

ABSTRACT
Antigen-binding fragments (Fab) and F(ab′)2 antibodies serve as alternative formats to full-length anti-bodies in therapeutic and immune assays. They provide the advantage of small size, short serum half-life, and lack of effector function. Several proteases associated with invasive diseases are known to cleave antibodies in the hinge-region, and this results in anti-hinge antibodies (AHA) toward the neoepitopes. The AHA can act as surrogate Fc and reintroduce the properties of the Fc that are otherwise lacking in antibody fragments. While this response is desired during the natural process of fighting disease, it is commonly unwanted for therapeutic antibody fragments. In our study, we identify a truncation in the lower hinge region of the antibody that maintains efficient proteolytic cleavage by IdeS protease. The resulting neoepitope at the F(ab′)2 C-terminus does not have detectable binding of pre-existing AHA, providing a practical route to produce F(ab′)2 in vitro by proteolytic digestion when the binding of pre-existing AHA is undesired. We extend our studies to the upper hinge region of the antibody and provide a detailed analysis of the contribution of C-terminal residues of the upper hinge of human IgG1, IgG2 and IgG4 to pre-existing AHA reactivity in human serum. While no pre-existing antibodies are observed toward the Fab of IgG2 and IgG4 isotype, a significant response is observed toward most residues of the upper hinge of human IgG1. We identify a T225L variant and the natural C-terminal D221 as solutions with minimal serum reactivity. Our work now enables the production of Fab and F(ab′)2 for therapeutic and diagnostic immune assays that have minimal reactivity toward pre-existing AHA.
KEYWORDS: Antibody engineering, epitope mapping, immunoglobulin G (IgG), immunogenicity, monoclonal antibody, protease
Introduction
Antibodies are composed of two antigen-binding fragments (Fab) that are connected by a flexible hinge-region to the Fc. While the Fab mediates recognition and binding of the antigen, the Fc mediates effector function by engagement with Fcγ receptors 1 and confers long serum half-life by binding to the salvage receptor, FcRn.2 In particular, the long in vivo half-life of IgG contributes to the success of antibodies as therapeutics because it enables less frequent dosing compared to other biotherapeutics. Consequently, the majority of approved therapeutic antibodies are full-length IgGs. Unlike IgG, the serum half-life of an isolated Fab is short,3 which can be useful for some indications. Three Fab products are approved by the US Food and Drug Administration. One therapeutic Fab molecule directed against platelet surface receptor GPIIb/IIIa (abciximab, ReoPro®) is commercially produced by proteolytic cleavage with papain,4 which is the original method of Fab production.5 With advances in molecular cloning, recombinant expression of antibody fragments became a more attractive route to generate Fab molecules as exemplified by 2 other approved Fab therapeutics, anti-vascular endothelial growth factor (VEGF) ranibizumab (Lucentis®) 6 and anti-dabigatran idarucizumab (Praxbind®) 7 and the approved pegylated Fab′, anti-tumor necrosis factor certolizumab pegol (Cimzia®).8 Fab molecules are advantageous when transient systemic activity that doesn't persist past dosing is desired, lack of Fc-mediated effects is wanted, or when administration and activity are localized to a peripheral compartment such as the eye.
Generating Fab molecules by proteolytic digestion defines the C-terminal sequence of the Fab heavy chain by the protease cleavage site. In turn, a Fab molecule typically includes a part of the upper hinge of the antibody. This upper hinge region of the antibody serves as the linker between Fab and Fc region, but has no structural or functional role in a Fab molecule. It can be considered as an unstructured appendix (Fig. 1A) as it is often not fully resolved in crystal structures of Fab molecules. In contrast to the proteolytic digestion as a production route, the recombinant expression of Fab molecules provides flexibility in defining the length of the included upper hinge region. In this study, we leverage these engineering benefits and study the binding of pre-existing anti-hinge antibodies (AHA) to residues of the upper hinge.
Figure 1.

Pre-existing human antibodies to the Fab of human IgG1, IgG2 and IgG4. (A) X-ray crystal structure of the Fab region (PDB: 1HZH) including the upper hinge; light chain (gray), heavy chain (blue), interchain disulfide (orange), and upper hinge (magenta). In the isolated Fab molecule the upper hinge is a protruding unstructured region without structural and functional role. The residues of the upper hinge are displayed in magenta to indicate the T225L mutation (black) that is perturbing binding to pre-existing AHA. Numbering of residues is according to EU numbering nomenclature. (B) Pooled human serum was incubated with human IgG1 Fab with different upper hinge lengths and ends. Binding pre-existing antibodies detected by anti-Fc ELISA. Truncating the Fab C-terminus to D221 (D) and the C-terminal variant T225L (DKTHL) greatly reduced binding of pre-existing antibodies to almost background. Strong response is observed toward T223 as the C-terminal residue (DKT), coinciding with the cleavage site of human neutrophil elastase. The mean value of the individual data points is represented by the horizontal line. (C) Three different Fabs were incubated with pooled human serum and binding of pre-existing antibodies detected by ELISA. Significant signal is observed for different Fabs with DKTHT C-terminus. Reduced binding of pre-existing antibodies to the D221 and T225L C-terminus is detected across different Fabs. Fab-1 includes the antibody variable domain used in (B) and all other AHA binding experiments throughout this study. Fab-2 and Fab-3 are antibodies toward different antigens. (D) Pooled human serum was incubated with human IgG2 Fab and IgG4 Fabs with different upper hinge lengths and binding antibodies detected by ELISA. No pre-existing antibodies can be detected to the upper hinge of human IgG2 and IgG4.
The apparent high affinity of an antibody is often enabled by bivalent target engagement, facilitating avidity. In contrast, the target engagement of a Fab is monovalent. This often leads to lower target affinity compared to the full-length IgG. By joining two Fabs to create a F(ab′)2, avidity can be restored while preserving critical properties of the Fab, such as short serum half-life. In addition, targeting multiple disease mediators by bispecific antibodies has become increasingly important for therapeutic antibody development.9 F(ab′)2 molecules can provide a natural scaffold to produce small bispecific antibody fragments. In contrast to the production of Fab, the recombinant expression of F(ab′)2 is not possible without fusion to a non-native homo- or heterodimerization domain.10,11 Hence, there are two main approaches to generate F(ab′)2 molecules: 1) chemical conjugation and 2) proteolytic digestion. For chemical conjugation, recombinantly generated Fab′ molecules are coupled by homo- or heterobifunctional crosslinkers.3,10,12,13 Analogous to the proteolytic digestion approach to produce Fab molecules, a number of proteases are known to cleave the intact antibody in the lower hinge region to produce F(ab′)2 molecules.14 Such proteolytic digestion results in F(ab′)2 molecules where the 2 Fab molecules are connected by the disulfide-bonds of the core-hinge. Pepsin is most widely used,15 but a highly specific IgG-degrading enzyme of Streptococcus pyogenes, IdeS, has been described more recently.16,17 The use of IdeS enables generation of highly homogenous product by eliminating the C-terminal heterogeneity observed from pepsin digest.3
Production of proteases against the antibody hinge region has been implicated as a mechanism by which pathogens and tumor cells attempt to evade the host immune response.14 However, resulting C-terminal neoepitopes are eventually recognized by the immune system and AHA are generated. Such autoantibodies to the upper-hinge region of the Fab and the lower-hinge region of the F(ab′)2 have been previously demonstrated.18-21 The pre-existing AHA titers from healthy individuals vary from donor to donor 20 and may represent past and current exposure to the neoepitopes derived from infection and inflammation. The AHA can act as surrogate Fc and restore effector function of the otherwise proteolytically inactivated antibody,22 but may introduce potential safety concerns to a Fab or F(ab′)2 molecule for therapeutic use.
In this work, we provide a detailed characterization of the specificity of pre-existing AHA toward the residues of the upper hinge of the Fabs of human IgG1, IgG2 and IgG4 isotypes. While we do not observe pre-existing antibodies to the upper hinge of IgG2 and IgG4 Fab, we found that the T225L variant and the C-terminal D221 can be used to avoid reactivity with pre-existing AHA for the IgG1 Fab. To avoid reactivity of F(ab′)2 molecules with pre-existing AHA while maintaining efficient proteolytic cleavage by IdeS for production, we engineer the lower hinge of the IgG. With this engineering solution, it is now possible to generate F(ab′)2 molecules that are devoid of reactivity with pre-existing AHA in human serum and potentially provide superior safety in a therapeutic setting by minimizing immune responses following drug treatment.
Results
Fab C-terminus determines response to pre-existing AHA
Reactivity of autoantibodies in human serum toward the upper hinge of Fab molecules was originally studied with a papain-cleaved antibody, abciximab.4 Papain cleavage leaves a C-terminal H224 on the Fab. A more comprehensive study was later carried out using biotinylated peptide analogs to dissect the contribution of individual C-terminal residues of the upper hinge.20 In this study, only minimal AHA reactivity toward the upper hinge residues K222 through H224 was observed. No signal was observed toward peptides with T225 as the C-terminal residue. The use of synthetic peptides may confound results because the Fab-tail spanning the upper hinge residues D221 to T225 (Fig. 1A) is presented outside of the context of the intact molecule. Thus, we decided to study the Fab-tail contribution for binding to pre-existing AHA in the setting of an intact Fab. The recombinant expression of Fab molecules in E. coli and mammalian cells allows production of molecules with defined C-terminal ends without the need of proteolytic cleavage. To ensure the integrity of the C-terminus, correct mass of the purified Fab was confirmed by intact mass-spectrometry (data not shown). The Fab molecules were coated on microtiter plates, and after incubation with pooled human donor serum, binding of pre-existing AHA was quantified by anti-Fc detection. In agreement with the previous study,20 T223 at the C-terminus (DKT) showed the highest reactivity of all upper hinge variants toward pre-existing AHA (Fig. 1B). Significant difference to the previous studies is observed with T225 at the C-terminus (DKTHT). This variant did not bind AHA as peptide; 20 however, we observed substantial AHA reactivity when tested as Fab. With D221 at the C-terminus (D), binding of AHA was reduced to almost background (Fig. 1B). Two other Fab molecules also had substantial AHA signals with T225 at the C-terminus and much reduced AHA signals with D221 at the C-terminus (Fig. 1C). The lower AHA signals seen in Fab-1 compared to the other 2 Fab molecules were at least partially due to a slightly lower coating efficiency (data not shown). Thus, terminating the Fab at D221 provides a solution to minimize recognition by pre-existing AHA while maintaining a natural antibody sequence.
As demonstrated by these experiments, the C-terminal Fab residue has a profound effect on AHA binding. Next, we wanted to determine if the binding by pre-existing antibodies could also be obviated by a single amino acid change at the C-terminus and provide an alternative route to the D221 to minimize reactivity toward AHA. We introduced a T225L change to place a non-germline residue at the Fab C-terminus, and tested for binding of AHA. The T225L variant was described earlier in an anti-VEGF Fab (ranibizumab).6 The variant perturbed binding of the pre-existing AHA (Fig. 1B), further highlighting the importance of the C-terminus for binding. To ensure that the reduced AHA binding to T225L was not due to reduced coating efficiency, we also used an antigen capture format to capture Fab molecules and observed a similar fold of AHA signal reduction (data not shown). In addition, the reduced AHA binding to T225L translated across different Fab molecules (Fig. 1C), further demonstrating that this is a broadly applicable tool.
We extended our studies to Fab molecules in IgG2 and IgG4 isotypes. While IgG1, IgG2 and IgG4 are commonly used for therapeutic antibodies, the use of IgG2 and IgG4 Fabs has not been leveraged for therapeutic development so far. Thus, we tested IgG2 and IgG4 Fabs with the C-terminus K218 and P225, respectively (Fig. 1D). In contrast to IgG1 Fab, IgG2 and IgG4 Fabs were not recognized by pre-existing AHA. While we can exclude prevalent reactivity toward IgG2 and IgG4 isotypes, reactivity present in a smaller specific subset of the general population is still possible since it has been previously demonstrated that the prevalence of AHA can differ between healthy donors and rheumatoid arthritis patients.23 The IgG2 does not have any upper hinge to consider possible pre-existing AHA binding to a different C-terminus, but the IgG4 upper hinge was considered for the C-terminus effect seen with IgG1. The length of the IgG4 isotype upper hinge is shorter compared to IgG1; however, since the cysteine involved in the heavy-light interchain disulfide is located in the center of the CH1 primary structure, we were able to include residues K218 and Y219 of the CH1 domain in our truncation experiments. These truncated IgG4 upper hinge Fabs displayed a signal similar to the intact upper hinge (Fig 1B).
All Fab molecules within the same isotype yielded similar expression levels in E. coli and Chinese hamster ovary (CHO) cells (data not shown), and no effect on thermal stability was observed (data not shown). However, overall thermal stability of IgG2 and IgG4 Fabs was decreased by about 6°C compared to IgG1 isotype. In conclusion, multiple Fab formats with minimal reactivity toward pre-existing AHA exist: IgG1-D221, IgG1-T225L, IgG2 and IgG4.
IgG1 with the lower hinge of IgG2 cannot be cleaved efficiently
Pre-existing AHA toward the lower hinge region of F(ab′)2 have been extensively described in the literature.14,24 Analogous to the AHA to the upper hinge of Fab molecules, these AHA can act as surrogate Fc or introduce assay artifacts. Thus, development of a F(ab′)2 format that impedes AHA binding is desirable. While AHA in serum are found toward F(ab′)2 of IgG1 isotype, it has not been possible to establish the existence of autoantibodies to the lower hinge of IgG2 isotype. Interestingly, the lack of such autoantibodies coincides with the inability of physiologically relevant human proteases to efficiently cleave IgG2 into a F(ab′)2.25 Inefficient cleavage of IgG2 has been observed using IdeS protease,25 an IgG specific endoprotease from Streptococcus pyrogens that cleaves after G236(Fig. 2A).17 In addition to the cleavage site in the antibody hinge region, it recognizes a second site in the Fc that contributes to its high specificity toward IgG.16,17 The inefficient cleavage of IgG2 antibodies could be caused by the exosite outside of the cleavage site. Thus, we grafted the lower hinge residues of IgG2 onto IgG1 to create an IgG1-2 chimera (Fig. 2B). We tested the cleavage efficiency at different IdeS:IgG ratios (Fig. 2C). While IgG1 wild-type is efficiently cleaved into F(ab′)2 at an IdeS:IgG ratio of 1:500, at least 50-fold higher protease concentration is necessary to achieve similar cleavage of the IgG1-2 chimera. We conclude that the sequence differences in the lower hinge at least partially contribute to the poor cleavage efficiency of IgG2 antibodies. Thus, the IgG1-2 chimera is not a practical strategy to generate F(ab′)2 molecules that do not bind to pre-existing AHA.
Figure 2.
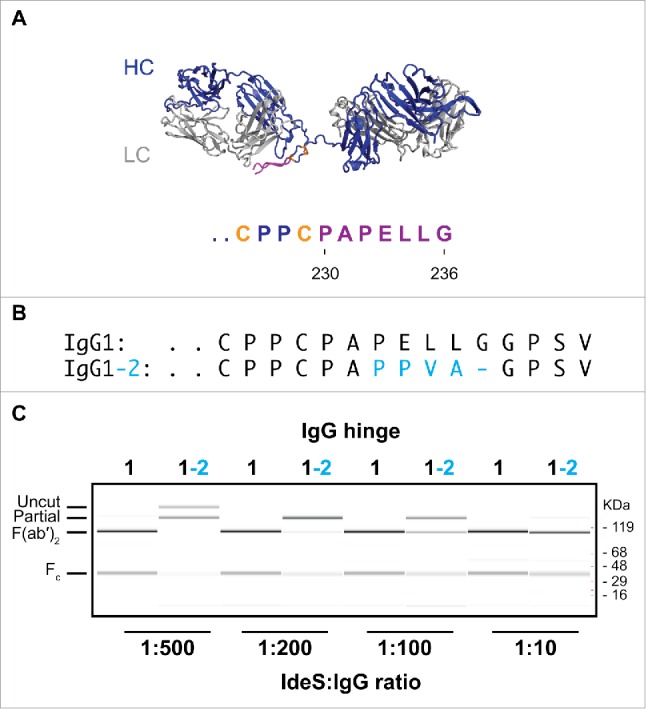
IgG1-2 chimera is inefficiently cleaved by IdeS. (A) Model of the F(ab′)2 region of antibody cAC10 modeled with MOE; light chain (gray), heavy chain (blue), interchain disulfide (orange), and lower hinge (magenta). The P1 position of IdeS is G236. Numbering of residues is according to EU numbering nomenclature. (B) Alignment of the lower hinge of IgG1 and the IgG1-2 chimera. Residues in cyan are IgG2 isotype residues introduced into the lower hinge of IgG2. (C) Cleavage efficiency of human IgG1 and IgG1-2 chimera. One mg/ml of IgG1 and IgG1-2 were incubated for 24 hours at 37°C with different IdeS amounts as indicated. Cleavage was analyzed by capillary electrophoresis. While IgG1 is efficiently cleaved into F(ab′)2 at an IdeS:IgG ratio of 1:500, IgG1-2 requires 50-fold higher IdeS concentrations for complete cleavage.
Characterizing the P1 and P2 positions for efficient IdeS cleavage
We demonstrated that binding of pre-existing AHA can be prevented by a single C-terminal T225L mutation with our Fab experiments. A similar strategy was employed for the F(ab′)2. As a first step, we identified residues in the P1 and P2 site that allow cleavage by IdeS protease. We generated a set of 76 Fab variants that included L235, L235V, L235I, or L235M at the P2 position for IdeS combined with any amino acid except cysteine in P1. The antibodies were purified and digested with IdeS at an IdeS:IgG ratio of 1:10 to identify variants that can be cleaved by IdeS (data not shown). Such high protease to antibody ratio was chosen to assess proteolysis without taking cleavage efficiency into account. Seven variants were identified to cleave by IdeS (Fig. 3A). While the P2 position tolerated all 4 residues tested, only 2 amino acids with very small side chains, the natural glycine and alanine, were accepted in the P1 position. We took this subset and investigated the cleavage efficiency at 3 different IdeS:IgG ratios: 1:500, 1:200, and 1:100 (Fig. 3B). The variant L235V in the P2 position demonstrated minimal change in cleavage efficiency compared to the wild-type sequence. All other variants were characterized by a dominant single cleavage at only one side of the hinge, leaving the other side of the antibody intact. While L235I and L235M variants with glycine in the P1 position could be completely cleaved at IdeS:IgG ratio of 1:200, all P2 variants with alanine in P1 needed significantly higher protease amounts. Based on our data, an efficient cleavage by IdeS requires glycine in the P1 position.
Figure 3.
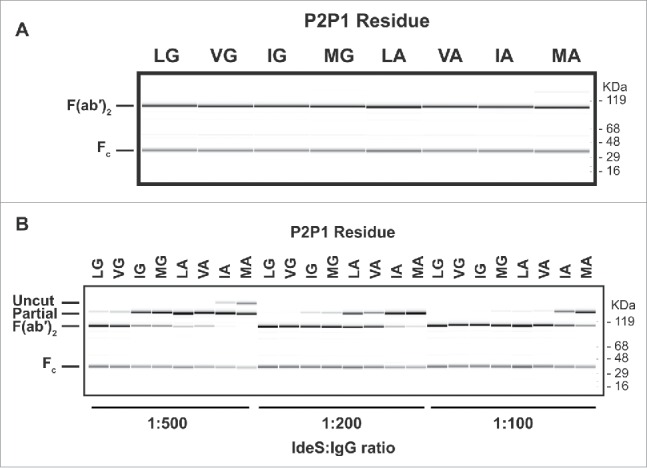
Cleavage of human IgG1 with variants in the P1 and P2 positions of IdeS. (A) Capillary electrophoresis of antibodies with variants at the P1 and P2 position were digested for 24 hours at 37°C with a 1:10 ratio of IdeS:IgG at 1mg/ml. The P1 and P2 residues are designated in 1-letter code. Leucine and glycine (L235G236) are the natural amino acids at these positions. All antibody variants can be completely cleaved into F(ab′)2 fragments. (B) The cleavage efficiency of the variants is assessed by the amount of F(ab′)2 produced at different IdeS:IgG ratios. While the variant VG is cleaved comparably to the wild-type sequence (LG), other variants require increased amounts of IdeS for complete digestion.
C-terminal F(ab′)2 variants do not prevent recognition by pre-existing AHA
IdeS can confound results of our pre-existing AHA assay with a false positive result (data not shown). This can be explained by binding of serum antibodies by IdeS and their subsequent recognition by the anti-Fc detection antibody. Therefore, we ensured that F(ab′)2 molecules used in the assay did not contain IdeS from the preceding proteolysis reaction. The removal of IdeS in the purified F(ab′)2 proteins was confirmed by SDS-PAGE followed by Coomassie staining and anti-IdeS immunoblot (Fig. 4A).
Figure 4.

P1 and P2 variants do not remove recognition by pre-existing AHA. (A) IdeS is efficiently removed during purification and cannot be detected in the purified F(ab′)2 variants by SDS-PAGE followed by Coomassie staining (upper panel) or immunoblot analysis with anti-IdeS antibodies (lower panel). (B) Pooled human serum was incubated with human IgG1 Fab with T225 C-terminus and F(ab′)2 generated by IdeS cleavage of antibodies with variants in P1 and P2 positions. The pre-existing AHA binding was detected by ELISA. The hinge variants reduce reactivity to levels comparable to Fab, but do not eliminate reactivity completely.
The purified P1 and P2 F(ab′)2 variants were then tested for their recognition by pre-existing AHA (Fig. 4B). While there was reduced signal compared to the wild-type, none of the antibodies with C-terminal modifications eliminated the reactivity with pre-existing antibodies in serum. Changes to the P2 position had only a modest effect. The G236A change in the P1 position reduced the signal to levels comparable to a Fab. Thus, it is not possible to design a lower hinge with an amino acid variant that retains cleavage by IdeS and concurrently eliminates the pre-existing antibody response toward the F(ab′)2.
Deletion in the lower hinge prevents recognition by pre-existing AHA while retaining IdeS cleavage
We employed a subsequent strategy to remove the epitope of the AHA by truncating the lower hinge instead. Leaving the P1 and P2 residues unchanged for their significance in cleavage efficiency, we started with a single and double residue deletion of the P3, P4, and P5 sites of IdeS to identify residues with a minimal effect in cleavage efficiency. Significantly poor cleavage efficiency was observed with deletion of the P3 position (Fig. 5A), thus this position was not considered for further studies. Antibodies with deletion of P4, P5, and a combination of both (ΔP45) were tested for binding to AHA (Fig. 5B). For these variants, a lower signal was observed compared to the wild-type hinge sequence. To further reduce the hinge recognition by pre-existing AHA, we extended the deletion to include the P6 and P7 residues. While the deletion of P4 through P7 sites (ΔP4567) resulted in modestly reduced cleavage efficiency, the deletion of P4 through P6 sites (ΔP456) had a cleavage efficiency comparable to wild-type at IdeS:IgG ratio of 1:200 (Fig. 5C). Both F(ab′)2 variants did not result in binding of pre-existing AHA (Fig. 5D). To ensure high cleavage specificity of IdeS is maintained, we analyzed the IdeS cleaved F(ab′)2 by ESI-TOF mass spectrometry. Only a single mass corresponding to the expected cleavage site after G236 17 was observed for the F(ab′)2 from the wild-type as well as the ΔP456 variant F(ab′)2 (Fig. 5E).
Figure 5.

Truncating the lower hinge eliminates pre-existing AHA binding. (A) IdeS cleavage of antibodies with deletions in the IdeS P3, P4, and P5 sites. While deletion of the IdeS P3 residue (L234) in the lower hinge severely affects cleavage efficiency, deletion of the P4 (E233) or P5 (P232) positions do not affect cleavage with IdeS compared to wild-type (WT). (B) Deletion of the P4 and P5 positions is not sufficient to avoid binding of pre-existing AHA. (C) IdeS cleavage of antibodies with deletions of the IdeS P4 through P6 (ΔP456) and P4 through P7 (ΔP4567) sites. While cleavage efficiency of the ΔP4567 variant is slightly reduced compared to the wild-type lower hinge sequence (WT), ΔP456 displays cleavage efficiency comparable to the wild-type at IdeS:IgG ratio of 1:200. (D) Pooled human serum was incubated with F(ab′)2 produced by IdeS digestion and binding antibodies detected by ELISA. F(ab′)2 lower hinge deletions ΔP456 and ΔP4567 are not recognized by pre-existing AHA. (E) IdeS cleaves the ΔP456 hinge variant with high specificity. After digestion of wild-type (WT) and ΔP456 hinge IgG, reduced F(ab′)2 was analyzed by mass spectrometry. Only a single heavy chain species corresponding to the expected molecular mass was observed.
The observed AHA binding signal of the ΔP456 variant F(ab′)2 is comparable to the 2 Fab C-terminal variants, Fab D221and Fab T225L (Fig. 6). The 5-fold higher AHA seen with F(ab′)2 compared to the Fab T225L (Fig. 6) could be explained by the potential bivalent binding of AHA to the F(ab′)2 and raises an even greater significance to avoid pre-existing AHA with F(ab′)2 molecules. To exclude the possibility that the reduced AHA reactivity seen in F(ab′)2 ΔP456 and Fab T225L was due to reduced coating efficiency, an antigen capture format was used to detect AHA. The OD readings (n = 4) were 2.2 ± 0.1, 0.34 ± 0.03, 1.3 ± 0.1, 0.36 ± 0.02 and 0.32 ± 0.01 on antigen-coated wells receiving F(ab′)2, F(ab′)2 ΔP456, Fab, Fab T225L and buffer at 1:30 serum dilution, respectively. Thus, the results confirmed that F(ab′)2 ΔP456 and Fab T225L had reduced AHA reactivity compared to the corresponding wild type molecules. With the ΔP456 variant, we now provide a solution to avoid pre-existing AHA response toward the lower hinge of F(ab′)2 while maintaining the possibility to produce the F(ab′)2 antibody fragment by the well-established route of proteolytic digestion.
Figure 6.
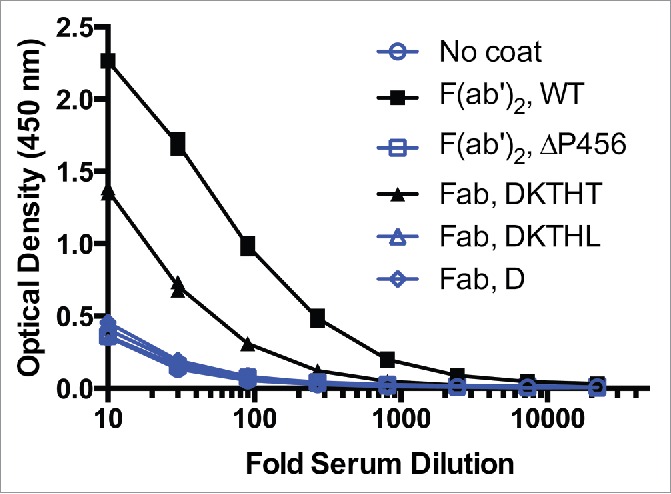
F(ab′)2 has stronger AHA binding than IgG1 Fab T225 in the AHA ELISA. Titration curves of F(ab′)2 and Fab variants in the AHA ELISA. F(ab′)2has 5-fold higher AHA reactivity than Fab T225. The dilutions corresponding to the OD (1.15) at the middle of the F(ab′)2 titration curves were 70 and 14 for F(ab′)2 and Fab, respectively. F(ab′)2, F(ab′)2 ΔP456, Fab T225, Fab T225L and Fab D221 were coated on the wells. Serial dilutions of pooled human serum were added to the wells and control wells were uncoated. Similar results were obtained in 4 other experiments. The data shown here and in Fig. 1B and Fig. 5D were collected from the same experiment.
F(ab′)2 ΔP456 has greatly reduced AHA mediated FcγRIIIa and C1q binding
It has been previously described that purified AHA antibodies can act as surrogate Fc and restore antibody-dependent cell-mediated cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC) function that was lost by IdeS generated F(ab′)2.20,22 To study if the reduced binding of AHA by our engineered F(ab′)2 variants is further reflected by reduced recruitment of Fcγ receptors and C1q, we employed a bridging experiment. IdeS generated F(ab′)2 was absorbed to the plate. After incubation with human serum to pre-bind AHA, binding of purified FcγRIIIa and C1q was analyzed.
As expected, we observed significant binding of FcγRIIIa and C1q for F(ab′)2 while little signal was detected for our ΔP456 F(ab′)2 variant, indicating that our engineering significantly reduced the risk of ADCC/CDC activation. The OD readings (n = 3) were 0.45 ± 0.05, 0.10 ± 0.02 and 0.09 ± 0.02 for FcγRIIIa binding and 0.98 ± 0.09, 0.158 ± 0.004 and 0.107 ± 0.009 for C1q binding on F(ab′)2, ΔP456 variant, and uncoated wells at 1:10 diluted serum, respectively. Further experiments are necessary to demonstrate ADCC/CDC function in cell-based assays or in vivo.
Discussion
Antibody fragments such as Fab and F(ab′)2 are attractive therapeutic formats when a short systemic half-life and an effector-silent molecule are concurrently desired. These fragments are also natural products of proteases associated with invasive diseases such as tumor cells and bacteria and are generated in an effort to evade immune surveillance. As a consequence, the C-terminal neoepitopes of Fab and F(ab′)2 are recognized by the immune system and result in AHA that can provide surrogate Fc.
In this study, we dissected the reactivity of pre-existing AHA toward the individual C-terminal residues spanning the upper hinge region of human IgG1, IgG2 and IgG4 isotypes. While it has been previously reported that pre-existing AHA toward neoepitopes in the lower hinge of IdeS-cleaved human IgG2 antibodies do not exist in serum of healthy human donors,25 reactivity toward the upper hinge of human isotypes was incompletely investigated so far. In our study, we could not detect pre-existing AHA in serum of healthy human donors toward the upper hinge of IgG2 and IgG4 isotypes. This may suggest that these isotypes are not the target of proteases of invasive diseases and may be explained by the effector-attenuated nature of these isotypes and the fact that removing the Fc region of these isotypes does not provide an advantage for tumors and bacteria. In contrast, IgG1 isotype seems to be the prime target for these proteases. Indeed, several proteases have been described to cleave in the upper hinge of human IgG1, including plasmin, human neutrophil elastase and LysC. Our study shows that pre-existing AHA exist toward all known cleavage sites of the upper hinge of human IgG1. The absence of AHA toward D221 may be a reflection of the inability of human proteases to cleave after this residue or the inability to raise antibodies toward this neoepitope. The highest reactivity was observed toward the C-terminal T223 Fab. Intriguingly, this C-terminus coincides with the cleavage point of human neutrophil elastase, a protease that is secreted by neutrophils and macrophages during inflammation to destroy bacteria and host tissue.26
Pre-existing AHA can rapidly recruit effector-function to a molecule that is designed to be effector-less. Using a Fab of IgG2 or IgG4 isotype can provide one strategy to supply a molecule without pre-existing antibody response. Alternatively, introducing a non-germline residue at the heavy chain C-terminus, such as the T225L variant or truncating the upper hinge to D221 is a strategy for the IgG1 isotype. While the T225L mutation eliminates the response toward pre-existing AHA, it implies that it can potentially elicit an immune response as well. This is further supported by a recent study with a domain antibody targeting tumor necrosis factor receptor 1.27 The addition of a C-terminal alanine was sufficient to reduce binding of pre-existing human anti-VH antibodies during screening in vitro; however, one subject was found to develop high levels of antibodies specific toward the modified C-terminus in a Phase 1 clinical trial. In addition, potential exopeptidase activity on a longer tail can eventually result in additional neoepitopes that can be recognized by other pre-existing AHA. Removing the unstructured upper hinge altogether as with the Fab-D221 further minimizes the risk of such secondary responses.
Our findings can also have implications on the design of studies in cynomolgus monkeys. While the lower hinge region is highly conserved between cynomolgus and human IgG, considerable differences exist in the upper hinge region 28 that will prevent cross-species reactivity. This may have an effect on toxicological studies in cynomolgus monkeys as liabilities from pre-existing AHA toward a human Fab cannot be addressed by these studies.
Beyond the therapeutic use, the Fab-D221 can also be considered for the recombinant expression of Fab for crystallographic studies since the unstructured nature of the upper hinge is commonly unresolved in crystal structures. Our experiments demonstrate that equal stability and expression can be achieved with a Fab-D221 construct. Eliminating unstructured areas may further improve crystallization outcomes.
Currently, the most efficient route to generate F(ab′)2 molecules is by proteolytic digest and proteases with high-specificity such as IdeS are preferred. As discussed earlier, pre-existing AHA also exist toward the lower hinge of F(ab′)2 molecules. The AHA titer toward IdeS-cleaved antibody is higher compared to a Fab. This may be due to the bivalent nature of the F(ab′)2 that provides an avidity-based binding component in the assay or a natural higher abundance of F(ab′)2 that leads to increased titers. We employed several strategies to remove the AHA reactivity while maintaining cleavage efficiency by IdeS.
Since the C-terminal residue has an important role in epitope recognition,22 we first carried out a strategy to change the C-terminal residue of the F(ab′)2 to remove binding activity of AHA. However, this was not possible due to the strict requirement of glycine in the P1-position for efficient cleavage by IdeS. Our selected set of mutations in the P2-site had only modest effect on AHA binding, further confirming the importance of the C-terminal residue for reactivity with AHA. The coinciding differences in AHA binding with the IdeS cleavage efficiency of the P1 and P2 variants perhaps explain the reason why the IgG2 isotype with valine at the P2 position and alanine at the P1 position is less susceptible to AHA response. The requirement for glycine in position P1 for efficient cleavage is accompanied by the high conservation of this residue within different isotypes and across species.
By deleting 3 residues in the lower hinge (ΔP456), we are able to maintain the cleavage efficiency of IdeS while removing pre-existing AHA recognition. It demonstrates that positions upstream of the P3 site have minor relevance for efficient cleavage. In addition to removing the reactivity toward pre-existing AHA, we speculate that truncating the lower hinge may dampen an immune response toward this epitope. Based on structural studies, an AHA binds the lower hinge in an extended conformation and the 5 C-terminal residues interact with the antibody complementarity-determining regions.29 By removing 3 residues from the lower hinge, only 4 residues remain after IdeS cleavage. This short sequence may not be sufficient for a robust immune response and reduce the likelihood of developing de novo antibodies toward the engineered hinge.
Pre-existing autoantibodies have also been demonstrated to decrease effector-function 30 in vitro and can also confound immunogenicity assays during drug development. While the majority of anti-therapeutic antibodies toward a humanized antibody are targeting the idiotype, rheumatoid factor, a low-affinity antibody toward the Fc region has been described as well. One way to eliminate artifacts by rheumatoid factor in the immunogenicity assay is to use an antibody fragment devoid of the Fc region. However, it is important to use fragments that are not recognized by other pre-existing antibodies. With our study, we now provide multiple Fab formats and a F(ab′)2 format that can fulfill these criteria.
In summary, by choosing the right antibody fragment, it is possible to evade recognition by pre-existing AHA. For Fab molecules, several options exist: 1) use of an IgG2 or IgG4 isotype, 2) mutation of the C-terminal residue (T225L), or 3) termination of the Fab with residue D221. Options are more limited for F(ab′)2 molecules due to the need for proteolytic digestion; however, we were able to identify a deletion in the lower hinge of IgG1 that maintains high cleavage efficiency and specificity, but removes reactivity with pre-existing AHA. We believe that using these formats in the future will enable further minimization of safety concerns with antibody fragments in a therapeutic setting and remove interference in assay deve-lopment.
Materials and methods
Plasmid construction and antibody expression
Antibodies were cloned by standard molecular biology techniques into E. coli expression vectors 10,31 or mammalian expression vectors 32 as previously described. E. coli expression was carried out as described in Simmons et al.31 IgG and Fab were expressed in 30 mL transient transfection cultures of CHO 33 or HEK293T 34 cells as previously described.
Cloning, expression, and purification of IdeS
IdeS was expressed as N-terminal glutathione S-transferase (GST) fusion protein. The mature sequence of IdeS from Streptococcus pyogenes MGAS1882 (Uniprot ID H8HDR0) was codon optimized for E. coli expression, synthesized by GeneArt™, and cloned by standard molecular biology techniques into an E. coli expression vector.31 IdeS was expressed using conditions described in Simmons et al.31 and purified using a glutathione sepharose column. Eluate fractions in 50 mM Tris-HCl, pH 8.0, 20 mM glutathione from glutathione sepharose column were concentrated and loaded onto a S200 column and eluted with 200 mM K2HPO4, pH 6.2, 250 mM KCl.
Antibody and Fab purification
After expression, the cells were pelleted by gravity. The supernatants were transferred to a 50 ml Falcon tube (Corning, Corning, NY, USA). Four hundred µl of 50% MabSelect Sure protein affinity slurry or Gamma Bind Plus slurry (GE Healthcare, Pittsburgh, PA, USA) was added to the supernatants for IgG and Fab purification, respectively. The mixture was incubated overnight at room temperature on an Innova 2000 platform shaker (New Brunswick Scientific, Enfield, CT, USA). Supernatants were removed and the resin transferred to a 96-well 2 ml filter plate with a 25 µm size membrane (Thompson Instrument, Oceanside, CA, USA). The resin was washed 3 times with 1 ml of 1x phosphate-buffered saline (PBS) pH 7.4 by centrifugation at 1,120 x g for 5 minutes using a Sorvall HT6 Centrifuge (Thermo Scientific, Waltham, MA, USA). For Fab purification, the resin was further washed with 0.2x PBS pH 5.0 before elution. The IgG was eluted using 50 mM phosphoric acid pH 2.9 and the eluate neutralized with 20x PBS pH 11.0 by centrifugation at 1,000 x g for 5 minutes. The Fabs were eluted using 10 mM sodium citrate pH 2.9 and neutralized with 0.3 M Tris pH 9.0. The eluted IgG and Fab was filtered through 0.2 µm 96-well filter plate (Orochem, Naperville, IL, USA) by centrifugation at 1,000 x g for 5 minutes using an Sorvall HT6 Centrifuge (Thermo Scientific, Waltham, MA, USA).
IdeS digestion of IgG hinge variants
IgG at 1 mg/ml was incubated with stated IdeS:IgG ratio (w/w) at 37°C for 24 hours. For scaled-up digestion to generate highly pure F(ab′)2 material for AHA assays, up to 1:10 IdeS:IgG ratio was used to drive complete digestion.
F(ab′)2 purification after IgG cleavage by IdeS
The IdeS cleaved sample was diluted with 25 mM sodium acetate, pH 4.4 (Buffer A) and loaded onto a 1 mL SP Sepharose High Performance strong cation exchange column (GE Healthcare, Pittsburgh, PA, USA) at 150 cm/hr (0.7 cm diameter, 10 cm bed height) equilibrated in Buffer A. The column was washed to baseline with Buffer A and F(ab′)2 eluted with a linear salt gradient from 0 to 0.5 M NaCl over 30 column volumes. The eluate was neutralized by addition of 3 M Tris pH 9.0 to adjust the pH to 7.0 and filtered through 0.22 µm Steriflip (EMD Millipore, Billerica, MA, USA). The SP eluted F(ab′)2 was further purified on a MonoS 5/50 GL strong cation exchange column (GE Healthcare, Pittsburgh, PA, USA) after diluting with Buffer A to lower the conductivity <5 mS/cm. The column was washed to baseline (<0 .05mAU) with Buffer A and the F(ab′)2 eluted using a salt gradient from 0 to 0.6 M NaCl over 40 column volumes. The eluted F(ab′)2 solution was neutralized with 3 M Tris pH 9.0 to adjust the pH to 7.0 and filtered through 0.22 µm steriflip (EMD Millipore, Billerica, MA, USA).
Mass spectrometry of Fab and F(ab′)2
Mass spectrometric data was acquired using an Agilent 6224 TOF LC-MS system (Agilent Technology, Santa Clara, CA, USA). F(ab′)2 was reduced with 100 mM dithiothreitol at 37°C for 20 minutes. The polypeptide chains were separated with a PLRP-S reversed phase column (Agilent Technology, Santa Clara, CA, USA). Intact masses of the reduced light and heavy chains were obtained by Maximum Entropy Deconvolution using MassHunter software (Qualitative Analysis B.03.01).
Analysis of protein by capillary electrophoresis
All samples were prepared by mixing 5 µl of sample volume with 7 µl of HT Protein Express Sample buffer and incubated for 5 minutes at 70°C. 32 µl of water was added to the samples and centrifuged at 1,000 x g for 5 minutes. The chip was prepared according to manufacturer's instructions provided in the LabChip GXII User Guide and samples were analyzed on a Caliper GX II microfluidic system (PerkinElmer Biotechnology, Waltham, MA, USA). Samples were analyzed on a Caliper GX II microfluidic system (PerkinElmer Biotechnology, Waltham, MA, USA). All reagents were obtained from PerkinElmer Biotechnology.
Pre-existing AHA enzyme-linked immunosorbent assay (ELISA)
MaxiSorp plates (384-well, Nunc, Thermo Fisher Scientific, Rochester, NY, USA) were coated with 1 µg/ml F(ab′)2or Fab in 50 mM carbonate, pH 9.6 at 4°C overnight. Plates were washed with 0.05% polysorbate 20 in PBS, pH 7.4 and then blocked with 0.5% BSA, 15 ppm proclin in PBS, pH 7.4. Pooled human serum from 25 female and 25 male individuals (Bioreclamation, Westbury, NY, USA; catalog # HMSRM, lot # BRH806182) were serially diluted in assay buffer (0.5% BSA, 0.05% polysorbate 20, 15 ppm proclin, in PBS, pH 7.4) and added to the plates. After a 2 hour incubation, bound pre-existing AHA was detected using horseradish peroxidase (HRP) conjugated goat anti-F(ab′)2 anti-human IgG Fc (Jackson ImmunoResearch, West Grove, PA) in assay buffer, followed by 3,3′,5,5′-tetramethyl benzidine (TMB, Moss Inc., Pasadena, MD, USA) as the substrate. The reaction was stopped with 1 M phosphoric acid and absorbance was read at 450 nm. The absorbance readings at 1:30 dilution were used for the figures to allow presentation of all samples. Comparable results were observed at a 1:10 serum dilution. To calculate the relative AHA response, the titration curve of F(ab′)2 was fitted with a 4 parameter curve fitting program (KaleidaGraph, Synerg Software, Reading, PA). MidOD (the average OD of the top and bottom OD readings) of the F(ab′)2 titration curve was determined. The dilutions of Fab DKTHT and F(ab′)2 corresponding to this midOD were calculated and used to calculate the relative AHA reactivity.
To assess binding of AHA to FcγRIIIa, human serum was added to F(ab′)2-coated wells and incubated for 2 hours as described above. After the plates were washed, soluble FcγRIIIa(V158)-His-GST (consisting of the extracellular domain fused with Gly-His6-glutathione-S-transferase at the carboxy-terminus) was added at 0.5 μg/ml. Bound FcγRIIIa(V158)-His-GST was detected using HRP-conjugated mouse anti-His antibody (Penta-His, Qiagen, Germantown, MD) followed by TMB as the substrate. To assess binding of AHA to human C1q, human serum was added to F(ab′)2-coated wells and incubated for 2 hours as described above. After the plates were washed, purified human C1q (Quidel, San Diego, CA) was added. Bound C1q was detected with goat anti-C1q antibody (Nordic Immunological Laboratories, Tilburg, The Netherlands) followed by rabbit anti-goat IgG-HRP (Jackson ImmunoResearch, West Grove, PA) and TMB as the substrate.
SDS-PAGE and immunoblots
For SDS-PAGE, 5 µg of purified F(ab′)2 variants and GST-IdeS were mixed with SDS-sample buffer, heated for 5 minutes at 95°C and spun for 1 minute at 16,000 relative centrifugal force. The samples were loaded onto a NuPAGE 4 –12% BisTris/MES gels (Invitrogen). For immunoblotting, 5 ng of protein samples were used for SDS-PAGE. Gels were transferred by iBlot (Invitrogen) onto nitrocellulose membranes, immunoblotted with anti-IdeS (Genovis, USA; catalog #A3-AF1-010, lot # A3AF1-7C17H) as primary antibody and IRDye800CW conjugated donkey anti-goat antibody (Li-COR, USA; catalog # 926-32214, lot # B80821-03) as the secondary antibody, and imaged with a LI-COR Odyssey Imager (Li-COR, USA). Odyssey Two-color protein molecular weight marker (LI-COR, USA) was used for immunoblots and pre-stained SeeBluePlus2 (Invitrogen, USA) was used for Coomassie stained gels.
Disclosure of potential conflicts of interest
All authors are employed by Genentech, which develops and markets drugs for profit.
Acknowledgments
We thank members of the Antibody Engineering Department at Genentech for technical support and/or advice and stimulating discussions. In particular, we thank Farzam Farahi, Sharon ViaJar, Avinash Gill, and Randall Brezski.
Author contributions
HSK and CS designed the experimental strategy, analyzed data, and wrote the manuscript. HSK performed molecular biology and proteolysis experiments. IK purified and characterized proteins. LZ and YGM designed and performed pre-existing antibody assays. JMV and YGM designed and performed the FcγRIIIa and C1q binding assays. All authors read and approved the final manuscript.
References
- 1.Desjarlais JR, Lazar GA. Modulation of antibody effector function. Exp Cell Res 2011; 317:1278-85; PMID:21459085; http://dx.doi.org/ 10.1016/j.yexcr.2011.03.018 [DOI] [PubMed] [Google Scholar]
- 2.Olafsen T. Fc engineering: serum half-life modulation through FcRn binding. Methods Mol Biol 2012; 907:537-56; PMID:22907373; http://dx.doi.org/ 10.1007/978-1-61779-974-7_31 [DOI] [PubMed] [Google Scholar]
- 3.Gadkar K, Pastuskovas CV, Le Couter JE, Elliott JM, Zhang J, Lee CV, Sanowar S, Fuh G, Kim HS, Lombana TN, et al.. Design and Pharmacokinetic characterization of novel antibody formats for ocular therapeutics. Invest Ophthalmol Vis Sci 2015; 56:5390-400; PMID:26275136; http://dx.doi.org/ 10.1167/iovs.15-17108 [DOI] [PubMed] [Google Scholar]
- 4.Curtis BR, Swyers J, Divgi A, McFarland JG, Aster RH. Thrombocytopenia after second exposure to abciximab is caused by antibodies that recognize abciximab-coated platelets. Blood 2002; 99:2054-9; PMID:11877279; http://dx.doi.org/ 10.1182/blood.V99.6.2054 [DOI] [PubMed] [Google Scholar]
- 5.Porter RR. The formation of a specific inhibitor by hydrolysis of rabbit antiovalbumin. Biochem J 1950; 46:479-84; PMID:15420176; http://dx.doi.org/ 10.1042/bj0460479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Wiesmann C, Fuh G, Li B, Christinger HW, McKay P, de Vos AM, Lowman HB. Selection and analysis of an optimized anti-VEGF antibody: crystal structure of an affinity-matured fab in complex with antigen. J Mol Biol 1999; 293:865-81; PMID:10543973; http://dx.doi.org/ 10.1006/jmbi.1999.3192 [DOI] [PubMed] [Google Scholar]
- 7.Pollack CV, Reilly PA, Eikelboom J, Glund S, Verhamme P, Bernstein RA, Dubiel R, Huisman MV, Hylek EM, Kamphuisen PW, et al.. Idarucizumab for Dabigatran Reversal. N Engl J Med 2015; 373:511-20; PMID:26095746; http://dx.doi.org/ 10.1056/NEJMoa1502000 [DOI] [PubMed] [Google Scholar]
- 8.Goel N, Stephens S. Certolizumab Pegol. MAbs 2010; 2:137-47; PMID:20190560; http://dx.doi.org/ 10.4161/mabs.2.2.11271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol 2015; 67:95-106; PMID:25637431; http://dx.doi.org/ 10.1016/j.molimm.2015.01.003 [DOI] [PubMed] [Google Scholar]
- 10.Carter P, Kelley RF, Rodrigues ML, Snedecor B. High level Escherichia coli expression and production of a bivalent humanized antibody fragment. Nature 1992; 10(2):163-7; PMID:1368228; http://dx.doi.org/1531669 10.1038/nbt0292-163 [DOI] [PubMed] [Google Scholar]
- 11.Kostelny SA, Cole MS, Tso JY. Formation of a bispecific antibody by the use of leucine zippers. J Immunol 1992; 148:1547-53; PMID:1531669 [PubMed] [Google Scholar]
- 12.Glennie MJ, McBride HM, Worth AT, Stevenson GT. Preparation and performance of bispecific F(ab′ gamma)2 antibody containing thioether-linked Fab′ gamma fragments. J Immunol 1987; 139:2367-75; PMID:2958547 [PubMed] [Google Scholar]
- 13.Ellerman D, Scheer JM. Generation of bispecific antibodies by chemical conjugation. In: Kontermann RE, editor. Springer-Verlag; Berlin Heidelberg; 2011; 47-63; http://dx.doi.org/ 10.1007/978-3-642-20910-9_3 [DOI] [Google Scholar]
- 14.Brezski RJ, Jordan RE. Cleavage of IgGs by proteases associated with invasive diseases: an evasion tactic against host immunity? MAbs 2010; 2:212-20; PMID:20400859; http://dx.doi.org/ 10.4161/mabs.2.3.11780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nisonoff A, Wissler FC, Lipman LN. Properties of the major component of a peptic digest of rabbit antibody. Science 1960; 132:1770-1; PMID:13729245; http://dx.doi.org/ 10.1126/science.132.3441.1770 [DOI] [PubMed] [Google Scholar]
- 16.Pawel-Rammingen von U, Johansson BP, Björck L. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J 2002; 21:1607-15; PMID:11927545; http://dx.doi.org/ 10.1093/emboj/21.7.1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vincents B, Pawel-Rammingen von U, Björck L, Abrahamson M. Enzymatic characterization of the streptococcal endopeptidase, IdeS, reveals that it is a cysteine protease with strict specificity for IgG cleavage due to exosite binding. Biochemistry 2004; 43:15540-9; PMID:15581366; http://dx.doi.org/ 10.1021/bi048284d [DOI] [PubMed] [Google Scholar]
- 18.Mellbye OJ, Natvig JB. Evidence for immune complexes containing antibody to the pepsin site of IgG in rheumatoid synovial fluids. Clin Exp Immunol 1971; 8:889-99; PMID:4933318 [PMC free article] [PubMed] [Google Scholar]
- 19.Terness P, Kohl I, Hübener G, Battistutta R, Moroder L, Welschof M, Dufter C, Finger M, Hain C, Jung M. The natural human IgG anti-F(ab′)2 antibody recognizes a conformational IgG1 hinge epitope. J Immunol 1995; 154:6446-52; PMID:7539020 [PubMed] [Google Scholar]
- 20.Brezski RJ, Luongo JL, Petrone D, Ryan MH, Zhong D, Tam SH, Schmidt AP, Kruszynski M, Whitaker BP, Knight DM, et al.. Human anti-IgG1 hinge autoantibodies reconstitute the effector functions of proteolytically inactivated IgGs. J Immunol 2008; 181:3183-92; PMID:18713989; http://dx.doi.org/ 10.4049/jimmunol.181.5.3183 [DOI] [PubMed] [Google Scholar]
- 21.Rispens T, de Vrieze H, de Groot E, Wouters D, Stapel S, Wolbink GJ, Aarden LA. Antibodies to constant domains of therapeutic monoclonal antibodies: anti-hinge antibodies in immunogenicity testing. J Immunol Methods 2012; 375:93-9; PMID:21986105; http://dx.doi.org/ 10.1016/j.jim.2011.09.011 [DOI] [PubMed] [Google Scholar]
- 22.Brezski RJ, Kinder M, Grugan KD, Soring KL, Carton J, Greenplate AR, Petley T, Capaldi D, Brosnan K, Emmell E, et al.. A monoclonal antibody against hinge-cleaved IgG restores effector function to proteolytically-inactivated IgGs in vitro and in vivo. MAbs 2014; 6:1265-73; PMID:25517311; http://dx.doi.org/24782178 10.4161/mabs.29825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van de Stadt LA, de Vrieze H, Derkse/auqn NIL, Brouwer M, Wouters D, van Schaardenburg D, Wolbink G, Rispens T. Antibodies to IgG4 hinge can be found in rheumatoid arthritis patients during all stages of disease and may exacerbate chronic antibody-mediated inflammation. Arthritis Rheumatol 2014; 66:1133-40; PMID:24782178; http://dx.doi.org/ 10.1002/art.38335 [DOI] [PubMed] [Google Scholar]
- 24.Van Schie KA, Wolbink GJ, Rispens T. Cross-reactive and pre-existing antibodies to therapeutic antibodies-Effects on treatment and immunogenicity. MAbs 2015; 7:662-71; PMID:25962087; http://dx.doi.org/ 10.1080/19420862.2015.1048411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brezski RJ, Oberholtzer A, Strake B, Jordan RE. The in vitro resistance of IgG2 to proteolytic attack concurs with a comparative paucity of autoantibodies against peptide analogs of the IgG2 hinge. MAbs 2011; 3:558-67; PMID:22123056; http://dx.doi.org/ 10.4161/mabs.3.6.18119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belaaouaj A, Kim KS, Shapiro SD. Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science 2000; 289:1185-8; PMID:10947984; http://dx.doi.org/ 10.1126/science.289.5482.1185 [DOI] [PubMed] [Google Scholar]
- 27.Cordy JC, Morley PJ, Wright TJ, Birchler MA, Lewis AP, Emmins R, Chen YZ, Powley WM, Bareille PJ, Wilson R, et al.. Specificity of human anti-variable heavy (VH) chain autoantibodies and impact on the design and clinical testing of a VH domain antibody antagonist of tumour necrosis factor-α receptor 1. Clin Exp Immunol 2015; 182:139-48; PMID:26178412; http://dx.doi.org/ 10.1111/cei.12680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacobsen FW, Padaki R, Morris AE, Aldrich TL, Armitage RJ, Allen MJ, Lavallee JC, Arora T. Molecular and Functional Characterization of Cynomolgus Monkey IgG Subclasses. J Immunol 2011; 186:341-9; PMID:21131427; http://dx.doi.org/ 10.4049/jimmunol.1001685 [DOI] [PubMed] [Google Scholar]
- 29.Malia TJ, Teplyakov A, Brezski RJ, Luo J, Kinder M, Sweet RW, Almagro JC, Jordan RE, Gilliland GL. Structure and specificity of an antibody targeting a proteolytically cleaved IgG hinge. Proteins 2014; 82:1656-67; PMID:24638881; http://dx.doi.org/ 10.1002/prot.24545 [DOI] [PubMed] [Google Scholar]
- 30.Jones JD, Shyu I, Newkirk MM, Rigby WF. A rheumatoid factor paradox: inhibition of rituximab effector function. Arthritis Res Ther 2013; 15:R20; PMID:23351360; http://dx.doi.org/ 10.1186/ar4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simmons LC, Reilly D, Klimowski L, Raju TS, Meng G, Sims P, Hong K, Shields RL, Damico LA, Rancatore P, et al.. Expression of full-length immunoglobulins in Escherichia coli: rapid and efficient production of aglycosylated antibodies. J Immunol Methods 2002; 263:133-47; PMID:12009210; http://dx.doi.org/ 10.1016/S0022-1759(02)00036-4 [DOI] [PubMed] [Google Scholar]
- 32.Eaton DL, Wood WI, Eaton D, Hass PE, Hollingshead P, Wion K, Mather J, Lawn RM, Vehar GA, Gorman C. Construction and characterization of an active factor VIII variant lacking the central one-third of the molecule. Biochem 1986; 25:8343-7; PMID:3030393; http://dx.doi.org/ 10.1021/bi00374a001 [DOI] [PubMed] [Google Scholar]
- 33.Wong AW, Baginski TK, Reilly DE. Enhancement of DNA uptake in FUT8-deleted CHO cells for transient production of afucosylated antibodies. Biotechnol Bioeng 2010; 106:751-63; PMID:20564613; http://dx.doi.org/ 10.1002/bit.22749 [DOI] [PubMed] [Google Scholar]
- 34.Bos AB, Duque JN, Bhakta S, Farahi F, Chirdon LA, Junutula JR, Harms PD, Wong AW. Development of a semi-automated high throughput transient transfection system. J Biotechnol 2014; 180:10-6; PMID:24704608; http://dx.doi.org/ 10.1016/j.jbiotec.2014.03.027 [DOI] [PubMed] [Google Scholar]