

ABSTRACT
DSTA4637A, a novel THIOMAB™ antibody antibiotic conjugate (TAC) against Staphylococcus aureus (S. aureus), is currently being investigated as a potential therapy against S. aureus infections. Structurally, TAC is composed of an anti-S. aureus antibody linked to a potent antibiotic, dmDNA31. The goal of the current study was to characterize the pharmacokinetics (PK) of TAC in mice, assess the effect of S. aureus infection on its PK, and evaluate its pharmacodynamics (PD) by measuring the bacterial load in various organs at different timepoints following TAC treatment. Plasma concentrations of 3 analytes, total antibody (TAb), antibody-conjugated dmDNA31 (ac-dmDNA31), and unconjugated dmDNA31, were measured in these studies. In non-infected mice (target antigen absent), following intravenous (IV) administration of a single dose of TAC, systemic concentration-time profiles of both TAb and ac-dmDNA31 were bi-exponential and characterized by a short distribution phase and a long elimination phase as expected for a monoclonal antibody-based therapeutic. Systemic exposures of both TAb and ac-dmDNA31 were dose proportional over the dose range tested (5 to 50 mg/kg). In a mouse model of systemic S. aureus infection (target antigen present), a single IV dose of TAC demonstrated PK behavior similar to that in the non-infected mice, and substantially reduced bacterial load in the heart, kidney, and bones on 7 and 14 d post dosing. These findings have increased our understanding of the PK and PK/PD of this novel molecule, and have shown that at efficacious dose levels the presence of S. aureus infection had minimal effect on TAC PK.
KEYWORDS: Antibody-drug conjugate, antibody-antibiotic conjugate, pharmacokinetics, pharmacodynamics, Staphylococcus aureus infections, THIOMAB™ antibody antibiotic conjugate
Abbreviations
- AAC
Antibody-antibiotic conjugate
- ADC
Antibody-drug conjugate
- AUCinf
time curve extrapolated to infinity
- ac-dmDNA31
antibody-conjugated dmDNA31
- Cmax
maximum concentration observed
- CFU
colony forming units
- DAR
drug-antibody ratio
- PK
Pharmacokinetics
- PD
Pharmacodynamics
- IV
Intravenous
- IgG
Immunoglobulin G
- S. aureus
Staphylococcus aureus
- TAC
THIOMAB™ antibody antibiotic conjugate
- Tab
TAC total antibody
- SCID
severe combined immunodeficiency
- VC
Valine-Citrulline
- Vss
volume of distribution at steady state
- t1/2λz
terminal half-life
- tmax
time of maximum concentration observed
Introduction
Invasive Staphyloccocus aureus (S. aureus) infection is the leading cause of death by an infectious agent, with a mortality rate of ∼20% in the USA.1,2 It is one of the leading contributors of hospital-acquired infections, including post-surgical wound infections.1,2 Reduced susceptibility to standard therapy such as vancomycin and nafcillin, and the failures associated with treatment-limiting adverse events caused by existing standard therapy3 warrant the development of novel anti-bacterial agents. Survival within the host phagocytic cells is one of the proposed mechanisms by which S. aureus escapes standard antibiotic treatment.4 Therefore, therapeutic approaches ablating intracellular S. aureus could provide a superior treatment option for the invasive infections caused by these bacteria.5
To target intracellular S. aureus, a novel antibody-antibiotic conjugate (AAC) strategy was utilized to produce a THIOMAB™ antibody antibiotic conjugate (TAC; DSTA4637A). TAC is composed of a monoclonal human immunoglobulin (Ig)G1 antibody (anti-S. aureus antibody) conjugated to a novel antibiotic via a protease cleavable valine-citrulline (VC) linker. The antibody was designed to specifically bind to the wall teichoic acid, a major cell wall component of S. aureus. The novel antibiotic, dmDNA31 (4-dimethylamino piperidino-hydroxybenzoxazino rifamycin), showed very potent bactericidal activity against S. aureus with an in vitro minimum inhibitory concentration (MIC) <10 nM.5 The proposed mechanism of action for TAC is illustrated in Fig. 1. It involves binding of TAC to S. aureus surface antigen resulting in opsonization of the bacteria. When TAC-opsonized S. aureus bacteria enter the host cell intracellular environment, host proteases in the phagolysosome such as cathepsins cleave the linker and the antibiotic is readily released in its active form.5 Since large numbers of TAC molecules are capable of binding to a single bacterium, the antibiotic can be released in the contained intracellular environment in concentrations sufficient to sustain bacterial elimination. Furthermore, it is hypothesized that when bacteria are released from pre-existing intracellular reservoirs into the extracellular space, the prolonged presence of TAC in the systemic circulation ensures immediate “tagging” of these bacteria for phagocytic uptake, thereby mitigating further spread of the infection.6 TAC has demonstrated potent killing of S. aureus both in in vitro and in vivo studies. More importantly, TAC has been shown to be efficacious in in vivo models where standard therapy, such as vancomycin, fails.5
Figure 1.
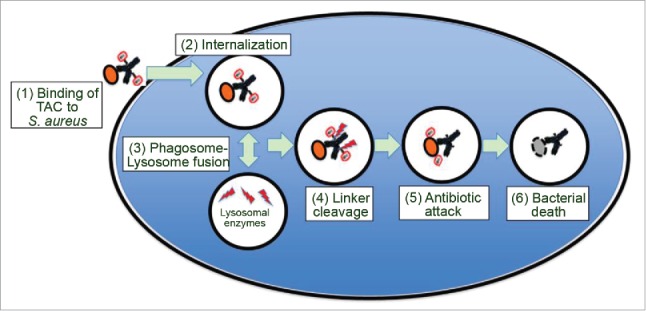
Model for the mechanism of action of TAC. As depicted in the model, (1) TAC binds to S. aureus bacteria, (2) TAC bound S. aureus bacteria are internalized by professional phagocytes or other host cells such as epithelial cells. After (3) phagosome-lysosome fusion, (4) lysosomal cathepsins cleave the linker, which (5) releases the active antibiotic dmDNA31 attacking the intracellular bacteria, resulting in (6) elimination of the bacteria.
In the current study, we investigated and characterized the PK of TAC in infected and non-infected mice to evaluate whether S. aureus infection will alter the PK of this molecule. We also characterized the PK of the unconjugated anti-S. aureus antibody to investigate the effect of conjugation on TAC PK. It is well known for antibody-drug conjugates (ADCs) that chemical or enzymatic activity may lead to deconjugation.7-9 Comparable to ADCs, TACs are complex and dynamically changing mixtures in vivo that require comprehensive bioanalytical strategies for PK characterization due to deconjugation and other potential biotransformations. Similar bioanalytical strategies that were utilized for ADCs8 were used in our studies with TAC. The characterization of PK for TAC included the quantification of 3 key analytes: TAC total antibody (TAb, measurement of all drug antibody ratios of TAC including fully conjugated, partially deconjugated, and fully deconjugated anti-S. aureus antibodies), antibody-conjugated dmDNA31 (ac-dmDNA31, measurement of dmDNA31 conjugated to the antibody), and unconjugated dmDNA31 (measurement of dmDNA31 that is not conjugated to the antibody through the VC linker) (Fig. 2).
Figure 2.
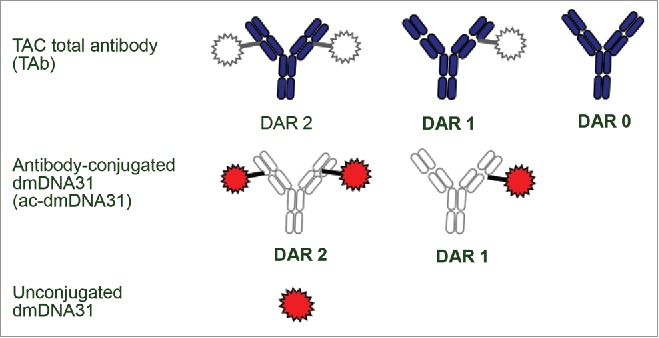
Three analytes measured for PK ccharacterization. The figure depicts the analyte mixtures for ac-dmDNA31 and TAC total antibody. The gray areas indicate parts of TAC structure that would not be measured by the respective assay. The colored areas indicate parts of TAC structure that would be determined by each assay. Catabolites of TAC other than unconjugated dmDNA31 may be present in circulation. DAR = drug-to-antibody ratio.
In addition to characterizing the PK, we also investigated the PD of TAC in a mouse model of systemic S. aureus infection by measuring the bacterial load in various organs following dosing this molecule in mice.
Results
PK of unconjugated anti-S. aureus antibody in non-infected mice
The PK of unconjugated anti-S. aureus antibody (i.e., naked antibody) following a single IV dose in non-infected severe combined immunodeficiency (SCID) mice at dose levels of 5, 25, and 50 mg/kg are shown in Fig. 3 and PK parameters tabulated in Table 1. In non-infected mice, the plasma concentration-time profile of unconjugated anti-S. aureus antibody was bi-exponential, characterized by a short distribution phase and a long elimination phase as expected for a monoclonal antibody. Systemic exposure of unconjugated anti-S. aureus antibody was dose proportional over the dose range of 5 to 50 mg/kg, which was expected given the absence of target antigen in non-infected mice. In non-infected mice, unconjugated anti-S. aureus antibody clearance ranged from 4.69 to 5.19 mL/day/kg, terminal half-life (t1/2λz) ranged from 16.4 to 18.0 days, and volume of distribution at steady-state (Vss) ranged from 111 to 139 mL/kg across all dose groups, which were similar to those for other monoclonal antibodies in the absence of target antigen.10
Figure 3.
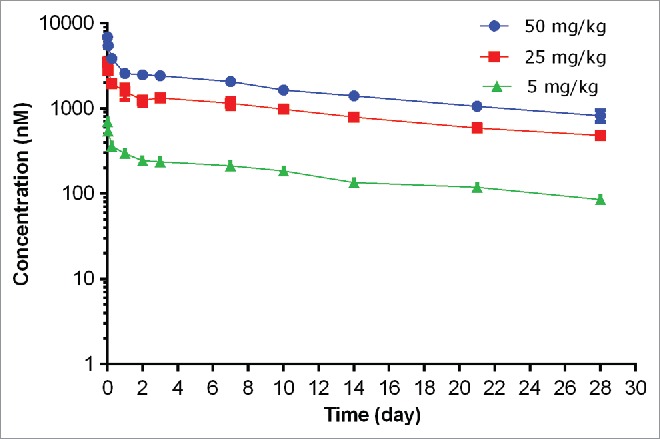
Plasma concentration−time profiles of unconjugated (naked) anti-S. aureus antibody following IV administration of unconjugated anti-S. aureus antibody in non-infected mice.
Table 1.
Non-compartmental PK parameters following a single IV administration of unconjugated (naked) anti-S. aureus antibody in non-infected mice and mice infected with S. aureus.
| Non-infected mice |
Mice infected with S. aureus |
|||||
|---|---|---|---|---|---|---|
| PK Parameters | 5 mg/kg | 25 mg/kg | 50 mg/kg | 5 mg/kg | 25 mg/kg | 50 mg/kg |
| Cmax (nM) | 698 | 3497 | 6911 | 739 | 4058 | 7869 |
| AUC0-inf (day • nM) | 6690 | 36470 | 65900 | 2160 | 16080 | 28190 |
| Clearance (mL/day/kg) | 5.11 | 4.69 | 5.19 | 15.8 | 10.6 | 12.1 |
| Vss (mL/kg) | 122 | 111 | 139 | 100 | 80.2 | 94.5 |
| t1/2λz (day) | 16.9 | 16.4 | 18.0 | 5.28 | 3.74 | 3.98 |
PK of unconjugated anti-S. aureus antibody in mice infected with S. aureus
The PK of anti-S. aureus antibody following a single IV dose in infected SCID mice at dose levels of 5, 25, and 50 mg/kg are shown in Fig. 4 and PK parameters are illustrated in Table 1. Following IV administration of 5, 25, or 50 mg/kg unconjugated anti-S. aureus antibody in infected mice, the antibody showed a bi-exponential plasma concentration-time profile with a short distribution phase followed by a long elimination phase (Fig. 4). In infected mice, the systemic clearance of unconjugated anti-S. aureus antibody was 10.6 to 15.8 mL/day/kg, which was 2-3-fold faster than that in the non-infected mice. The t1/2λz ranged from 3.74 to 5.28 days, and Vss ranged from 80.2 to 100 mL/kg across all dose groups (Table 1).
Figure 4.
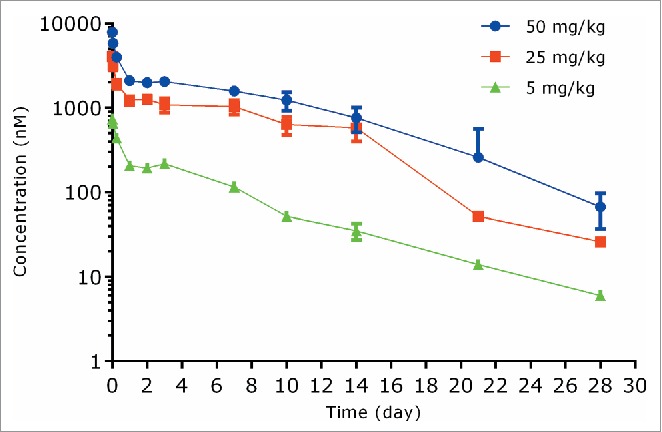
Plasma concentration−time profiles of unconjugated (naked) anti-S. aureus antibody following IV administration of unconjugated anti-S. aureus antibody in mice infected with S. aureus.
In the heart and kidneys, treatment with 50 mg/kg of unconjugated anti-S. aureus antibody did not lead to a statistically significant difference (geometric mean difference < 1 log10) in the colony forming units (CFU) count compared to the control group on Day 3 (data not shown).
PK of TAC in non-infected mice
The PK of TAC following a single IV dose in non-infected SCID mice at dose levels of 5, 25, and 50 mg/kg are shown in Fig. 5 and PK parameters are shown in Table 2. Following IV administration of a single dose of 5, 25, or 50 mg/kg TAC, the plasma concentration-time profiles of TAC TAb were similar to the PK profiles of a typical monoclonal antibody-based therapeutic (Fig. 5A).11
Figure 5.
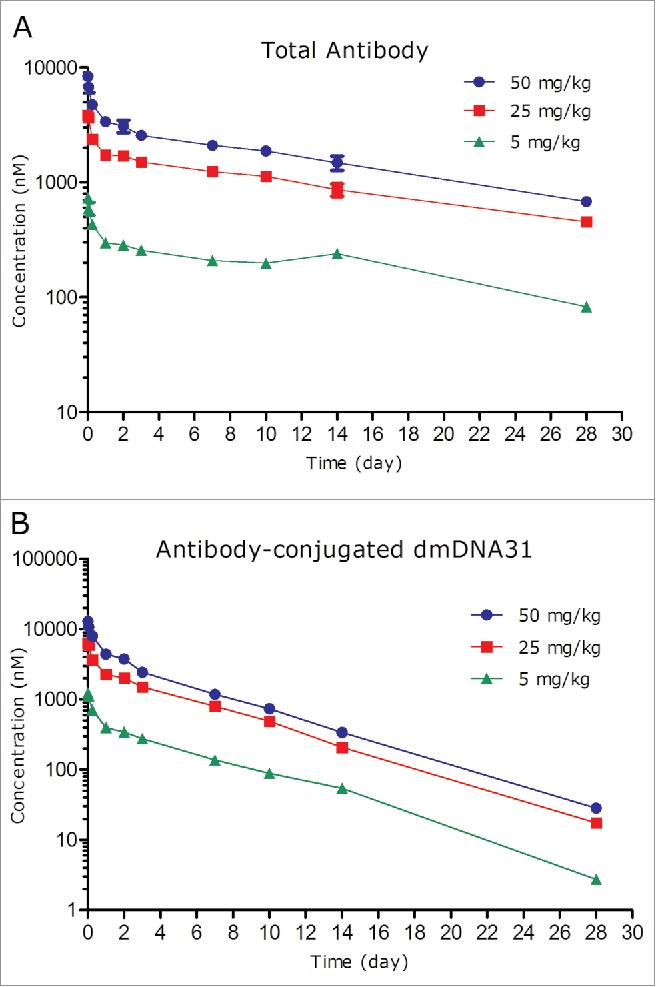
Plasma concentration-time profiles of TAC total antibody (TAb) (A) and antibody-conjugated dmDNA31 (ac-dmDNA31) (B) following IV administration of TAC in non-infected mice.
Table 2.
Non-compartmental PK parameters of TAC TAb and ac-dmDNA31 following a single IV administration of TAC in non-infected mice.
| TAC Total Antibody |
Antibody conjugated dmDNA31 |
|||||
|---|---|---|---|---|---|---|
| PK Parameters | 5 mg/kg | 25 mg/kg | 50 mg/kg | 5 mg/kg | 25 mg/kg | 50 mg/kg |
| Cmax (nM) | 734 | 3830 | 8370 | 1230 | 6320 | 12900 |
| AUC0-inf (day • nM) | 7530 | 37290 | 59880 | 3180 | 16800 | 29000 |
| Clearance (mL/day/kg) | 4.53 | 4.59 | 5.72 | 19.9 | 18.9 | 21.8 |
| Vss (mL/kg) | 95.7 | 91.4 | 97.6 | 109 | 97.1 | 103 |
| t1/2λz (day) | 16.1 | 14.3 | 12.4 | 3.82 | 3.84 | 3.89 |
The systemic exposure of TAC TAb [maximum concentration observed (Cmax), and area under the plasma concentration−time curve extrapolated to infinity (AUC0-inf)] was dose proportional over the dose range of 5 to 50 mg/kg, which is expected given the absence of target antigen in non-infected mice. Following IV administration of 5 to 50 mg/kg TAC, the corresponding TAb systemic clearance ranged from 4.53 to 5.72 mL/day/kg; t1/2λz ranged from 12.4 to 16.1 days, and Vss ranged from 91.4 to 97.6 mL/kg (Table 2).
The concentrations of ac-dmDNA31 reached the peak immediately after administration of TAC and also emulated a bi-exponential disposition pattern (Fig. 5B). The clearance of ac-dmDNA31 following 5 to 50 mg/kg TAC administration ranged from 18.9 to 21.8 mL/day/kg, which was about 4 times faster than that of the TAC TAb (Table 2). The t1/2λz of ac-dmDNA31 following 5 to 50 mg/kg TAC administration ranged from 3.82 to 3.89 days, which was about 3 times shorter than that of the TAC TAb. The Vss of ac-dmDNA31 ranged from 97.1 to 109 mL/kg, which was comparable to that of the TAC Tab.
Unconjugated dmDNA31 plasma concentrations after administration of TAC were low across all groups (Table 3). The highest group mean, Cmax (1.41 nM) was observed at the 50 mg/kg group.
Table 3.
Non-compartmental PK parameters of unconjugated dmDNA31 following a single IV administration of TAC in non-infected mice.
| PK Parameter* | TAC 5 mg/kg* | TAC 25 mg/kg | TAC 50 mg/kg |
|---|---|---|---|
| Cmax (nM) | Below LLOQ | 0.662 | 1.41 |
| tmax (day) | NA | 0.00694 | 0.00694 |
Plasma concentrations of unconjugated dmDNA31 following a 5 mg/kg TAC were all below lower limit of quantitation (LLOQ) and therefore were not included for PK analysis
PK of TAC in mice infected with S. aureus
The PK values of TAC following a single IV dose in infected SCID mice at dose levels of 25 and 50 mg/kg are shown in Fig. 6 and PK parameters are illustrated in Table 2. The plasma concentration-time profiles of TAC TAb at 25 or 50 mg/kg were also bi-exponential in mice infected with S. aureus and consistent with the typical PK profiles of monoclonal antibody-based therapeutic (Fig. 6A). The systemic exposure of TAC TAb (Cmax and AUC0-inf) was dose proportional over the dose range of 25 to 50 mg/kg in mice infected with S. aureus. Systemic clearance of TAC TAb following IV administration of 25 or 50 mg/kg TAC was 5.78 and 6.37 mL/day/kg, respectively; t1/2λz was 14.0 and 13.8 days, respectively (Table 4).
Figure 6.
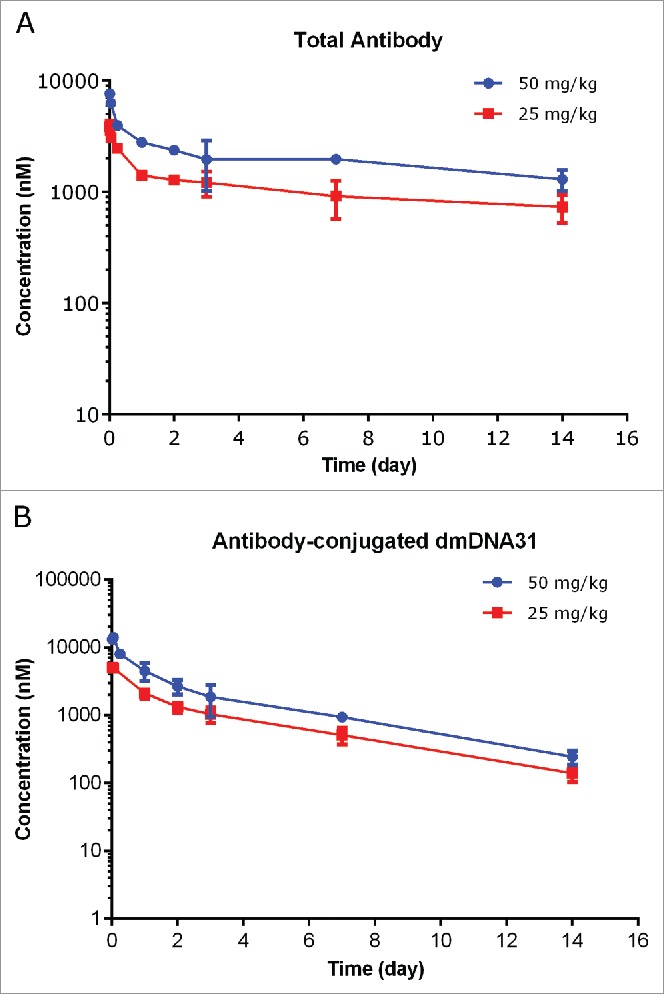
Plasma concentration−time profiles of TAC total antibody (TAb) (A) and antibody-conjugated dmDNA31 (ac-dmDNA31) (B) following IV administration of TAC in mice infected with S. aureus.
Table 4.
Non-compartmental PK parameters of TAC TAb and ac-dmDNA31 following a single IV administration of TAC in mice infected with S. aureus.
| TAC Total Antibody |
Antibody-conjugated dmDNA31 |
|||
|---|---|---|---|---|
| PK Parameters | 25 mg/kg | 50 mg/kg | 25 mg/kg | 50 mg/kg |
| Cmax (nM) | 3850 | 7640 | 5060 | 13100 |
| AUC0-inf (day • nM) | 29560 | 53720 | 12630 | 24390 |
| Clearance (mL/day/kg) | 5.78 | 6.37 | 26.6 | 27.5 |
| Vss (mL/kg) | 115 | 123 | 114 | 110 |
| t1/2λz (day) | 14.0 | 13.8 | 3.81 | 3.73 |
Plasma concentration-time profiles of ac-dmDNA31 showed similar bi-exponential disposition pattern of TAC total antibody (Fig. 6B). The clearance of ac-dmDNA31 following IV administration of 25 or 50 mg/kg TAC was 26.6 and 27.5 mL/day/kg, respectively, which was about 4 times faster than that of the TAC TAb; t1/2λz of ac-dmDNA31 was 3.81 and 3.73 days, respectively, which was about 3 times shorter than that of TAC TAb (Table 4). Unconjugated dmDNA31 concentrations following administration of TAC were low, with the highest group mean Cmax of 1.69 nM (Table 5).
Table 5.
Non-compartmental PK parameters of unconjugated dmDNA31 following a single IV administration of TAC mice infected with S. aureus.
| PK Parameter | TAC 25 mg/kg | TAC 50 mg/kg |
|---|---|---|
| Cmax (nM) | 1.19 | 1.69 |
| tmax (day) | 3.00 | 3.00 |
PD of TAC in mice infected with S. aureus
The PD values of TAC, measured by the bacterial load in various organs (i.e., CFU count), following a single IV dose in infected SCID mice at dose levels of 25 and 50 mg/kg are shown in Fig. 7. Mice treated with TAC at both dose levels showed substantial reduction in CFU in kidneys, heart, and bones compared to the phosphate-buffered saline (PBS)-treated control group on 7 and 14 d post dose. On Day 7, a single dose of TAC at 25 or 50 mg/kg led to 4 – 6.5 log10 reduction of the numbers of CFU (geometric mean) in kidneys, heart, and bones compared to the control group. The decrease in the numbers of CFU was statistically significant in the heart (50 mg/kg) and bones (25 and 50 mg/kg). On day 14, a 2 – 7.6 log10 decrease of the numbers of CFU (geometric mean) compared to the control group was observed in the kidneys, heart, and bones. The reduction of CFU was statistically significant in the kidneys (25 and 50 mg/kg), heart (50 mg/kg), and bones (50 mg/kg) compared to controls. Over 50% of the TAC-treated animals had CFU cleared to the level below the detection limit (250 CFU/organ for heart and kidney, and 167 CFU/organ for bones) in the kidneys, heart and bones on days 7 and 14 (Fig. 7).
Figure 7.
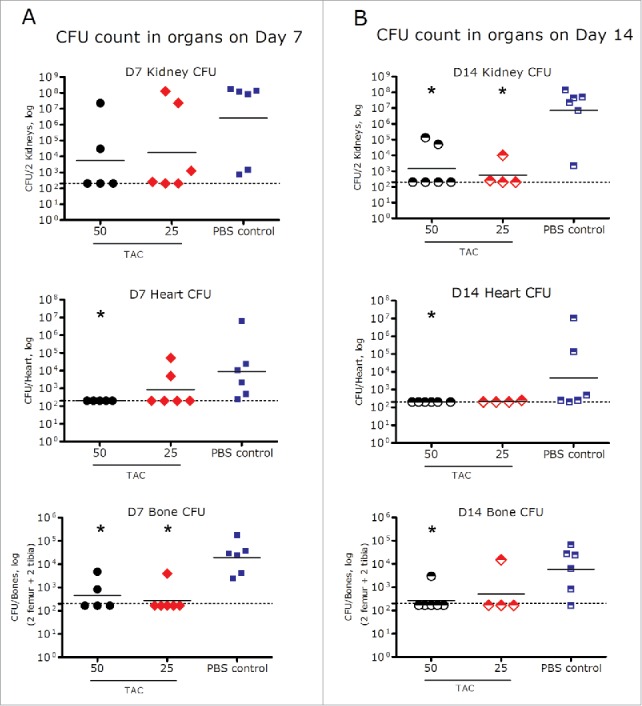
Bacterial load in heart, kidneys, and bones on day 7 (A) and day 14 (B) after a single IV administration of TAC or PBS control in mice infected with S. aureus. *p < 0.05 compared to PBS control (Mann-Whitney test). Dots represent the CFU of the organs from individual infected animals; black dashes represent the group geometric mean. Dash line: Limit of detection (250 CFU/organ for heart and kidney, and 167 CFU/organ for bones).
Discussion
S. aureus is a bacterial pathogen that can cause life-threatening conditions, including bacteremia and osteomyelitis.1,2 Here, we conducted a series of PK and PK/PD studies of TAC and the anti-S. aureus antibody from which TAC was derived in both non-infected and infected mice. These studies characterized PK of TAC in non-infected mice, evaluated the effect of conjugation on the PK of TAC by comparing it to the unconjugated anti-S. aureus antibody, assessed the effect of infection on the PK of TAC, and examined the PD of the TAC in infected mice.
In non-infected mice, both unconjugated anti-S. aureus antibody and TAC TAb demonstrated expected the PK behavior of a monoclonal antibody, characterized by short distribution, relatively slow clearance, and a long half-life.12,13,14 In non-infected mice, TAC total antibody clearance was similar to that of the unconjugated anti-S. aureus antibody, suggesting a minimal effect of the dmDNA31 conjugation on the total antibody clearance. In mice, the PK properties of TAC TAb were generally consistent with the PK properties of other THIOMAB™-drug conjugates.15 Conjugation of dmDNA31 to unconjugated anti-S. aureus antibody extended the half-life of dmDNA31 from ∼3-4 hours (unpublished data) to ∼4 days.16 As a result, TAC offers the advantage of requiring less frequent dosing compared to other small molecule antibiotics because ac-dmDNA31 can act as a reservoir for dmDNA31 due to its longer half-life. The Vss of ac-dmDNA31 was similar to that of the TAC TAb, indicating that the distribution of ac-dmDNA31 was primarily governed by the distribution of the antibody.
In our studies, we observed that clearance of TAC TAb was comparable in mice infected with S. aureus (in the presence of the antigen) and in the non-infected mice over the tested dose range of 25-50 mg/kg. Clearance of ac-dmDNA31 in the infected mice was marginally increased compared to non-infected mice, which could be due to a minor increase in deconjugation caused by the uptake of TAC-bacteria complexes. The PD data indicated that, at the above mentioned dose levels (25-50 mg/kg), there was substantial reduction of bacterial load in the kidneys, heart, and bones. Our data illustrated that infection by S. aureus in mice had minimal effect on the PK of TAC at these efficacious dose levels, suggesting that when target antigen levels were significantly reduced by TAC, target-mediated clearance did not play a major role in clearing TAC. Although doses lower than 25 mg/kg of TAC were not tested in infected mice, comparison with the unconjugated antibody provided information on possible target-mediated clearance of TAC at lower, potentially non-efficacious doses. The clearance of the unconjugated anti-S. aureus antibody was ∼2 to 3-fold faster in mice infected with S. aureus than that in non-infected mice. Unconjugated anti-S. aureus antibody treatment did not significantly reduce the number of CFU in the kidneys and heart compared to the control mice (data not shown). Therefore, the enhanced clearance of unconjugated anti-S. aureus antibody in the mice infected with S. aureus could be attributed to the specific binding of unconjugated anti-S. aureus antibody to target antigen, the internalization of unconjugated anti-S. aureus antibody-opsonized bacteria into the intracellular environment such as a phagolysosome and host proteases, and the resulting increased catabolism of unconjugated anti-S. aureus antibody in the intracellular environment.5 Interestingly, even though the clearance of unconjugated anti-S. aureus antibody increased in the infected mice, it was still approximately linear in the dose range of 5 to 50 mg/kg, suggesting that the binding of unconjugated anti-S. aureus antibody to its target was not saturated in the tested dose range.
The PK studies of TAC in non-infected and infected mice also revealed the in vivo stability of the linker. The change of TAC TAb concentration over time is a function of typical antibody clearance pathways, while the change in ac-dmDNA31 concentration is a product of both antibody clearance and release of dmDNA31 from TAC. In non-infected and infected mice, systemic Cmax of unconjugated dmDNA31 concentrations (Table 3 and Table 5) in plasma following administration of TAC were substantially lower than ac-dmDNA31 (Table 2 and 4), suggesting that dmDNA31 was slowly released from TAC and rapidly cleared from systemic circulation. Our data also demonstrated that S. aureus infection had a limited effect on the linker stability of TAC, as evident by the comparable clearance and half-life of ac-dmDNA31, and comparable systemic Cmax of unconjugated dmDNA31 in non-infected and infected mice at the tested dose range.
TAC showed potent antibacterial activity in the systemic mouse model of S. aureus infection. USA300 was chosen as a representative S. aureus strain to assess the efficacious dose range due to its high prevalence in the United States.17 TAC showed potent antibacterial activity at both 25 and 50 mg/kg in mice infected with S. aureus. The antibacterial activity of TAC was observed in multiple organs, such as kidneys, heart, and bones. The substantial reduction of bacterial load in multiple organs on Days 7 and 14 after a single dose of the TAC implied that it adequately distributed throughout the tissues that were tested, and led to sufficient and prolonged tissue exposures to eliminate the bacteria. The prolonged antibacterial activity may be attributed to the extended half-life of ac-dmDNA31 after a single administration of the TAC.
In conclusion, we characterized the PK of TAC in mice and assessed the effect of drug conjugation and bacterial infection on its PK. TAC showed typical monoclonal antibody PK behavior in both non-infected mice and mice infected with S. aureus. The observed data indicated a minimal impact of conjugation on the PK of the unconjugated anti-S. aureus antibody in non-infected mice. At efficacious doses levels (25-50 mg/kg), infection by S. aureus in mice showed minimal effect on the PK of TAC. TAC also showed strong and prolonged antibacterial activity at 25 and 50 mg/kg in mice infected with S. aureus.
Materials and methods
THIOMAB™ antibody antibiotic conjugate
The anti-S. aureus TAC for in vivo PK and PK/PD studies was generated at Genentech Inc. (South San Francisco, CA). The TAC molecule is composed of a human IgG1 monoclonal antibody, a protease cleavable linker (maleimido-caproyl-valine-citrulline-para-aminobenzyl), and a novel antibiotic, dmDNA31 (4-dimethylamino piperidino-hydroxybenzoxazino rifamycin). dmDNA31 was conjugated to unconjugated anti-S. aureus antibody at the cysteine residues to yield an average antibiotic to unconjugated anti-S. aureus antibody ratio of ∼2.
PK and PK/PD studies
All in vivo PK and PK/PD studies were approved by the Institutional Animal Care and Use Committee at Genentech, Inc. and were conducted in compliance with the regulations of the Association for Assessment and Accreditation of Laboratory Animal Care.
For the PK study using TAC or unconjugated anti-S. aureus antibody in non-infected mice, female SCID mice (6-8 weeks old) received a single IV dose of 5, 25, or 50 mg/kg of TAC or unconjugated anti-S. aureus antibody via the tail vein injection (n = 15/group). Blood samples were collected from 3 mice in each dosing group at each of the following time points: 10 minutes, 1 and 6 hour, 1, 2, 3, 7, 10, 14, 21, and 28 d. The sample collection was done via retro-orbital bleeds or cardiac puncture. Samples were processed to collect plasma for measuring TAC or unconjugated anti-S. aureus antibody concentrations as described in the bioanalysis section.
For the PK study of unconjugated anti-S. aureus antibody in infected mice, female SCID mice were infected with S. aureus by administration of 100 μL PBS containing USA300 NRS384 strain (BEI Resources; Catalog # NRS384) at 1 × 107 CFU per mouse through IV tail injection on day −1. One day post-infection (study day 0), a single IV bolus dose of 5, 25, or 50 mg/kg of anti-S. aureus antibody, or 50 mg/kg control IgG was administered via the tail vein. Blood samples were collected from 3 mice in each dosing group at each of the following time points: 10 min, 1 and 6 hours, 1, 2, 3, 7, 10, 14, 21, and 28 d. The collection was done via retro-orbital bleeds or cardiac puncture. Samples were processed to collect plasma for measuring unconjugated anti-S. aureus antibody concentrations as described in the bioanalysis section. Tissue samples from heart and kidney were collected from 6 animals in each group on day 3 post-dose (n = 6 mice/group). The CFU counts were assessed in these tissues at various time points.
For the PK/PD study of TAC, female SCID mice were infected with S. aureus by administration of 100 μL USA300 NRS384 strain (BEI Resources) at 1 × 107 CFU per mouse through IV tail injection on day −1. One day post-infection (study day 0), a single IV bolus dose of 25 or 50 mg/kg TAC, or 200 μl/mouse of PBS for control group was administered via the tail vein. Blood samples were collected from 3 mice in each dosing group at each of the following time points: 10 minutes, 1 and 6 hours, 1, 2, 3, 7, 10, and 14 d. The blood collection was done via retro-orbital bleeds or cardiac puncture. Samples were processed to collect plasma for measuring TAC concentrations as described in the bioanalysis section. Tissue samples from heart, kidney, tibia, and femur were collected from 6 animals in each group on Days 7 and 14 postdose (n = 6 mice/group). CFU was assessed in these tissues.
Bioanalysis of plasma samples
TAC total antibody and unconjugated anti-S. aureus antibody assay
TAC total antibody and unconjugated anti-S. aureus antibody concentrations were determined in plasma using an enzyme-linked immunosorbent assay (ELISA). The ELISA format used donkey anti-human IgG (Jackson ImmunoResearch, West Grove, PA, Cat 709-006-098) coated on microtiter plates as the capture reagent. Goat anti-human IgG conjugated to horseradish peroxidase (HRP) (Jackson ImmunoResearch, Cat 109-036-098) was used as the detection reagent. The assay had a lower limit of quantitation (LLOQ) of 200 ng/mL in mouse plasma, with a 1:100 minimum dilution.
Antibody-conjugated dmDNA31 assay
Antibody-conjugated dmDNA31 concentrations were determined in plasma samples using a hybrid binding liquid chromatography mass spectrometry (LC-MS/MS) assay. Antibody-conjugated drug was measured using Protein A affinity capture chromatography from plasma followed by enzyme-mediated release of dmDNA31 and electrospray-ionization LCMS/MS for detection. The assay had a LLOQ of 0.244 nM (i.e., 0.226 ng/mL) in mouse plasma.
Unconjugated dmDNA31 assay
The unconjugated dmDNA31 concentrations were determined in plasma samples using a LC-MS/MS assay. For unconjugated dmDNA31 analysis, 25 μL of plasma samples were protein precipitated with 100 μL methanol containing 10 nM piperizino as the internal standard. Supernatants were analyzed using LC-MS/MS with a QTRAP® 5500 mass spectrometer (AB Sciex). The LC-MS/MS assay had a lower limit of quantification of 0.391 nM (i.e., 0.362 ng/mL) in mouse plasma.
Tissue sample for PD (CFU) measurements
Heart, both kidneys, and bones (tibia and femur) were collected at selected time points from each mouse as described above following end point euthanasia at different time points. Heart and kidneys were homogenized in gentleMACSTM Dissociator (Miltenyi Biotec, Inc; Catalog # 130-093-235). Tissue homogenates in a 10-fold serial dilution were spread on blood agar plates (Teknova) and incubated under aerobic conditions at 37°C for 14-18 hours. The total bacterial burden in the organs was determined by counting colonies in the highest dilution with non-overlapping colonies followed by multiplying the colony count with the dilution factor; the final product being expressed as CFU per organ(s).
Femurs and tibias were dissected from muscle, and bones from each leg were placed in a 2 mL screwcap tube containing 2.8 mm ceramic beads (Omni International; Catalog #19628) with 1 mL cold PBS. Bones were homogenized in an Omni Bead Ruptor 24 homogenizer (Omni International; Catalog # 19-040) for 2 cycles of 30 seconds each at 6.95 m/s. The samples were immediately placed on ice to eliminate sample heating, followed by a brief mixing by vortex. The sample homogenates were finally centrifuged for 10 minutes to pellet debris. The homogenate (∼200 μL) of each leg was combined into a single tube per animal and thoroughly mixed by vortexing the sample. The resulting homogenate were 10-fold serially diluted in sterile PBS + 0.05% Tween and were plated on blood agar plates in triplicate followed by overnight incubation at 37°C under aerobic conditions. The total bone bacterial burden was determined by counting colonies in the highest dilution and multiplying this number with the dilution factor and total volume, the final product was expressed as CFU/bones.
PK data analysis
TAC total antibody and antibody-conjugated dmDNA31 plasma concentration−time data were analyzed using a non-compartmental model using Phoenix WinNonlin (Pharsight Corporation Princeton, NJ). The following PK parameters were obtained for TAC total antibody and antibody-conjugated dmDNA31: maximum concentration observed (Cmax), area under the plasma concentration−time curve extrapolated to infinity (AUC0-inf), clearance, volume of distribution at steady state (VSS), and terminal half-life (t1/2λz). The unconjugated dmDNA31 plasma concentration−time data were also analyzed using a non-compartmental model using Phoenix WinNonlin. The following PK parameters were obtained: Cmax and time of maximum concentration observed (tmax). A naïve pooled approach was used to provide one estimate for each treatment group.
Disclosure of potential conflicts of interest
All authors are employees of Genentech, a member of the Roche Group, and hold financial interest in Hoffman-La Roche.
Acknowledgments
The authors would like to thank Saileta Prabhu, Donna Dambach, Mark Velligan, Wouter Hazenbos, and Anshin BioSolutions for reviewing and editing this manuscript. The anti-S. aureus antibody was discovered in collaboration with Symphogen. The linker was licensed from Seattle Genetics.
References
- 1.van Hal SJ, Jensen SO, Vaska VL, Espedido BA, Paterson DL, Gosbell IB. Predictors of mortality in Staphylococcus aureus Bacteremia. Clin Microbiol Rev 2012; 25:362-86; PMID:22491776; http://dx.doi.org/ 10.1128/CMR.05022-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeLeo FR, Chambers HF. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J Clin Invest 2009; 119:2464-74; PMID:19729844; http://dx.doi.org/ 10.1172/JCI38226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Youngster I, Sauk J, Pindar C, Wilson RG, Kaplan JL, Smith MB, Alm EJ, Gevers D, Russell GH, Hohmann EL. Fecal microbiota transplant for relapsing Clostridium difficile infection using a frozen inoculum from unrelated donors: a randomized, open-label, controlled pilot study. Clin Infect Dis 2014; 58:1515-22; PMID:24762631; http://dx.doi.org/ 10.1093/cid/ciu135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thwaites GE, Gant V. Are bloodstream leukocytes Trojan Horses for the metastasis of Staphylococcus aureus? Nat Rev Microbiol 2011; 9:215-22; PMID:21297670; http://dx.doi.org/ 10.1038/nrmicro2508 [DOI] [PubMed] [Google Scholar]
- 5.Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, DePalatis L, Raab H, Hazenbos WL, Hiroshi Morisaki J, et al.. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015; PMID:26536114; http://dx.doi.org/ 10.1038/nature16057 [DOI] [PubMed] [Google Scholar]
- 6.Zhou CLS, Gutierrez J, Rosenberger C, Ljumanovic N, Baruch A, Carrasco M, Saad O, Mariathasan S, Kamath A. Thiomab Antibiotic Conjugate (TAC): PKPD of a novel platform for invasive staphylococcus aureus infections. Interscience Conference of Antimicrobial Agents and Chemotherapy (ICAAC) 2015 [Google Scholar]
- 7.Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, et al.. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol 2012; 30:184-9; PMID:22267010; http://dx.doi.org/ 10.1038/nbt.2108 [DOI] [PubMed] [Google Scholar]
- 8.Kaur S, Xu K, Saad OM, Dere RC, Carrasco-Triguero M. Bioanalytical assay strategies for the development of antibody-drug conjugate biotherapeutics. Bioanalysis 2013; 5:201-26; PMID:23330562; http://dx.doi.org/ 10.4155/bio.12.299 [DOI] [PubMed] [Google Scholar]
- 9.Gorovits B, Alley SC, Bilic S, Booth B, Kaur S, Oldfield P, Purushothama S, Rao C, Shord S, Siguenza P. Bioanalysis of antibody-drug conjugates: American association of pharmaceutical scientists antibody-drug conjugate working group position paper. Bioanalysis 2013; 5:997-1006; PMID:23641692; http://dx.doi.org/ 10.4155/bio.13.38 [DOI] [PubMed] [Google Scholar]
- 10.Deng R, Jin F, Prabhu S, Iyer S. Monoclonal antibodies: what are the pharmacokinetic and pharmacodynamic considerations for drug development? Expert Opin Drug Metab Toxicol 2012; 8:141-60; PMID:22248267; http://dx.doi.org/ 10.1517/17425255.2012.643868 [DOI] [PubMed] [Google Scholar]
- 11.Deng R, Iyer S, Theil FP, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? MAbs 2011; 3:61-6; PMID:20962582; http://dx.doi.org/ 10.4161/mabs.3.1.13799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, et al.. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 2008; 26:925-32; PMID:18641636; http://dx.doi.org/ 10.1038/nbt.1480 [DOI] [PubMed] [Google Scholar]
- 13.Wang W, Yang JJ, Liu L, Chen ZC, Gao Y, Gao JE, An LG, Sun QH. Generation and characterization of monoclonal antibody against HSD11B1. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2008; 24:38-40; PMID:18177616 [PubMed] [Google Scholar]
- 14.Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos 2011; 39:1469-77; PMID:21610128; http://dx.doi.org/ 10.1124/dmd.111.039453 [DOI] [PubMed] [Google Scholar]
- 15.Sukumaran S, Gadkar K, Zhang C, Bhakta S, Liu L, Xu K, Raab H, Yu SF, Mai E, Fourie-O'Donohue A, et al.. Mechanism-based pharmacokinetic/pharmacodynamic model for Thiomab drug conjugates. Pharm Res 2015; 32:1884-93; PMID:25446772; http://dx.doi.org/ 10.1007/s11095-014-1582-1 [DOI] [PubMed] [Google Scholar]
- 16. http://www.lymepa.org/Antibiotics_spreadsheet_2004-1215.pdf [Google Scholar]
- 17.Tenover FC, Sinner SW, Segal RE, Huang V, Alexandre SS, McGowan JE Jr, Weinstein MP. Characterisation of a Staphylococcus aureus strain with progressive loss of susceptibility to vancomycin and daptomycin during therapy. Int J Antimicrob Agents 2009; 33:564-8; PMID:19233622; http://dx.doi.org/ 10.1016/j.ijantimicag.2008.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]