total cell lysates were prepared by resuspending cell pellets in lysis buffer (50 mm Tris/HCl pH 8, 50 mm NaCl, 0.5% Nonidet P40, 5 mm EDTA, 1 mm Na3VO4, and 1 mm Pefabloc (Roche)) and incubation on ice for 5 min. Cell debris was removed by centrifugation at 15,700 × g for 1 min, and the supernatant transferred.
Immunofluorescence and Statistical Data Analysis
HeLa or CAL-62 cells grown on cover slips were washed in PBS or cytoskeleton buffer (134 mm NaCl, 5 mm KCl, 1.1 mm Na2HPO2.2H2O, 0.4 mm KH2PO4, 5.5 mm glucose, 4 mm NaHCO3, 10 mm MES, 2 mm EGTA, 2 mm MgCl2), fixed in 3% paraformaldehyde, permeabilized in 0.2% Triton X-100, and blocked in 10% BSA. Proteins of interest were labeled with primary antibodies as indicated (1:250). Secondary antibodies were Alexa Fluor 488-, 594- or 555-conjugated IgGs (Invitrogen) (1:200). Blue 4′,6-diamidino-2-phenylindole (DAPI) staining was used to stain the nuclei. MitoTracker® Red CMXRos was added to live cells according to the manufacturer's instructions (Life technologies, M-7512) at a final concentration of 50 nm. Rhodamine phalloidin was added to fixed cells to visualize actin according to the manufacturer's instructions (Life technologies, R415). In cases when double labeling with mouse primary antibodies was performed, samples were incubated with goat anti-mouse IgG (H + L) Fab fragments (1:100) after an incubation of the first mouse primary antibody. This incubation was followed by incubation of with donkey anti-goat Alexa secondary antibody and then in blocking buffer (PBS, 10% BSA, 5% goat serum). Next, the second primary mouse antibody was added to the samples, followed by goat anti-mouse Alexa secondary antibody. Between all incubations the samples were washed in 0.1% Triton X-100. Images were acquired using a Leica DMI 6000b microscope or a Leica TCS SP5 confocal microscopy. Where indicated, microscopic recordings were processed by de-convolution with 10 iterations using 3D Blind settings from 12–18 z-planes (Leica software). Z-stacks and Z-stack projections were handled using the Fiji Image Processing Software. Brightness and contrast were adjusted using the Fiji Image Processing Software. The data for quantification after hNAA30 knockdown are shown as the mean of at least 100 cells counted in three independent samples.
Data for quantification of phenotypes after Naa30 knockdown were analyzed using SPSS or Prism statistical software package. Student's t test was used to compare the mean percentage of defined phenotypes in siNAA30 versus siCTR samples. Significance level was set to p < 0.05 for all analyses.
RESULTS
COFRADIC Analysis of hNAA30 Knockdown Cells Reveals Novel Human NatC Substrates
We characterized the in vivo substrate specificity of the human NatC complex by comparing the Nt-acetylomes of A-431 cells treated with sihNAA30 versus siControl treated cells (siCTR) (Fig. 1A). Here a knockdown efficiency of more than 95% could be observed (Fig. 1B). Interestingly, Naa35 and Naa38 were also significantly depleted upon sihNAA30-mediated knockdown (Fig. 1 and supplemental Table S1). Next to total cell lysates, samples enriched for cytoplasmic and organellar fractions were analyzed (Fig. 1A). A GO-enrichment analysis pointed to a significant enrichment of organelle proteins in the organelle versus cytosolic enriched fractions and thus efficient fractionation (supplemental Fig. S1).
Following LC-MS/MS analysis, we identified 1874 unique N termini with a minimum amino acid length of seven originating from 1754 human proteins that were in compliance with the rules of Nt-acetylation (37) and initiator methionine (iMet) processing, thereby being proxies of translation initiation (supplemental Table S1). Of these, 1855 started at position 1 or 2, whereas 19 started beyond position 2, the latter indicative of alternative translation initiation events (16, 38).
In total, 46 N termini displayed a >10% reduction in their degree of Nt-acetylation upon hNAA30 knockdown, a cut-off value where the false discovery rate when studying differences in the degree of Nt-acetylation was found to be less than 1% (see Experimental Procedures). These N termini all started with an iMet as reflected by their Swiss-Prot database annotated start position of 1. Based on the sihNAA30 dependent reduction in their degree of Nt-acetylation, the identification of the previously characterized substrate hArl8b (20, 26) and the substrate specificity profile observed, we consider these N termini as direct Naa30 substrates and they thus constitute part of the in vivo substrate repertoire of the human NatC complex (Table I and supplemental Table S1B) (14). Representative MS spectra of sihNAA30 affected substrate N termini of the cell differentiation protein RCD1 homolog and the hydroxysteroid dehydrogenase-like protein 2 (HSDL2) identified in the total cell lysate setup are shown in Fig. 2A and 2B.
Table I. NatC type N-termini: their Nt-acetylation status and Naa30 dependency of Nt-acetylation. All types of N-termini identified in this study matching previously reported NatC substrate specificities (ML-, MY-, MF-, MI-) (22–24) as well as the previously uncharacterized hNatC substrate specificities identified in this study (MM-, MH-, MV-, and MK-) are listed. In all cases, the numbers of uniquely identified N-termini, the number of N-termini for which the degree of Nt-Acetylation could be univocally calculated/determined and their corresponding number and proportion of complete and partial acetylation is indicated. Whenever a reduced degree of acetylation could be observed upon sihNAA30 knockdown the corresponding number and proportion of affected N-termini are indicated.
| NatC substrate type | Identified | Nt-Ac determined | Full + partial Nt-Ac (%) | sihNAA30 affected |
|---|---|---|---|---|
| Met-Met | 17 | 16 | 16 (100%) | 1 (6%) |
| Met-His | 3 | 3 | 3 (100%) | 1 (33%) |
| Met-Leu | 50 | 48 | 43 (90%) | 18 (42%) |
| Met-Tyr | 9 | 9 | 8 (89%) | 0 (0%) |
| Met-Val | 41 | 40 | 34 (85%) | 8 (24%) |
| Met-Phe | 24 | 23 | 19 (83%) | 5 (26%) |
| Met-Ile | 12 | 12 | 8 (67%) | 0 (0%) |
| Met-Lys | 52 | 50 | 30 (60%) | 13 (43%) |
| Total | 208 | 201 | 161 (80%) | 46 (29%) |
Fig. 2.

The effect of Naa30 knockdown on the Nt-acetylation states of individual proteins and the Nt-acetylome. A, Representative MS spectrum of the N terminus originating from the cell differentiation protein RCD1 homolog (1MHSLATAAPVPTTLAQVDR19). The peptide was fully acetylated in the control setup while being only partially acetylated (83%) in the sihNAA30 knockdown sample. The total concentration of the N terminus remained unaffected in the two samples analyzed (ratio of 1). B, Representative MS spectrum of the N terminus of the hydroxysteroid dehydrogenase-like protein 2 (1MLPNTGR7). The peptide was partially acetylated in both control (32%) and knockdown (3%) samples. The total concentration of the N terminus is significantly lowered upon sihNAA30 knockdown (p ≤ 0.01; ratio of 0.5). C, The scatterplot displays the correlation of the determined degrees of Nt-acetylation when comparing the siCTR (X-axis) and the sihNAA30(Y-axis) N-terminome data sets (N termini with start position 1 or 2) of the total lysate samples (n = 1065). The circles falling outside the ±10% boundaries (dashed lines) represent N termini displaying a significant reduction in the degree of Nt-acetylation upon sihNAA30 knockdown (2). The circles are color coded according to their indicated NAT substrate specificity profiles (see also NAT type column in supplemental Table S1). sihNAA30 knockdown causes a decrease in the levels of Nt-Ac as compared with the siCTR setup only for specific N termini in the NAT type categories of “NatC” or “Other” N termini.
Many of the here identified substrates are previously unidentified NatC substrates not belonging to the class of predicted NatC-type N termini (holding Met-Leu, Met-Ile, Met-Phe, and Met-Trp N termini) (22–24). Indeed, our data reveal that human NatC in vivo additionally acetylates N termini starting with Met-Lys, Met-Val, Met-His, and Met-Met N termini (Table I and supplemental Table S1B). Of note, as siRNA-mediated knockdown does not lead to a full depletion of the target protein, mostly suboptimal (i.e. not fully acetylated) NatC-type N termini displayed reduced Nt-acetylation (Fig. 2C), an observation previously also seen for hNatA and hNatB substrates (1, 25). Further, no Met-Trp and Met-Ile N termini were identified among the Naa30 substrates.
hNAA30 Substrates Are Not Enriched for Organellar Localization
As described above, several studies have linked NatC to organelle traffic. In yeast, it was previously shown that proteins harboring a signal or transit peptide are highly protected against removal of their initiator methionine as they have a higher frequency of bulky and basic amino acids (Leu, Ile, Trp, Lys and Arg) at position 2 (10). This was also evident when searching the human Swiss-Prot database for proteins holding a transit or signal sequence at their N terminus (supplemental Fig. S2). These proteins typically have Arg, Leu, Lys and Trp as their second amino acid, which partly overlaps with our revised NatC substrate specificity profile (Table I). Further, these signal peptides typically encompass around 16 to 30 amino acids, including at least one positively charged residue, followed by 6 to 12 hydrophobic residues. Despite this indication for organelle localization of several human NatC substrates, we did not find a significant enrichment for organellar proteins by gene ontology analysis among the identified NatC substrates (data not shown). More specifically, organelle-localized substrates constitute about 20% of the human NatC substrates identified, whereas the majority is cytosolic, nuclear or cytoskeleton associated (supplemental Fig. S1). Of note however is that the rapid removal of protein signal sequences accompanying organellar protein transport may largely account for this (see also below for the identification of mature/processed mitochondrial protein N termini). Furthermore, whenever protein N termini were identified in multiple fractions (i.e. total lysate, organelle and/or cytosol enriched fraction from siCTR and/or sihNAA30 setups) no notable differences in their degree of Nt-acetylation could be observed. This indicates that Nt-acetylation is not critically involved in steering the localization of proteins with multiple subcellular localization patterns identified in our study (supplemental Table S1).
hNAA30 Knockdown Negatively Impacts Mitochondrial Protein Levels
With the proteomics data at hand, we also monitored possible differences in protein levels based on comparing the MS-signal intensities of N-terminal peptides identified in the siCTR and sihNAA30 samples that were identified in the total lysate sample (supplemental Table S1B). In total, 43 unique protein N termini displayed significantly altered steady state levels (p ≤ 0.05). Note that only 9 of 43 proteins displayed an elevated protein level upon Naa30 depletion, whereas the remaining 34 displayed decreased protein levels. Among these 43 regulated proteins, eight proteins were here identified as NatC substrates, and all eight displayed significantly reduced protein levels upon Naa30 depletion in the total lysate setup. Interestingly, proteins with decreased levels upon Naa30 depletion were enriched for mitochondria-localized proteins. Indeed, when searching for enriched GO terms indicative of protein subcellular localization among regulated proteins, GO terms such as mitochondrial part (and related terms) were found significantly enriched (p value = 4.44 × 10−6) using GOrilla.
To more globally assess changes in the mitochondrial proteome, we additionally considered the expression ratios of proteolytic N termini indicative of mitochondrial protein processing events (i.e. in vivo Nt-free N termini starting beyond position 2 originating from mitochondrial proteins, and thus serving as proxies of N-terminal signal peptide removal upon mitochondrial protein import among others) (supplemental Table S1). Interestingly, clearly distinct profiles could be observed when comparing the ratio density plots (log2) of database annotated N termini versus N termini indicative of mitochondrial processing events (Fig. 3A), the latter showing two distinct populations. Upon closer inspection, we observed that the expression ratios of mitochondrial matrix proteins were specifically affected as processed mitochondrial matrix proteins displayed significantly reduced expression levels as determined by a Chi-squared test of Independency (p value < 0.00001). Additionally, NatC type substrates appeared enriched in the category of mitochondrial matrix proteins whereas NatB type substrates were underrepresented (Fig. 3B). Overall, these findings point to a general clearance of mitochondria, likely by mitophagy (39), upon hNAA30 knockdown, bringing along the selectively reduced levels of mitochondrial matrix proteins, a class enriched in NatC substrates. Given these data we hypothesized that mitochondria were impaired in hNAA30 knockdown cells.
Fig. 3.
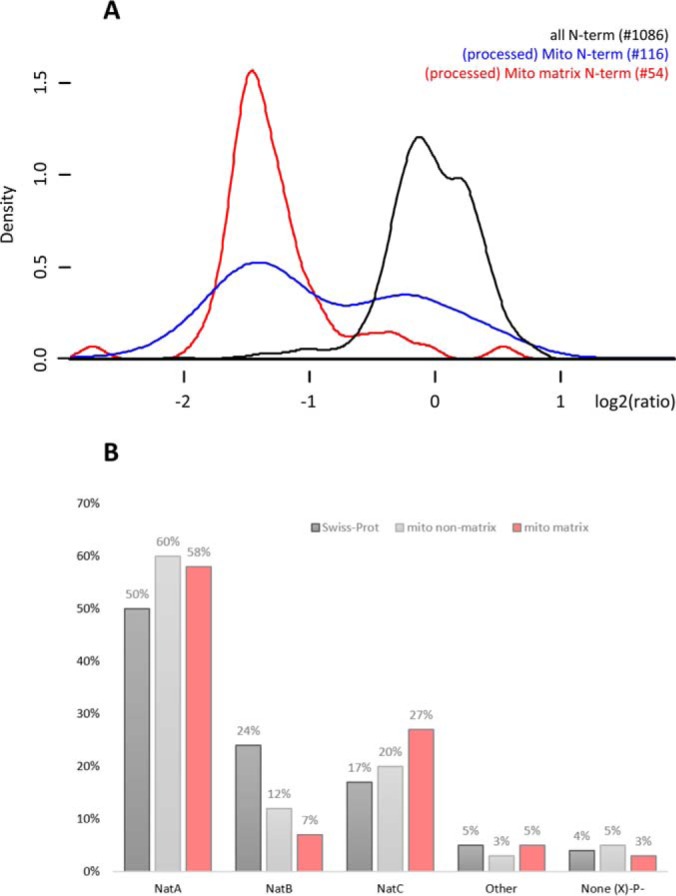
hNAA30 knockdown impacts mitochondrial matrix protein levels, a category of proteins enriched for NatC type substrates. A, Density plots of log2 transformed sihNAA30/siCTR ratios of all unique N termini (1149), (processed) mitochondrial N termini (116) and (processed) mitochondrial matrix protein N termini (54) identified in the total lysate setup. B, Bar chart of the NAT-type substrate specificity distribution of all initiator methionine (iMet) starting Swiss-Prot protein entries (25744), mitochondrial proteins excluding mitochondrial matrix proteins (761) and mitochondrial matrix proteins (304).
hNAA30 Knockdown Induces Mitochondrial Fragmentation
To investigate the effect of NatC depletion on mitochondria in more detail, we compared the mitochondrial morphology of sihNAA30 and siCTR treated HeLa cells. Mitochondria were visualized by immunofluorescence microscopy and staining cells for the cytochrome c oxidase subunit IV (COX IV), which is localized to the inner mitochondrial membrane. In untreated and siCTR treated HeLa cells, mitochondria are typically organized in a thread-like continuous network that spans the entire cell and appear to be present in a higher density close the nucleus and more dispersed toward the cell perimeter (Fig. 4). In CAL-62 cells mitochondria are typically clustered at one nuclear side and not spanning the entire cell. Occasionally they have a similar mitochondrial network arrangement as observed in HeLa cells (Fig. 4). In cells where apoptosis has been induced by staurosporin treatment, the mitochondrial network appears to be reduced in complexity with swollen parts, and perinuclear localization (supplemental Fig. S3). Interestingly, in sihNAA30 treated HeLa cells we observed a large percentage of cells with altered mitochondrial morphology. These cells have lost their longer thread-like mitochondria, which are instead present as fragmented, smaller units that are mostly circular in shape. In addition, they do not span the entire cell, but are usually concentrated around the nucleus (Fig. 4A, 4B). A cell was counted as fragmented when it displayed an alteration in morphology as compared with the morphology of the vast majority of the cells in untreated and siCTR treated cells. This change in morphology was defined as a high degree of fragmentation present as smaller discontinuous units of mitochondria, usually concentrated around the nucleus, and loss of a continuous thread-like mitochondrial network. Because anaplastic thyroid carcinoma CAL-62 cells contain a non-functional form of p53 (30), we also depleted Naa30 in these cells, and investigated the morphology of mitochondria by COX IV staining. Naa30-depletion induced redistribution of mitochondria toward the nucleus and an increased fragmentation and clearance after Naa30 depletion (Fig. 4C–4E). Overall, we conclude that depletion of Naa30 leads to mitochondrial fragmentation, perinuclear redistribution and clearance, independent of p53-induced apoptosis. In both cell types, the difference in mitochondrial fragmentation between siCTR and sihNAA30 treated cells was found to be statistically significant (p < 0.05, student t test) (Fig. 4B, 4D). The mitochondrial fragmentation and clearance observed in HeLa and CAL-62 cells upon hNAA30 knockdown supports the proteomics data derived from A-431 cells and suggest that the mitochondrial effects observed are not cell line specific. Western blot analysis was performed for each experiment to confirm hNAA30 knockdown efficiency on Naa30 protein levels (Fig. 4F).
Fig. 4.

sihNAA30-treated cells display fragmented mitochondria. HeLa (A, B) and CAL-62 (C, D, E) cells depleted for Naa30 were immunostained for the mitochondrial marker COX IV-mouse (A) or COX IV-rabbit (C, E). Micrographs were taken with a confocal and Z-stacks were Z-projected to visualize COX IV. DAPI (blue) was used to visualize the nuclei (A, C) in overlay with COX-IV (red). The confocal micrograph in (E) was taken with a 6.5 zoom and deconvoluted. White bars correspond to 10 μm. The percentages of cells with perinuclear and fragmented mitochondria were calculated for each sample (B, D). Data are mean and standard deviations from three independent experiments (n = 100–150). The difference between siCTR and sihNAA30 treated cells was statistically significant (p < 0.05, student t test). (F) Immunoblots of cell lysates from HeLa cells treated with nontargeting siRNA (siCTR) or sihNAA30. Cells were harvested 72 h post siRNA transfection. Blots were probed with anti-Naa30 to assess levels of endogenous Naa30. β-tubulin was used as loading control.
hNAA30 Knockdown Does Not Cause Global Disturbances in Organelle Homeostasis
Because mitochondrial morphology is strongly dependent on ER-induced fission (27) and microtubular (40) and actin cytoskeletal changes (41) we assessed if the observed mitochondrial effect was caused by a more generalized abrogated cellular architecture and thus investigated the implication of Naa30 perturbation on organelle organization in general. More specifically, we investigated the morphology and distribution patterns of several other organelles as well as cytoskeletal integrity. Overall, we could not observe any global changes in morphology and distribution of the ER, microtubules, actin cytoskeleton, early endosomes and peroxisomes (supplemental Fig. S6), indicating that Naa30 depletion does not globally affect organellar structures or cytoskeletal rearrangements.
hNAA30 Knockdown Induces Functional Impairment of the Mitochondria
To investigate if mitochondria were still functional upon hNAA30 knockdown, we stained siCTR and sihNAA30 treated HeLa cells with the vital mitochondrial dye MitoTracker Red CMXRos. Accumulation of this dye depends on the mitochondrial membrane potential generated across the inner mitochondrial membrane during ATP production (42). Loss of membrane potential and thus loss of MitoTracker Red CMXRos staining can be used as a parameter to assess mitochondrial impairment (43). In sihNAA30 treated cells, MitoTracker Red CMXRos typically did not stain fragmented mitochondria, but rather shows a more diffuse cytoplasmic staining (Fig. 5), which indicates that mitochondria in Naa30 depleted cells are functionally impaired as compared with siCTR treated cells. In fact, note that all counted sihNAA30 treated cells with fragmented mitochondria demonstrated reduced or loss of CMXRos staining.
Fig. 5.

MitoTracker Red CMXRos staining reveals non-functional mitochondria in sihNAA30 treated cells. Confocal micrographs of siRNA treated HeLa cells stained with MitoTracker Red CMXRos. All cells displaying a phenotype of fragmented mitochondria are not or only partially stained by MitoTracker Red CMXRos. At least 100 cells in three independent parallels were observed. DAPI was used to visualize nuclei. White bar indicates 10 μm.
Loss of Nt-acetylation was previously shown to affect the subcellular localization of a few substrates (6–9). We therefore tested whether sihNAA30 treatment affected subcellular localization of the here-identified mitochondrial Naa30 substrates metaxin-1 (MTX-1), cytochrome b-c1 complex subunit Rieske (UCRI), mitochondrial 28S ribosomal protein S36 (MRPS36), and the hydroxysteroid dehydrogenase-like protein 2 (HSDL2). MTX-1, UCRI and MRPS36 and HSDL2 all displayed a strong co-distribution with the mitochondrial marker COX IV in both siCTR and sihNAA30 treated cells (supplemental Fig. S4). Even though mitochondrial morphology was altered by sihNAA30 treatment, the codistribution patterns of MTX-1, UCRI, MRPS36, and HSDL2 with COX IV were not altered. In line with previous data on HSDL2 localization data, the observed dual cytoplasmic and peroxisomal staining pattern of HSDL2 appeared not to be affected by sihNAA30 treatment (supplemental Fig. S5) (44, 45), and these observations were additionally confirmed in CAL-62 cells (data not shown).
DISCUSSION
We here investigated the substrate repertoire of human NatC, which was now found to include proteins harboring Met-Leu, Met-Ile, Met-Phe, Met-Trp, Met-Val, Met-Met, Met-His, and Met-Lys N termini. This reveals a substrate specificity overlap of human NatC with NatE and NatF, which have been demonstrated to acetylate some of these N-terminal sequences (2, 46, 47) and might indicate a redundancy between these NATs. However, differences in the subcellular localization and functional partners might direct these NATs to different substrate pools in vivo. Indeed, a recent study revealed that NatF (Naa60) localizes to organellar membranes and specifically Nt-acetylates transmembrane proteins harboring a cytosolic N terminus (48). In contrast to the identified NatF substrates, in the current data set there was no enrichment for transmembrane proteins among identified NatC substrates (48). Furthermore, NatE (Naa50) acts in physical association with NatA, and was suggested to have a role in the Nt-acetylation of non-iMet cleaved N termini of the NatA-class (e.g. Met-Thr-, Met-Ala-, Met-Ser- etc.) (49). Interestingly, several of the NatC substrates detected do not match previous types of NatC-type substrate N termini (i.e. Met-Leu, Met-Trp, Met-Ile, Met-Phe) (Table I) (20, 50). In this study we find that human NatC in vivo acetylates Met-Lys, Met-His, Met-Met, and Met-Val in addition to Met-Leu and Met-Phe, the latter two specificities matching previously annotated NatC type substrate N termini (Table I). Unexpectedly, we could not detect any changes in the levels of Nt-acetylation annotated Met-Ile (Table I) N termini. Besides, no Met-Trp N termini were identified in our study, the latter possibly because of the scarcity of detectable (mature) N termini (1.3% of the human proteome) (2). When considering those type of protein N termini displaying the observed NatC substrate specificity profile (i.e. putative Naa30 substrate N termini starting with Met-Phe, Met-His, Met-Lys, Met-Leu, Met-Met, Met-Val), 35% of the putative candidate NatC substrates were identified in this study as genuine NatC substrates (i.e. for NatC type N termini of which the degree of Nt-acetylation could univocally be assigned in both setups) (i.e. 29 of 82 proteins in total lysates). Interestingly, none of the N termini affected by hNAA30 knockdown harbor an acidic amino acid at position 3 or 4 (Met-x-(x)-Glu/Asp, where x = Leu/Lys/Val/Met/Phe). This could indicate that the presence of an acidic residue at these positions steers NatC-mediated Nt-acetylation. In addition, these N termini are typically Nt-acetylated to a higher degree as compared with their nonacidic residue containing counterparts. Previous N-terminomics studies demonstrated that higher eukaryotes have an increased level of Nt-acetylation as compared with lower eukaryotes, which is partially attributed to the lack or very rare occurrence of Nt-acetylation of Met-Lys in yeast (2). Thus, Nt-acetylation of Met-Lys and potentially other types of N termini by human NatC, together with NatF and perhaps NatE, likely contributes to the increased levels of Nt-acetylation in higher eukaryotes. Further, as knockdown of NAA30 resulted not only in the depletion of Naa30 but also Naa35 and Naa38, thereby reflecting the effects of depleting the entire NatC complex, this indicates a vital role for Naa30 in maintaining Naa35 and Naa38 protein levels.
Several studies have pointed toward a possible role of NatC in the subcellular localization of proteins and organelle organization. Although gene ontology analysis of human NatC substrates did not reveal any pronounced enrichment for organelle localization, we observed a significant enrichment of mitochondrial proteins among the proteins with significantly altered protein levels (supplemental Table S1C). This particular data set is only based on statistically significant differences of MS-intensities from N termini (typically a single peptide per protein), but it seems to be accurate enough to provide clues for further investigation. Indeed, the majority of proteins with reduced levels were significantly overrepresented by mitochondrial proteins hinting to the fact that hNAA30 knockdown affects mitochondria. Moreover, (mature) mitochondrial matrix proteins displayed significantly reduced expression levels as compared with mitochondrial nonmatrix proteins. Although degradation of outer membrane proteins of depolarized mitochondria is mediated by proteasome-dependent degradation, degradation of matrix proteins largely depends on mitophagy (39, 51). Besides, previous studies demonstrated that mitochondrial complex III—of which all components were here found to be downregulated—acts as a specific positive regulator of autophagy (52) and that coenzyme Q deficiency triggers mitophagy (53). We also noticed the up-regulation of the autophagy protein Ambra 1 (activating molecule in BECN1 regulated autophagy), a factor critically involved in mitochondrial clearance (54, 55) and its up-regulation, together with the observation of mitochondrial depolarization in hNAA30 knockdown cells leads us to speculate that the loss of Nt-acetylation of one or more NatC substrates causes mitochondrial damage and mitophagy. Mitophagy degrades damaged mitochondria by autophagosomal delivery to lysosomes (56), a process often accompanied by mitochondrial fission facilitating degradation of mitochondria (57). In line, immunofluorescence studies revealed a clear effect of Naa30 depletion on mitochondria as they were more fragmented in hNAA30 knockdown cells. With respect to differences in protein levels, only the data set of the total lysate sample was considered because the large differences in mitochondrial morphology in sihNAA30 treated cells might have perturbed subcellular fractionation, and thus extraction of proteins in the fractionated samples.
Naa30 depletion was previously reported to be involved in p53-dependent apoptosis (20). Mitochondrial fragmentation and clearance is associated with cell death (58) and it has been reported that treatment with pan-caspase inhibitors during apoptosis will selectively remove mitochondria from cells (59). However, in this study we verified the mitochondrial phenotype in CAL-62 cells which express mutated, nonfunctional p53 and without the addition of pan-caspase inhibitor (Fig. 4). Further, in contrast to apoptotic cells, mitochondrial fragmentation appears limited in sihNAA30 treated cells and no cells with fragmented nuclei were observed. Thus, the observed effects are not likely to be dependent on apoptosis.
By means of immunofluorescence studies, we could not observe any obvious changes in subcellular localization for the mitochondrial substrates investigated, indicating that Nt-acetylation by NatC is not a general determinant of subcellular localization, observations in agreement with a previous study on yeast NatC substrates (60). Eight NatC substrates were found to display reduced protein levels upon Naa30 depletion (supplemental Table S1C), which could indicate that they are targeted for degradation by the N-end rule pathway (12, 13, 61). Three of the substrates with reduced protein level are mitochondrial proteins that appear to be cleared from the cell (UCRI, MRPS36 and HSDL2, supplemental Fig. S4), whereas the nonmitochondrial fraction of UCRI and HSDL2 appear to be unaffected.
Here, we show that the human NatC complex Nt-acetylates a broad range of cellular proteins, and that depletion of the catalytic subunit Naa30 affects mitochondrial morphology and function. However, the molecular mechanisms involved still remain elusive.
Supplementary Material
Acknowledgments
We thank Professor Johan R. Lillehaug for valuable discussions.
Footnotes
Author contributions: P.V., J.V., K.G., and T.A. designed research; P.V., T.V.K., K.K.S., V.J., and L.M.M. performed research; P.V., T.V.K., K.K.S., V.J., and G.M. analyzed data; P.V., T.V.K., and K.K.S. wrote the paper.
* This work was supported by the Fund for Scientific Research Flanders (Belgium) (project number G.0269.13N (to P.V.D.)), the Norwegian Cancer Society (to T.V.K., J.E.V., K.K.S. and T.A.), the Bergen Research Foundation (to T.A.), the Research Council of Norway (Grants 197136 and 230865), and the Western Norway Regional Health Authority (to J.E.V. and T.A.).
 This article contains supplemental material.
This article contains supplemental material.
Authors declare no competing interests.
1 The abbreviations used are:
- Nt
- N-terminal
- Ac-CoA
- Acetyl coenzyme A
- COFRADIC
- combined fractional diagonal chromatophagy
- Ctr/CTR
- control
- DAPI
- 4′,6-diamidino-2- phenylindole
- GNAT
- GCN5-related N-acetyltransferase superfamily
- iMet
- initiator methionine
- NAA#
- N-alpha acetyltransferase # (gene/protein)
- NAT
- N-terminal acetyltransferase
- z-VAD-fmk
- carbobenzoxy-VAD (O-methyl)- fluoromethylketone.
REFERENCES
- 1. Arnesen T., Van Damme P., Polevoda B., Helsens K., Evjenth R., Colaert N., Varhaug J. E., Vandekerckhove J., Lillehaug J. R., Sherman F., and Gevaert K. (2009) Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. USA 106, 8157–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Van Damme P., Hole K., Pimenta-Marques A., Helsens K., Vandekerckhove J., Martinho R. G., Gevaert K., and Arnesen T. (2011) NatF Contributes to an evolutionary shift in protein N-terminal acetylation and is important for normal chromosome segregation. PLoS Genet. 7, e1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brown J. L., and Roberts W. K. (1976) Evidence that approximately eighty percent of the soluble proteins from ehrlich ascites cells are IV-acetylated. J. Biol. Chem. 251, 1009–1014 [PubMed] [Google Scholar]
- 4. Bienvenut W. V., Sumpton D., Martinez A., Lilla S., Espagne C., Meinnel T., and Giglione C. (2012) Comparative large-scale characterisation of plant vs. mammal proteins reveals similar and idiosyncratic N-alpha acetylation features. Mol. Cell. Proteomics 11, M111.015131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scott D. C., Monda J. K., Bennett E. J., Harper J. W., and Schulman B. a. (2011) N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science 334, 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Setty S. R. G., Strochlic T. I., Tong A. H. Y., Boone C., and Burd C. G. (2004) Golgi targeting of ARF-like GTPase Arl3p requires its Nalpha-acetylation and the integral membrane protein Sys1p. Nat. Cell Biol. 6, 414–419 [DOI] [PubMed] [Google Scholar]
- 7. Behnia R., Panic B., Whyte J. R. C., and Munro S. (2004) Targeting of the Arf-like GTPase Arl3p to the Golgi requires N-terminal acetylation and the membrane protein Sys1p. Nat. Cell Biol. 6, 405–413 [DOI] [PubMed] [Google Scholar]
- 8. Behnia R., Barr F. a., Flanagan J. J., Barlowe C., and Munro S. (2007) The yeast orthologue of GRASP65 forms a complex with a coiled-coil protein that contributes to ER to Golgi traffic. J. Cell Biol. 176, 255–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murthi A., and Hopper A. K. (2005) Genome-wide screen for inner nuclear membrane protein targeting in Saccharomyces cerevisiae: roles for N-acetylation and an integral membrane protein. Genetics 170, 1553–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Forte G. M. a., Pool M. R., and and Stirling C. J. (2011) N-Terminal Acetylation Inhibits Protein Targeting to the Endoplasmic Reticulum. PLos Biol. 9, e1001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Holmes W. M., Mannakee B. K., Gutenkunst R. N., and Serio T. R. (2014) Loss of amino-terminal acetylation suppresses a prion phenotype by modulating global protein folding. Nat. Commun. 5, 4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hwang C. S., Shemorry A., and Varshavsky A. (2010) N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327, 973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shemorry A., Hwang C.-S., and Varshavsky A. (2013) Control of Protein Quality and Stoichiometries by N-Terminal Acetylation and the N-End Rule Pathway. Mol. Cell 50, 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Starheim K. K., Gevaert K., and Arnesen T. (2012) Protein N-terminal acetyltransferases: when the start matters. Trends Biochem. Sci. 37, 152–161 [DOI] [PubMed] [Google Scholar]
- 15. Helbig A. O., Rosati S., Pijnappel P. W. W. M., Breukelen B. V., Timmers M. H. T. H., Mohammed S., Slijper M., and Heck A. J. R. (2010) Perturbation of the yeast N-acetyltransferase NatB induces elevation of protein phosphorylation levels Perturbation of the yeast N-acetyltransferase NatB induces elevation of protein phosphorylation levels. BMC Genomics 11, 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Helsens K., Van Damme P., Degroeve S., Martens L., Arnesen T., Vandekerckhove Jl., and Gevaert K. (2011) Bioinformatics Analysis of a Saccharomyces cerevisiae N-Terminal Proteome Provides Evidence of Alternative Translation Initiation and Post-Translational N-Terminal Acetylation. J. Proteome Res. 10, 3578–3589 [DOI] [PubMed] [Google Scholar]
- 17. Lange P. F., Huesgen P. F., Nguyen K., and Overall C. M. (2014) Annotating N termini for the human proteome project: N termini and Nalpha-acetylation status differentiate stable cleaved protein species from degradation remnants in the human erythrocyte proteome. J. Proteome Res. 13, 2028–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tercero J. C., and Wickner R. B. (1992) MAK3 encodes an N-acetyltransferase whose modification of the L-A gag NH2 terminus is necessary for virus particle assembly. J. Biol. Chem. 267, 20277–20281 [PubMed] [Google Scholar]
- 19. Polevoda B., and Sherman F. (2001) NatC Nalpha-terminal acetyltransferase of yeast contains three subunits, Mak3p, Mak10p, and Mak31p. J. Biol. Chem. 276, 20154–20159 [DOI] [PubMed] [Google Scholar]
- 20. Starheim K. K., Gromyko D., Evjenth R., Ryningen A., Varhaug J. E., Lillehaug J. R., and Arnesen T. (2009) Knockdown of human N alpha-terminal acetyltransferase complex C leads to p53-dependent apoptosis and aberrant human Arl8b localization. Mol. Cell. Biol. 29, 3569–3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Warnhoff K., Murphy J. T., Kumar S., Schneider D. L., Peterson M., Hsu S., Guthrie J., Robertson J. D., and Kornfeld K. (2014) The DAF-16 FOXO Transcription Factor Regulates natc-1 to Modulate Stress Resistance in Caenorhabditis elegans, Linking Insulin/IGF-1 Signaling to Protein N-Terminal Acetylation. PLoS Genetics 10, e1004703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Polevoda B., Norbeck J., Takakura H., Blomberg A., and Sherman F. (1999) Identification and specificities of N-terminal acetyltransferases from Saccharomyces cerevisiae. EMBO J. 18, 6155–6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tercero J. C., Dinman J. D., and Wickner R. B. (1993) Yeast MAK3 N-acetyltransferase recognizes the N-terminal four amino acids of the major coat protein (gag) of the L-A double-stranded RNA virus. J. Bacteriol. 175, 3192–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kimura Y., Takaoka M., Tanaka S., Sassa H., Tanaka K., Polevoda B., Sherman F., and Hirano H. (2000) N(alpha)-acetylation and proteolytic activity of the yeast 20 S proteasome. J. Biol. Chem. 275, 4635–4639 [DOI] [PubMed] [Google Scholar]
- 25. Van Damme P., Lasa M., Polevoda B., Gazquez C., Elosegui-Artola A., Kim D. S., De Juan-Pardo E., Demeyer K., Hole K., Larrea E., Timmerman E., Prieto J., Arnesen T., Sherman F., Gevaert K., and Aldabe R. (2012) N-terminal acetylome analyses and functional insights of the N-terminal acetyltransferase NatB. Proc. Natl. Acad. Sci. USA 109, 12449–12454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hofmann I., and Munro S. (2006) An N-terminally acetylated Arf-like GTPase is localised to lysosomes and affects their motility. J. Cell Sci. 119, 1494. [DOI] [PubMed] [Google Scholar]
- 27. Archer S. L. (2013) Mitochondrial Dynamics - mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 369, 2236–2251 [DOI] [PubMed] [Google Scholar]
- 28. Nunnari J., and Suomalainen A. (2012) Mitochondria: In sickness and in health. Cell 148, 1145–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Helbig A. O., Gauci S., Raijmakers R., van Breukelen B., Slijper M., Mohammed S., and Heck A. J. R. (2010) Profiling of N-acetylated protein termini provides in-depth insights into the N-terminal nature of the proteome. Mol. Cell. Proteomics 9, 928–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gromyko D., Arnesen T., Ryningen A., Varhaug J. E., and Lillehaug J. R. (2010) Depletion of the human Nα-terminal acetyltransferase A induces p53-dependent apoptosis and p53-independent growth inhibition. Int. J. Cancer 127, 2777–2789 [DOI] [PubMed] [Google Scholar]
- 31. Staes A., Impens F., Van Damme P., Ruttens B., Goethals M., Demol H., Timmerman E., Vandekerckhove J., and Gevaert K. (2011) Selecting protein N-terminal peptides by combined fractional diagonal chromatography. Nat. Protocols 6, 1130–1141 [DOI] [PubMed] [Google Scholar]
- 32. Van Damme P., Van Damme J., Demol H., Staes A., Vandekerckhove J., and Gevaert K. (2009) A review of COFRADIC techniques targeting protein N-terminal acetylation. BMC Proc. 3, S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Van Damme P., Hole K., Gevaert K., and Arnesen T. (2015) N-terminal acetylome analysis reveals the specificity of Naa50 (Nat5) and suggests a kinetic competition between N-terminal acetyltransferases and methionine aminopeptidases. Proteomics 15, 2436–2446 [DOI] [PubMed] [Google Scholar]
- 34. Martens L., Vandekerckhove J., and Gevaert K. (2005) DBToolkit: processing protein databases for peptide-centric proteomics. Bioinformatics 21, 3584–3585 [DOI] [PubMed] [Google Scholar]
- 35. Helsens K., Colaert N., Barsnes H., Muth T., Flikka K., Staes A., Timmerman E., Wortelkamp S., Sickmann A., Vandekerckhove J. l., Gevaert K., and Martens L. (2010) ms-lims, a simple yet powerful open source laboratory information management system for MS-driven proteomics. Proteomics 10, 1261–1264 [DOI] [PubMed] [Google Scholar]
- 36. Wang R., Fabregat A., Rios D., Ovelleiro D., Foster J. M., Cote R. G., Griss J., Csordas A., Perez-Riverol Y., Reisinger F., Hermjakob H., Martens L., and Vizcaino J. A. (2012) PRIDE Inspector: a tool to visualize and validate MS proteomics data. Nat. Biotech. 30, 135–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Goetze S., Qeli E., Mosimann C., Staes A., Gerrits B., Roschitzki B., Mohanty S., Niederer E. M., Laczko E., Timmerman E., Lange V., Hafen E., Aebersold R., Vandekerckhove J., Basler K., Ahrens C. H., Gevaert K., and Brunner E. (2009) Identification and functional characterization of N-terminally acetylated proteins in Drosophila melanogaster. PLoS Biology 7, e1000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Damme P. V., and Gawron D. (2014) N-terminal proteomics and ribosome profiling provide a comprehensive view of the alternative translation initiation landscape in mice and men. Mol. Cell. Proteomics 13, 1245–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Abeliovich H., Zarei M., Rigbolt K. T. G., Youle R. J., and Dengjel J. (2013) Involvement of mitochondrial dynamics in the segregation of mitochondrial matrix proteins during stationary phase mitophagy. Nat. Commun. 4, 2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karbowski M., and Youle R. J. (2003) Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differentiation 10, 870–880 [DOI] [PubMed] [Google Scholar]
- 41. Boldogh I. R., and Pon L. A. (2006) Interactions of mitochondria with the actin cytoskeleton. Biochim. Biophys. Acta 1763, 450–462 [DOI] [PubMed] [Google Scholar]
- 42. Griffiths E. J. (2000) Mitochondria–potential role in cell life and death. Cardiovasc. Res. 46, 24–27 [DOI] [PubMed] [Google Scholar]
- 43. Poot M., Zhang Y. Z., Kramer J. A., Wells K. S., Jones L. J., Hanzel D. K., Lugade A. G., Singer V. L., and Haugland R. P. (1996) Analysis of mitochondrial morphology and function with novel fixable fluorescent stains. J. Histochem. Cytochem. 44, 1363–1372 [DOI] [PubMed] [Google Scholar]
- 44. Kowalik D., Haller F., Adamski J., and Moeller G. (2009) In search for function of two human orphan SDR enzymes: Hydroxysteroid dehydrogenase like 2 (HSDL2) and short-chain dehydrogenase/reductase-orphan (SDR-O). J. Steroid Biochem. Mol. Biol. 117, 117–124 [DOI] [PubMed] [Google Scholar]
- 45. Gronemeyer T., Wiese S., Ofman R., Bunse C., Pawlas M., Hayen H., Eisenacher M., Stephan C., Meyer H. E., Waterham H. R., Erdmann R., Wanders R. J., and Warscheid B. (2013) The Proteome of Human Liver Peroxisomes: Identification of Five New Peroxisomal Constituents by a Label-Free Quantitative Proteomics Survey. PLoS ONE 8, e57395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Van Damme P., Evjenth R., Foyn H., Demeyer K., De Bock P.-J., Lillehaug J. R., Vandekerckhove J., Arnesen T., and Gevaert K. (2011) Proteome-derived peptide libraries allow detailed analysis of the substrate specificities of N{alpha}-acetyltransferases and point to hNaa10p as the posttranslational actin N{alpha}-acetyltransferase. Mol. Cell. Proteomics 10, M110.004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Evjenth R., Hole K., Karlsen O. a., Ziegler M., Arnesen T., and Lillehaug J. R. (2009) Human Naa50p (Nat5/San) displays both protein N alpha- and N epsilon-acetyltransferase activity. J. Biol. Chem. 284, 31122–31129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aksnes H., Van Damme P., Goris M., Starheim K. K., Marie M., Støve S. I., Hoel C., Kalvik T. V., Hole K., Glomnes N., Furnes C., Ljostveit S., Ziegler M., Niere M., Gevaert K., and Arnesen T. (2015) An organellar nα-acetyltransferase, naa60, acetylates cytosolic N termini of transmembrane proteins and maintains Golgi integrity. Cell Reports 10, 1362–1374 [DOI] [PubMed] [Google Scholar]
- 49. Van Damme P., Hole K., Gevaert K., and Arnesen T. (2015) N-terminal acetylome analysis reveals the specificity of Naa50 (Nat5) and suggests a kinetic competition between N-terminal acetyltransferases and methionine aminopeptidases. Proteomics 15, 2436–2446 [DOI] [PubMed] [Google Scholar]
- 50. Polevoda B., Arnesen T., and Sherman F. (2009) A synopsis of eukaryotic Nalpha-terminal acetyltransferases: nomenclature, subunits and substrates. BMC Proceedings 3, S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yoshii S. R., Kishi C., Ishihara N., and Mizushima N. (2011) Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286, 19630–19640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ma X., Jin M., Cai Y., Xia H., Long K., Liu J., Yu Q., and Yuan J. (2011) Mitochondrial electron transport chain complex III is required for antimycin A to inhibit autophagy. Chem. Biol. 18, 1474–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rodríguez-Hernández Á., Cordero M. D., Salviati L., Artuch R., Pineda M., Briones P., Gómez Izquierdo L., Cotán D., Navas P., and and Sánchez-Alcázar J. A. (2009) Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy 5, 19–32 [DOI] [PubMed] [Google Scholar]
- 54. Van Humbeeck C., Cornelissen T., Hofkens H., Mandemakers W., Gevaert K., De Strooper B., and Vandenberghe W. (2011) Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 31, 10249–10261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Strappazzon F., Vietri-Rudan M., Campello S., Nazio F., Florenzano F., Fimia G. M., Piacentini M., Levine B., and Cecconi F. (2011) Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J. 30, 1195–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Youle R. J., and Narendra D. P. (2011) Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Twig G., Elorza A., Molina A. J. A., Mohamed H., Wikstrom J. D., Walzer G., Stiles L., Haigh S. E., Katz S., Las G., Alroy J., Wu M., Py B. F., Yuan J., Deeney J. T., Corkey B. E., and Shirihai O. S. (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wakabayashi T. (1999) Structural changes of mitochondria related to apoptosis: swelling and megamitochondria formation. Acta Biochim.Polonica 46, 223–237 [PubMed] [Google Scholar]
- 59. Xue L., Fletcher G. C., and Tolkovsky A. M. (2001) Mitochondria are selectively eliminated from eukaryotic cells after blockade of caspases during apoptosis. Curr. Biol. 11, 361–365 [DOI] [PubMed] [Google Scholar]
- 60. Aksnes H., Osberg C., and Arnesen T. (2013) N-Terminal Acetylation by NatC Is Not a General Determinant for Substrate Subcellular Localization in Saccharomyces cerevisiae. PLoS ONE 8, e61012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kim H.-K., Kim R.-R., Oh J.-H., Cho H., Varshavsky A., and Hwang C.-S. (2013) The N-Terminal Methionine of Cellular Proteins as a Degradation Signal. Cell 156, 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.