

Abstract
Hepatic stellate cells are of mesenchymal cell type located in the space of Disse. Upon liver injury, HSCs transactivate into myofibroblasts with increase in expression of fibrillar collagen, especially collagen I and III, leading to liver fibrosis. Previous studies have shown mTOR signaling is activated during liver fibrosis. However, there is no direct evidence in vivo. The aim of this study is to examine the effects of conditional deletion of TSC1 in mesenchymal on pathogenesis of liver fibrosis. Crossing mice bearing the floxed TSC1 gene with mice harboring Col1α2-Cre-ER(T) successfully generated progeny with a conditional knockout of TSC1 (TSC1 CKO) in collagen I expressing mesenchymal cells. TSC1 CKO and WT mice were subjected to CCl4, oil or CCl4+ rapamycin treatment for 8 weeks. TSC1 CKO mice developed pronounced liver fibrosis relative to WT mice, as examined by ALT, hydroxyproline, histopathology, and profibrogenic gene. Absence of TSC1 in mesenchymal cells induced proliferation and prevented apoptosis in activated HSCs. However, there were no significant differences in oil-treated TSC1 CKO and WT mice. Rapamycin, restored these phenotypic changes by preventing myofibroblasts proliferation and enhancing their apoptosis. These findings revealed mTOR overactivation in mesenchymal cells aggravates CCl4− induced liver fibrosis and the rapamycin prevent its occurance.
Liver fibrosis is a major cause of morbidity and mortality worldwide due to chronic viral hepatitis, alcoholic hepatitis, and nonalcoholic steatohepatitis. Fibrosis formation is a wound-healing response in response to liver injury that is characterized by the accumulation of extracellular matrix (ECM)1. Myofibroblasts (MF) are defined primarily by their ability to produce ECM and contractile activity2. Hepatic stellate cells (HSCs) were found to be the major source of myofibroblasts in a mouse model of CCl4− induced liver fibrosis3. The transformation of quiescent vitamin A-rich HSCs into proliferative, contractile, fibrogenic myofibroblasts following liver injury has launched an era of astonishing progress in understanding the mechanistic basis of hepatic fibrosis progression and regression. Moreover, this simple paradigm has yielded a remarkably broad appreciation of myofibroblast function, not only in liver injury, but also hepatic development, regeneration, xenobiotic responses, intermediary metabolism, and immunoregulation4. At the same time, countless studies have used tissue immunohistochemistry to identify HSCs, while the most prominent proteins analyzed to date include α-smooth muscle actin (α-SMA), desmin, and glial fibrillary acidic protein (GFAP)5.
The mammalian target of rapamycin (mTOR) nucleates two distinct multi-protein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 has five components that include mTOR, regulatory-associated protein of mTOR (RAPTOR), target of rapamycin complex subunit LST8 (mLST8), proline-rich AKT1 substrate 1 (PRAS40), and DEP domain-containing mTOR-interacting protein (DEPTOR), while mTORC2 has six components that include mTOR, RPTOR-independent companion of mTOR (RICTOR), target of rapamycin complex 2 subunit MAPKAP1 (mSIN1), protein observed with Rictor-1 (Protor-1), mLST8, and DEPTOR6. The tuberous sclerosis complex (TSC), which comprises TSC1 and TSC2, is involved in the negative regulation of mTORC1 activity7. Loss of TSC1 causes cells and tissues to display constitutive mTORC1 activation. In response to numerous intracellular and extracellular stimuli, mTORC1 phosphorylates eukaryotic initiation factor 4E-binding protein-1 (4E-BP1) and S6 kinase 1 (S6K1), which exerts an essential role in increasing translation of a subset of mRNAs and accelerating growth and proliferation6,8. Under certain circumstances, inhibition of mTOR activity by rapamycin accelerates apoptosis9,10. Induction of mutations of mTOR can contribute to apoptotic resistance and might contribute to cellular transformation11.
Activation of the mTOR pathway has been reported in several fibrotic diseases. The mTOR pathway plays an important role in cardiac fibrosis and rapamycin is a potential therapeutic treatment that can be used to attenuate cardiac fibrosis12. The rapamycin analogue SDZ RAD was shown to promote dramatic inhibitory effects on collagen accumulation in the lungs in a bleomycin model of pulmonary fibrosis13, thus suppression of mTOR may be a viable treatment for pulmonary fibrosis14. mTORC1 signaling promotes the activation of kidney fibroblasts and contributes to the development of interstitial fibrosis15. There is growing evidence to support the idea that the mTOR signaling pathway plays a key role in liver fibrosis16. Activation of HSCs is regarded as a critical step in the pathogenesis of liver fibrosis3. HSCs are a resident mesenchymal cell type located in the subendothelial space of Disse that are interposed between the sinusoidal endothelium and hepatocytes4. Based on the findings of these studies, we speculate that activation of mTOR signaling in mesenchymal cells might also play a role in liver fibrosis.
To date, the specific effect of mTOR on liver fibrosis has not yet been reported. Therefore, the aim of the present study was to investigate the function of mTOR overactivation in mesenchymal cells in CCl4− induced liver fibrosis and to reveal possible mechanisms underlying its participation in fibrotic diseases.
Results
Generation of TSC1–conditional knockout mice in the mesenchymal compartment
Homozygous floxed TSC1 mice (TSC1fl/fl) were cross-bred with mice harboring a Cre-ER(T) recombinase gene to generate Cre/TSC1 heterozygous mice. The second cross generated [TSC1fl/fl, Col1α2-CreER(T)+/0] mice. Genomic DNA from tail tissue samples of these mice was genotyped by PCR analysis to confirm the presence of the TSC1 and Cre genes (Fig. 1A). To delete the TSC1 gene in the mesenchymal compartments, mice with genotype [TSC1fll/fl, Col1α2-CreER(T)+/0] were treated with tamoxifen as described in the Materials and Methods section and were referred to as TSC1 CKO mice. Mice with genotype [TSC1fl/fl,Col1α2-CreER(T)0/0] under the same treatment were referred to as WT mice. Deficiency of TSC1 expression in the mesenchymal compartment was verified by western blot and immunofluorescence analyses. As shown in Figs 1B and 5B, expression levels of p-S6 (s235/236), mTOR, and the S6K1 downstream effector protein were strongly increased in TSC1 CKO mice.
Figure 1. Generation of TSC1 conditional knockout (CKO) mice in the mesenchymal compartment.

Generation of TSC1 conditional knockout (CKO) mice in the mesenchymal compartment A: Mouse genotype was detected by PCR. Genomic DNA samples from the indicated strains of mice were analyzed using primers specific for wild-type (WT) and floxed (fl) TSC1, or Cre-ER (T) mice. The PCR products and 100-bp DNA ladder were separated by agarose gel electrophoresis. The bands corresponding to the WT (193 bp) and fl (230 bp) TSC1 alleles are indicated, and confirm the correct TSC1 genotype for each strain. The correct genotype with respect to Cre-ER(T) (500 bp) gene is confirmed in the respective strain. B: TSC1 knockout efficiency was detected by western blotting after daily injection of tamoxifen for 8 days. Liver tissue samples from WT and TSC1 CKO mice were analyzed for p-S6 (s235/236). The phosphorylation status of S6 was elevated, indicating TSC1 knock-down in the livers of TSC1 CKO mice. S6 and GAPDH were included as loading controls.
Figure 2. TSC1 deletion augments CCl4− induced liver fibrosis in mice.

Tamoxifen-treated control [wild-type (WT)] and TSC1 conditional knockout (CKO) mice received intraperitoneal injections of either CCl4 or oil, as indicated. H&E staining (A) and Sirius Red staining (B) were performed 8 weeks later. TSC1 CKO mice had more severe liver fibrosis than WT mice. All were imaged at 100x or 400x magnification. (C) The Sirius Red staining area in the liver was calculated by Image-Pro Plus 6.0. in six different images taken at 100x magnification on each slide. (D) The liver samples were analyzed for hydroxyproline content. (E) Liver injury was determined by serum ALT in WT and TSC1 CKO mice. The results are presented as the mean ± SEM derived from three independent experiments. NS, p > 0.05; *p < 0.05; #p < 0.01 for TSC1 CKO vs. WT mice.
Figure 3. Increased α-SMA expression due to loss of TSC1.

(A) Liver tissue samples from oil- or CCl4− treated mice for 8 weeks were analyzed for α-SMA protein expression by western blot analysis. The expression of α-SMA was significantly increased in CCl4− treated TSC1 CKO mice. (B) IHC staining was performed with α-SMA in oil- and CCl4− treated mouse liver tissues. (C) α-SMA staining area in the liver was calculated by Image-Pro Plus 6.0. in six different images taken at 400x magnification on each slide. NS, p > 0.05; *p < 0.05 for TSC1 CKO vs. WT mice.
Figure 4. Increased expression of profibrogenic markers in TSC1 CKO mice.
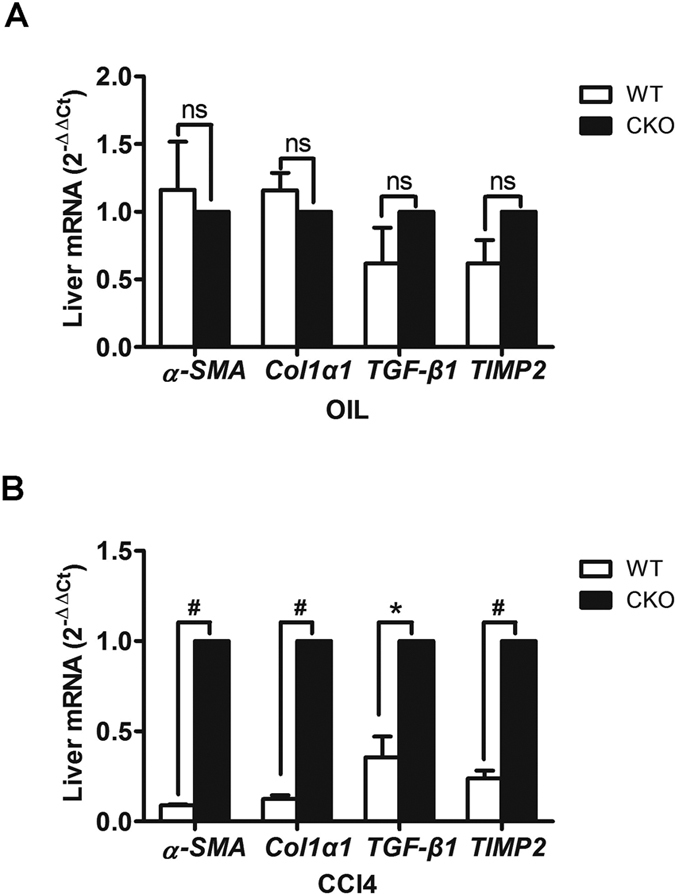
(A) Expression levels of α-SMA, Col1α1, TGF-β1, and TIMP2 were assessed by RT-PCR 8 weeks after treatment with oil. (B) Expression of α-SMA, Col1α1, TGF-β1, and TIMP2 were assessed by RT-PCR 8 weeks after treatment with CCl4. Expression was normalized to the levels of GAPDH and results are presented as the mean ± SEM derived from three independent experiments. NS, p > 0.05; *p < 0.05; #p < 0.01 for TSC1 CKO vs. WT mice.
Figure 5. Increased myofibroblasts in TSC1 CKO Mice.
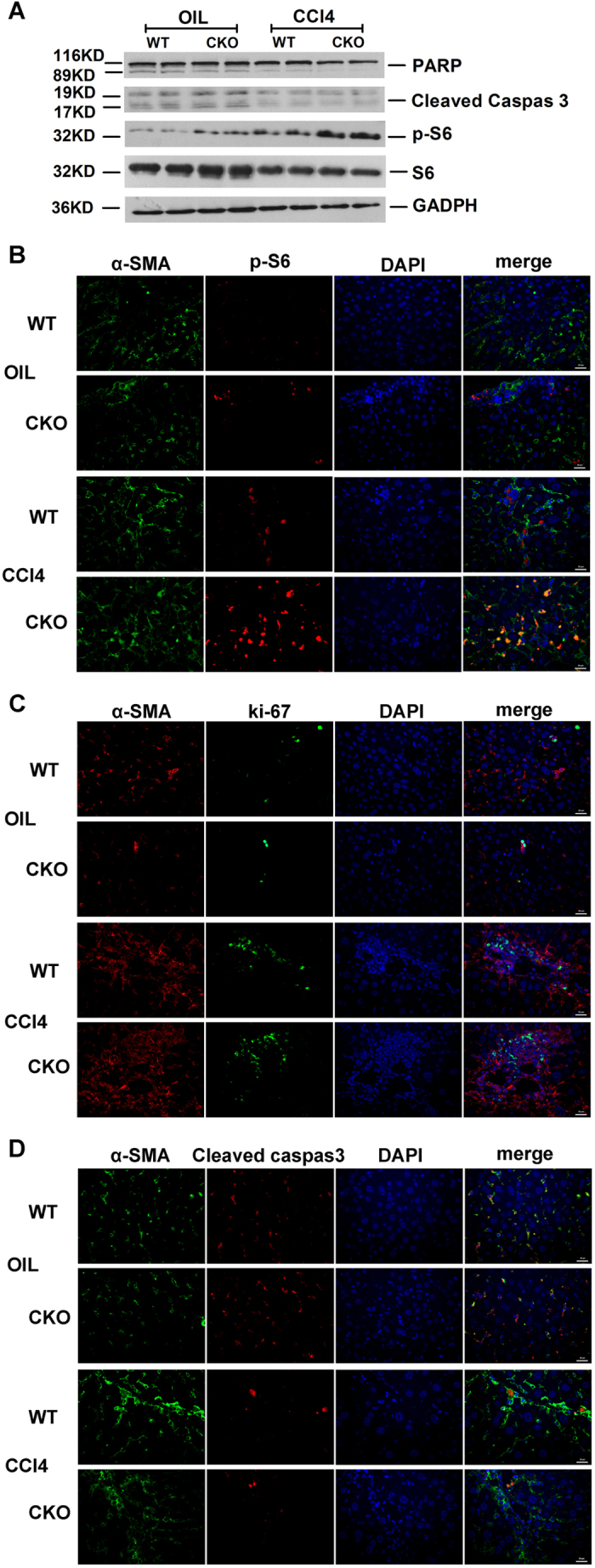
(A) Liver tissue samples from oil- or CCl4− treated mice were analyzed for PARP, cleaved caspase-3, p-S6, and S6 protein by western blot analysis. Immunofluorescence (IF) staining were performed to evaluate the impact of TSC1 deletion on myofibroblasts proliferation and apoptosis. (B) Sections were stained with α-SMA (green) and p-S6 (red) antibodies. There were increased both α-SMA and pS6 positive cells in TSC1 CKO mice administered CCl4. (C) Sections were stained with α-SMA (red) and ki67 (green) antibodies. Both α-SMA- and ki67-positive cells were increased in TSC1 CKO mice following CCl4 treatment. (D) Sections were stained with α-SMA (green) and cleaved caspase-3 (red) antibodies. Both α-SMA- and cleaved caspase-3-positive cells decreased in the CCl4− treated TSC1 CKO mice. Tissues were counterstained with DAPI (blue) to detect nuclei and imaged at 400x magnification.
TSC1 deletion augments CCl4− induced liver fibrosis in mice
To evaluate the effect of TSC1 deficiency in mesenchymal cells on liver fibrosis in vivo, TSC1 CKO and WT mice were treated with CCl4, and the fibrotic response was evaluated. A histologic examination of liver sections using H&E and Sirius Red staining was performed, which showed that TSC1 CKO mice exhibited greater degrees of lobular architecture damage and severe bridging necrosis than WT mice (Fig. 2A). TSC1 CKO mice presented exacerbated fibrosis by the morphometric assessment of the total area positively stained with Sirius Red (Fig. 2B,C) and by higher liver hydroxyproline(HYP) content (174.09 ± 4.12 μg/mg vs. 199.78 ± 8.41 μg/mg, P < 0.05; Fig. 2D). Meanwhile, TSC1 CKO mice exhibited higher serum ALT concentrations relative to WT mice (350.00 ± 5.77 U/L vs. 496.67 ± 8.82 U/L, P < 0.01; Fig. 2E). In comparison, there were no apparent differences in HYP (112.94 ± 9.74 μg/mg vs. 118.92 ± 5.07 μg/mg, P = 0.62) and ALT (30.00 ± 1.00 U/L vs. 31.67 ± 1.45 U/L, P = 0.40) detected after oil treatment between WT and TSC1 CKO mice. Collectively, these results demonstrated that TSC1 deletion in the mesenchymal compartment augments CCl4− induced liver fibrosis in mice.
Increased α-SMA expression due to loss of TSC1
The results of the above analyses showed that loss of TSC1 resulted in exacerbation in CCl4− induced hepatic fibrosis. Consistent with enhanced liver fibrosis and collagen production, α-SMA positive staining was increased in CCl4− treated TSC1 CKO mice (Fig. 3B,C). Western blot analysis confirmed significant CCl4− induced increases in α-SMA protein expression in TSC1 CKO mice (Fig. 3A). However, there were no apparent differences in α-SMA expression detected after oil treatment between WT and TSC1 CKO mice. These results suggest that TSC1 deletion from mesenchymal cells promotes the development of liver fibrosis owing to the increasing number of α-SMA positive cells.
Increased expression of profibrogenic markers in TSC1 CKO mice
RT-PCR was performed to quantify the expression of profibrogenic markers. As shown in Fig. 4A, levels of the profibrogenic markers α-SMA, Col1α1, TGF-β1, and TIMP2 were unchanged between the two genotypes following treatment with oil. In CCl4− induced animals, there was a trend towards increased levels of α-SMA(11.2-fold; P < 0.01), Col1α1 (8.3-fold; P < 0.01), TGF-β1(2.8-fold; P = 0.03), and TIMP2(4.2-fold; P < 0.01) in TSC1 CKO mice, as compared with WT mice (Fig. 4B). These results strengthened the evidence for the vulnerability of TSC1 CKO mice to CCl4− induced fibrosis.
Increased myofibroblasts in TSC1 CKO mice
To further confirm the importance of mTOR activity in myofibroblast proliferation in CCl4− induced liver fibrosis, protein expression was monitored by western blot and immunofluorescence analyses. As expected, p-S6 (s235/236) expression was upregulated in myofibroblasts from CCl4− treated mice, but was higher in the TSC1 CKO mice than WT mice (Fig. 5A). Further detection of apoptotic proteins showed that expression levels of the cleaved form of caspase-3 and PARP were reduced in CCl4− treated mice, but was lower in the TSC1 CKO mice than the WT mice (Fig. 5A). Myofibroblast formation was measured by immunofluorescence analysis with α-SMA antibody. Consistent with the western blotting results, increased phosphorylation of S6 with CCl4 treatment, at least partially, caused the number of myofibroblasts to increase in TSC1 CKO mice (Fig. 5B). Following CCl4 treatment, ki67 and α-SMA double-positive cells, which are recognized as proliferative myofibroblasts, were significantly increased to a greater extent in TSC1 CKO mice than in WT mice (Fig. 5C). Meanwhile, the abundance of cleaved caspase-3 and α-SMA double-positive cells, which are recognized as apoptotic myofibroblasts, was decreased in TSC1 CKO mice, as compared with WT mice (Fig. 5D). Intriguingly, expression of α-SMA, ki67, and cleaved caspase-3 was not appreciably altered in oil group mice, indicating that under quiescent conditions, loss of TSC1 in cells expressing collagen type I was not accompanied by an increase in the abundance of myofibroblasts in the liver.
Rapamycin attenuates CCl4− Induced Liver Fibrosis and reverses phenotypes in TSC1 CKO mice
It is known from the literature that the rapamycin has inhibitory effects on liver fibrosis17, and this is consistent with our results. In CCl4+ rapamycin-treated CKO or WT mice, there was less severe liver histology injury and lower serum ALT concentrations compared with the mice merely treated with CCl4 (Fig. 6A,B). CCl4+ rapamycin-treated mice presented attenuated fibrosis by the morphometric assessment of the total area positively stained with Sirius Red (Fig. 6A,C). Immunohistochemical staining for α-SMA showed that α-SMA-positive staining was decreased in CCl4+ rapamycin-treated mice (Fig. 6A,D). As shown in Fig. 6E, western blot analyses indicated that α-SMA and p-S6 protein expression were lower in the CCl4+ rapamycin-treated mice in comparison with CCl4− treated mice, but the expression levels of the cleaved form of caspase-3 and PARP were upregulated in CCl4+ rapamycin-treated mice. Consistent with the western blotting results, decreased phosphorylation of S6 with rapamycin treatment, at least partially, caused the number of myofibroblasts to decrease (Fig. 6F). Ki67 and α-SMA double-positive cells were also significantly decreased to a greater extent in CCl4+ rapamycin-treated mice (Fig. 6G). Meanwhile, the number of cleaved caspase-3 and α-SMA double-positive cells were increased in CCl4+ rapamycin-treated mice, as compared with CCl4− treated mice (Fig. 6H). Intriguingly, there were no significant differences between the WT and the TSC1 CKO in CCl4+ rapamycin-treated mice.
Figure 6. Rapamycin attenuates CCl4− Induced Liver Fibrosis and reverses phenotypes in TSC1 CKO mice.
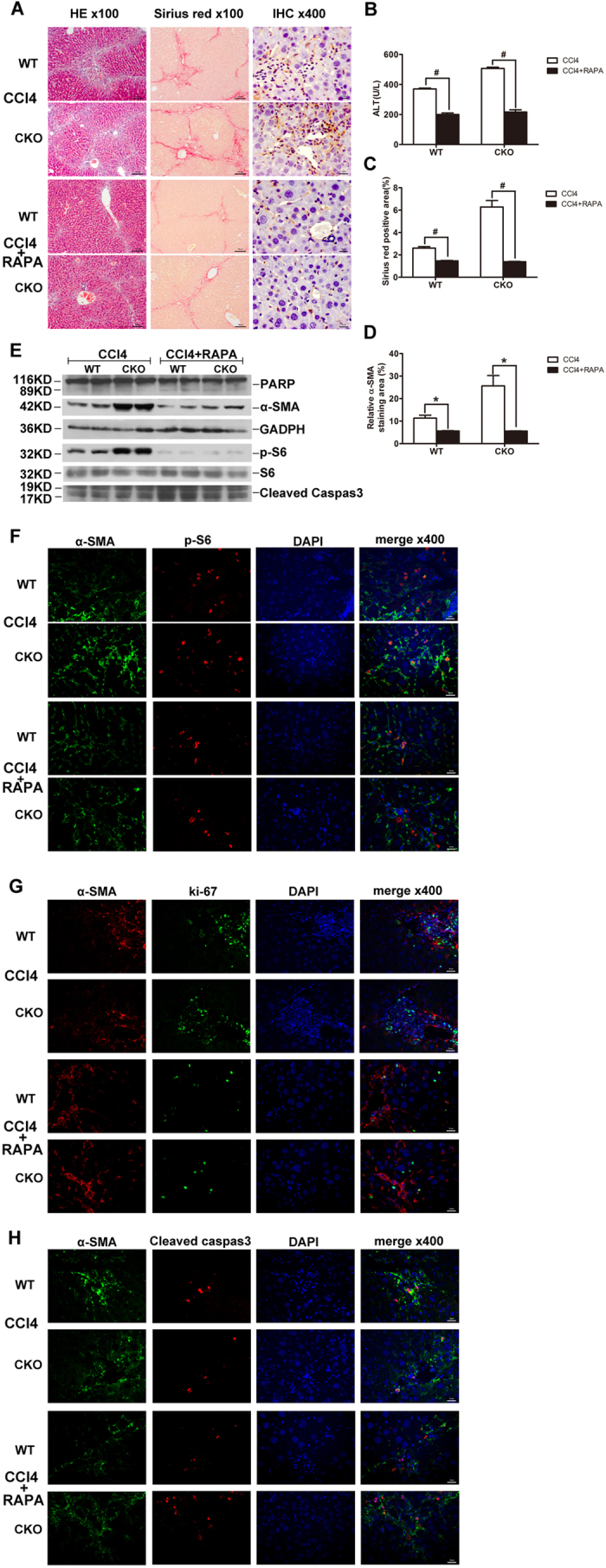
(A) Liver tissue samples from CCl4− or CCl4+ rapamycin- treated mice for 8 weeks were analyzed for histological analysis. As shown in figure H&E staining and Sirius Red staining, CCl4+ rapamycin- treated mice had less severe liver fibrosis than CCl4− treated mice. All were imaged at 100x magnification. IHC staining was performed with α-SMA in CCl4− or CCl4+ rapamycin- treated mice liver tissues. (B) Liver injury was determined by serum ALT in CCl4− and CCl4+ papamycin-treated mice. (C) The Sirius Red staining area in the liver was calculated by Image-Pro Plus 6.0. in six different images taken at 100x magnification on each slide. (D) a-SMA staining area in the liver was calculated by Image-Pro Plus 6.0. in six different images taken at 400x magnification on each slide. (E) Liver tissue samples from CCl4− or CCl4+ rapamycin- treated mice were analyzed for PARP, cleaved caspase-3, p-S6, and S6 protein by western blot analysis. Immunofluorescence(IF) staining were performed to evaluate the impact of rapamycin on myofibroblasts proliferation and apoptosis. (F) Sections were stained with α-SMA (green) and p-S6 (red) antibodies. There were decreased α-SMA and pS6 double positive cells in mice administered CCl4+ rapamycin. (G) Sections were stained with α-SMA (red) and ki67 (green) antibodies.α-SMA- and ki67 double positive cells were decreased in mice following CCl4+ rapamycin treatment. (H) Sections were stained with α-SMA (green) and cleaved caspase-3 (red) antibodies. α-SMA- and cleaved caspase-3 double positive cells increased in the CCl4+ rapamycin-treated mice. Tissues were counterstained with DAPI (blue) to detect nuclei and imaged at 400x magnification. #p < 0.01; *p < 0.05 for CCl4− vs. CCl4+ rapamycin-treated mice.
Thus, TSC1 deficiency in the mesenchymal compartment resulted in a significant increase in the abundance of myofibroblasts, thereby contributing to the noted exacerbation of liver fibrosis. Conversely, rapamycin can alleviated CCl4− induced liver fibrosis by inhibiting myofibroblast proliferation and inducing apoptosis, and rescued the specific phenomenon by TSC1 deficiency in the mesenchymal compartment.
Discussion
Fibrosis is a pathological process characterized by excessive accumulation of connective tissue components in organs and tissues. Liver fibrosis develops as a consequence of liver injury and activation of HSCs, which represents the imbalance between ECM production and degradation. Previous studies have reported that mTOR was involved in the process of liver fibrosis in human tissues and animal models of CCl4− mediated fibrosis18,19. Understanding the importance of mTOR signaling in different cell compartments is critical for future studies to develop effective drugs targeting this signaling pathway with minimal off-target or negative adverse effects. In this study, the role of aberrant mTOR activity in the mesenchymal compartment in the pathogenesis of liver fibrosis in a conditional TSC1 knockout mouse model was investigated, which showed that TSC1 deletion in mesenchymal cells aggravated liver fibrosis and was associated with myofibroblast proliferation and apoptosis.
Growing evidence supports the hypothesis that mTOR overactivation is involved in the pathogenesis of fibrotic diseases, including liver fibrosis16,20,21,22. We specifically focused on the role of mesenchymal mTOR overactivation in liver fibrosis. Our data further indicated that the mTOR signaling pathway was activated in CCl4− mediated liver fibrosis (Fig. 5A,B), which is consistent with a previous study that reported mTOR activation in liver fibrosis in an animal model19. After obtaining evidence supporting our hypothesis that mTOR overactivation was involved in the process of liver fibrosis, we further demonstrated that mTOR activation resulting in increased myofibroblast accumulation in the liver could be a major mechanism underlying fibrosis formation.
When the Cre-ER(T) recombinase was activated by tamoxifen, TSC1 expression was selectively absent only in mesenchymal cells. HSCs, first described by Kupffer in the 19th century, have emerged in the past 25 years as remarkably versatile mesenchymal cells4. The mTOR signaling pathway may be activated in HSCs in TSC1 CKO mice (Fig. 1C). As previously reported using other methodologies, HSCs are the major source of myofibroblasts (>87%) in CCl4− induced liver injury23.
Myofibroblasts are the major source of fibrogenic cytokines and ECM24. Increasing myofibroblast accumulation in TSC1 CKO mice in response to CCl4 treatment is thought to contribute to the noted significant increase in liver fibrosis. This result is also consistent with a previous study in which rapamycin appeared to inhibit proliferation and differentiation of corneal myofibroblasts in vitro25.
mTOR regulates renal myofibroblast activity and proliferation, and also plays an important role in preventing apoptosis of activated HSCs and promoting progression of fibrosis26. Apoptosis of HSCs may be involved in the termination of this response27. In a microarray study of human HSCs, cells became senescent and switched from a primarily fibrogenic to an inflammatory phenotype, with decreased proliferation and gene expression, and increased apoptosis28. PI3K/AKT as the upstream of the mTOR signaling pathway, inhibition of PI3K/Akt signaling pathway has been shown to induce HSCs apoptosis and attenuate liver fibrosis in vitro29. These findings suggest that reduced myofibroblast proliferation and increased myofibroblast apoptosis could effectively reduce fibrosis.
Interestingly, the CCl4− treated TSC1 CKO mice exhibited obvious differences in liver fibrogenesis relative to control mice. However, there were no significant differences in the oil group between TSC1 CKO and WT mice. These results imply that the effect of mTOR pathway activation in mesenchymal cell in normal mice is balanced by tissue homeostasis. In the experimental hepatic fibrosis model, after liver cell injury, mTOR pathway activation in mesenchymal cell enhanced the wound healing response.
In summary, mTOR overactivation in the mesenchymal compartment augmented liver fibrosis induced by CCl4. The mechanism likely involved activation of the mTOR pathway, which induced myofibroblast proliferation and prevented apoptosis. Although these results confirmed the importance of mesenchymal mTOR activity on myofibroblast proliferation in vivo, additional and/or alternate mechanisms cannot be excluded, thus additional future studies are warranted. Inhibition of mesenchymal mTOR overactivation and the induction of myofibroblasts present potential treatments for liver fibrosis.
Methods
Mice
All animal experiments are approved by the Southern Medical University Animal Care and Use Committee. All animals received humane care and the study protocols were conducted in compliance with the institution’s guidelines. Mice importing, transporting, housing and breeding were conducted according to the recommendations of “The use of non-human primates in research.” Col1α2CreERT and TSC1fl/fl mice were both purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Col1α2CreERT mice express a tamoxifen-inducible Cre recombinase driven by the mouse Col1α2, collagen, type1, alpha 2, promoter. The transgene insert contains a fusion product involving Cre recombinase and a mutant form of the mouse estrogen receptor ligand binding domain. Cre (C57Bl/6) transgenic mice were crossed with mice having both TSC1 alleles floxed (TSC1fl/fl) (129S4/SvJae) to generate mice heterozygous for both alleles. Mice heterozygous for both alleles were backcrossed with TSC1fl/fl mice to yield Col1α2CreERT/TSC1fl/fl mice.
To selectively delete TSC1 in Cre expressing cells, Col1α2CreERT /TSC1fl/fl, mice (6–8 weeks old) received daily intraperitoneal injections of the tamoxifen suspension (0.1 ml of diluted stock) for 8 days (TSC1 CKO), while TSC1 wild-type (WT) mice, as controls, received the same treatment. After administration of the tamoxifen treatment regimen, 8–12-week-old male TSC1 CKO and WT mice were induced by intraperitoneal injection with 20%CCl4 (5 μl/g body weight; Sigma Chemicals, Heidelberg, Germany) dissolved in olive oil for 8 weeks. The control group received the same volume of olive oil only. In the CCl4+ rapamycin group, after treatment with CCl4, the mice also received rapamycin (1 mg/kg body weight/day; Sigma-Aldrich) by intraperitoneal injection daily until sacrifice.
Hydroxyproline Assay
The hydroxyproline assay was used to estimate the percentage of degraded collagen30. To assess the extent of fibrosis, the collagen content of liver homogenates after alkaline hydrolysis was assayed using a colorimetric assay (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Alanine transaminase (ALT)
Serum was obtained by cardiac puncture from anesthetized mice following overnight fasting for detection of ALT levels using an IDEXX Catalyst Dx Biochemical Analyzer (IDEXX Laboratories, Westbrook, ME, USA).
Histological analysis
For histological analysis, liver samples were fixed for 24 h in 4% paraformaldehyde, embedded in paraffin, sliced into 5 μm-thick sections, and stained with hematoxylin and eosin (H&E)or Sirius Red, as previously described31.
For immunohistochemical analysis, mouse liver sections were deparaffinized, rehydrated, and incubated with α-SMA antibody (1:100; Abcam, Cambridge, UK) overnight at 4 °C. The same concentration of normal mouse IgG served as a negative control. The bound antibodies were then visualized using diaminobenzidine as a chromogen and the slides were counterstained with hematoxylin. All sections were observed and photographed using an Olympus BX51 microscope (Olympus Corporation, Tokyo, Japan). The area of positive staining was measured in six different images taken at 400x magnification on each slide and quantified using Image Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA).
For immunofluorescent staining, the sections were incubated with antibodies against phospho-S6 ribosomal protein (Ser235/236) (1:100; Cell Signaling Technology, Boston, MA, USA), α-SMA (1:100; Abcam), Ki67 (1:200; GeneTex, Inc., Irvine, CA, USA), and cleaved caspase-3 (1:200; Cell Signaling Technology). After incubation with the primary antibodies, the sections were then washed with phosphate-buffered saline and incubated with appropriate fluorescent secondary antibodies (1:200; Invitrogen Corporation, Carlsbad, CA, USA). Sections were mounted using 4′,6-diamidino-2-phenylindole (DAPI) and imaged by fluorescent microscopy.
Western blot analysis
Protein extracts were separated by gel electrophoresis, transferred to nitrocellulose membranes, and subsequently incubated with antibodies against mouse phospho-S6 ribosomal protein (Ser235/236) (1:2000; Cell Signaling Technology), S6 (1:3000; Santa Cruz Biotechnology Inc., Dallas, TX, USA), α-SMA (1:3000; Abcam), PRAP (1:1000; Cell Signaling Technology), cleaved caspase-3 (1:2000; Cell Signaling Technology), or glyceraldehyde 3-phosphate dehydrogenase (1:3000; Santa Cruz Biotechnology Inc.). Thereafter, the blots were washed three times with TBST (50 mM Tris, 150 mM NaCl, 0.05% Tween 20, adjusted to pH 7.6 with HCl) for 5 min and then incubated with horseradish peroxidase-conjugated secondary antibodies, and visualized using an enhanced chemiluminescence kit (PerkinElmer, Inc., Waltham, MA, USA).
Real-time polymerase chain reaction (RT-PCR) analysis
Total RNA was isolated using TRI reagent (Sigma Chemicals) and cDNA was generated using Primescript RT master mix (Takara Bio, Inc., Ōtsu, Japan). Duplicate PCR amplifications were carried out using a Light Cycler LC480 Real-Time PCR System (Roche, Basel, Switzerland) with the SYBR Green PCR Kit (Takara Bio, Inc.). mRNA concentrations were calculated using the 2−ΔΔCt method. The primers used in this study were produced by Sangon Biotech Co., Ltd. (Shangai, China) and the sequences are listed in Table 1.
Table 1. Primer sequences and accession numbers for primers used for RT-PCR.
| Gene | Accession No. | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|---|
| α-SMA | NM_007392.4 | GTCCCAGACATCAGGGAGTAA | TCGGATACTTCAGCGTCAGGA |
| col1α1 | NM_007742.3 | GCTCCTCTTAGGGGCCACT | CCACGTCTCACCATTGGGG |
| TGF-β1 | NM_011577.4 | CTCCCGTGGCTTCTAGTGC | GCCTTAGTTTGGACAGGATCTG |
| TIMP2 | NM_011594.1 | TCAGAGCCAAAGCAGTGAGC | GCCGTGTAGATAAACTCGATGTC |
Statistical analysis
All data are expressed as the mean ± standard error of the mean (SEM) from three individual experiments. The data for each group were analyzed using the t-test with SPSS ver. 20.0 software (IBM-SPSS, Inc., Chicago, IL, USA). A probability (p) value of < 0.05 was considered statistically significant.
Additional Information
How to cite this article: Lanlan, S. et al. mTOR Overactivation in Mesenchymal cells Aggravates CCl4 − Induced liver Fibrosis. Sci. Rep. 6, 36037; doi: 10.1038/srep36037 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Acknowledgments
The work was supported by the National Natural Science Foundation of China (No. 81301525), and Natural Science Foundation of Guangdong Province (No. 2014A030313297).
Footnotes
Author Contributions Designed research: M.D., C.H.J. and L.L.S. Performed research: L.L.S., L.Z., Y.D., Y.F., S.Z.C., W.Q.N., X.Y.D., H.Y.W., W.F.X. and Z.G.C. Analyzed data: all authors. Wrote the paper: L.L.S., C.H.J., H.Z., X.C.B. and M.D. Revised manuscripts: L.L.S., Y.D. and Y.F.
References
- Hernandez-Gea V. & Friedman S. L. Pathogenesis of liver fibrosis. Annu Rev Pathol 6, 425–456, doi: 10.1146/annurev-pathol-011110-130246 (2011). [DOI] [PubMed] [Google Scholar]
- Takuwa Y., Ikeda H., Okamoto Y., Takuwa N. & Yoshioka K. Sphingosine-1-phosphate as a mediator involved in development of fibrotic diseases. Biochimica et biophysica acta 1831, 185–192, doi: 10.1016/j.bbalip.2012.06.008 (2013). [DOI] [PubMed] [Google Scholar]
- Bataller R. & Brenner D. A. Liver fibrosis. The Journal of clinical investigation 115, 209–218, doi: 10.1172/jci24282 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman S. L. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 88, 125–172, doi: 10.1152/physrev.00013.2007 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis 21, 311–335, doi: 10.1055/s-2001-17550 (2001). [DOI] [PubMed] [Google Scholar]
- Laplante M. & Sabatini D. M. mTOR signaling at a glance. J Cell Sci 122, 3589–3594, doi: 10.1242/jcs.051011 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X. et al. Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nature cell biology 4, 699–704, doi: 10.1038/ncb847 (2002). [DOI] [PubMed] [Google Scholar]
- Hay N. & Sonenberg N. Upstream and downstream of mTOR. Genes & development 18, 1926–1945, doi: 10.1101/gad.1212704 (2004). [DOI] [PubMed] [Google Scholar]
- Thimmaiah K. N. et al. Insulin-like growth factor I-mediated protection from rapamycin-induced apoptosis is independent of Ras-Erk1-Erk2 and phosphatidylinositol 3′-kinase-Akt signaling pathways. Cancer Res 63, 364–374 (2003). [PubMed] [Google Scholar]
- Vignot S., Faivre S., Aguirre D. & Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO 16, 525–537, doi: 10.1093/annonc/mdi113 (2005). [DOI] [PubMed] [Google Scholar]
- Edinger A. L. & Thompson C. B. An activated mTOR mutant supports growth factor-independent, nutrient-dependent cell survival. Oncogene 23, 5654–5663, doi: 10.1038/sj.onc.1207738 (2004). [DOI] [PubMed] [Google Scholar]
- Yu S. Y., Liu L., Li P. & Li J. Rapamycin inhibits the mTOR/p70S6K pathway and attenuates cardiac fibrosis in adriamycin-induced dilated cardiomyopathy. The Thoracic and cardiovascular surgeon 61, 223–228, doi: 10.1055/s-0032-1311548 (2013). [DOI] [PubMed] [Google Scholar]
- Simler N. R. et al. The rapamycin analogue SDZ RAD attenuates bleomycin-induced pulmonary fibrosis in rats. The European respiratory journal 19, 1124–1127 (2002). [DOI] [PubMed] [Google Scholar]
- Gui Y. S. et al. mTOR Overactivation and Compromised Autophagy in the Pathogenesis of Pulmonary Fibrosis. PloS one 10, e0138625, doi: 10.1371/journal.pone.0138625 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L. et al. Rheb/mTORC1 signaling promotes kidney fibroblast activation and fibrosis. Journal of the American Society of Nephrology: JASN 24, 1114–1126, doi: 10.1681/asn.2012050476 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsenker E. et al. Potent antifibrotic activity of mTOR inhibitors sirolimus and everolimus but not of cyclosporine A and tacrolimus in experimental liver fibrosis. J Hepatol 55, 388–398, doi: 10.1016/j.jhep.2010.10.044 (2011). [DOI] [PubMed] [Google Scholar]
- Kim Y. J. et al. Inhibitory effects of rapamycin on the different stages of hepatic fibrosis. World journal of gastroenterology 20, 7452–7460, doi: 10.3748/wjg.v20.i23.7452 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villamil F. G. et al. Fibrosis progression in maintenance liver transplant patients with hepatitis C recurrence: a randomised study of everolimus vs. calcineurin inhibitors. Liver international: official journal of the International Association for the Study of the Liver 34, 1513–1521 (2014). [DOI] [PubMed] [Google Scholar]
- Li J. et al. Antifibrotic effects of luteolin on hepatic stellate cells and liver fibrosis by targeting AKT/mTOR/p70S6K and TGFbeta/Smad signalling pathways. Liver international: official journal of the International Association for the Study of the Liver 35, 1222–1233, doi: 10.1111/liv.12638 (2015). [DOI] [PubMed] [Google Scholar]
- Gao X. M. et al. Inhibition of mTOR reduces chronic pressure-overload cardiac hypertrophy and fibrosis. J Hypertens 24, 1663–1670, doi: 10.1097/01.hjh.0000239304.01496.83 (2006). [DOI] [PubMed] [Google Scholar]
- Chen G. et al. Rapamycin ameliorates kidney fibrosis by inhibiting the activation of mTOR signaling in interstitial macrophages and myofibroblasts. PLoS One 7, e33626, doi: 10.1371/journal.pone.0033626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki A. et al. Treatment with rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models of systemic sclerosis. Arthritis Rheum 62, 2476–2487, doi: 10.1002/art.27498 (2010). [DOI] [PubMed] [Google Scholar]
- Iwaisako K. et al. Origin of myofibroblasts in the fibrotic liver in mice. Proceedings of the National Academy of Sciences of the United States of America 111, E3297–E3305, doi: 10.1073/pnas.1400062111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B. et al. The myofibroblast: one function, multiple origins. The American journal of pathology 170, 1807–1816, doi: 10.2353/ajpath.2007.070112 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani B. Y. et al. Rapamycin inhibits the production of myofibroblasts and reduces corneal scarring after photorefractive keratectomy. Investigative ophthalmology & visual science 54, 7424–7430, doi: 10.1167/iovs.13-12674 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winbanks C. E. et al. Role of the phosphatidylinositol 3-kinase and mTOR pathways in the regulation of renal fibroblast function and differentiation. The international journal of biochemistry & cell biology 39, 206–219, doi: 10.1016/j.biocel.2006.08.004 (2007). [DOI] [PubMed] [Google Scholar]
- Fischer R., Schmitt M., Bode J. G. & Haussinger D. Expression of the peripheral-type benzodiazepine receptor and apoptosis induction in hepatic stellate cells. Gastroenterology 120, 1212–1226, doi: 10.1053/gast.2001.23260 (2001). [DOI] [PubMed] [Google Scholar]
- Schnabl B., Purbeck C. A., Choi Y. H., Hagedorn C. H. & Brenner D. Replicative senescence of activated human hepatic stellate cells is accompanied by a pronounced inflammatory but less fibrogenic phenotype. Hepatology 37, 653–664, doi: 10.1053/jhep.2003.50097 (2003). [DOI] [PubMed] [Google Scholar]
- Wang Y., Jiang X. Y., Liu L. & Jiang H. Q. Phosphatidylinositol 3-kinase/Akt pathway regulates hepatic stellate cell apoptosis. World J Gastroenterol 14, 5186–5191 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamall I. S., Finelli V. N. & Que Hee S. S. A simple method to determine nanogram levels of 4-hydroxyproline in biological tissues. Anal Biochem 112, 70–75 (1981). [DOI] [PubMed] [Google Scholar]
- Chen W. B. et al. GGPPS deficiency aggravates CCl4− induced liver injury by inducing hepatocyte apoptosis. FEBS Lett 589, 1119–1126, doi: 10.1016/j.febslet.2015.03.015 (2015). [DOI] [PubMed] [Google Scholar]