

Summary
Chronic hepatitis B, C, and D virus (HBV, HCV, and HDV) infections are the leading causes of liver disease and cancer worldwide. Recently, the solute carrier and sodium taurocholate co-transporter NTCP has been identified as a receptor for HBV and HDV. Here, we uncover NTCP as a host factor regulating HCV infection. Using gain- and loss-of-function studies, we show that NTCP mediates HCV infection of hepatocytes and is relevant for cell-to-cell transmission. NTCP regulates HCV infection by augmenting the bile-acid-mediated repression of interferon-stimulated genes (ISGs), including IFITM3. In conclusion, our results uncover NTCP as a mediator of innate antiviral immune responses in the liver, and they establish a role for NTCP in the infection process of multiple viruses via distinct mechanisms. Collectively, our findings suggest a role for solute carriers in the regulation of innate antiviral responses, and they have potential implications for virus-host interactions and antiviral therapies.
Keywords: NTCP, hepatitis C virus, solute carrier, innate immune responses, viral entry, host factor, signaling, antiviral therapy, hepatitis B virus, hepatitis D virus
Graphical Abstract
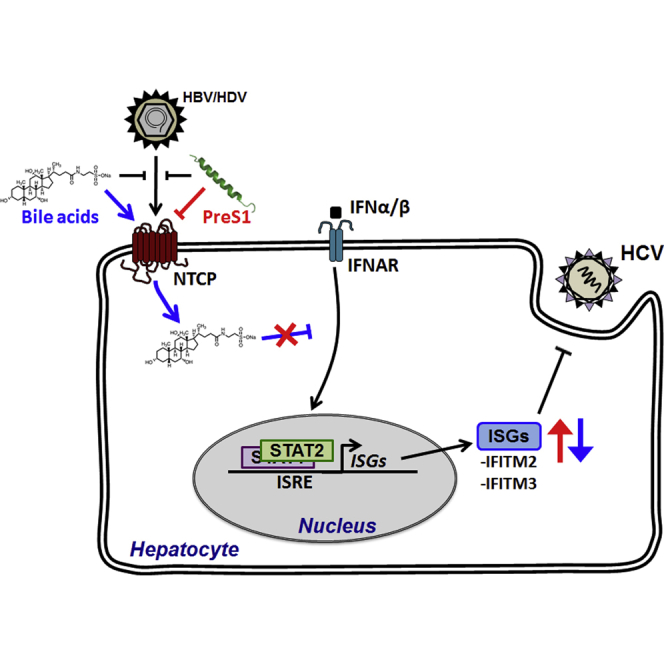
Highlights
-
•
NTCP is involved in hepatocyte infection by multiple viruses via distinct mechanisms
-
•
NTCP facilitates HCV infection by modulating innate antiviral responses
-
•
Solute carrier NTCP is a regulator of antiviral immune responses in the liver
-
•
This function is relevant for infection and therapies for hepatotropic viruses
Verrier et al. identify the sodium taurocholate co-transporter NTCP as a host factor regulating HCV infection. NTCP-mediated bile acid transport regulates innate responses targeting HCV infection. NTCP is a mediator of innate immunity and plays a role in the infection of multiple hepatotropic viruses.
Introduction
Hepatitis B and C viruses (HBV and HCV) are the leading causes of chronic liver disease worldwide (El-Serag, 2012). With ∼400 million individuals chronically infected with HBV or HCV and at risk for severe liver disease, these viruses are a major global health burden (El-Serag, 2012, Wedemeyer et al., 2015). Although HBV and HCV differ in their genomic organization and life cycles, these viruses exclusively infect human hepatocytes (Baumert et al., 2014), suggesting that liver-specific factors are important for the life cycle of both viruses.
Viral entry pathways contribute to HBV and HCV liver tropism. HCV requires a number of host proteins, including cluster of differentiation 81 (CD81), scavenger receptor BI (SR-BI), claudin-1 (CLDN1), occludin (OCLN), and accessory factors, such as epidermal growth factor receptor (EGFR), to enter hepatocytes (Baumert et al., 2014, Lupberger et al., 2011, Martin and Uprichard, 2013, Zeisel et al., 2013b, Zona et al., 2013). Some of these host factors interact directly with HCV glycoproteins, whereas others contribute to HCV internalization by promoting co-receptor associations and inducing intracellular signaling pathways (Kim et al., 2013, Lupberger et al., 2011). While key host factors mediating HCV entry are well characterized (Zeisel et al., 2013a), it is only partially understood how cell entry is regulated. Furthermore, the role of innate immune responses targeting HCV entry is poorly understood.
The sodium taurocholate co-transporting polypeptide (NTCP), a bile acid transporter expressed at the hepatocyte basolateral membrane (Claro da Silva et al., 2013), was identified previously as a receptor for HBV and hepatitis D virus (HDV) (Ni et al., 2014, Yan et al., 2012). HDV can use HBV envelope proteins to assemble infectious particles and is widely used as a surrogate model to study HBV entry (Sureau, 2010). Exogenous NTCP expression in NTCP-lacking human hepatoma cell lines (such as Huh7 and HepG2) renders these cells susceptible to HBV/HDV entry (Ni et al., 2014, Yan et al., 2012). The natural ligands of NTCP (i.e., conjugated and hydrophilic bile acids) compete with HBV/HDV for NTCP binding and inhibit viral infection (Yan et al., 2014). In contrast, bile acids have been shown to enhance HCV replication (Chang and George, 2007, Chhatwal et al., 2012), although the mechanisms are not yet defined. Furthermore, whether bile acid transporters such as NTCP play a role in infection of alternative hepatotropic viruses, such as HCV, is unclear. In this study, we tested the role of NTCP in HCV infection, and we identified the mechanisms by which a solute carrier affects infectivity of three major hepatotropic viruses.
Results
NTCP Overexpression Enhances HCV Infection
To investigate the effect of NTCP expression on HCV infection, we transduced the Huh7.5.1 cell line (Zhong et al., 2005) to express human NTCP. Huh7.5.1-NTCP cells expressed significantly higher NTCP mRNA and protein levels (Figure 1A) and surface levels of NTCP (Figure S1A) than parental Huh7.5.1 cells. To confirm that NTCP is functional as a viral host factor in these cells, Huh7.5.1-NTCP cells were infected with recombinant HDV. Huh7.5.1-NTCP cells supported HDV infection, as demonstrated by the presence of HDV RNA and delta antigen (HDAg) 7 days post-inoculation (Figure 1B). Moreover, HDV infection was inhibited by the HBV preS1-derived peptide (Figure 1B), which binds to NTCP and prevents HBV/HDV entry (Schieck et al., 2013).
Figure 1.

NTCP Expression Modulates HCV Infection
(A) NTCP expression in Huh7.5.1-NTCP cells compared to parental Huh7.5.1 cells evaluated by qRT-PCR and western blot. qRT-PCR results are expressed as means ± SD. NTCP expression was normalized by GAPDH expression (log) from three independent experiments performed in triplicate (n = 9).
(B) Functional evaluation of HDV infection in Huh7.5.1-NTCP cells using northern blot and immunofluorescence (IF). Huh7.5.1 and Huh7.5.1-NTCP cells were treated with an HBV-derived preS1 peptide (PreS1) or with a control peptide (Ctrl) (200 nM) and infected with recombinant HDV for 7 days. HDV RNA was detected by northern blot. HDV+ corresponds to ∼2 × 107 HDV RNA genome equivalents extracted from HDV particles produced in Huh7 cells. NI, non-infected cells. One experiment is shown. For IF analysis, cells were fixed 7 days after infection and stained with HDAg-specific antibodies purified from HBV/HDV co-infected patients. HDV-positive cells are visualized in red. Cell nuclei were stained with DAPI (blue). One experiment is shown.
(C and D) HCVcc and HCVpp infection of NTCP-overexpressing cells. Huh7.5.1 and Huh7.5.1-NTCP cells were infected with HCVcc (C, Luc-Jc1 and Luc-JcR2A; D, HCVpp (genotypes 1b, 2a, 3a, and 4), VSVpp, and MLVpp). Infection was assessed after 72 hr by measuring luciferase activity. Results are expressed as means ± SEM percentage virus infection relative to parental Huh7.5.1 cells from three independent experiments (one performed in triplicate, one in quintuplicate, and one in hextuplicate; n = 14) (C) or three independent experiments performed in triplicate (n = 9, except MLVpp: two independent experiments performed in triplicate, n = 6).
(E–G) NTCP overexpression increases HCV cell-to-cell transmission. HCV-electroporated Huh7.5.1 were transduced or not (Ctrl) with lentiviruses expressing NTCP. (E) NTCP expression was assessed by qRT-PCR. Results are expressed as means ± SD relative NTCP expression (log) compared to NTCP expression in non-transduced cells (set at 1) from one experiment performed in triplicate (n = 3). Cells were then co-cultured with GFP-expressing Huh7.5.1 cells in the presence of neutralizing antibody AP33. (F and G) After NS5A staining, HCV cell-to-cell transmission was assessed by flow cytometry as the percentage of GFP- and NS5A-positive cells. One experiment is shown (F) and results are expressed as means ± SD percentage infected cells from two independent experiments (one performed in hextuplicate and one in nonuplicate; n = 15) (G).
To assess if NTCP expression affects HCV infection, we used cell culture-derived HCV (HCVcc) and lentiviral particles pseudotyped with HCV E1E2 glycoproteins (HCVpp) of different genotypes. Expression of NTCP significantly enhanced HCVcc (Jc1 and JcR2A) infection and HCVpp (genotypes 1b, 2a, 3a, and 4) entry compared to the parental cells (normalized as 100%) (Figures 1C and 1D). Interestingly, entry of vesicular stomatitis virus pseudoparticles (VSVpp) and murine leukemia virus pseudoparticles (MLVpp) was not significantly affected by NTCP expression under these conditions (Figure 1D). The growth of Huh7.5.1 and Huh7.5.1-NTCP cells after seeding was similar, indicating that our observations were unrelated to effects on cell proliferation (Figure S1B). Furthermore, the effect of NTCP on HCV entry/infection was independent of the infectivity levels of different HCVpp and HCVcc strains (Figures S1C and S1D).
Since HCV can infect cells by direct cell-to-cell transmission (Meredith et al., 2013, Timpe et al., 2008), we evaluated the role of NTCP in this mode of viral dissemination. HCV-replicating Huh7.5.1 cells were co-cultured with GFP-expressing Huh7.5 cells (Lupberger et al., 2011, Xiao et al., 2014) transduced or not to express NTCP (Figure 1E). In this assay, the presence of an anti-envelope E2 antibody inhibits cell-free infection, allowing the specific assessment of cell-to-cell transmission. NTCP expression significantly increased the percentage of GFP-positive HCV-infected cells, suggesting that NTCP also plays a role in cell-to-cell spread (Figures 1F and 1G).
Silencing NTCP Expression Inhibits HCV Infection
We confirmed the role of NTCP in HCV infection by silencing NTCP expression in Huh7.5.1-NTCP cells using a small interfering RNA (siRNA) targeting NTCP, prior to infection with HCVcc. Following NTCP silencing in Huh7.5.1-NTCP cells, total protein expression was decreased by ∼80%, which was reflected by decreased surface expression (Figure 2A). This corresponded to a significant decrease in HCVcc infection (Figure 2B). To rule out potential off-target effects, we tested multiple NTCP-targeting small hairpin RNA (shRNA) sequences in Huh7.5.1-NTCP cells. We confirmed that a decrease in NTCP expression (by shNTCP2 and shNTCP3) (Figure 2C) correlated with a decrease in HCVcc infection (Figure 2D). Furthermore, silencing NTCP expression with shNTCP2 decreased protein expression by >80% (Figure 2E), which significantly decreased entry of HCVpp into Huh7.5.1-NTCP cells (Figure 2F), confirming the relevance of NTCP for HCV entry.
Figure 2.

Silencing NTCP Expression Inhibits HCV Infection in Human Hepatocytes
(A and B) NTCP silencing in hepatoma cell lines. Huh7.5.1-NTCP cells were transfected with siRNA control (siCtrl) or siRNA targeting NTCP (siNTCP). siRNA efficacy was assessed 72 hr after transfection by western blot and IF (scale bar, 10 μm) (A), and cell viability was assessed using PrestoBlue reagent (B). 3 days after NTCP silencing, Huh7.5.1-NTCP cells were infected with HCVcc (Luc-Jc1). Infection was assessed after 72 hr by measuring luciferase activity. Results are expressed as means ± SEM percentage cell viability compared to cells treated with siCtrl from three independent experiments (one performed in sextuplicate and two performed in octuplicate; n = 22) and means ± SEM percentage HCVcc infection from four independent experiments performed in quadruplicate (n = 16) (B).
(C–F) Effect of shNTCP silencing in hepatoma cells. Huh7.5.1-NTCP cells were transduced with lentiviruses expressing shRNAs targeting NTCP (shNTCP1, shNTCP2, or shNTCP3) or control shRNA (shCtrl). shRNA efficacy was assessed 72 hr after transduction by western blot (C) or by western blot and IF (scale bar, 10 μm) (E). Cell viability was assessed using PrestoBlue reagent (D). Results are expressed as means ± SEM percentage cell viability from three independent experiments performed in octuplicate (n = 24). 3 days after NTCP silencing, Huh7.5.1-NTCP cells were infected with HCVcc (Luc-Jc1) (D and F), HCVpp (genotype 1b) (F), or VSVpp (F). Infection was assessed after 72 hr by measuring luciferase activity. Results are expressed as means ± SEM percentage HCVcc infection from three independent experiments (two performed in quadruplicate and one in sextuplicate; n = 14) (D), or they are expressed as means ± SEM percentage pseudoparticle entry from three independent experiments performed in triplicate (n = 9) (F) and as means ± SEM percentage HCVcc infection from three independent experiments performed in quadruplicate (n = 12) (F).
NTCP Modulates HBV/HDV and HCV Infection via Distinct Mechanisms
To assess whether NTCP interacts directly with HCV glycoproteins, we measured binding of recombinant soluble HCV E2 (sE2) to cells. We observed no significant difference in sE2 binding to NTCP-expressing or parental cells (Figure 3A). However, sE2 treatment of Huh7.5.1 cells inhibited HCVcc infection (Figure 3B), confirming the functionality of the sE2 protein. These findings indicate that NTCP is not involved in E2 binding. Furthermore, NTCP expression did not affect the expression of canonical HCV entry factors in Huh7.5.1-NTCP cells compared to the parental cells (Figure 3C).
Figure 3.

Distinct Roles of NTCP in HDV/HBV and HCV Infections
(A) Flow cytometry analysis of HCV glycoprotein sE2 binding to Huh7.5.1 (Ctrl) and Huh7.5.1-NTCP (NTCP). Results are expressed as means ± SD percentage sE2 binding compared to control cells from three independent experiments (two performed in triplicate and one in quadruplicate; n = 10).
(B) sE2 binding inhibits HCVcc infection. Huh7.5.1 cells were treated with soluble sE2 and then infected with HCVcc (Luc-Jc1). Infection was assessed after 72 hr by measuring luciferase activity. Results are expressed as means ± SD percentage HCVcc infection from three independent experiments performed in triplicate (n = 9).
(C) Flow cytometry analysis of HCV entry factor expression in Huh7.5.1-NTCP cells. Results are expressed as percentage protein expression compared to parental Huh7.5.1 cells (set at 100%) from four independent experiments performed in duplicate (n = 8).
(D) Effect of HBV preS1-derived peptide on HDV, HCVpp (genotype 1b), and VSVpp entry into Huh7.5.1-NTCP cells. Huh7.5.1-NTCP cells were treated for 1 hr with preS1 or Ctrl peptide (200 nM). For HDV, cells were then infected with recombinant HDV for 7 days. Total RNA was purified and HDV RNA was detected by qRT-PCR. Results are expressed as percentage HDV RNA level compared to non-treated Huh7.5.1-NTCP cells from three independent experiments performed in triplicate (n = 9). For HCVpp (genotype 1b) and VSVpp, cells were infected with pseudoparticles and infection was assessed after 72 hr by luciferase activity. Results are expressed as means ± SD percentage viral entry from three independent experiments performed in quintuplicate (n = 15).
(E–G) Time-dependent inhibition of viral entry by preS1 treatment. Cells were treated with preS1 or Ctrl peptide for 24, 48, and 72 hr prior to infection with HCVpp (genotype 1b), VSVpp, or HDV. Infection was assessed as described above. (E) Results are expressed as means ± SD percentage HCVpp entry from five independent experiments performed in triplicate (n = 15). (F) Results are expressed as means ± SD percentage VSVpp entry from four independent experiments performed in triplicate (n = 12). (G) Results are expressed as means ± SD percentage HDV infection from three independent experiments performed in duplicate (n = 6).
(H) Treatment with preS1 inhibits HCVcc infection. Cells were treated with preS1 or Ctrl peptide for 72 hr prior to infection with HCVcc (Luc-Jc1). Infection was assessed after 72 hr by measuring luciferase activity. Results are expressed as means ± SD HCVcc infection (relative light unit, RLU) from three independent experiments performed in triplicate (n = 9).
We next tested the effect of the HBV preS1-derived peptide, which is known to bind to NTCP and inhibit HBV/HDV entry (Ni et al., 2014). Although preS1 inhibited HDV infection of Huh7.5.1-NTCP cells after 1 hr pre-treatment, it had no effect on HCVpp entry under these conditions (Figure 3D), indicating that the HBV preS1-binding domain of NTCP is not required to promote HCV entry. We next evaluated whether the bile acid transporter function of NTCP is important for HCV entry. Huh7.5.1-NTCP cells were treated with the HBV preS1-derived peptide (König et al., 2014, Slijepcevic et al., 2015), which inhibits bile acid uptake in vitro (IC50, 4 nM) (Ni et al., 2014), for 24, 48, and 72 hr prior to the addition of HDV, HCVpp, or VSVpp (Figures 3E–3G). PreS1 treatment (200 nM) significantly inhibited HCVpp entry in a time-dependent manner, with a maximal inhibition of ∼70% after 72 hr (Figure 3E). Under these conditions, VSVpp entry also was reduced by preS1, although to a lesser extent (Figure 3F), suggesting a general effect on viral infection. In contrast, HDV infection was impaired by preS1 regardless of the treatment duration (Figure 3G). HCVcc infection also was decreased by 72-hr preS1 treatment (Figure 3H), confirming that our observations are relevant for the infectious virus and the full HCV life cycle. Taken together, our data suggest that long-term preS1 inhibition of NTCP-mediated bile acid transport perturbs cellular physiology to modulate HCV entry.
PreS1-Mediated Inhibition of NTCP Induces Interferon-Stimulated Gene Expression
Since preS1 only affected HCV entry after long-term treatment (Figure 3), we hypothesized that the bile acid transport activity of NTCP induces metabolic or transcriptional changes that regulate virus infection. To test this hypothesis, we evaluated the effect of preS1-NTCP binding on gene expression in hepatoma cells. We performed genome-wide microarray analyses of Huh7.5.1-NTCP cells that were treated with preS1 or a scrambled peptide (200 nM) for 48 hr. Gene set enrichment analysis (GSEA) indicated that preS1 treatment induces the expression of genes involved in bile acid and fatty acid metabolism, as previously described (Oehler et al., 2014). Unexpectedly, preS1 treatment also induced the expression of genes involved in innate immunity, such as IFNα responses and the Jak/Stat-signaling pathway (Table 1; Figure 4A). Interestingly, IFITM2 and IFITM3, which encode two restriction factors targeting HCV entry (Narayana et al., 2015), were among the preS1-induced genes (Figure 4B).
Table 1.
Hallmark Gene Sets Significantly Induced, Positive NESa, or Repressed, Negative NES, after preS1 Treatment in Huh7.5.1-NTCP Cells Shown in Figure 4
| Gene Set | NESa | p value | FDRb |
|---|---|---|---|
| Bile acid metabolism | 2.32 | <0.001 | <0.001 |
| Coagulation | 2.25 | <0.001 | <0.001 |
| Xenobiotic metabolism | 2.20 | <0.001 | <0.001 |
| Fatty acid metabolism | 2.20 | <0.001 | <0.001 |
| Heme metabolism | 2.04 | <0.001 | <0.001 |
| Adipogenesis | 1.91 | <0.001 | <0.001 |
| Peroxisome | 1.90 | <0.001 | <0.001 |
| Interferon alpha response | 1.90 | <0.001 | <0.001 |
| Oxidative phosphorylation | 1.90 | <0.001 | <0.001 |
| Estrogen response late | 1.89 | <0.001 | <0.001 |
| Angiogenesis | 1.72 | 0.002 | 0.003 |
| Cholesterol homeostasis | 1.68 | 0.004 | 0.005 |
| Hypoxia | 1.61 | <0.001 | 0.010 |
| Interferon gamma response | 1.60 | <0.001 | 0.009 |
| Reactive oxygen species pathway | 1.59 | 0.013 | 0.010 |
| Estrogen response early | 1.58 | <0.001 | 0.011 |
| Myogenesis | 1.57 | 0.002 | 0.012 |
| IL2 STAT5 signaling | 1.54 | <0.001 | 0.013 |
| IL6 JAK STAT3 signaling | 1.54 | 0.008 | 0.014 |
| Complement | 1.49 | 0.003 | 0.021 |
| MTORC1 signaling | −1.50 | 0.005 | 0.012 |
| Unfolded protein response | −1.82 | <0.001 | <0.001 |
| G2M checkpoint | −2.01 | <0.001 | <0.001 |
| E2F targets | −2.02 | <0.001 | <0.001 |
| MYC targets V2 | −2.54 | <0.001 | <0.001 |
| MYC targets V1 | −2.64 | <0.001 | <0.001 |
Normalized enrichment score.
False discovery rate.
Figure 4.

Binding of HBV-Derived preS1 Peptide to NTCP Increases ISG Expression
Huh7.5.1-NTCP cells were treated with preS1 or control peptide for 48 hr. Cells were then lysed and total RNA was extracted and purified. Total gene expression was analyzed by genome-wide microarray. Three independent biological replicates per condition from one experiment were analyzed.
(A) Schematic representation of Hallmark gene sets that are significantly induced (red, including bile acid metabolism and IFNα response) or repressed (blue) following preS1 treatment, based on Table 1. p < 0.05 and FDR < 0.05 were considered statistically significant.
(B) List of individual IFNα response genes that are significantly (p < 0.05) overexpressed following preS1 treatment. Individual Z scores for each sample are presented. Negative Z score (blue) and positive Z score (red) correspond to repression and induction of the indicated genes, respectively.
(C and D) IFITM2 and IFITM3 overexpression inhibits HCV infection. Huh7.5.1-NTCP cells were transfected with an empty vector, pCMV-HA-hIFITM2, or pCMV-HA-hIFITM3 for 3 days. (C) Expression of transduced proteins as assessed by anti-HA western blot is shown. (D) Transduced cells were then infected for 3 days with HCVcc (Luc-Jc1). Infection was assessed after 72 hr by measuring luciferase activity. Results are expressed as means ± SD percentage HCVcc infection compared to control cells (set at 100%) from three independent experiments performed in triplicate (n = 9).
(E) IFITM3 protein expression in hepatoma cells. IFITM3 protein expression was assessed by western blot in Huh7.5.1 and Huh7.5.1-NTCP cells. One experiment is shown.
(F) IFITM3, PARP9, and CXCL10 expression in hepatoma cells. Basal expression of IFITM3, PARP9, and CXCL10 mRNA was quantified by qRT-PCR in Huh7.5.1 and Huh7.5.1-NTCP cells. Results are expressed as means ± SD percentage gene expression compared to expression levels in Huh7.5.1 cells (set at 100%) from three independent experiments performed in triplicate (n = 9).
To confirm that IFITMs function as restriction factors in Huh7.5.1-NTCP cells, we exogenously overexpressed IFITM2 and IFITM3 (Figure 4C). Indeed, IFITM2 and IFITM3 overexpression in Huh7.5.1-NTCP cells inhibited HCVcc infection (Figure 4D). However, as endogenous IFITM2 expression in Huh7.5.1 and Huh7.5.1-NTCP cells was low and at the limit of detection, we focused on IFITM3 in further functional studies. Notably, IFITM3 expression was decreased at the protein level by ∼60% in Huh7.5.1-NTCP cells compared to parental cells (Figure 4E), suggesting that NTCP modulates IFITM3 expression. To confirm that the changes in gene expression were directly related to NTCP and not to off-target effects of preS1, we selected IFITM3 and two other genes involved in IFNα responses (PARP9 and CXCL10) which were induced by preS1 treatment (Figure 4B), and we compared their expression levels in Huh7.5.1 and Huh7.5.1-NTCP cells. As expected, Huh7.5.1-NTCP cells had decreased mRNA expression of IFITM3, PARP9, and CXCL10 compared to parental cells (Figure 4F), confirming the specific role of NTCP in the suppression of these genes. These data support previous findings that preS1 binds with high specificity to NTCP without off-target effects (Bogomolov et al., 2016).
Bile Acid Transport through NTCP Modulates the Expression of Interferon-Stimulated Genes to Affect HCV Infection
The gene expression analyses implied that NTCP facilitates HCV entry by altering the expression of interferon-stimulated genes (ISGs). Given that the physiological function of NTCP is to transport bile acids, and bile acids are known to affect ISG expression in hepatocytes (Graf et al., 2010, Podevin et al., 1999), we hypothesized that bile acid transport by NTCP regulates the expression of ISGs and viral infection. Since IFITM3 functions as an HCV restriction factor in our model system (Figure 4D), we selected IFITM3 as a representative ISG for functional studies to probe the link between NTCP and the IFN response. To ensure that IFN responses are indeed functional in Huh7.5.1-NTCP cells, we treated cells with Poly(I:C) and IFNα2, and we evaluated ISG induction by the expression of IFITM3. Poly(I:C) stimulation of Huh7.5.1-NTCP cells significantly increased IFITM3 expression (Figure S2A). Moreover, STAT1 phosphorylation (Figure S2B) and IFITM3 mRNA expression (Figure S2C) were markedly induced after IFNα2 treatment. This induction was repressed by a specific antibody targeting the type I IFN receptor (IFNAR) (Figures S2B and S2C).
We then evaluated the effect of NTCP on IFN responses in these cells. The mere presence of NTCP decreased mRNA expression of IFITM3 by ∼50% compared to parental cells (Figure 5A). The addition of bile acid (100 μM sodium taurocholate) to Huh7.5.1-NTCP cells decreased IFITM3 mRNA expression even further, whereas blocking bile acid transport with the preS1 peptide restored IFITM3 mRNA expression to the levels observed in Huh7.5.1 cells (Figure 5A). These findings suggest that the effect of NTCP on ISG expression and HCV infection is dependent on bile acid. Indeed, the addition of supplementary bile acid in the cell culture medium dose-dependently increased HCVcc infection in Huh7.5.1-NTCP cells, but not in parental cells (Figure 5B).
Figure 5.

Bile Acid Uptake Enhances HCV Infection by Decreasing ISG Expression
(A) The impact of bile acid and preS1 treatment on IFITM3 expression. Huh7.5.1-NTCP cells were treated with the bile acid (BA) sodium taurocholate (100 μM) or with 200 nM preS1 for 72 hr. IFITM3 expression was then quantified by qRT-PCR. Results are expressed as means ± SD percentage IFITM3 expression compared to untreated (Ctrl) Huh7.5.1-NTCP cells (set at 100%) from three independent experiments performed in duplicate (n = 6).
(B) The impact of bile acid on HCVcc infection. Huh7.5.1 and Huh7.5.1-NTCP cells were treated 0, 25, or 100 μM sodium taurocholate for 72 hr and then infected with HCVcc (Luc-Jc1). Results are expressed as means ± SD percentage HCVcc infection compared to untreated (Ctrl) Huh7.5.1 and Huh7.5.1-NTCP cells (both set at 100%) from three independent experiments performed in triplicate (n = 9).
(C and D) Effect of preS1-mediated inhibition of bile acid uptake on IFITM3 protein expression and HCVcc infection. Huh7.5.1 and Huh7.5.1-NTCP cells were treated with sodium taurocholate (100 μM) in the presence or absence of preS1 peptide for 72 hr. (C) IFITM3 protein expression was assessed by western blot. One experiment is shown. (D) Cells were then infected by HCVcc (Jc1) for 3 days. Results are expressed as means ± SD percentage HCVcc infection compared to Huh7.5.1 and Huh7.5.1-NTCP cells treated with control peptide (both set at 100%) from three independent experiments performed in triplicate (n = 9).
(E and F) Silencing of IFITM3 expression increases HCV infection in a bile acid-dependent manner. Huh7.5.1-NTCP cells were transfected with siRNA control (siCtrl) or siRNA targeting IFITM3 (siIFITM3) and then treated for 72 hr in the absence or presence of BA (100 μM) in the presence of either preS1 or Ctrl peptide (200 nM). (E) Then 3 days after transfection, silencing efficacy was assessed by measuring expression of IFITM3 mRNA by qRT-PCR. Results are expressed as means ± SD percentage IFITM expression from four independent experiments performed in triplicate (n = 12). (F) Cells were then infected with HCVcc Luc-Jc1. Infection was assessed after 72 hr by measuring luciferase activity. Results are expressed as means ± SD percentage HCVcc infection compared to cells treated with siCtrl (set at 100% for each condition) from four independent experiments performed in triplicate (n = 12).
We next evaluated the effect of preS1 treatment on IFITM3 protein expression and HCV infection in the presence of bile acids. Reflecting our observations of the mRNA level (Figure 5A), IFITM3 protein expression was decreased in Huh7.5.1-NTCP cells compared to Huh7.5.1 cells in the presence of bile acid (Figure 5C). However, treatment of Huh7.5.1-NTCP cells with preS1 (to block bile acid uptake) under these conditions induced a 2-fold increase in IFITM3 protein expression, effectively restoring it to the level observed in Huh7.5.1 cells (Figure 5C). Furthermore, treatment of Huh7.5.1-NTCP cells with the preS1 peptide in the presence of bile acid inhibited HCVcc infection (Figure 5D). We did not observe these effects in Huh7.5.1 cells (Figures 5C and 5D), confirming that the bile acid-mediated effect is dependent on NTCP.
Interestingly, when we silenced IFITM3 expression (Figure 5E) in the presence of bile acid, we observed an increase in HCV infection (Figure 5F). However, this effect was no longer observed in the presence of preS1 to block bile acid uptake into the cells or in the absence of bile acid (Figure 5F). These results suggest that bile acid-mediated suppression of other ISGs (which may otherwise compensate the absence of an individual ISG) is necessary to observe a functional effect of IFITM3 silencing on HCV infection. Differences in bile acid levels in cell culture medium also may explain why IFITM3 was not identified as an HCV restriction factor in previous screens (Brass et al., 2009).
NTCP-Mediated Bile Acid Transport Affects ISG Expression and HCV Entry into Primary Human Hepatocytes
For validation in a more physiological context, we investigated the role of NTCP during HCV entry into primary human hepatocytes (PHHs). First, we silenced NTCP expression in PHHs using an NTCP-targeting siRNA. Following transfection of siNTCP, the expression of NTCP protein was reduced by ∼50% (Figure 6A). The decrease in NTCP expression correlated with a significant decrease in the entry of HCVpp genotype 1b (Figure 6B).
Figure 6.

NTCP-Mediated Bile Acid Uptake Modulates ISG Expression and HCV Entry in PHHs
(A and B) Silencing NTCP expression in PHHs. PHHs were transfected with siRNA control (siCtrl) or siRNA targeting NTCP (siNTCP). 3 days after transfection, siRNA efficacy was assessed by western blot (A), and cell viability was assessed using PrestoBlue reagent (B). Results are expressed as means ± SEM percentage cell viability compared to cells treated with siCtrl from two independent experiments performed in quintuplicate (n = 10) (B). 3 days after NTCP silencing, PHHs were incubated with HCVpp (genotype 1b). Infection was assessed after 72 hr by measuring luciferase activity. Results are expressed as means ± SEM percentage HCVpp entry from three independent experiments performed in triplicate (n = 9) (B).
(C and D) Bile acids modulate the expression of ISGs in PHHs. PHHs from a single donor were treated with the bile acid (BA) sodium taurocholate (500 μM) in the presence or absence of the preS1 peptide (400 nM) for 48 hr. Cells were then lysed and total RNA was extracted and purified. Total gene expression was analyzed by genome-wide microarray. Three independent biological replicates per condition were analyzed. (C) Individual IFNα response genes that are significantly (p < 0.05) repressed following BA treatment are shown. Individual Z scores for each sample are presented. Negative Z score (blue) and positive Z score (red) correspond to repression and induction of the indicated genes, respectively. (D) Effect of preS1 treatment on the expression of IFNα response genes presented in (C). Individual Z scores for each sample are presented.
(E) Effect of bile acid and preS1 treatment on HCVpp infection in PHHs. PHHs were treated with increasing concentrations (0, 100, and 500 μM) of BA in the presence of either preS1 or Ctrl peptide (400 nM) for 72 hr and then infected with HCVpp (genotype 1b). Infection was assessed after 72 hr by measuring luciferase activity. Results are expressed as means ± SEM percentage HCVpp entry compared to untreated PHHs in the presence of the control peptide (set at 100%) from four independent experiments performed in triplicate (n = 12).
(F) PreS1 inhibition of HCVpp entry is dependent on the IFN-signaling pathway in PHHs. PHHs were treated with 500 μM BA in the presence of preS1 or a scrambled control peptide (400 nM), with or without treatment with an antibody targeting the type I IFN receptor (IFNAR) for 72 hr. Cells were then infected with HCVpp (genotype 1b) and infection was assessed after 72 hr by measuring luciferase activity. Results are expressed as means ± SEM percentage HCVpp entry compared to PHHs treated with the control peptide and an IgG control (set at 100%) from three independent experiments (one performed in triplicate and two in quintuplicate; n = 13).
We next evaluated whether NTCP-mediated bile acid transport affects ISG expression in PHHs. We treated PHHs with bile acid in the presence or absence of preS1 and performed genome-wide microarray analyses. GSEA showed that bile acid treatment of PHHs suppressed the expression of genes involved in the IFNα response (normalized enrichment score [NES] −2.11; p value < 0.001; false discovery rate [FDR] < 0.001) (Figure 6C) as well as other immune-related pathways. However, treatment of PHHs with preS1 under these conditions induced the expression of genes involved in the IFNα response (NES 1.5; p value = 0.014; FDR = 0.035) to restore their expression levels to normal conditions (i.e., PHHs in the absence of bile acid) (Figure 6D). These findings are consistent with our microarray analyses in Huh7.5.1-NTCP cells (Figure 4), where expression of genes involved in IFNα responses was regulated by treatment with preS1. Interestingly, IFITM1, IFITM2, and IFITM3 were among the genes suppressed by bile acid treatment of PHHs (Figure 6C), and their expression could be rescued by the addition of preS1 (Figure 6D). Moreover, the effects we observed in PHHs were dependent on the presence of bile acid, as treatment with preS1 in the absence of bile acid did not have a major impact on the expression of these genes (data not shown).
We next evaluated the functional effect of these changes in ISG expression on HCVpp entry into PHHs. Treatment of PHHs with bile acid increased HCVpp genotype 1b entry, and the addition of preS1 under these conditions restored the level of infection to that observed in the absence of bile acid (Figure 6E). Interestingly, the concentration of bile acid required to see a robust effect for HCVpp entry into PHHs was higher than for HCVcc infection of Huh7.5.1-NTCP cells (Figure 5B), which may reflect different bile acid uptake efficiency in PHHs or that the effect is more potent when the full HCV life cycle is measured and ISGs targeting different steps of the viral infection cycle are involved. Finally, to confirm that the activity of preS1 is linked to IFN responses, we tested the effect of preS1 in the presence of an antibody blocking the type I IFN receptor. In the presence of this antibody, preS1 no longer inhibited HCVpp entry into PHHs (Figure 6F), suggesting that the inhibitory effect of preS1 against HCV entry is indeed mediated by the IFN signal transduction cascade and resulting IFN responses in PHHs.
Discussion
NTCP was recently described as a major receptor for HBV and HDV entry. Here we show that NTCP also plays a role in HCV infection. Exogenous expression of NTCP in Huh7.5.1 hepatoma cells increased HCV infection, whereas silencing of NTCP expression reduced HCV entry (Figures 1 and 2). Using microarray analyses in cell lines (Figure 4) and PHHs (Figure 6), we discovered that NTCP-mediated bile acid transport regulates innate antiviral responses, thereby inhibiting HCV infection and uncovering a role for NTCP as a regulator of antiviral immunity.
Innate antiviral responses mediated by ISGs have been shown to target multiple steps during viral infection, including entry (Liu et al., 2011, Smith et al., 2014). IFITM1, IFITM2, and IFITM3, which belong to a group of five IFN-induced transmembrane proteins (Smith et al., 2014), have been shown to exert broad antiviral activity against a range of viruses, including VSV, HIV, dengue virus, influenza virus, and Zika virus (Savidis et al., 2016, Smith et al., 2014). Interestingly, IFITM2 and IFITM3 were recently reported to restrict HCV infection at a late entry step by targeting HCV for lysosomal degradation following endocytosis (Narayana et al., 2015). IFITM1 was shown to interact with HCV co-receptors at tight junctions to disrupt the HCV entry process by alternative mechanisms (Wilkins et al., 2013).
Given that bile acids have been shown to modulate cellular antiviral responses (Graf et al., 2010, Podevin et al., 1999), we hypothesized that NTCP affects the induction of ISG expression via its bile acid transport activity. Indeed, the expression of ISGs (including IFITM1, IFITM2, and IFITM3) in PHHs was suppressed by the addition of bile acid (Figure 6C). However, expression of these ISGs in PHHs was restored by addition of the preS1 peptide, which blocks NTCP-mediated bile acid uptake (Figure 6D). Furthermore, modulation of the expression of IFITM3 by bile acid or preS1 treatment had a clear functional effect on HCV infection (Figures 5 and 6). Our findings are consistent with a previous report showing that bile acids affect HCV replication (Chhatwal et al., 2012), probably by similar mechanisms as those we describe here. In this study, we selected IFITM3 as a representative ISG for functional characterization, but the expression of other ISGs is also affected by bile acids (Figure 6), likely contributing further to the overall effect on HCV infection. Since ISGs broadly restrict viral infection, this is consistent with the time-dependent effect of preS1 on VSVpp entry that we observed (Figure 3).
Our data suggest that NTCP facilitates HCV infection by modulating bile acid transport and ISG expression. ISGs also restrict HCV cell-to-cell transmission (Meredith et al., 2014), consistent with our finding that NTCP contributes to HCV cell-to-cell spread (Figures 1E–1G). It should be noted that NTCP expression did not appear to affect HCV entry in a recent Huh7 cell line model (Meredith et al., 2016). Huh7.5.1 cells are derived from Huh7.5 cells, which differ from Huh7 cells by a single point mutation in the retinoic acid-inducible gene-I (Bartenschlager and Pietschmann, 2005, Zhong et al., 2005). This may explain the differences observed between the two cell lines. Moreover, differences in bile acid concentrations in cell culture medium may be responsible for differences in the effect of NTCP on cell entry of HCV (Figure 5C). However, our loss-of-function studies and microarray analyses in PHHs (Figure 6) clearly demonstrate the impact of NTCP on HCV infection in primary cells with physiological innate immune responses.
For HBV/HDV infection, viral entry requires direct interaction with NTCP (Ni et al., 2014, Yan et al., 2012). Furthermore, HBV binding to NTCP may interfere with the physiological function of NTCP (i.e., bile acid uptake), and NTCP ligands can abrogate HBV/HDV infection (Oehler et al., 2014, Yan et al., 2014). In contrast, NTCP modulates HCV entry independently of direct binding mechanisms. We confirmed the inhibitory effect of the HBV preS1-derived peptide on HDV infection following 1-hr treatment (Figure 3D), but we did not see a corresponding effect on HCV infection under these conditions. Furthermore, the effect of NTCP on HCV infection does not appear to involve HCV E2 or binding factors CD81 and SR-BI (Pileri et al., 1998, Scarselli et al., 2002) (Figures 3A and 3B). NTCP expression did not modulate the expression of canonical entry factors CLDN1 and OCLN or EGFR either (Figure 3C).
Other mechanisms may contribute to the effects of NTCP on viral entry and infection that we observed. Indeed, modulation of NTCP bile acid transport activity by preS1 affected IFN responses, but also bile acid metabolism and cholesterol homeostasis in Huh7.5.1-NTCP cells as well as other pathways (Table 1). In particular, modulation of bile acid metabolism and cholesterol homeostasis by preS1 would alter cellular cholesterol pools, which has been shown to affect the activation of type I IFN responses (York et al., 2015) and may potentially contribute to the effect on HCV infection that we observed. Furthermore, modulation of signaling and transcription factor targets (Table 1) may have additional effects on viral replication and translation.
Our results demonstrate that NTCP, acting by distinct mechanisms, is relevant for the three major viruses causing chronic hepatitis and liver disease. This finding could contribute to the development of antiviral strategies targeting NTCP. Targeting viral cell entry with receptor antagonists, antibodies, peptides, and receptor kinase inhibitors has provided perspectives to prevent and treat chronic hepatitis B and C infections (Colpitts et al., 2015). Myrcludex B, a preS1-derived peptide targeting NTCP (Ni et al., 2014, Volz et al., 2013), has been shown to protect against HBV infection (Petersen et al., 2008), to modulate viral spread in animal models (Volz et al., 2013), and to decrease HDV viral load in patients in a phase II clinical trial (Bogomolov et al., 2016). Here we show that preS1-NTCP binding enhances the expression of ISGs. These data suggest that myrcludex B may inhibit HBV infection by a dual mode of action, by interfering with viral binding and potentially increasing ISG expression. NTCP-targeting agents in clinical development also may inhibit HCV infection, which is of interest particularly for patients with HBV/HDV/HCV co-infections.
NTCP is a member of the solute carrier (SLC) family of proteins, a group of membrane proteins having crucial roles in many physiological functions (César-Razquin et al., 2015). However, these proteins are relatively uncharacterized. Here we uncover NTCP as a mediator of innate antiviral responses in hepatocytes and establish a role for NTCP in the entry process of multiple viruses. Our data uncover an important role of SLCs in virus-host interactions by linking their function to the regulation of innate immune responses. Moreover, bile acid transport through SLCs profoundly affects liver gene expression (Table 1).
Overall, we have identified NTCP as a regulator of innate immune responses in the human liver. These findings improve the understanding of virus-host interactions in the human liver, and they may open perspectives for the development of broad antiviral therapies targeting hepatotropic viruses that cause chronic liver disease and cancer.
Experimental Procedures
Cell Lines
The sources for 293T and Huh7.5.1 cells have been described (Lupberger et al., 2011). Huh7.5.1 cells were seeded in six-well plates at 50% confluency 1 day prior to transduction with human NTCP-expressing VSVpp (GeneCopoeia). Cells were incubated in DMEM (Gibco) containing 10% fetal bovine serum (Dutscher). After 3 days, the cells were expanded and selected for NTCP expression with 1.8 μg/mL puromycin.
PHHs
Liver tissue from patients undergoing surgical resection was obtained with informed consent from all patients. The respective protocols were approved by the Ethics Committee of the University of Strasbourg Hospitals (CPP 10-17). PHHs were isolated from liver resection tissue and cultured in William’s E medium (Sigma) (Krieger et al., 2010, Lupberger et al., 2011).
Viral Infection
Lentiviral pseudoparticles expressing HCV envelope glycoproteins (HCVpp strains HCV-J [1b], JFH1 [2a], NIH S52 [3a], and UKN4.21.16 [4]), VSVpp, MLVpp, as well as cell culture-derived HCVcc (Luc-Jc1 and Luc-JcR2A) were generated as described (Fofana et al., 2010, Lupberger et al., 2011). Huh7.5.1 cells and PHHs were infected as described (Krieger et al., 2010, Lupberger et al., 2011). Pseudoparticle entry and HCVcc infection were assessed by measuring luciferase activity 72 hr post-infection (Krieger et al., 2010, Lupberger et al., 2011). HDV production and infection have been described (Verrier et al., 2016) (see the Supplemental Experimental Procedures).
Gene Expression Analyses in Huh7.5.1-NTCP Cells and PHHs Treated with Bile Acids and preS1 Peptide
Huh7.5.1-NTCP cells were treated with preS1 or a control peptide (200 nM) for 48 hr. Alternatively, PHHs were treated with bile acids (500 μM) in presence of preS1 or a control peptide (400 nM) for 48 hr. Total RNA from three biological replicates per condition from one experiment was then extracted using the ReliaPrep Kit (Promega), and 200 ng was subjected to genome-wide transcriptome profiling using HumanHT-12 beadarray (Illumina), following the manufacturer’s protocol. Raw scanned data were normalized by cubic spline algorithm using Illumina normalizer module of GenePattern genomic analysis toolkit (http://software.broadinstitute.org/cancer/software/genepattern), as previously described (Hoshida et al., 2008). Modulated molecular pathways were determined using GSEA (Subramanian et al., 2005).
Statistical Analysis
Each experiment was performed at least two times in an independent manner. The number of independent experiments as well as the total number of biological replicates (n) are indicated in the figure legends. Statistical analyses were performed using Mann-Whitney U test by comparing values from every biological replicate per study (indicated by “n” in the figure legends); p < 0.05 (∗), p < 0.01 (∗∗), and p < 0.001 (∗∗∗) were considered statistically significant. Significant p values are indicated by asterisks in the individual figures. For microarray analyses, two-tailed unpaired Student’s t test was performed by comparing the values from three biological replicates (p < 0.05 was considered statistically significant).
Author Contributions
M.B.Z. and T.F.B. designed and supervised research. T.F.B. initiated the study. E.R.V., C.C.C., C.B., L.H., L.Z., F.X., C.T., E.C., R.G., and C.S. performed experiments. E.R.V., C.C.C., C.B., L.H., L.Z., F.X., C.T., E.C., R.G., C.S., Y.H., M.B.Z., and T.F.B. analyzed data. P.P. and F.-L.C. contributed key reagents. E.R.V., C.C.C., J.A.M., M.B.Z., and T.F.B. wrote the paper. E.R.V. and C.C.C. contributed equally to this work as first authors. M.B.Z. and T.F.B. contributed equally to this work as senior authors.
Acknowledgments
We thank R. Bartenschlager (University of Heidelberg), C. Rice (Rockefeller University), T. Wakita (University of Tokyo), and J. Ball (University of Nottingham) for plasmids for HCVcc and HCVpp production; J. Taylor (Fox Chase Cancer Center) for HDV plasmid; F.V. Chisari (The Scripps Research Institute) for Huh7.5.1 cells; A. Patel (MRC Virology Unit) for Huh7.5-GFP cells and AP33; H. Hang and J. Yount (Rockefeller University) for IFITM plasmids; and D. Trono (Ecole Polytechnique Fédérale de Lausanne) and T. Pietschmann (Twincore) for lentiviral expression constructs. We also thank our colleagues J. Lupberger, H. El Saghire, N. van Renne, R. Tawar, and S. Durand. This work was supported by INSERM, the University of Strasbourg, the European Union (Infect-ERA hepBccc, ERC-2014-AdG-671231-HEPCIR, INTERREG-IV-Rhin Supérieur-FEDER-Hepato-Regio-Net, FP7 HepaMab, and EU H2020 Hep-CAR), Agence Nationale de Recherches sur le Sida et les hépatites virales (ANRS; 2012/239, 2012/318, 2013/108), and the French Cancer Agency (ARC IHU201301187). Research in the J.A.M. laboratory was funded by the Medical Research Council, the National Institute for Health Research (NIHR) Birmingham Liver Biomedical Research Unit, EU FP7 PathCO, and EU H2020 Hep-CAR. This work has been published under the framework of the LABEX ANR-10-LAB-28 and benefits from a funding from the state managed by the French National Research Agency as part of the Investments for the future program. C.C.C. acknowledges fellowships from the Canadian Institutes of Health Research (201411MFE- 338606-245517) and the Canadian Network on Hepatitis C.
Published: October 25, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and two figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.09.084.
Contributor Information
Mirjam B. Zeisel, Email: mirjam.zeisel@unistra.fr.
Thomas F. Baumert, Email: thomas.baumert@unistra.fr.
Accession Numbers
The accession number for the datasets reported in this paper is GEO: GSE85092.
Supplemental Information
References
- Bartenschlager R., Pietschmann T. Efficient hepatitis C virus cell culture system: what a difference the host cell makes. Proc. Natl. Acad. Sci. USA. 2005;102:9739–9740. doi: 10.1073/pnas.0504296102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumert T.F., Meredith L., Ni Y., Felmlee D.J., McKeating J.A., Urban S. Entry of hepatitis B and C viruses - recent progress and future impact. Curr. Opin. Virol. 2014;4:58–65. doi: 10.1016/j.coviro.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Bogomolov P., Alexandrov A., Voronkova N., Macievich M., Kokina K., Petrachenkova M., Lehr T., Lempp F.A., Wedemeyer H., Haag M. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J. Hepatol. 2016;65:490–498. doi: 10.1016/j.jhep.2016.04.016. [DOI] [PubMed] [Google Scholar]
- Brass A.L., Huang I.C., Benita Y., John S.P., Krishnan M.N., Feeley E.M., Ryan B.J., Weyer J.L., van der Weyden L., Fikrig E. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- César-Razquin A., Snijder B., Frappier-Brinton T., Isserlin R., Gyimesi G., Bai X., Reithmeier R.A., Hepworth D., Hediger M.A., Edwards A.M., Superti-Furga G. A call for systematic research on solute carriers. Cell. 2015;162:478–487. doi: 10.1016/j.cell.2015.07.022. [DOI] [PubMed] [Google Scholar]
- Chang K.O., George D.W. Bile acids promote the expression of hepatitis C virus in replicon-harboring cells. J. Virol. 2007;81:9633–9640. doi: 10.1128/JVI.00795-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatwal P., Bankwitz D., Gentzsch J., Frentzen A., Schult P., Lohmann V., Pietschmann T. Bile acids specifically increase hepatitis C virus RNA-replication. PLoS ONE. 2012;7:e36029. doi: 10.1371/journal.pone.0036029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claro da Silva T., Polli J.E., Swaan P.W. The solute carrier family 10 (SLC10): beyond bile acid transport. Mol. Aspects Med. 2013;34:252–269. doi: 10.1016/j.mam.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colpitts C.C., Verrier E.R., Baumert T.F. Targeting viral entry for treatment of hepatitis B and C virus infections. ACS Infect. Dis. 2015;1:420–427. doi: 10.1021/acsinfecdis.5b00039. [DOI] [PubMed] [Google Scholar]
- El-Serag H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–1273. doi: 10.1053/j.gastro.2011.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fofana I., Krieger S.E., Grunert F., Glauben S., Xiao F., Fafi-Kremer S., Soulier E., Royer C., Thumann C., Mee C.J. Monoclonal anti-claudin 1 antibodies prevent hepatitis C virus infection of primary human hepatocytes. Gastroenterology. 2010;139:953–964. doi: 10.1053/j.gastro.2010.05.073. [DOI] [PubMed] [Google Scholar]
- Graf D., Haselow K., Münks I., Bode J.G., Häussinger D. Inhibition of interferon-α-induced signaling by hyperosmolarity and hydrophobic bile acids. Biol. Chem. 2010;391:1175–1187. doi: 10.1515/BC.2010.108. [DOI] [PubMed] [Google Scholar]
- Hoshida Y., Villanueva A., Kobayashi M., Peix J., Chiang D.Y., Camargo A., Gupta S., Moore J., Wrobel M.J., Lerner J. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N. Engl. J. Med. 2008;359:1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Ishida H., Yamane D., Yi M., Swinney D.C., Foung S., Lemon S.M. Contrasting roles of mitogen-activated protein kinases in cellular entry and replication of hepatitis C virus: MKNK1 facilitates cell entry. J. Virol. 2013;87:4214–4224. doi: 10.1128/JVI.00954-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König A., Döring B., Mohr C., Geipel A., Geyer J., Glebe D. Kinetics of the bile acid transporter and hepatitis B virus receptor Na+/taurocholate cotransporting polypeptide (NTCP) in hepatocytes. J. Hepatol. 2014;61:867–875. doi: 10.1016/j.jhep.2014.05.018. [DOI] [PubMed] [Google Scholar]
- Krieger S.E., Zeisel M.B., Davis C., Thumann C., Harris H.J., Schnober E.K., Mee C., Soulier E., Royer C., Lambotin M. Inhibition of hepatitis C virus infection by anti-claudin-1 antibodies is mediated by neutralization of E2-CD81-claudin-1 associations. Hepatology. 2010;51:1144–1157. doi: 10.1002/hep.23445. [DOI] [PubMed] [Google Scholar]
- Liu S.Y., Sanchez D.J., Cheng G. New developments in the induction and antiviral effectors of type I interferon. Curr. Opin. Immunol. 2011;23:57–64. doi: 10.1016/j.coi.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupberger J., Zeisel M.B., Xiao F., Thumann C., Fofana I., Zona L., Davis C., Mee C.J., Turek M., Gorke S. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D.N., Uprichard S.L. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. USA. 2013;110:10777–10782. doi: 10.1073/pnas.1301764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith L.W., Harris H.J., Wilson G.K., Fletcher N.F., Balfe P., McKeating J.A. Early infection events highlight the limited transmissibility of hepatitis C virus in vitro. J. Hepatol. 2013;58:1074–1080. doi: 10.1016/j.jhep.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Meredith L.W., Farquhar M.J., Tarr A.W., McKeating J.A. Type I interferon rapidly restricts infectious hepatitis C virus particle genesis. Hepatology. 2014;60:1891–1901. doi: 10.1002/hep.27333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith L.W., Hu K., Cheng X., Howard C.R., Baumert T.F., Balfe P., van de Graaf K.F., Protzer U., McKeating J.A. Lentiviral hepatitis B pseudotype entry requires sodium taurocholate co-transporting polypeptide and additional hepatocyte-specific factors. J. Gen. Virol. 2016;97:121–127. doi: 10.1099/jgv.0.000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayana S.K., Helbig K.J., McCartney E.M., Eyre N.S., Bull R.A., Eltahla A., Lloyd A.R., Beard M.R. The interferon-induced transmembrane proteins, IFITM1, IFITM2, and IFITM3 inhibit hepatitis C virus entry. J. Biol. Chem. 2015;290:25946–25959. doi: 10.1074/jbc.M115.657346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Y., Lempp F.A., Mehrle S., Nkongolo S., Kaufman C., Fälth M., Stindt J., Königer C., Nassal M., Kubitz R. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology. 2014;146:1070–1083. doi: 10.1053/j.gastro.2013.12.024. [DOI] [PubMed] [Google Scholar]
- Oehler N., Volz T., Bhadra O.D., Kah J., Allweiss L., Giersch K., Bierwolf J., Riecken K., Pollok J.M., Lohse A.W. Binding of hepatitis B virus to its cellular receptor alters the expression profile of genes of bile acid metabolism. Hepatology. 2014;60:1483–1493. doi: 10.1002/hep.27159. [DOI] [PubMed] [Google Scholar]
- Petersen J., Dandri M., Mier W., Lütgehetmann M., Volz T., von Weizsäcker F., Haberkorn U., Fischer L., Pollok J.M., Erbes B. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat. Biotechnol. 2008;26:335–341. doi: 10.1038/nbt1389. [DOI] [PubMed] [Google Scholar]
- Pileri P., Uematsu Y., Campagnoli S., Galli G., Falugi F., Petracca R., Weiner A.J., Houghton M., Rosa D., Grandi G., Abrignani S. Binding of hepatitis C virus to CD81. Science. 1998;282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- Podevin P., Rosmorduc O., Conti F., Calmus Y., Meier P.J., Poupon R. Bile acids modulate the interferon signalling pathway. Hepatology. 1999;29:1840–1847. doi: 10.1002/hep.510290617. [DOI] [PubMed] [Google Scholar]
- Savidis G., Perreira J.M., Portmann J.M., Meraner P., Guo Z., Green S., Brass A.L. The IFITMs inhibit Zika virus replication. Cell Rep. 2016;15:2323–2330. doi: 10.1016/j.celrep.2016.05.074. [DOI] [PubMed] [Google Scholar]
- Scarselli E., Ansuini H., Cerino R., Roccasecca R.M., Acali S., Filocamo G., Traboni C., Nicosia A., Cortese R., Vitelli A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002;21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieck A., Schulze A., Gähler C., Müller T., Haberkorn U., Alexandrov A., Urban S., Mier W. Hepatitis B virus hepatotropism is mediated by specific receptor recognition in the liver and not restricted to susceptible hosts. Hepatology. 2013;58:43–53. doi: 10.1002/hep.26211. [DOI] [PubMed] [Google Scholar]
- Slijepcevic D., Kaufman C., Wichers C.G., Gilglioni E.H., Lempp F.A., Duijst S., de Waart D.R., Elferink R.P., Mier W., Stieger B. Impaired uptake of conjugated bile acids and hepatitis b virus pres1-binding in na(+) -taurocholate cotransporting polypeptide knockout mice. Hepatology. 2015;62:207–219. doi: 10.1002/hep.27694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S., Weston S., Kellam P., Marsh M. IFITM proteins-cellular inhibitors of viral entry. Curr. Opin. Virol. 2014;4:71–77. doi: 10.1016/j.coviro.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureau C. The use of hepatocytes to investigate HDV infection: the HDV/HepaRG model. Methods Mol. Biol. 2010;640:463–473. doi: 10.1007/978-1-60761-688-7_25. [DOI] [PubMed] [Google Scholar]
- Timpe J.M., Stamataki Z., Jennings A., Hu K., Farquhar M.J., Harris H.J., Schwarz A., Desombere I., Roels G.L., Balfe P., McKeating J.A. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology. 2008;47:17–24. doi: 10.1002/hep.21959. [DOI] [PubMed] [Google Scholar]
- Verrier E.R., Colpitts C.C., Bach C., Heydmann L., Weiss A., Renaud M., Durand S.C., Habersetzer F., Durantel D., Abou-Jaoudé G. A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology. 2016;63:35–48. doi: 10.1002/hep.28013. [DOI] [PubMed] [Google Scholar]
- Volz T., Allweiss L., Ben MBarek M., Warlich M., Lohse A.W., Pollok J.M., Alexandrov A., Urban S., Petersen J., Lütgehetmann M., Dandri M. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J. Hepatol. 2013;58:861–867. doi: 10.1016/j.jhep.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Wedemeyer H., Dore G.J., Ward J.W. Estimates on HCV disease burden worldwide - filling the gaps. J. Viral Hepat. 2015;22(Suppl 1):1–5. doi: 10.1111/jvh.12371. [DOI] [PubMed] [Google Scholar]
- Wilkins C., Woodward J., Lau D.T., Barnes A., Joyce M., McFarlane N., McKeating J.A., Tyrrell D.L., Gale M., Jr. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology. 2013;57:461–469. doi: 10.1002/hep.26066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao F., Fofana I., Heydmann L., Barth H., Soulier E., Habersetzer F., Doffoël M., Bukh J., Patel A.H., Zeisel M.B., Baumert T.F. Hepatitis C virus cell-cell transmission and resistance to direct-acting antiviral agents. PLoS Pathog. 2014;10:e1004128. doi: 10.1371/journal.ppat.1004128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H., Zhong G., Xu G., He W., Jing Z., Gao Z., Huang Y., Qi Y., Peng B., Wang H. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife. 2012;1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H., Peng B., Liu Y., Xu G., He W., Ren B., Jing Z., Sui J., Li W. Viral entry of hepatitis B and D viruses and bile salts transportation share common molecular determinants on sodium taurocholate cotransporting polypeptide. J. Virol. 2014;88:3273–3284. doi: 10.1128/JVI.03478-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York A.G., Williams K.J., Argus J.P., Zhou Q.D., Brar G., Vergnes L., Gray E.E., Zhen A., Wu N.C., Yamada D.H. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell. 2015;163:1716–1729. doi: 10.1016/j.cell.2015.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel M.B., Felmlee D.J., Baumert T.F. Hepatitis C virus entry. Curr. Top. Microbiol. Immunol. 2013;369:87–112. doi: 10.1007/978-3-642-27340-7_4. [DOI] [PubMed] [Google Scholar]
- Zeisel M.B., Lupberger J., Fofana I., Baumert T.F. Host-targeting agents for prevention and treatment of chronic hepatitis C - perspectives and challenges. J. Hepatol. 2013;58:375–384. doi: 10.1016/j.jhep.2012.09.022. [DOI] [PubMed] [Google Scholar]
- Zhong J., Gastaminza P., Cheng G., Kapadia S., Kato T., Burton D.R., Wieland S.F., Uprichard S.L., Wakita T., Chisari F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zona L., Lupberger J., Sidahmed-Adrar N., Thumann C., Harris H.J., Barnes A., Florentin J., Tawar R.G., Xiao F., Turek M. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe. 2013;13:302–313. doi: 10.1016/j.chom.2013.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.