

Abstract
The manner in which mitochondria take up and store Ca2+ remains highly debated. Recent experimental and computational evidence has suggested the presence of at least two modes of Ca2+ uptake and a complex Ca2+ sequestration mechanism in mitochondria. But how Mg2+ regulates these different modes of Ca2+ uptake as well as mitochondrial Ca2+ sequestration is not known. In this study, we investigated two different ways by which mitochondria take up and sequester Ca2+ by using two different protocols. Isolated guinea pig cardiac mitochondria were exposed to varying concentrations of CaCl2 in the presence or absence of MgCl2. In the first protocol, A, CaCl2 was added to the respiration buffer containing isolated mitochondria, whereas in the second protocol, B, mitochondria were added to the respiration buffer with CaCl2 already present. Protocol A resulted first in a fast transitory uptake followed by a slow gradual uptake. In contrast, protocol B only revealed a slow and gradual Ca2+ uptake, which was approximately 40 % of the slow uptake rate observed in protocol A. These two types of Ca2+ uptake modes were differentially modulated by extra-matrix Mg2+. That is, Mg2+ markedly inhibited the slow mode of Ca2+ uptake in both protocols in a concentration-dependent manner, but not the fast mode of uptake exhibited in protocol A. Mg2+ also inhibited Na+-dependent Ca2+ extrusion. The general Ca2+ binding properties of the mitochondrial Ca2+ sequestration system were reaffirmed and shown to be independent of the mode of Ca2+ uptake, i.e. through the fast or slow mode of uptake. In addition, extra-matrix Mg2+ hindered Ca2+ sequestration. Our results indicate that mitochondria exhibit different modes of Ca2+ uptake depending on the nature of exposure to extra-matrix Ca2+, which are differentially sensitive to Mg2+. The implications of these findings in cardiomyocytes are discussed.
Keywords: Mitochondria, Cardiac, Calcium uptake, Calcium sequestration, Calcium efflux
Introduction
Mitochondria have the capacity to take up and sequester large amounts of Ca2+ (Vasington and Murphy 1962). For decades it was believed that mitochondrial Ca2+ was a necessary means to maintain proper energy balance in the heart. However, recent mitochondrial Ca2+ uniporter (MCU) knockout studies in mice have contested the importance of Ca2+ in mitochondrial energy homeostasis (Pan et al. 2013). Cardiac specific MCU knockout studies using a mouse model reveal that cardiac mitochondrial Ca2+ is only essential during high stress conditions, but not for routine activities (Kwong et al. 2015; Luongo et al. 2015; Wu et al. 2015). Whereas the role of mitochondria as a Ca2+ sink against cytosolic Ca2+ overload is well recognized (Szabadkai and Duchen 2008; Nicholls 2005; Rasola and Bernardi 2011), their role in shaping the cytosolic Ca2+ transients during physiological conditions is still debated (Boyman et al. 2014; Lu et al. 2013). Moreover, the precise mechanisms and consequences by which mitochondria take up, extrude, and sequester cytosolic Ca2+ remain obscure.
It has been suggested that mitochondrial Ca2+ uptake consists of different components that result in different matrix free Ca2+ dynamics. One possible component constitutes the rapid and beat-to-beat changes in the concentration of matrix free Ca2+ ([Ca2+]m) (Dedkova and Blatter 2013; Huser et al. 2000). This fast mode of Ca2+ uptake is proposed to be responsible for regulating ATP production (Dedkova and Blatter 2013; Carafoli 2010; Tarasov et al. 2012) by altering activities of Ca2+-sensitive dehydrogenases and other Ca2+-sensitive processes. However, this idea, as noted above, is controversial based on recent data obtained from MCU knockout mice (Pan et al. 2013; Kwong et al. 2015). Another possible component that has been proposed is the slow mode of Ca2+ uptake, which functions as a low-pass filter of the cytosolic Ca2+ transient while gradually accumulating matrix Ca2+ (Dedkova and Blatter 2013; Sedova et al. 2006). It is also thought to be a significant source of Ca2+ uptake during periods of sustained elevation of cytosolic Ca2+ (Carafoli 2010; Dorn and Maack 2013), such as in ischemia and reperfusion (IR) injury (Varadarajan et al. 2001).
The MCU is the primary pathway for Ca2+ uptake (Baughman et al. 2011; De Stefani et al. 2011) and may possess multiple conductance modes (Kirichok et al. 2004; Wei et al. 2012; Csordas et al. 2013). The recent identities of the MCU channel and its regulatory proteins (e.g. MICU1 and MICU2) involved in modulating mitochondrial Ca2+ uptake are now characterized as sensors of extra-matrix [Ca2+] ([Ca2+]e) that stimulate or inhibit MCU, respectively. This would allow for a rapid response of mitochondria to Ca2+ signals generated in the cytoplasm (Marchi and Pinton 2014). Previous studies suggest that there exist additional potential Ca2+ uptake modes: the mitochondrial ryanodine-like receptor (Beutner et al. 2001; Tewari et al. 2014) and the rapid-mode (RaM) of Ca2+ uptake (Buntinas et al. 2001; Gunter et al. 1998; Sparagna et al. 1995). These fast modes of Ca2+ uptake were postulated, but not conclusively shown, to be responsible for transduction of physiological Ca2+ transients to match oxidative phosphorylation with energetic demands (Huser et al. 2000; O’Rourke and Blatter 2009).
Pharmacological inhibitors and/or modeling approaches have been used to identify the different modes of Ca2+ uptake, but the conclusions drawn from these studies have been disputed. For example, recent studies show relatively little mitochondrial Ca2+ uptake during physiological Ca2+ transients (Boyman et al. 2014; Lu et al. 2013), which implies that in the physiological setting, MCU might play only a negligible role in shaping physiological Ca2+ dynamics in the beating heart. Moreover, when mitochondria are loaded with sufficient Ca2+, changes in matrix free Ca2+ are difficult to detect due to the higher buffering power associated with higher mitochondrial Ca2+ loads (Bazil et al. 2013; Blomeyer et al. 2013). Regardless, the MCU becomes most relevant when cytosolic [Ca2+] is abnormally elevated, such as during IR, oxidative stress, sarcoplasmic reticulum stress, or Ca2+ overload (Varadarajan et al. 2001; An et al. 2001a; Rhodes et al. 2003; An et al. 2001b).
Excess Ca2+ uptake by mitochondria is sequestered in a phosphate-dependent reaction that forms amorphous calcium phosphate (CaPi) granules (Chalmers and Nicholls 2003; Greenawalt et al. 1964; Kristian et al. 2007). The exact composition of these CaPi granules is unknown, but they are presumed to consist of mixtures of Ca3(PO4)2, Ca8H2(PO4)6, and Ca10(PO4)6(OH)2 (Thomas and Greenawalt 1968). The properties of this sequestration system are a current area of interest because the buffering system alters both Ca2+ uptake and release dynamics (Blomeyer et al. 2013), and may regulate mitochondrial permeability transition (mPTP) (Szabadkai and Duchen 2008; Wei et al. 2012), which in turn may regulate mitochondrial Ca2+ transients (Sareen et al. 2007). In the heart and other excitatory tissues, the mitochondrial Na+/Ca2+ exchanger (mNCE) is the primary pathway for Ca2+ extrusion (Boyman et al. 2013; Cai and Lytton 2004; Palty et al. 2004). Therefore, [Ca2+]m is primarily maintained by a balance of Ca2+ uptake facilitated by the MCU, Ca2+ extrusion by the mNCE, and Ca2+ buffering by the sequestration system. The sequestration system acts as a Ca2+ reservoir so that the activities of MCU and mNCE establish a set point that helps maintain sub-lethal thresholds of cytosolic Ca2+ during Ca2+ loading conditions.
Thus, the modes of mitochondrial Ca2+ uptake and their regulation remain obscure. Interestingly, in a recent study, we reported that mitochondrial Ca2+ uptake displays two different profiles of matrix Ca2+ transient when CaCl2 was added to mitochondrial suspension: a fast uptake of Ca2+ followed by a slower and more gradual uptake as matrix Ca2+ plateaus to a steady state (Boelens et al. 2013). Addition of Mg2+ to the respiration buffer appeared to have a differential effect on the two phases of the Ca2+ transient, with the slow phase attenuated more than the fast uptake phase. However, the impact of Mg2+ on mitochondria buffering of Ca2+ is not known. In the present study, we used a non-pharmacological approach to distinguish a slow and fast mode of mitochondrial Ca2+ uptake and determine the impact of extra-matrix Mg2+ on the modes of Ca2+ uptake and the mitochondrial buffering capacity or sequestration power of Ca2+. To further delineate the two modes of Ca2+ uptake, mitochondria were exposed to two different protocols based on the order by which mitochondria or CaCl2 were first added to the respiration buffer with and without added MgCl2.
Methods
Mitochondrial isolation
All experiments conformed to the Guide for the Care and Use of Laboratory Animals, and were approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee. Mitochondria from guinea pig hearts were isolated as previously described (Blomeyer et al. 2013; Boelens et al. 2013; Aldakkak et al. 2013; Haumann et al. 2010). Briefly, guinea pigs (250–350 g) were anesthetized with intraperitoneal injection of 30 mg ketamine, and 700 units of heparin for anticoagulation. Hearts were excised and minced in ice-cold isolation buffer containing 200 mM mannitol, 50 mM sucrose, 5 mM KH2PO4, 5 mM MOPS, 1 mM EGTA and 0.1 % bovine serum albumin (BSA). Buffer pH was adjusted to 7.15 with KOH. The minced pieces were suspended in 2.65 ml ice-cold buffer with 5 U/ml protease (from Bacillus licheniformis) and homogenized at low speed for 30 s. Afterwards, 17 ml of ice-cold isolation buffer was added, and the suspension was again homogenized for 30 s and centrifuged at 8000 g for 10 min. The supernatant was discarded, and the pellet was re-suspended in 25 ml of ice-cold isolation buffer and centrifuged at 900 g for 10 min. The supernatant was recovered and centrifuged once more at 8000 g to yield the final mitochondrial pellet, which was re-suspended in isolation buffer and kept on ice (4 °C) for experiments after fluorescent dye loading to measure matrix Ca2+ transients.
The mitochondrial protein concentration was measured using the Bradford method (1976) and diluted with isolation buffer to a protein concentration of 5 mg/ml and incubated with the appropriate fluorescent dye or the vehicle (DMSO). Incubated mitochondria were re-suspended in 25 ml of ice-cold isolation buffer and re-centrifuged at 8000 g. Subsequently, the dye-loaded pellet was re-suspended in cold isolation buffer, and the protein concentration was measured again using the Bradford method and diluted finally to 12.5 mg/ml. The final mitochondrial suspension was kept in packed ice (4 °C), and all subsequent experiments were conducted within 6 h after the last step of the isolation procedure.
Experimental groups and protocols
Isolated mitochondria were exposed to five different concentrations of CaCl2 and three different concentrations of MgCl2. In addition, each of these groups was exposed to two different Ca2+ loading protocols, resulting in 30 groups overall. The only difference between the two experimental protocols (A and B) was the order of addition of mitochondria and CaCl2 to the respiration buffer containing pyruvic acid. Extra-matrix (buffer) free Ca2+ ([Ca2+]e) and matrix free Ca2+ ([Ca2+]m) were monitored under identical experimental conditions for both protocols, except for the order of addition of CaCl2 or mitochondria to the buffer. For the NADH and ΔΨm measurements, the number of experimental groups was reduced to eight for each experimental protocol (A and B). Each experiment was conducted with mitochondria pooled from 2 hearts and repeated 3–4 times on different days. Respiration buffer, which was adjusted to pH 7.15 with KOH, contained 130 mM KCl, 5 mM K2HPO4, 20 mM MOPS, 0.1 % BSA, and 40 μM EGTA (Blomeyer et al. 2013; Haumann et al. 2010). The mitochondrial concentration for all experiments was 0.5 mg/mL. The pH of the respiratory buffer remains at 7.15 after the addition of CaCl2 and other substances, which was verified using a self-calibrating pH meter at the end of each experiment.
All experiments were conducted at room temperature (25 °C) using the cuvette-based fluorescence spectrophotometer described in our previous studies (Blomeyer et al. 2013; Boelens et al. 2013; Haumann et al. 2010; Agarwal et al. 2012, 2014; Aldakkak et al. 2011). Pyruvic acid (PA, 0.5 mM), cyclosporine A (CsA, 0.5 μM) and MgCl2 (0, 0.5, 1.0 mM) were always present in the buffer. Ruthenium red (RR, 1 μM) was added at 300 s after all of the Ca2+ bolus was taken up. The volume of the experimental buffer in the cuvette containing no added EGTA, and before adding any substance was 910 μL. The volume of added substances (PA, MgCl2, CaCl2, CsA, NaCl) to the cuvette was 10 μL each and the volume of the mitochondrial suspension added to the cuvette was 40 μL resulting in a total volume of 1000 μl for each experiment in protocols A and B. At a final volume of 1000 μL, the dilution factor after the addition of mitochondria was 25 times. Since the initial concentration of mitochondria following isolation was 12.5 mg/mL, the final mitochondrial suspension was 0.5 mg/mL in each experiment. The addition of a 40 μL volume of mitochondrial suspension, which had a concentration of 1 mM EGTA from the original mitochondrial suspension (12.5 mg/mL), resulted in a final EGTA concentration of approximately 40 μM for the mitochondrial suspension in the cuvette. The experimental buffer had a [Ca2+]e of approximately 50 nM that was similar to [Ca2+]m before adding any CaCl2.
The protocols (A and B) used to assess differences in [Ca2+] dynamics are shown in Fig.1. In protocol A at t = 30 s, 40 μL mitochondrial suspension was added to the experimental buffer. At t = 60 s, 0, 10, 20, 30 or 40 μM total CaCl2 (10 μL of concentrated solution) was added to the mitochondrial suspension. In protocol B at t = 30 s, 0, 10, 20, 30 or 40 μM total CaCl2 (10 μL of concentrated solution) was added first to the experimental buffer followed by addition of 40 μL mitochondrial suspension at t = 60 s. A high-speed magnetic stirring bar was turned on at the onset of each experiment to ensure prompt and rapid continuous mixing of the cuvette contents. With this approach, the effect of high initial localized boluses of [CaCl2] on mitochondria in protocol A was minimized. In both protocols at t = 300 s, 1 μM RR was added to block the MCU and to prevent Ca2+ re-uptake after its release via the NCE. At t = 360 s, 10 mM NaCl was added to induce Ca2+ efflux. The respiration buffer including the added PA was Na+-free, except for the NaCl added at the end of each protocol to elicit Ca2+ release via mNCE activation. To avoid differences in buffer volume, the vehicle (deionized H2O) was used for 0 mM MgCl2 and 0 μM CaCl2. Some additional experiments (not shown) were conducted in the presence of the mNCE inhibitor, CGP-37,157 (25 μM; Tocris Bioscience, Minneapolis, MN), to verify that the observed Na+-induced Ca2+ efflux was due only to mNCE activity. All chemicals were obtained from Sigma-Aldrich (St. Louis, MO) unless noted otherwise.
Fig. 1.
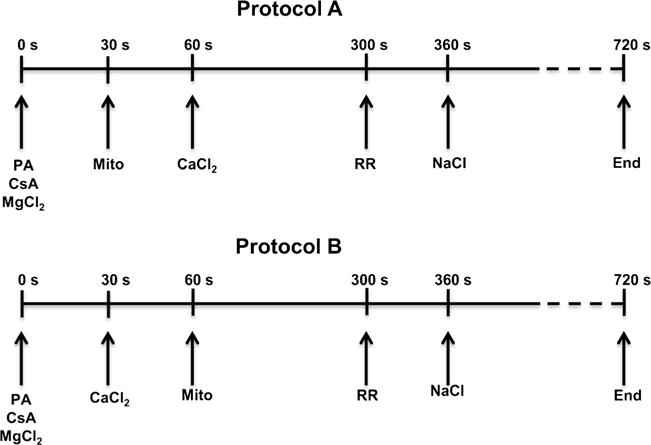
Timelines show the two experimental protocols used to characterize and quantify mitochondrial Ca2+ handling (influx, efflux, sequestration) in isolated guinea pig cardiac mitochondria. In protocol A, mitochondria were added to the experimental buffer before CaCl2 was added. In protocol B, mitochondria were added to respiration buffer with CaCl2 already present. All other additions were identical between protocols. 40 μM EGTA was present in all the experimental buffers; 0.5 μM CsA was added to all mitochondrial suspensions. Inset axes are the same as the main figure panels
Assessment of mitochondrial functional integrity
Before and after fluorescence measurements were made using the PTI spectrofluorometer, mitochondrial functional integrity/viability was assessed by measuring the respiratory control index (RCI) with a Clark type O2 electrode (MT200A, Strathkelvin Instruments, Glasgow, UK). The RCI of mitochondria energized with 0.5 mM PA was calculated by dividing the rate of state 3 respiration (250 μM ADP) by the rate of state 4 respiration. Before the start of fluorescent measurements, RCI values were above 12 and at the end of the measurements (after approximately 6 h) RCI values were revaluated and remained above 8. This strong RCI after 6 h indicates that the mitochondria were viable and well coupled for the duration of the experiments.
Fluorescence measurements
Fluorescence spectrophotometry (Qm-8, Photon Technology International, Birmingham, NJ) was used to assess levels of [Ca2+]e and [Ca2+]m, ΔΨm, and NADH. Fura-4F (Invitrogen™, Eugene, OR) was used to measure either [Ca2+]m (AM form) or [Ca2+]e (penta-potassium salt). For [Ca2+]m measurements, mitochondria were incubated for 30 min at room temperature (25 °C) with Fura-4F AM (5 μM) dissolved in DMSO. A final spin and resuspension of mitochondria were performed to remove any residual dye. To measure [Ca2+]e, Fura-4F penta-potassium (1 μM) was added to the respiration buffer. ΔΨm was measured using the lipophilic dye TMRM (1 μM, Invitrogen™, Eugene, OR) in a ratiometric excitation approach (Scaduto and Grotyohann 1999). NADH was measured by its autofluorescence (Blomeyer et al. 2013; Boelens et al. 2013; Haumann et al. 2010). To ensure identical conditions as for the [Ca2+]m measurement, mitochondria were incubated with the appropriate concentration of DMSO for 30 min at 25 °C before measuring [Ca2+]e, ΔΨm, and NADH. [Ca2+]e and ΔΨm were measured by directly adding the appropriate dye to the respiration buffer.
Measurement of free Ca2+
The Ca2+ measurements in this study represent the free matrix and extra-matrix Ca2+ for both protocols whereas the added CaCl2 (10–40 μM) represents the total amount of CaCl2 added to the cuvette. The molecular dye Fura-4F was used to measure concentrations of both matrix and extra-matrix free Ca2+. The vendor specified dissociation constant (Kd) of Fura-4F is 770 nM. However, when loaded in mitochondria, we determined a Kd of 890 nM. At an emission wavelength (λem) of 510 nm, the peak of the excitation wavelength (λex) of Fura-4F shifted from 380 nm to 340 nm on binding Ca2+ to the dye molecules. Measuring Ca2+ at these wavelengths was not influenced by background noise (e.g. NADH autofluorescence), so a background subtraction was unnecessary. For calibration, ratios were obtained when all Fura-4F had become bound to Ca2+ (Rmax) and when no Ca2+ was bound to Fura-4F (Rmin). To determine these ratios, experiments were conducted in PA energized mitochondria using 500 nM CsA, 500 μM CaCl2 for Rmax, and A23187 (Ca2+-ionophore) in the presence of 2.5 mM EGTA for Rmin. The [Ca2+]m was calculated using the following equation (Grynkiewicz et al. 1985):
| (1) |
Kd is 890 nM, Sf2 is the signal intensity of free Fura-4F measured at 380 nm, and Sb2 is the signal intensity of Ca2+-saturated Fura-4F measured at 380 nm. Their values were obtained from the Rmax and Rmin experiments, which were done for each preparation. For details, see Supplemental Figs. S1 and S2 for the fluorescence spectra in the presence of various amounts of Ca2+ and calibration of Fura-4F, respectively. Calibration of the Fura-4F penta-potassium signal in the presence of mitochondria was slightly different from the one used for Fura-4F AM. Rmin was measured in respiration buffer with added 100 μM EGTA without added CaCl2 to chelate all the Ca2+, while Rmax was determined in the respiration buffer containing 1 mM CaCl2. Ca2+ fluorescent measurements and calculation of [Ca2+]e were conducted in the same manner as described above for [Ca2+]m. Free [Mg2+] was not measured in this study. Our previous study showed that the calculated buffer [Mg2+] after adding MgCl2 was nearly identical to the [Mg2+] measured using Mag-Fura-2 fluorescence (Boelens et al. 2013).
Measurement of NADH
NADH autofluorescence signals were measured at λex of 350 nm and λem of 456 nm. For calibration, the NADH pool was either fully oxidized (0 %) with the respiratory uncoupler carbonyl cyanide 3-chlorophenylhydrazone (CCCP, 4 μM), or fully reduced (100 %) with the complex I blocker rotenone (10 μM).
Measurement of ΔΨm
Fluorescence changes using TMRM were detected by two λex (546 and 573 nm) and one λem (590 nm). Using the ratio of the emission at both excitation wavelengths (573/546) has the advantage of a higher dynamic range when compared to a single wavelength technique (Scaduto and Grotyohann 1999). At the end of each protocol at t = 720 s, CCCP (4 μM) was added to fully depolarize mitochondria.
Statistical analyses
The data from four hearts were pooled separately for each of the four fluorescence measurements. Data were transferred from PTI FelixGX (Version 3) into Microsoft® Excel® (2007). IBM® SPSS® (Version 19) was used to execute statistical analysis, which was performed using one-way analysis of variance followed by post hoc Tukey’s range test to examine differences among individual groups. Changes were considered statistically significant when the p-value was set at ≤0.05. Data for analyses were collected at the times given below and are presented as means ± SEM. For some analyses, results are presented as means ± standard deviation. Statistical tests were performed to compare changes in [Ca2+]m averaged from 60 to 70 s of all 30 possible treatment combinations for the five [CaCl2], three [MgCl2], and two protocol effects.
Calculation of mitochondrial Ca2+ buffering power
Initial rates of Ca2+ flux for both the uptake and extrusion phases were derived from the data and converted from d[Ca2+]e/dt to d[Ca2+]tot/dt using the method outlined in Bazil et al. (2013). Briefly, non-linear trend lines were fitted to the data shown in Figs. 2 and 4, and the analytical derivatives were used to compute the rate of change in free Ca2+. Trend line fits to the extra-matrix and matrix dynamics are shown in Supplemental Figs. S4 and S5, respectively. The extra-matrix rates were converted to total Ca2+ flux by using the known buffering power of EGTA. For simplicity, all Ca2+ uptake rates were assumed to be through the MCU and all Ca2+ release rates were assumed to be via the NCE. To compute the mitochondrial Ca2+ buffering power, the approach described in Bazil et al. (2013) was used. Specifically, the buffering power was calculated using the Ca2+ extrusion phase of the free Ca2+ dynamics profiles. The calculated Ca2+ buffering power was expressed as:
| (2) |
Fig. 2.
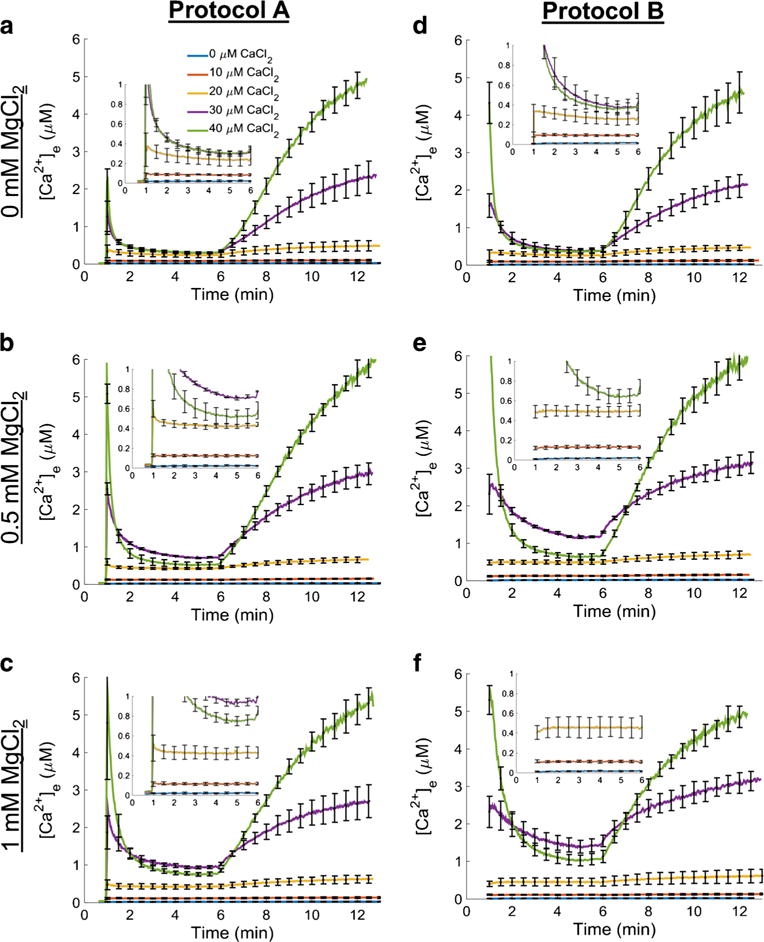
Extra-matrix free Ca2+ ([Ca2+]e) dynamics. Ca2+ uptake and Ca2+ release for each combination of CaCl2 and MgCl2 concentrations are shown using the protocol depicted in Fig. 1. CaCl2 was added to respiring mitochondrial suspension (left column; protocol A) or mitochondria were added to the buffer containing a given CaCl2 concentration (right column; protocol B) at 60 s followed by ruthenium red (RR) at 300 s and NaCl at 360 s. The results for protocol A are shown in the left column and the results for protocol B are shown in the right column. Each row corresponds to the buffer MgCl2 indicated on the left of each row. Insets show [Ca2+]e dynamics for 0, 10, and 20 μM CaCl2 in more detail. The axes are the same as the axes in the main figure panels. Error bars signify standard error of the mean
Fig. 4.
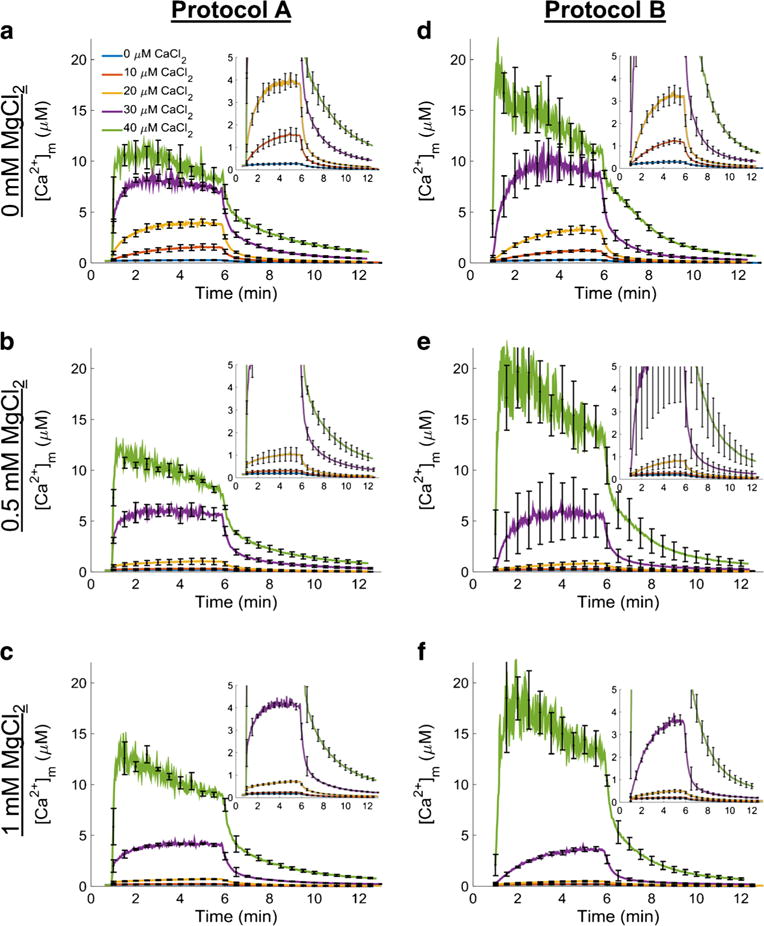
Matrix free Ca2+ ([Ca2+]m) dynamics. Ca2+ uptake and Ca2+ release for each combination of CaCl2 and MgCl2 concentrations are shown using the protocol depicted in Fig. 1. CaCl2 was added to the respiring mitochondrial suspension (left column; protocol A) or mitochondria were added to the buffer containing a given CaCl2 concentration (right column; protocol B) at 60 s followed by ruthenium red (RR) at 300 s and NaCl at 360 s. The results for protocol A are shown in the left column and the results for protocol B in the right column. In protocol B when CaCl2 was 40 μM, the fluorescent signal was close to Rmax, so the calculated [Ca2+]m is likely an overestimation of the true value of [Ca2+]m. Each row corresponds to the buffer MgCl2 indicated on the left of each row. Insets show [Ca2+]m dynamics for 0, 10, and 20 μM CaCl2 in more detail. The axes are the same as the axes in the main figure panels. Error bars signify standard error of the mean
Results
Changes in extra-matrix ionized Ca2+
The dynamics of extra-matrix free Ca2+ for each protocol (A and B) and experimental conditions are shown in Fig. 2. Fluorescence ratios were converted to concentrations using Eq. 1. Each panel consists of five different traces representing one concentration of CaCl2 added (0, 10, 20, 30, or 40 μM) at one of three fixed MgCl2 concentrations (0, 0.5, or 1.0 mM) in the presence of 40 μM EGTA. The dynamics of lower CaCl2 concentrations (0, 10, and 20 μM) are illustrated in more detail in the insets that show axes with the same labels seen in the main figure panels. Panels a–c show the Ca2+ dynamics profile using Protocol A, whereas panels d–f depict the Ca2+ dynamics profile using protocol B.
On a gross scale, the rates of decrease in [Ca2+]e (Ca2+uptake rates), magnitude of uptake before addition of RR, and the rates of increase in [Ca2+]e (Ca2+ efflux rates) after addition of NaCl, were similar in each protocol for the same [CaCl2] and [MgCl2] combination (Fig. 2). And as expected, Ca2+ uptake was attenuated when MgCl2 was included in the buffer. However, distinct differences in the rates of Ca2+ uptake between each protocol were revealed by careful analysis. Double exponential functions fit to the Ca2+ uptake dynamics for protocol A revealed that the overall dynamics consisted of two time scales as shown in Fig. 3. These trend lines were fit between 60 and 150 s of the early phase of the uptake as shown in Supplemental Fig. S4. A single exponential function was sufficient to characterize the Ca2+ uptake dynamics for protocol B for all [CaCl2] and [MgCl2] combinations except for 40 μM CaCl2 (data not shown). For this concentration of CaCl2, a double exponential function was a better fit with similar time constants for the 40 μM CaCl2 condition in protocol A. For additions of CaCl2 of 10 μM and below, the dynamics of [Ca2+]e were at or below detectable limits and were not used in the analysis. Furthermore, adding 40 μM CaCl2 in the presence of 0.5 or 1.0 mM MgCl2 in either protocol induced a faster Ca2+ uptake rate compared to 30 μM CaCl2, which resulted in lower levels of [Ca2+]e before addition of RR and NaCl.
Fig. 3.
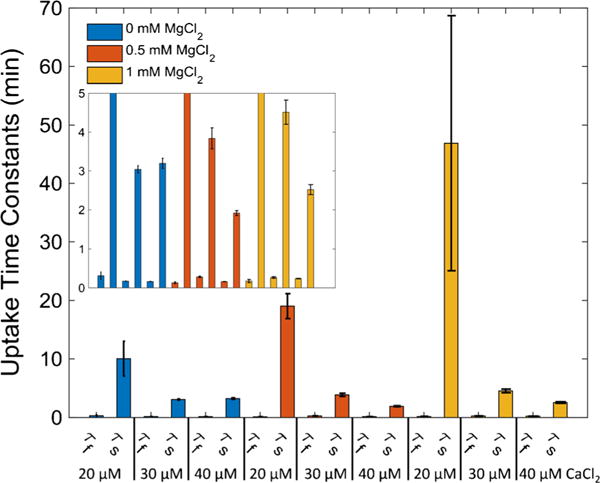
Two modes of Ca2+ uptake. For CaCl2 concentrations of 20 μM and greater, a double exponential function was fit between 65 s and 150 s to the [Ca2+]e dynamics observed in protocol A (Fig. 2). The fit time constants show that there was a fast and slow component of Ca2+ uptake associated with each bolus of CaCl2 administered. The inset axes labels are the same as the main figure panel. Error bars signify propagated standard deviations
Changes in matrix ionized Ca2+
Using the same protocols (A and B) shown in Fig. 1, CaCl2 was added and [Ca2+]m was measured. As with the extra-matrix Ca2+, the fluorescent ratios were converted to concentrations using Eq. 1. Fura-4F AM loading and washout was confirmed by ensuring that [Ca2+]m did not change after adding EGTA to the buffer (not shown). The results are displayed in Fig. 4 with a similar layout of the figure panels (a–f) shown in Fig. 2. Note that a very high [Ca2+]m was attained because CsA had been added to the buffer in all experiments. Panels a–c and d–f show [Ca2+]m dynamics at 0, 0.5 and 1.0 mM MgCl2 using protocol A and protocol B, respectively. Matrix free Ca2+ dynamics were quantitatively and qualitatively different for each protocol and CaCl2/MgCl2 combination. For protocol A (Fig. 4, panels a–c), adding CaCl2 (10, 20, 30, or 40 μM) induced an abrupt and rapid uptake of Ca2+. Adding lower [CaCl2] (10 and 20 μM, see insets) caused smaller increases in peak [Ca2+]m compared to the higher [CaCl2] (30 and 40 μM). Nonetheless, even at lower [CaCl2] (10 and 20 μM), there was an abrupt increase in Ca2+ uptake when CaCl2 was added to mitochondria. Note that in protocol A the Ca2+ uptake at any [CaCl2] showed an initial fast Ca2+ uptake phase followed by a slower gradual Ca2+ uptake phase. This fast uptake was not observed in protocol B.
Figure 5 shows the measured [Ca2+]m recorded over a few seconds (averaged from 60 to 70 s) for protocols A and B. For the 20 and 30 μM CaCl2 additions across all MgCl2 conditions using protocol A, there were statistically significant differences in [Ca2+]m when compared to the same CaCl2 and MgCl2 combination using protocol B (p ≤ 0.05). The presence of added MgCl2 in the respiration buffer attenuated the maximal [Ca2+]m in a concentration-dependent manner at each [CaCl2] except for the 40 μM CaCl2. For protocol B (Fig. 4, panels d–f), mitochondrial Ca2+ uptake was slower and more gradual at the lower [CaCl2] (10, 20, and 30 μM) as shown in the insets of Figs. 2 and 4. However, in the 40 μM CaCl2 group, adding mitochondria to respiration buffer with CaCl2 already present (protocol B) showed a similar fast Ca2+ uptake rate as observed in protocol A. After adding 40 μM CaCl2 in protocol B, the fluorescent signal was close to Rmax; thus the calculated [Ca2+]m was very noisy and likely not representative of the true value of [Ca2+]m. For protocol A and during exposure to low CaCl2 (10–30 μM), the [Ca2+]m attained during the slow uptake, but not the initial fast uptake phase, was significantly different among the MgCl2 additions for a given [CaCl2]. Overall, a comparison of these protocols shows that there exists a Mg2+-insensitive, fast component of Ca2+ uptake that appears only when CaCl2 is added to buffer already containing mitochondria, or when the [CaCl2] is high enough. The addition of RR (1 μM) completely inhibited Ca2+ uptake in both protocols A and B. Furthermore, at 40 μM CaCl2 the similarity in the rate of uptake between the two protocols is a verification to the equivalent rapidity of mixing of substances added to the cuvette.
Fig. 5.
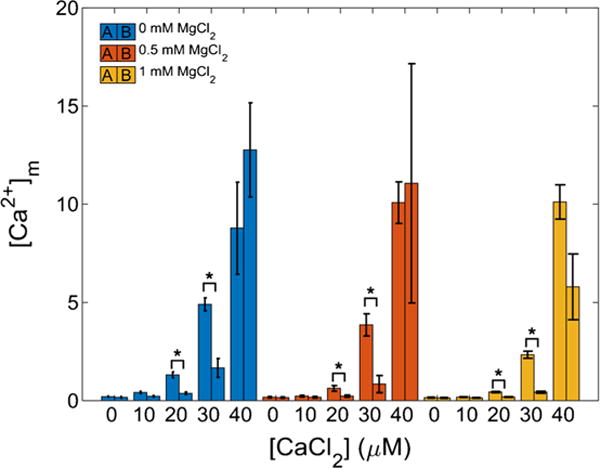
Ca2+ uptake by mitochondria. The amount of Ca2+ uptake by mitochondria depends on method of CaCl2 delivery. The bar plots show [Ca2+]m just after addition of CaCl2 to mitochondria (protocol A, left bar) or after addition of mitochondria to buffer containing CaCl2 (protocol B, right bar). These data correspond to a time of approximately 65 s. For all 20 and 30 μM CaCl2 conditions, the rate of Ca2+ uptake in protocol A was significantly different from that of protocol B (p ≤ 0.05) for most of the [CaCl2] and even in the presence of Mg2+. Error bars signify standard error of the mean
Changes in ΔΨm and NADH
In parallel studies, ΔΨm and NADH were measured to confirm that the differences observed in Ca2+ uptake dynamics were not due to changes in bioenergetics. Experimental conditions were simplified to consist of 0 and 1.0 mM MgCl2 and 0 and 40 μM CaCl2 combinations. Figure 6, panels a and b, and panels c and d, summarize changes in ΔΨm and NADH, respectively during protocol A and protocol B. Note that the starting point for the two variables in the figure were slightly different, with the start point at 30 and 60 s for protocols A and B, respectively. There were no significant differences observed in either ΔΨm or NADH between the two protocols. In addition, ΔΨm and NADH were not significantly affected by adding 40 μM CaCl2 (protocol A and B) in either the presence or absence of 1 mM MgCl2, which indicates that mitochondrial bioenergetics was not affected by the nature of mitochondrial exposure to CaCl2 (protocol A vs. protocol B) or MgCl2.
Fig. 6.
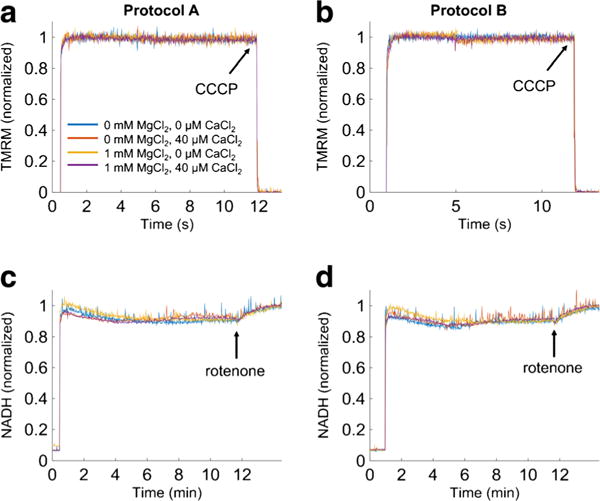
ΔΨm and NADH dynamics. The bioenergetics responses during protocols A and B were monitored in parallel using the ΔΨm sensitive dye TMRM and NADH autofluorescence. Traces are individual recordings. In averaged data there were no significant differences between the two MgCl2 and CaCl2 groups. The increases in signals mark the addition of mitochondria at 30 s (protocol A) and at 60 s (protocol B). The number of experimental groups was reduced to consist of only 0 and 1 mM MgCl2 and 0 and 40 μM CaCl2
Mitochondrial Ca2+ buffering power
The Ca2+ uptake dynamics obtained under protocols A and B that are shown in Figs. 2 and 4 were used to calculate the mitochondrial Ca2+ buffering power (βCa2+m) as a function of [Ca2+]m using Eq. 2 and shown in Fig. 7. At low [Ca2+]m, βCa2+m was nearly 3000:1 (total matrix [Ca2+]:free matrix [Ca2+]) but rose to 10,000:1 as [Ca2+]m was increased beyond 5 μM. The calculated βCa2+m values for [Ca2+]m below 500 nM were highly variable due to the [Ca2+]m measurement limitations discussed above. When MgCl2 was present, βCa2+m no longer increased as much and began to decrease as [Ca2+]m increased further in a concentration-dependent manner. Based on a two component-buffering model for matrix Ca2+, this decrease in βCa2+m is likely due to an effect of Mg2+ to reduce the Ca2+ binding capacity for the class 2 buffers. The effect of [Mg2+]e on the sequestration parameters is summarized in Table 1. Although higher [Ca2+]m were attained with protocol A due to increased MCU activity triggered by the activation of both fast and slow modes of Ca2+ uptake, βCa2+m was invariant with respect to either protocol A or B and was uniquely set both by [Ca2+]m and by [Mg2+]e.
Fig. 7.
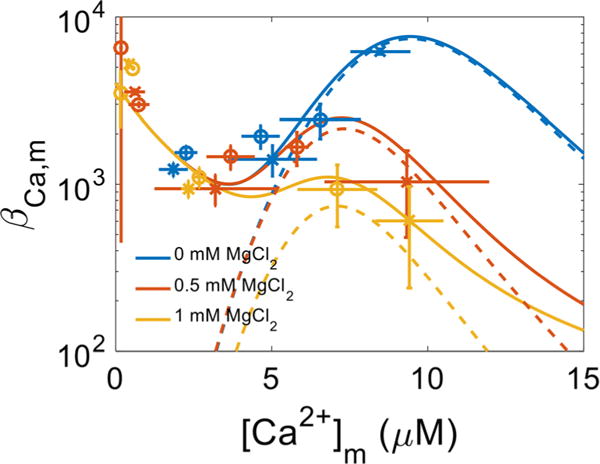
Mitochondrial Ca2+ buffering power. The Ca2+ sequestration system consists of at least two classes of buffers that bind Ca2+ with a differential affinity and capacity. Class 1 buffers are of the prototypical type whereby a single Ca2+ ion binds to a single site in an uncooperative manner. Class 2 buffers are atypical and bind multiple Ca2+ ions in a cooperative fashion. Class 1 buffers are not affected by Mg2+; however, in the presence of Mg2+, both the binding capacity and affinity of the Class 2 buffers are compromised. The blue, yellow and red colors correspond to the added 0 mM MgCl2, 0.5 mM MgCl2 and 1 mM MgCl2 conditions, respectively. The circles (O) and x’s (x) represent rates obtained using the data from Protocols A and B, respectively. The lines correspond to model simulations of the two classes of Ca2+ buffers using Eq. 2 with the parameters listed in Table 1. Error bars signify propagated standard deviations. Dashed lines represent contributions to mitochondrial buffering power from the class 2 buffers only
Table 1.
Mitochondrial Ca2+ sequestration system model parameters
| Parameter | Definition | Value | ||
|---|---|---|---|---|
| Class 1 | ||||
| [BCa,1]tot | Total Ca2+ buffer | 12 mM | ||
| KCa,1 | Buffer Ca2+ affinity | 3 μM | ||
| n1 | Hill coefficient | 1 | ||
| Class 2 | 0 Mg2+ | 0.5 mM Mg2+ | 1 mM Mg2+ | |
| [BCa,2]tot | Total Ca2+ buffer | 8 mM | 1.8 mM | 0.6 mM |
| KCa,2 | Buffer Ca2+ affinity | 10 μM | 7.75 μM | 7.5 μM |
| n2 | Hill coefficient | 6 |
Mg2+ attenuates both Ca2+ uptake and efflux
The data shown in Figs. 2 and 4 were also used to calculate the rates for the slow mode Ca2+ uptake by MCU and extrusion by mNCE under the experimental conditions used in this study. Note that these transport rates were calculated using JCa2+,tot = (d[Ca2+]m/dt)*βCa2+,m within 10 s of CaCl2 or NaCl-induced perturbations. Mg2+ was found to attenuate both the slow mode of Ca2+ uptake by MCU (Fig. 8a) and the release by mNCE (Fig. 8b). The slow mode Ca2+ uptake rates in protocol B were about 40 % of the slow mode Ca2+ uptake rates in protocol A for all three extra-matrix MgCl2 levels. The extrusion rates were independent of the Ca2+ loading protocol, i.e. protocol A vs. protocol B. These rates were estimated from the [Ca2+]e dynamics by averaging the rates between 75 and 90 s and 360–390 s for Ca2+ uptake and extrusion, respectively.
Fig. 8.
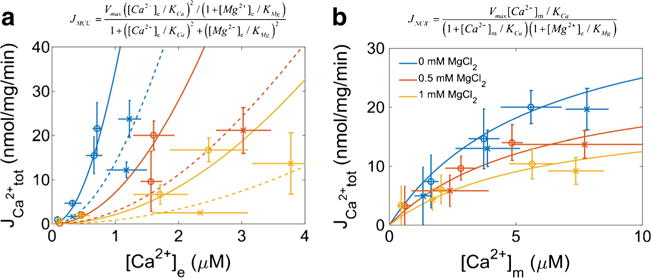
Mg2+ inhibition of Ca2+ uptake and extrusion. Panel 8a shows slow mode of Ca2+ uptake (via MCU) was attenuated by extra-matrix Mg2+ in the physiological concentration range. The model parameters for the simplified MCU model are: Vmax, 900 nmol/mg/min; KCa, 6 μM; and KMg, 0.3 mM. Solid lines correspond to slow mode of MCU rates observed during protocol A, and dotted lines correspond to the slow mode of MCU rates observed during protocol B. The rates of Ca2+ uptake in protocol B were approximately 40 % of the rates observed in protocol A. Panel 8b shows buffer Mg2+ in the physiological range used in this study also affected the rate of Ca2+ efflux (via mNCE). The model parameters for the simplified NCE are: Vmax, 40 nmol/mg/min; KCa, 5 μM; and KMg, 1 mM. The blue, yellow and red colors correspond to the 0 mM MgCl2, 0.5 mM MgCl2 and 1 mM MgCl2 conditions, respectively. Extra-matrix [Na+] was assumed constant (10 mM) and thus not included in the equation. The circles (O) and x’s (x) represent rates obtained using the data from Protocols A and B, respectively. The lines correspond to model simulations with the equations given above their respective panels. Error bars signify propagated standard deviations
Discussion
Mitochondrial Ca2+ uptake modes
In our examination of factors that affect mitochondrial Ca2+ handling (influx, efflux and sequestration), we measured extra-matrix and matrix free Ca2+ dynamics in isolated cardiac mitochondria using various combinations of [CaCl2] and [MgCl2] under two different Ca2+ loading protocols (Fig. 1). The order of addition, i.e. mitochondria before CaCl2 or mitochondria after CaCl2, resulted in significant differences in mitochondrial Ca2+ uptake dynamics. In protocol A, mitochondria displayed a fast Ca2+ uptake profile at each [CaCl2] that was followed by a slower and more gradual Ca2+ uptake profile when CaCl2 was added first as a bolus and mitochondria after that in the presence or absence of MgCl2. These two modes are evident from the time constant analyses summarized in Fig. 3 and the degree of Ca2+ uptake depicted in Fig. 5. In protocol B, when mitochondria were exposed to the buffer containing CaCl2 already present with or without MgCl2, Ca2+ uptake was slower and more gradual at CaCl2 concentrations below 40 μM. Therefore, when compared to protocol A, only the slower mode of Ca2+ uptake was apparent at the lower [CaCl2] in protocol B. These differences in Ca2+ uptake between the two protocols were not due to differences in ΔΨm as indicated in Fig. 4a, b. This finding is further supported by the data of Supplemental Fig. S3, which shows that in the absence of PA before adding CaCl2 first or mitochondria first, the different uptake modes remained evident as in the original two main protocols in which PA was present. The presence of extra-matrix MgCl2 markedly attenuated the slow, but not the fast, component of Ca2+ uptake and altered mitochondrial Ca2+ buffering (sequestration) power (Figs. 7 and 8). This is also evident by the decreasing effect of Mg2+ on the slow time constants shown in Fig. 3. Also, note that the fast time constants were relatively insensitive to Mg2+, which further supports the Mg2+ independent nature of the fast uptake shown in Fig. 5. These two potential modes of Ca2+ uptake are consistent with our recent computational modeling report on the dynamics of Ca2+ uptake (Tewari et al. 2014). Interestingly, the analysis also revealed that the slow mode was inhibited in protocol B reaching only 40 % of the rate observed during protocol A regardless of [Mg2+]e as shown in Fig. 8a (compare dashed lines vs. solid lines). We suggest that there are two distinct modes of Ca2+ uptake that occur at physiological levels of cytosolic Ca2+ and Mg2+. One mode is a fast, Mg2+-insensitive Ca2+ uptake pathway that could be modeled as a RyR-type channel (Tewari et al. 2014) and the other is a slower Mg2+-sensitive Ca2+ uptake pathway (Pradhan et al. 2011), that is also modulated by the mode of Ca2+ delivery. Moreover, the Mg2+-insensitive nature of the fast mode we found in the present study is compatible with the data on Mg2+-insensitive RaM of Ca2+ uptake reported by Gunter et al. (1998). Our findings are also consistent with the attenuating effect of Mg2+ on the slow mode of Ca2+ uptake reported earlier by our group (Boelens et al. 2013).
The focus of this study was not to characterize the regulatory action of the molecular components of the MCU complex; however, it suffices to note that the differences in uptake could be attributed to a differential regulation of MCU during exposure to different CaCl2 with and without MgCl2. It is possible that the marked differences in the two modes of Ca2+ uptake we found are attributable to the MCU complex and its regulatory subunits, e.g. MICU1 and MICU2 (Marchi and Pinton 2014; Patron et al. 2014; Mallilankaraman et al. 2012). MICU1 appears to act in response to high cytosolic [Ca2+] by stimulating Ca2+ uptake by MCU, whereas MICU2 appears to inhibit MCU functions at a lower cytosolic [Ca2+] (Patron et al. 2014; Kamer and Mootha 2014). However, major controversies abound in these observations, with conflicting results reported (Marchi and Pinton 2014; Patron et al. 2014; Mallilankaraman et al. 2012). Some of the discrepancies might be due to the composition of the buffer. For example, in one report Mg2+, an allosteric modulator of MCU activity was present, whereas in another study it was absent (Csordas et al. 2013; Mallilankaraman et al. 2012). And while incontrovertible evidence of MCU inhibition by Mg2+ exists (see, for example, Kirichok et al. 2004), the molecular components responsible remain unidentified. The ability of the Ca2+ delivery method to unmask different modes of MCU activity could be attributed to its effect on the state of the EF-hands located on the MICU1 and/or MICU2 regulatory subunits. For example, if the MICU1 and MICU2 subunits act in conjunction as a Ca2+-sensitive break mechanism (Patron et al. 2014; Kamer and Mootha 2014), a rapid change in Ca2+ near the subunits may temporarily release the break and result in a brief, rapid uptake of Ca2+. That said, the present study is focused on the observation of a fast and slow mode of Ca2+ uptake that is dependent on the manner by which mitochondria are exposed to a bolus of CaCl2, and not specifically on the regulation by MICU1, MICU2, and other regulatory proteins. Future elaborate studies in the contribution of these proteins to these two modes of Ca2+ in the presence and absence of Mg2+, and the impact on the mitochondrial Ca2+ buffering capacity should be undertaken.
It is evident that mitochondria took up Ca2+ in a quick and robust manner when CaCl2 was added to the mitochondrial suspension (protocol A). In contrast, the matrix free Ca2+ dynamics data showed that when mitochondria were added to respiration buffer with CaCl2 already present, the fast uptake mode was abolished for all CaCl2 concentrations except for the highest [CaCl2] (40 μM). Note that the order of adding 40 μM CaCl2 and mitochondria to the respiration buffer resulted in similar Ca2+ uptake profile for both protocols. This indicates that the rapid mixing of the cuvette content assured near instantaneous homogenous mixture, and that the difference in Ca2+ uptake between the two protocols was mostly due to the different modes of uptake in mitochondria and not to an uneven mix of the added CaCl2. The order of adding mitochondria or CaCl2 also did not affect the ΔΨ or redox state indicating that mitochondria were fully energized in the presence of PA when mitochondria were added before or after adding CaCl2. These observations provide further evidence that the method of exposure to Ca2+, and not bioenergetic differences such as ΔΨ, is the regulatory mechanism behind the vastly different uptake modes observed in our study. In addition, this difference in Ca2+ uptake was more apparent when MgCl2 was included in the respiration buffer of either Ca2+ loading protocol. This difference in the mode of Ca2+ uptake was also not dependent on the mitochondrial redox state because the NADH autofluorescence data (Fig. 6) showed no observable difference between the two protocols (A and B), in the presence or absence of added CaCl2 or MgCl2.
Based on these observations, we can conclude that Mg2+ regulates bulk Ca2+ uptake by attenuating mainly the slow component of the MCU-mediated Ca2+ uptake in a concentration-dependent manner. This conclusion is supported by our previous findings (Boelens et al. 2013) and recent computational models (Tewari et al. 2014; Bazil and Dash 2011) and indicates that the MCU is capable of operating in multiple conductance modes. In addition, the data revealed another intriguing aspect of the Ca2+-dependent regulation of mitochondrial Ca2+ uptake. As shown in Fig. 2, the 40 μM CaCl2 bolus resulted in a lower [Ca2+]e at the time of RR addition compared to the 30 μM CaCl2 bolus for either protocol. In other words, a higher bolus of CaCl2 resulted in more Ca2+ being taken up by mitochondria, which was inhibited by Mg2+ in a concentration-dependent manner. We speculate that this phenomenon is due to differential regulation or modulation of MCU conductance/open probability times by other components of the protein complex (e.g. MICU1 and MICU2) (Kamer and Mootha 2014; Harrington and Murphy 2015).
The Mg2+-sensitivity of the MCU complex likely plays a role in heart failure, ischemia and reperfusion injury, and other related events in the myocardium (for review see Douban et al. 1996; Kolte et al. 2014; Levitsky and Takahashi 2013). For example, cytosolic [Mg2+] is known to rise during ischemia due to net ATP hydrolysis. This rise in [Mg2+] would help mitigate mitochondrial Ca2+ loading via MCU, but the Ca2+ buffering system would be compromised, which could counter the Mg2+ effect and contribute to mitochondrial Ca2+ overload. In heart failure, the net decrease in cellular Mg2+ content would lead to more mitochondrial Ca2+ uptake via the MCU. Based on our results reported herein, the decrease in cytosolic [Mg2+] levels observed in these pathophysiological conditions, e.g. heart failure, would likely result in elevated [Ca2+]m. This would likely induce Ca2+-mediated mitochondrial dysfunction, and if the local cytosolic [Ca2+] is high, also possibly trigger mPTP opening with concomitant cell death. Indeed, it has been reported that the cardiovascular consequences of Mg2+ deficiency in clinical studies include multifocal necrosis with Ca2+ accumulation in mitochondria in a pattern suggestive of myocardial infarction (Seelig 1989).
MgCl2 and mitochondrial mNCE-induced Ca2+ efflux
The well-known effect of Mg2+ to attenuate Ca2+ uptake and its extrusion is demonstrated in greater detail by our data. The rate of increase in [Ca2+]m significantly decreased as a function of [Mg2+]e with an apparent Ki of 0.25 mM (Fig. 8a). This value is similar to that of our previously determined computational model of Ca2+ uptake (Pradhan et al. 2011). At 1 mM [Mg2+]e, close to the physiological extra-matrix level, the MCU complex was inhibited by greater than 15-fold at the peak of a 1 μM cytosolic Ca2+ transient. Despite this, [Ca2+]m reached close to 1 μM with addition of 20 μM CaCl2 (Fig. 4c) due to the rapid uptake mode by the MCU complex. Incidentally, [Mg2+]e also inhibits Na+-dependent Ca2+ efflux (Wingrove and Gunter 1986), which is consistent with some of our current findings (Fig. 8b). Here, it is shown that Mg2+ with a Ki of 1 mM inhibited the rate of decrease in [Ca2+]m. Moreover, the maximal rate of Ca2+ efflux was reduced to half when [Mg2+]e was increased from 0 mM to 1 mM. So not only is Ca2+ uptake slowed when there is an increase in [Mg2+]e, but also the rate of Ca2+ extrusion is impeded. Whereas it appears to be clear that [Mg2+]e regulates the slower phase of Ca2+ uptake, the Mg2+-insensitive rapid component of the MCU complex can lead to net Ca2+ uptake when mitochondria are presented with sufficiently high, pulse-like Ca2+ transients.
Mitochondrial Ca2+ buffering
The data from this study also corroborates previous reports that the mitochondrial Ca2+ sequestration system consists of at least two classes of buffers (Wei et al. 2012; Bazil et al. 2013; Blomeyer et al. 2013) (Fig. 7) and challenges recent findings (Wei et al. 2012; Tewari et al. 2014) that suggest the manner in which Ca2+ is loaded into mitochondria (fast vs. slow) modulates the degree of Ca2+ buffering. In our prior study, we used indo-1 to monitor [Ca2+] in both the extra-matrix and matrix compartments and mitochondrial Ca2+ buffering (Blomeyer et al. 2013). In contrast, in the current study we used Fura-4F because it can register higher [Ca2+] than indo-1 without saturating. Consequently, we also added CsA to the experimental buffer to prevent mPTP opening induced by high [Ca2+]m. As a result of using a different Ca2+ indicator dye and different experimental conditions, we observed somewhat different buffering parameters between the current and previous studies (Bazil et al. 2013; Blomeyer et al. 2013) that can be explained by the different extent of dye loading, cation binding properties between each type of Ca2+ fluorescence dye, and experimental conditions. Despite these differences, the general properties of the sequestration system identified in this study are identical to our previous reports (Bazil et al. 2013; Blomeyer et al. 2013). That is, the class 1 buffers are of the prototypical type and consist of binding sites that bind a single Ca2+ ion, whereas the class 2 buffers are capable of binding multiple Ca2+ ions at a single site in a cooperative fashion.
There is strong supporting evidence that the class 2 buffers consist of complexes formed by the annexin class of proteins in conjunction with acidic phospholipids in the membrane that promote or facilitate the formation of CaPi granules on the inner leaflets of the inner mitochondrial membrane (IMM) (for discussion, see Bazil et al. 2013). In our present study, we show that Mg2+ also modulates this class of Ca2+ buffers but does not regulate the class 1 buffers (Fig. 7). The mechanisms by which Mg2+ alters Ca2+ sequestration is unclear but may involve two mechanisms: i) extra-mitochondrial Mg2+ alters the class 2 buffer affinity for Ca2+ or ii) matrix Mg2+ encumbers the formation of CaPi complexes in the matrix on the surface of the IMM. We propose that latter is more likely. Based on earlier reports by Brierley et al. (1962, 1963, 1964), Mg2+ is actively loaded into energized mitochondria in a Pi-dependent and -independent manner. Therefore, Pi levels might modulate Mg2+ uptake during Ca2+ loading which would result in an increase in matrix Mg2+ content. However, Boelens et al. (2013) reported that increasing buffer MgCl2 did not change [Mg2+]m appreciably over time. In that study, Ca2+ uptake was absent, and the internal mechanisms regulating matrix Mg2+ were already in homeostasis. A plausible hypothesis is that Mg2+ enters mitochondria concomitantly with Ca2+ and interferes with CaPi granule formation. This is consistent with the observation that Mg2+ prevents and slows the spontaneous transition of CaPi from an amorphous state to a crystalline state (Wu et al. 2008, 2009). Nevertheless, our results demonstrate that when [Mg2+]e is elevated, the ability of mitochondria to sequester large amounts of Ca2+ is hindered.
Summary and perspective
The mechanism by which isolated mitochondria take up and sequester Ca2+ is a complex function not only of how much Ca2+ mitochondria are exposed to, but also the experimental conditions. In one scenario, when added CaCl2 exceeds a threshold, a rapid, Mg2+-insensitive mode of Ca2+ uptake is activated. In another scenario, when mitochondria are added to the buffer with low concentrations of EGTA and CaCl2 already present, the rapid mode of Ca2+ uptake is not activated, that is unless the buffer concentration of CaCl2 is sufficiently high. Thus, the MCU appears to consist of multiple conductance modes, which are differentially modulated by extra-matrix Mg2+.
Mitochondrial Ca2+ homeostasis plays important roles in cellular physiology, regulation of cellular bioenergetics, reactive oxygen species signaling and Ca2+ signaling. Agonist-induced intracellular Ca2+ signals can be rapidly transduced to the matrix because mitochondria can exist in close apposition to sites of Ca2+ release where local cytosolic [Ca2+] can be higher than in the rest of the cytoplasm (Mallilankaraman et al. 2012). The physiological implications of the different modes of Ca2+ uptake in this study are unclear but allude to a process whereby mitochondria can rapidly accumulate and sequester Ca2+ to prevent catastrophic cytosolic Ca2+ overload, mPTP opening and cell death. Therefore, a better understanding of the Ca2+ dynamics and how it is modulated by physiological [Mg2+] could provide new insights into the potential molecular mechanisms that regulate Ca2+ fluxes in and out of mitochondria under both physiological and pathophysiological conditions.
Supplementary Material
Acknowledgments
This work was funded by NIH grants R01-HL095122, P01-GM066730, and K99-HL121160.
Footnotes
This work was presented in podium form at the 2014 Biophysical Society meeting: Blomeyer CA, Bazil JN, Stowe DF, Dash RK, and Camara AKS. The potential for another calcium uptake mode in cardiac mitochondria. Biophysical Society Meeting, San Francisco, Feb. 15–19, 2014. [Biophys J 106(2), p.242a]
Electronic supplementary material The online version of this article (doi:10.1007/s10863-016-9644-1) contains supplementary material, which is available to authorized users.
References
- Agarwal B, Camara AK, Stowe DF, Bosnjak ZJ, Dash RK. Enhanced charge-independent mitochondrial free Ca(2+) and attenuated ADP-induced NADH oxidation by isoflurane: implications for cardioprotection. Biochim Biophys Acta. 2012;1817(3):453–465. doi: 10.1016/j.bbabio.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal B, Dash RK, Stowe DF, Bosnjak ZJ, Camara AK. Isoflurane modulates cardiac mitochondrial bioenergetics by selectively attenuating respiratory complexes. Biochim Biophys Acta. 2014;1837(3):354–365. doi: 10.1016/j.bbabio.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldakkak M, Camara AK, Heisner JS, Yang M, Stowe DF. Ranolazine reduces Ca2+ overload and oxidative stress and improves mitochondrial integrity to protect against ischemia reperfusion injury in isolated hearts. Pharmacol Res. 2011;64(4):381–392. doi: 10.1016/j.phrs.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldakkak M, Stowe DF, Dash RK, Camara AK. Mitochondrial handling of excess Ca2+ is substrate-dependent with implications for reactive oxygen species generation. Free Radic Biol Med. 2013;56:193–203. doi: 10.1016/j.freeradbiomed.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Varadarajan SG, Camara A, Chen Q, Novalija E, Gross GJ, et al. Blocking Na(+)/H(+) exchange reduces [Na(+)](i) and [Ca(2+)](i) load after ischemia and improves function in intact hearts. Am J Physiol Heart Circ Physiol. 2001a;281(6):H2398–H2409. doi: 10.1152/ajpheart.2001.281.6.H2398. [DOI] [PubMed] [Google Scholar]
- An J, Varadarajan SG, Novalija E, Stowe DF. Ischemic and anesthetic preconditioning reduces cytosolic [Ca2+] and improves Ca(2+) responses in intact hearts. Am J Physiol Heart Circ Physiol. 2001b;281(4):H1508–H1523. doi: 10.1152/ajpheart.2001.281.4.H1508. [DOI] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476(7360):341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazil JN, Dash RK. A minimal model for the mitochondrial rapid mode of Ca2+ uptake mechanism. PLoS One. 2011;6(6):e21324. doi: 10.1371/journal.pone.0021324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazil JN, Blomeyer CA, Pradhan RK, Camara AK, Dash RK. Modeling the calcium sequestration system in isolated guinea pig cardiac mitochondria. J Bioenerg Biomembr. 2013;45(3):177–188. doi: 10.1007/s10863-012-9488-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner G, Sharma VK, Giovannucci DR, Yule DI, Sheu SS. Identification of a ryanodine receptor in rat heart mitochondria. J Biol Chem. 2001;276(24):21482–21488. doi: 10.1074/jbc.M101486200. [DOI] [PubMed] [Google Scholar]
- Blomeyer CA, Bazil JN, Stowe DF, Pradhan RK, Dash RK, Camara AK. Dynamic buffering of mitochondrial Ca2+ during Ca2+ uptake and Na + -induced Ca2+ release. J Bioenerg Biomembr. 2013;45(3):189–202. doi: 10.1007/s10863-012-9483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boelens AD, Pradhan RK, Blomeyer CA, Camara AK, Dash RK, Stowe DF. Extra-matrix Mg2+ limits Ca2+ uptake and modulates Ca2+ uptake-independent respiration and redox state in cardiac isolated mitochondria. J Bioenerg Biomembr. 2013;45(3):203–218. doi: 10.1007/s10863-013-9500-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyman L, Williams GS, Khananshvili D, Sekler I, Lederer WJ. NCLX: the mitochondrial sodium calcium exchanger. J Mol Cell Cardiol. 2013;59:205–213. doi: 10.1016/j.yjmcc.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyman L, Chikando AC, Williams GS, Khairallah RJ, Kettlewell S, Ward CW, et al. Calcium movement in cardiac mitochondria. Biophys J. 2014;107(6):1289–1301. doi: 10.1016/j.bpj.2014.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brierley GP, Bachmann E, Green DE. Active transport of inorganic phosphate and magnesium ions by beef heart mitochondria. Proc Natl Acad Sci U S A. 1962;48:1928–1935. doi: 10.1073/pnas.48.11.1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley G, Murer E, Bachmann E, Green DE. Studies on ion transport. II. The accumulation of inorganic phosphate and magnesium ions by heart mitochondria. J Biol Chem. 1963;238:3482–3489. [PubMed] [Google Scholar]
- Brierley GP, Murer E, O’Brien RL. Studies on ion transport. Vi. The accumulation of Mg2+ by heart mitochondria in the absence of inorganic phosphate. Biochim Biophys Acta. 1964;88:645–647. [PubMed] [Google Scholar]
- Buntinas L, Gunter KK, Sparagna GC, Gunter TE. The rapid mode of calcium uptake into heart mitochondria (RaM): comparison to RaM in liver mitochondria. Biochim Biophys Acta. 2001;1504(2–3):248–261. doi: 10.1016/s0005-2728(00)00254-1. [DOI] [PubMed] [Google Scholar]
- Cai X, Lytton J. Molecular cloning of a sixth member of the K + -dependent Na+/Ca2+ exchanger gene family, NCKX6. J Biol Chem. 2004;279(7):5867–5876. doi: 10.1074/jbc.M310908200. [DOI] [PubMed] [Google Scholar]
- Carafoli E. The fateful encounter of mitochondria with calcium: how did it happen? Biochim Biophys Acta. 2010;1797(6–7):595–606. doi: 10.1016/j.bbabio.2010.03.024. [DOI] [PubMed] [Google Scholar]
- Chalmers S, Nicholls DG. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J Biol Chem. 2003;278(21):19062–19070. doi: 10.1074/jbc.M212661200. [DOI] [PubMed] [Google Scholar]
- Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab. 2013;17(6):976–987. doi: 10.1016/j.cmet.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476(7360):336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedkova EN, Blatter LA. Calcium signaling in cardiac mitochondria. J Mol Cell Cardiol. 2013;58:125–133. doi: 10.1016/j.yjmcc.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW, 2nd, Maack C. SR and mitochondria: calcium cross-talk between kissing cousins. J Mol Cell Cardiol. 2013;55:42–49. doi: 10.1016/j.yjmcc.2012.07.015. [DOI] [PubMed] [Google Scholar]
- Douban S, Brodsky MA, Whang DD, Whang R. Significance of magnesium in congestive heart failure. Am Heart J. 1996;132(3):664–671. doi: 10.1016/s0002-8703(96)90253-7. [DOI] [PubMed] [Google Scholar]
- Greenawalt JW, Rossi CS, Lehninger AL. Effect of active accumulation of calcium and phosphate ions on the structure of rat liver mitochondria. J Cell Biol. 1964;23:21–38. doi: 10.1083/jcb.23.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260(6):3440–3450. [PubMed] [Google Scholar]
- Gunter TE, Buntinas L, Sparagna GC, Gunter KK. The Ca2+ transport mechanisms of mitochondria and Ca2+ uptake from physiological-type Ca2+ transients. Biochim Biophys Acta. 1998;1366(1–2):5–15. doi: 10.1016/s0005-2728(98)00117-0. [DOI] [PubMed] [Google Scholar]
- Harrington JL, Murphy E. The mitochondrial calcium uniporter: mice can live and die without it. J Mol Cell Cardiol. 2015;78:46–53. doi: 10.1016/j.yjmcc.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haumann J, Dash RK, Stowe DF, Boelens AD, Beard DA, Camara AK. Mitochondrial free [Ca2+] increases during ATP/ADP antiport and ADP phosphorylation: exploration of mechanisms. Biophys J. 2010;99(4):997–1006. doi: 10.1016/j.bpj.2010.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huser J, Blatter LA, Sheu SS. Mitochondrial calcium in heart cells: beat-to-beat oscillations or slow integration of cytosolic transients? J Bioenerg Biomembr. 2000;32(1):27–33. doi: 10.1023/a:1005556227425. [DOI] [PubMed] [Google Scholar]
- Kamer KJ, Mootha VK. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 2014;15(3):299–307. doi: 10.1002/embr.201337946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427(6972):360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- Kolte D, Vijayaraghavan K, Khera S, Sica DA, Frishman WH. Role of magnesium in cardiovascular diseases. Cardiol Rev. 2014;22(4):182–192. doi: 10.1097/CRD.0000000000000003. [DOI] [PubMed] [Google Scholar]
- Kristian T, Pivovarova NB, Fiskum G, Andrews SB. Calcium-induced precipitate formation in brain mitochondria: composition, calcium capacity, and retention. J Neurochem. 2007;102(4):1346–1356. doi: 10.1111/j.1471-4159.2007.04626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, et al. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 2015;12(1):15–22. doi: 10.1016/j.celrep.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitsky DO, Takahashi M. Interplay of Ca(2+) and Mg (2+) in sodium-calcium exchanger and in other Ca(2+)-binding proteins: magnesium, watchdog that blocks each turn if able. Adv Exp Med Biol. 2013;961:65–78. doi: 10.1007/978-1-4614-4756-6_7. [DOI] [PubMed] [Google Scholar]
- Lu X, Ginsburg KS, Kettlewell S, Bossuyt J, Smith GL, Bers DM. Measuring local gradients of intramitochondrial [Ca(2+)] in cardiac myocytes during sarcoplasmic reticulum Ca(2+) release. Circ Res. 2013;112(3):424–431. doi: 10.1161/CIRCRESAHA.111.300501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 2015;12(1):23–34. doi: 10.1016/j.celrep.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012;151(3):630–644. doi: 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Pinton P. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol. 2014;592(Pt 5):829–839. doi: 10.1113/jphysiol.2013.268235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondria and calcium signaling. Cell Calcium. 2005;38(3–4):311–317. doi: 10.1016/j.ceca.2005.06.011. [DOI] [PubMed] [Google Scholar]
- O’Rourke B, Blatter LA. Mitochondrial Ca2+ uptake: tortoise or hare? J Mol Cell Cardiol. 2009;46(6):767–774. doi: 10.1016/j.yjmcc.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Ohana E, Hershfinkel M, Volokita M, Elgazar V, Beharier O, et al. Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J Biol Chem. 2004;279(24):25234–25240. doi: 10.1074/jbc.M401229200. [DOI] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15(12):1464–1472. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, et al. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell. 2014;53(5):726–737. doi: 10.1016/j.molcel.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan RK, Qi F, Beard DA, Dash RK. Characterization of Mg2+ inhibition of mitochondrial Ca2+ uptake by a mechanistic model of mitochondrial Ca2+ uniporter. Biophys J. 2011;101(9):2071–2081. doi: 10.1016/j.bpj.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasola A, Bernardi P. Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium. 2011;50(3):222–233. doi: 10.1016/j.ceca.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Rhodes SS, Ropella KM, Audi SH, Camara AK, Kevin LG, Pagel PS, et al. Cross-bridge kinetics modeled from myoplasmic [Ca2+] and LV pressure at 17 degrees C and after 37 degrees C and 17 degrees C ischemia. Am J Physiol Heart Circ Physiol. 2003;284(4):H1217–H1229. doi: 10.1152/ajpheart.00816.2002. [DOI] [PubMed] [Google Scholar]
- Sareen D, Darjatmoko SR, Albert DM, Polans AS. Mitochondria, calcium, and calpain are key mediators of resveratrol-induced apoptosis in breast cancer. Mol Pharmacol. 2007;72(6):1466–1475. doi: 10.1124/mol.107.039040. [DOI] [PubMed] [Google Scholar]
- Scaduto RC, J, Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J. 1999;76(1 Pt 1):469–477. doi: 10.1016/S0006-3495(99)77214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedova M, Dedkova EN, Blatter LA. Integration of rapid cytosolic Ca2+ signals by mitochondria in cat ventricular myocytes. Am J Physiol Cell Physiol. 2006;291(5):C840–C850. doi: 10.1152/ajpcell.00619.2005. [DOI] [PubMed] [Google Scholar]
- Seelig M. Cardiovascular consequences of magnesium deficiency and loss: pathogenesis, prevalence and manifestations–magnesium and chloride loss in refractory potassium repletion. Am J Cardiol. 1989;63(14):4G–21G. doi: 10.1016/0002-9149(89)90213-0. [DOI] [PubMed] [Google Scholar]
- Sparagna GC, Gunter KK, Sheu SS, Gunter TE. Mitochondrial calcium uptake from physiological-type pulses of calcium. A description of the rapid uptake mode. J Biol Chem. 1995;270(46):27510–27515. doi: 10.1074/jbc.270.46.27510. [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda) 2008;23:84–94. doi: 10.1152/physiol.00046.2007. [DOI] [PubMed] [Google Scholar]
- Tarasov AI, Griffiths EJ, Rutter GA. Regulation of ATP production by mitochondrial Ca(2+) Cell Calcium. 2012;52(1):28–35. doi: 10.1016/j.ceca.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari SG, Camara AK, Stowe DF, Dash RK. Computational analysis of Ca2+ dynamics in isolated cardiac mitochondria predicts two distinct modes of Ca2+ uptake. J Physiol. 2014;592(Pt 9):1917–1930. doi: 10.1113/jphysiol.2013.268847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RS, Greenawalt JW. Microincineration, electron microscopy, and electron diffraction of calcium phosphate-loaded mitochondria. J Cell Biol. 1968;39(1):55–76. doi: 10.1083/jcb.39.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadarajan SG, An J, Novalija E, Smart SC, Stowe DF. Changes in [Na(+)](i), compartmental [Ca(2+)], and NADH with dysfunction after global ischemia in intact hearts. Am J Physiol Heart Circ Physiol. 2001;280(1):H280–H293. doi: 10.1152/ajpheart.2001.280.1.H280. [DOI] [PubMed] [Google Scholar]
- Vasington FD, Murphy JV. Ca2+ ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J Biol Chem. 1962;237:2670–2677. [PubMed] [Google Scholar]
- Wei AC, Liu T, Winslow RL, O’Rourke B. Dynamics of matrix-free Ca2+ in cardiac mitochondria: two components of Ca2+ uptake and role of phosphate buffering. J Gen Physiol. 2012;139(6):465–478. doi: 10.1085/jgp.201210784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingrove DE, Gunter TE. Kinetics of mitochondrial calcium transport. II. A kinetic description of the sodium-dependent calcium efflux mechanism of liver mitochondria and inhibition by ruthenium red and by tetraphenylphosphonium. J Biol Chem. 1986;261(32):15166–15171. [PubMed] [Google Scholar]
- Wu LN, Genge BR, Wuthier RE. Analysis and molecular modeling of the formation, structure, and activity of the phosphatidylserine-calcium-phosphate complex associated with biomineralization. J Biol Chem. 2008;283(7):3827–3838. doi: 10.1074/jbc.M707653200. [DOI] [PubMed] [Google Scholar]
- Wu LN, Genge BR, Wuthier RE. Differential effects of zinc and magnesium ions on mineralization activity of phosphatidylserine calcium phosphate complexes. J Inorg Biochem. 2009;103(7):948–962. doi: 10.1016/j.jinorgbio.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, Chen B, et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun. 2015;6:6081. doi: 10.1038/ncomms7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.