

ABSTRACT
Inhibitors of tumor necrosis factor-α converting enzyme (TACE) have potential as therapeutics for various diseases. Many small molecule inhibitors, however, exhibit poor specificity profiles because they target the highly conserved catalytic cleft of TACE. We report for the first time the molecular interaction of a highly specific anti-TACE antagonistic antibody (MEDI3622). We characterized the binding of MEDI3622 using mutagenesis, as well as structural modeling and docking approaches. We show that MEDI3622 recognizes a unique surface loop of sIVa-sIVb β-hairpin on TACE M-domain, but does not interact with the conserved catalytic cleft or its nearby regions. The exquisite specificity of MEDI3622 is mediated by this distinct structural feature on the TACE M-domain. These findings may aid the design of antibody therapies against TACE.
KEYWORDS: ADAM17, antibody homology modeling, epitope mapping, mutagenesis, protein docking, TACE
Introduction
TACE (tumor necrosis factor-α converting enzyme, also known as ADAM17) is a membrane-bound metalloprotease responsible for ectodomain shedding of pathologically significant substrates.1 It was initially identified as a convertase for tumor necrosis factor-α, a immunomodulatory and proinflammatory cytokine.2,3 It has also been shown to catalyze the release of various epidermal growth factors (EGF),4,5 including amphiregulin, heparin-binding EGF-like growth factor, epiregulin, and epigen, which are key drivers of tumorigenesis. Therefore, TACE is an attractive target for treatments of autoimmune diseases and cancers.
Extensive efforts have been made to develop TACE inhibitors. Many small molecules antagonists against TACE have been reported.6,7 They all bind the catalytic site of TACE, which is remarkably conserved across a disintegrin and metalloprotease (ADAM), ADAM with thrombospondin type-1 motif (ADAMTS), and matrix metalloproteinases (MMPs).8-11 Such structural conservation of the catalytic active site has posed great challenges for the development specific TACE inhibitors.12 In fact, early trials of TACE small molecule inhibitors have failed because of their off-target toxicities.
Antibodies can overcome this specificity obstacle because they can probe diverse epitopes and recognize subtle structural differences. Exploration of unique features of the TACE ectodomain could aid development of TACE inhibitory antibodies with exquisite specificity. A mature TACE extracellular domain (ECD) contains a globular metalloprotease catalytic domain (also known as M-domain) and disulfide-dependent disintegrin (D-) and cysteine rich (C-) domains. The catalytic M-domain has an oblate ellipsoid shape, it and contains a small catalytic cleft at its flat side.8 Proteolytic activity of TACE is solely localized in the M-domain. The role of TACE non-catalytic extracellular domains, including D- and C-domains in enzyme-substrate interactions, has not been well understood. Based on studies involving short peptide-based substrates, the non-catalytic domains were shown not to directly interact with peptide substrates, but may regulate catalytic activity and affect substrate binding through steric hindrance.13 Structural features of TACE ectodomain besides the conserved catalytic cleft may provide unique antagonistic epitopes for TACE-specific therapeutics, thus overcoming the previously mentioned specificity challenges.
A specific TACE inhibitory antibody, MEDI3622 (IgG1, κ), that efficiently inhibited shedding of TACE substrates, such as EGFR-ligands and TNF-α, was recently reported.14 MEDI3622 was shown to induce tumor regression or stasis in multiple EGFR-dependent tumor models.14 The antibody was discovered by screening human single-chain Fv (scFv) phage libraries using the entire TACE ECD, including the catalytic M- and non-catalytic domains. However, the manner in which MEDI3622 binds to TACE and achieves such an exquisite specificity and potent inhibitory activity was not determined.
To understand MEDI3622 biological activity at a molecular level, we characterized the epitope and binding mode of MEDI3622. We mapped its epitope using chimeric variants of TACE and ADAM10, which is most closely related to TACE in the ADAM family. We also modeled the 3-dimensional structure of MEDI3622 and performed protein docking guided by the epitope. Furthermore, we validated the model of the MEDI3622/TACE complex by performing alanine mutagenesis for the interface residues. Our studies revealed that MEDI3622 binds a unique surface loop of TACE M-domain. This work provides the molecular basis for MEDI3622s exquisite specificity and inhibition mechanism, which is distinct from known TACE antagonists. These findings may aid the discovery and design of anti-TACE biologic therapies.
Results
MEDI3622 recognizes TACE M-domain
To map the epitope of MEDI3622 at a domain level, we constructed 2 chimeric TACE/ADAM10 variants by swapping their M- and non-catalytic domains (Fig. 1A). ADAM10 was chosen because it is the closest homolog to TACE, but not recognized by MEDI3622.14 The chimeric variants were constructed to encode full-length proteins with transmembrane and cytoplasmic domains, and were expressed on the surface of human embryonic kidney (HEK) 293F cells. Protein expression and binding of MEDI3622 to the variants were assessed using flow cytometry. Both variants expressed well as monitored using anti-TACE polyclonal and anti-ADAM10 antibodies (Fig. 1B). MEDI3622 did not recognize the loss-of-function (LoF) variant in which the TACE M-domain was replaced with its ADAM10 counterpart (LoF_M) (Fig. 1B). Moreover, MEDI3622 bound well to the gain-of-function (GoF) variant, in which the TACE M-domain was grafted into ADAM10 (KI_M). We further compared the binding kinetics of MEDI3622 to soluble M-domain and entire TACE ECD using surface plasmon resonance (SPR) ProteOn biosensors. MEDI3622 bound to the M-domain with a KD of 2.3 + 0.4 nM, comparable to the KD of 2.0 + 0.3 nM to the entire ECD. Therefore, the epitope of MEDI3622 is localized solely to the TACE M-domain.
Figure 1.
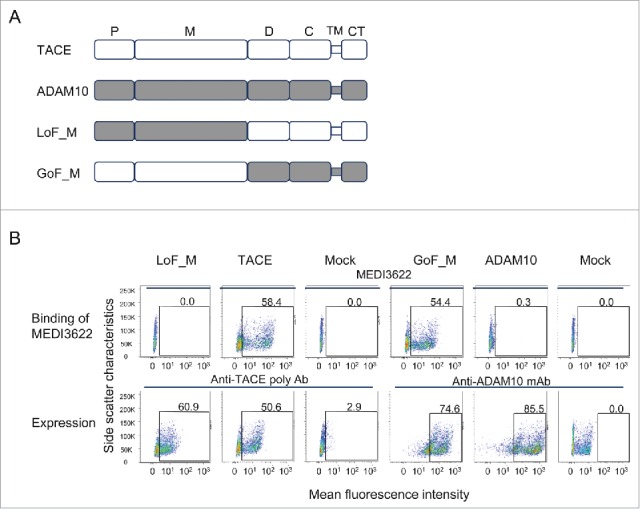
The epitope of MEDI3622 is located in TACE M-domain. (A) Schematic representations of the full-length TACE/ADAM10 chimeric variants. P, M, D, C, TM, and CT stand for prodomain (which is cleaved in mature proteins), metalloproteinase domain, disintegrin-like domain, Cys-rich domain, transmembrane domain, and cytoplasmic domain, respectively. The prodomain and metalloproteinase domain of TACE were replaced with the ADAM10 counterparts to construct the variant LoF_M, and vice versa for the variant GoF_M. (B) Characterization of MEDI3622s binding to full-length TACE chimeric variants by FACS. Protein expression was monitored using anti-TACE polyclonal antibody or anti-ADAM10 monoclonal antibody. The y axis represents side scatter characteristics, and the x axis represents the mean fluorescence intensity. Mock control staining was used to set gates; numbers in gates reflect the percentage of positive cells. MEDI3622 did not bind to the variant LoF_M that encodes the ADAM10 M-domain, but recognized the variant GoF_M when grafting the TACE M-domain into ADAM10.
Refining the epitope in TACE M-domain
To define the exact site bound by MEDI3622, a series of truncated TACE LoF variants were constructed to encode only the M-domain without the non-catalytic ECD domains. From N- to C- terminus, 10 short segments covering the entire TACE M domain were replaced with their ADAM10 counterparts as shown in Fig. 2A. All variants were expressed as soluble proteins with a 6×His tag and captured on SPR biosensors using an anti-His polyclonal antibody for characterization. Their expression levels were monitored using the anti-TACE polyclonal antibody. All variants were detectable at a level comparable to (variants LoF_D, E, F, G, H, I, J), or 40% lower than (variants LoF_A, B, C), the wild type TACE M-domain (data not shown). To assess MEDI3622 binding to the variants, a solution of MEDI3622 was passed over the sensors and its binding signal normalized by each variant's expression level. MEDI3622 bound to most variants at a similar (LoF_G, H, I, and J) or reduced (LoF_A, B, C, D, and F) level compared to the wild type TACE (Fig. 2B). Notably, the binding of MEDI3622 was substantially decreased when the segment E (P366-N381) of TACE, corresponding to the sIVa-sIVb region, was replaced by ADAM10 (LoF_E), which suggests that this region is crucial for the interaction with MEDI3622 (Fig. 2B).
Figure 2.

Determination of the epitope of MEDI3622 at a molecular level. (A) Amino acid alignment of the M-domains of human TACE and ADAM10. Their identical and similar amino acids are shown as green and blue, respectively. Secondary structural elements are shown according to the crystal structure of TACE M-domain,8 with arrows for β-strands, solid boxes for helices and lines for loops. Segments denoted as A-J with dotted lines were swapped between TACE and ADAM10 to construct chimeric variants. (B) Binding characterization of MEDI3622 to TACE/ADAM10 chimeric variants using SPR. Binding was calculated as % binding compared to wild-type TACE after normalization of expression levels using the following formula: [(Response TACE variants MEDI3622/Response TACE wildtype MEDI3622)/(Response TACE variants polyAb/Response TACE wildtype polyAb)] *100. Replacing the segment E with the ADAM10 residues abolished the binding of MEDI3622 (LoF_E), while grafting it to ADAM10 led to the recognition of MEDI3622 (GoF_E). Results represent the means of 3 independent experiments with error bars indicating standard deviations. (C) Determination of MEDI3622s binding to full-length TACE variants by FACS. Expression levels of TACE and its variants were monitored using anti-TACE polyclonal antibody. The y axis represents side scatter characteristics, and the x axis represents the mean fluorescence intensity. Mock control staining was used to set gates; numbers in gates reflect the percentage of positive cells. MEDI3622 did not recognize the full-length TACE chimeric variant encoding for the segment E of ADAM10.
Since LoF variants may show lack of binding to MEDI3622 due to improper folding, we further employed KI variants to confirm our data. To verify the importance of segment E, we constructed a KI variant by grafting it into ADAM10 (KI_E). Additionally, we built a KI variant (KI_A-F) encoding a larger E-containing segment, spanning from segment A to F, to account for LoF variants LoF_A, B, C, D, and F decreased binding to MEDI3622. MEDI3622 recognized both KI variants encoding the segment E of TACE. Interestingly, grafting additional segments of A, B, C, D and F (KI_A-F) did not further increase the binding of MEDI3622 compared to KI_E variant. Thus, these segments do not directly contribute to the interaction with MEDI3622. The reduced binding observed in their LoF variants is likely due to perturbed protein folding and conformation in the E region. In summary, essentially all the binding energy for MEDI3622 resides in TACE segment E, encompassing sIVa-sIVb P366-N381.
We also confirmed the identity of the MEDI3622 epitope using full-length membrane-bound TACE proteins. We replaced TACE segment E with its ADAM10 counterpart in the context of full-length TACE consisting of the entire ECD, transmembrane, and cytoplasmic domains. Variants were expressed on the surface of HEK293F cells and characterized using flow cytometry. As expected, MEDI3622 lost binding to the full-length variant LoF_Efl, in which the segment E was replaced by ADAM10 (Fig. 2C). Binding of MEDI3622 was not affected when segments D and J, which were randomly selected as controls, were replaced (Fig. 2C). Taken together, we determined that the epitope of MEDI3622 spans amino acids P366-N381 and consists of 2 β strands (sIVa and sIVb) and a short loop.
Structure modeling of the MEDI3622/TACE complex
To gain further insights on the binding mode of MEDI3622, computational modeling was applied to simulate the complex of MEDI3622/TACE. The three-dimensional structure of the variable domains (Fv) of MEDI3622 (amino acid sequences shown in Fig. 3A) was predicted by homology modeling using Discovery Studio. Standard antibody modeling procedures were applied to predict the framework structure and complementarity-determining region (CDR) conformations of MEDI3622 using the following homology templates: 3UC0 for variable light (VL) (94% identity), 4KFZ for variable heavy (VH) (98% identity), 1HEZ for VL/VH interface (93% identity), 4KMT for CDR1L, 2L and 3L (83%, 100%, and 100% identity, respectively), 3S34 for CDR1H (88% identity), 3R1G for CDR2H (75% identify) and 3EYO for CDR3H (42% identity). Side-chain minimization was performed on all the non-conserved residues to further refine the model. The top-ranked model was selected and validated using Ramachandran plots (98% Ramachandran favored residues). The model structure of MEDI3622 Fv shows that CDR1L, CDR2L, CDR3L, CDR1H and CDR2H adopt canonical conformations15 of L1-11A, L2-7A, L3-9A, H1-10A and H2-10B, respectively (Fig. 3B). Along with a short CDR3H loop (10 residues), the 6 CDR loops form a relatively flat antigen binding site surface (Fig. 3B).
Figure 3.

Structure prediction of MEDI3622. (A) Amino acid sequences of MEDI3622 VH and VL domains. CDRs are shown in bold and underlined according to Chothia definition.29 (B) Modeled 3-dimensional structure of MEDI3622 variable domains, with VH in orange and VL in beige. CDRs are shown in blue. The CDR1L, 2L, 3L, 1H, and 2H of MEDI3622 adopt canonical conformations L1-11A, L2-7A, L3-9A, H1-10A and H2-10B, respectively, and form a relative flat antigen-binding surface with a short CDR3H.
We then docked the refined model of MEDI3622 Fv to the crystal structure of TACE M-domain (PDB 3E8R)16 using protein docking tools integrated in Discovery Studio. After examining top ranked poses in large clusters, a final model consistent with epitope mapping results was selected. The model complex structure offers additional information. In particular, it suggested that MEDI3622 mainly interacts with the sIVa strand of the sIVa-sIVb β-hairpin of TACE M-domain. In the “standard” orientation, the M-domain has a shape of an oblate ellipsoid with a notch of the catalytic cleft in its side, creating an upper main molecular body and a lower small subdomain (Fig. 4A). The sIVa-sIVb β-hairpin, the epitope of MEDI3622, projects from the central β-sheet of the upper large subdomain of M-domain. The model indicated that MEDI3622 “clutches” this “ear-like” sIVa-sIVb β-hairpin surface loop from the lateral side of M-domain, without interacting with the major globular body of the TACE M-domain (Fig. 4A). MEDI3622 appears to interact primarily with the exterior side of the β-hairpin, with residues Y369Y370S371 of the sIVa strand located in the center of the binding interface; the interior side of the β-hairpin, sIVb strand remains peripheral to the interface in the model. Although a detailed binding interface analysis requires a crystal structure, this model complex offered new insights on the binding mode of MEDI3622.
Figure 4.
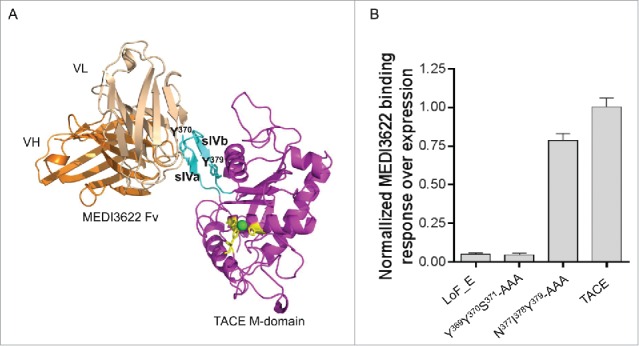
Proposed binding mode of MEDI3622. (A) Three-dimensional model of MEDI3622 bound to TACE. The identified epitope of MEDI3622 (P366-N381) (cyan) adopts a β-hairpin loop conformation projecting from the central body of M-domain (magenta). TACE residues Y370 and Y379 (cyan) along with conserved histidines H405, H409, H415 (yellow) are shown in sticks. The Zn ion is shown as a green sphere. MEDI3622 (VH in orange, VL in beige) binds to TACE through interacting with the sIVa-sIVb β-hairpin loop, with the sIVa strand (Y369Y370S371) located in the center of the binding interface. (B) Binding of MEDI3622 to TACE alanine mutants. Two stretches of amino acids were mutated into alanines, including Y369Y370S371 (sIVa strand, the center of the binding interface) and N377I378Y379 (sIVb strand, peripheral to the binding interface). The alanine variants were expressed as soluble proteins and their binding profiles with MEDI3622 were characterized using SPR. Binding was calculated as % binding compared to wild type TACE after normalization of expression levels using the following formula: [(Response TACE variants MEDI3622/Response TACE wildtype MEDI3622)/(Response TACE variants polyAb/Response TACE wildtype polyAb)] *100. Binding of MEDI3622 to TACE was abolished when mutating residues Y369Y370S371 to alanines, while its binding was retained upon mutating residues N377I378Y379. Results represent the means of 3 independent experiments with error bars indicating standard deviations.
Validation of the model of MEDI3622/TACE complex
We further performed alanine mutations to validate the model of MEDI3622/TACE complex. Based on the model complex structure, 2 stretches of amino acids of the sIVa-sIVb β-hairpin were mutated to alanines, including Y369Y370S371 (sIVa strand) in the center of the binding interface and N377I378Y379 (sIVb strand) peripheral to the binding interface. These alanine variants were expressed as soluble proteins and characterized using SPR. Mutation of the interface residues Y369Y370S371 abolished the binding of MEDI3622 to TACE to a degree comparable to replacing the entire β-hairpin loop (segment E) of TACE with ADAM10 (LoF_E) (Fig. 4B). On the other hand, MEDI3622 retained its binding upon alanine mutations of residues N377I378Y379. These results confirm the accuracy of the docking model of MEDI3622/TACE complex and further identified the critical residues that dominant contribution to the free energy of binding.
Unique inhibition mechanism of MEDI3622
We propose that MEDI3622 inhibits TACE proteolysis through a mechanism that is distinct compared to that of small molecule TACE inhibitors and its natural protein inhibitor TIMP-3 (tissue inhibitor of metalloproteinases 3). These inhibitors interact with the conserved catalytic cleft located on the surface of the ellipsoid-shaped M domain.7,8,17,18 The catalytic cleft contains a catalytic zinc (Zn) ion at its center, coordinated by 4 highly conserved residues (H405, E406, H409, H415) typical of a Zn-binding motif (HEXXHXXGXXHD where X denotes any amino acid). In proximity to the catalytic Zn and coordinating histidines, several conservative binding pockets (known as S1, S1′, S2, S2′, and S3′) exist in the catalytic cleft. Small molecule and peptide-based TACE inhibitors bind to these pockets and blocks the recognition of TACE substrates.7,8,16,17 Similarly, the protein inhibitor TIMP-3 occupies the entrance space to the catalytic cleft, through interacting with the catalytic cleft regions, including the sIV bulge-edge strand, S1′ wall-forming segment, and residues near the S1′ and S3′ pockets.18 In contrast, MEDI3622 only binds to the projecting surface loop of sIVa-sIVb, without interacting with the catalytic cleft or its nearby regions. As seen in Fig. 5A–B, no overlap of the MEDI3622 epitope and the footprints of small molecule-based inhibitors and TIMP-3 is observed when the MEDI3622/M-domain model complex is superimposed with the M-domains bound with small molecule inhibitors (PDB 3LE919 and 3O6420) or the N-terminal domain of TIMP-3 (PDB 3CKI18). Furthermore, we confirmed that MEDI3622 and TIMP-3 bound concurrently to TACE (Fig. 5C). Taken together, our results show that MEDI3622 interacts with TACE differently from small molecule TACE inhibitors and TIMP-3.
Figure 5.
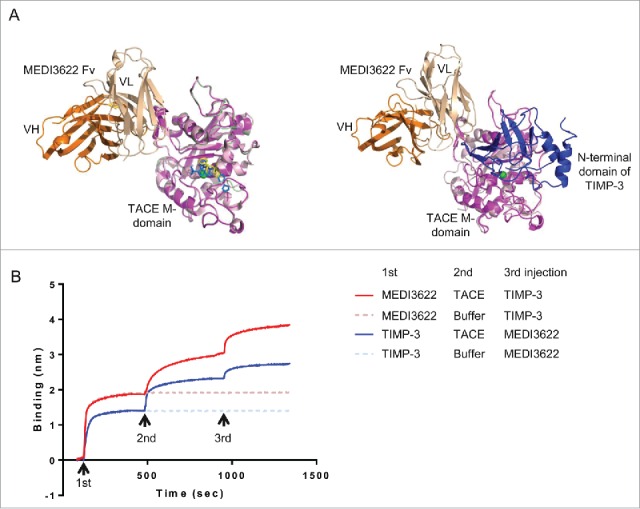
Unique binding mode of MEDI3622. (A) The binding mode of MEDI3622 appears different from TACE small molecule and natural protein inhibitors upon structure alignments. (Left) The model of the MEDI3622 (VH orange and VL beige)/TACE (magenta) complex was superimposed to crystal structures of TACE M-domains complexed with small molecule inhibitors (PDB 3LE919 and 3O6420) through the common TACE M-domains. The M-domains for 3LE9 and 3O64 were shown in light pink and pale green and their bound small molecule inhibitors were displayed in yellow and blue sticks, respectively. (Right) The model of the MEDI3622/TACE complex was aligned with the crystal structure of TACE M-domain (light pink) bound to the N-terminal domain of TIMP-3 (blue) (PDB 3CKI). (B) Concurrent binding of MEDI3622 and TIMP-3 to TACE. The concurrent binding of MEDI3622 and TIMP-3 to TACE was assessed using a “sandwich”-like binding assay by Octet. Streptavidin Octet sensors were immobilized with biotinylated MEDI3622 or TIMP-3 (1st response), then incubated with TACE ECD or buffer (2nd response), and lastly followed with TIMP-3 or MEDI3622 (3rd response).
Discussion
This study revealed a unique inhibitory epitope on TACE M-domain. Small molecule TACE inhibitors and TIMP-3 interact with the conserved catalytic cleft on TACE M-domain. The highly conserved nature of this catalytic cleft poses challenges for the development of specific TACE inhibitors that do not cross-react with other metalloproteinases. MEDI3622, however, binds to the sIVa-sIVb β-hairpin, a projecting surface loop on TACE M-domain that is peripheral to the catalytic cleft. Unlike the conserved catalytic cleft and central body of the M-domain, this surface loop is a unique structural feature for TACE. Superimposing the M-domain structure of TACE with other ADAMs, ADAMTSs, and MMPs, it becomes obvious that this β-hairpin loop is exclusive to TACE (Fig. 6). The exquisite specificity of MEDI3622 is due to its ability to bind this distinct structural feature.
Figure 6.
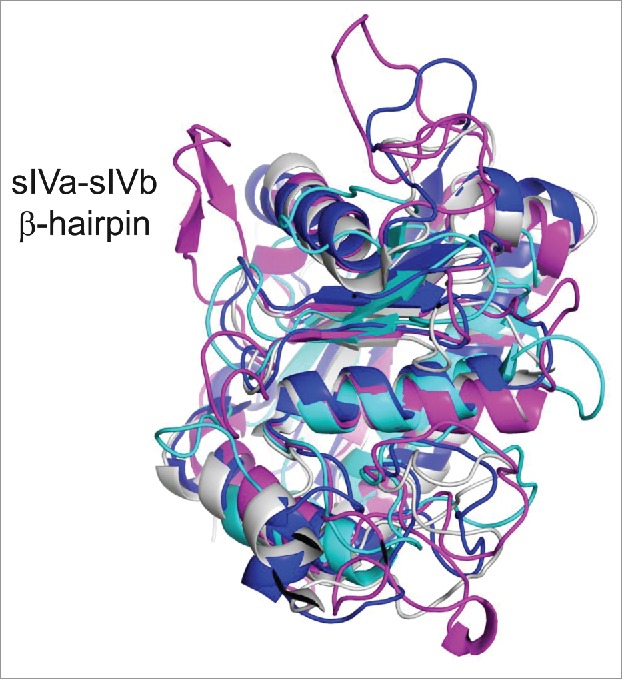
Structure alignment of the M-domain structures of TACE and other ADAMs, ADAMTSs and MMPs. Structural alignment of M-domain structures shows that the epitope of MEDI3622, the surface loop of sIVa-sIVb β-hairpin in the TACE M-domain, is a structural feature unique to TACE. The M-domain is shown in magenta for TACE (PDB 3E8R),16 blue for ADAMTS-5 (PDB 3LJT),30 cyan for MMP-9 (PDB 5CUH)31 and gray for ADAM22 (PDB 3G5C).32
The binding mode of MEDI3622 also differs from previously reported anti-TACE inhibitory mAbs, D1(A12) and A9.21,22 Tape et al identified the D1(A12) antibody using a “2-step” phage display approach involving first the isolation of an inhibitory antibody that bound TACE non-catalytic domains through VH, then the shuffling of its VL to introduce simultaneous binding to the M-domain through VL. The binding of D1(A12) thus requires both TACE non-catalytic domains and the catalytic M-domain. The A9 anti-TACE mAb was discovered from the same phage-panning campaign as D1(A12), but possesses mouse and human cross-reactivity compared to D1(A12). Their epitopes remain to be determined. MEDI3622 was discovered by panning scFv phage libraries using the entire TACE ECD composed of the M-domain and non-catalytic domains. Interestingly, MEDI3622 binds only the small surface loop of sIVa-sIVb β-hairpin on M-domain, regardless of the multidomain topology of TACE.
The inhibitory activity of MEDI3622 is likely mediated by competitive or non-competitive inhibition of protein substrates of TACE via steric hindrance. Although the recognition of natural protein substrates by TACE is not yet well understood, it seems that the cleavage site recognition is less critical in promoting TACE cleavage and substrate specificity compared to the general importance of the active site in mediating protease substrate recognition.23-25 Inspection of substrate cleavage sites for TACE failed to reveal a well-defined consensus sequence, despite tendencies for specific residues to be present at several positions. It is conceivable that specific complementary interactions between TACE and its substrates besides the catalytic active site are important. For instance, the surface loops of TACE M-domain may be involved in or regulate substrates recognition and specificity, although they are peripheral to the catalytic cleft. Given the large footprint of an antibody, the binding of MEDI3622 to the surface loop of sIVa-sIVb β-hairpin potentially blocks the binding of natural macromolecular substrates via steric hindrance.
In conclusion, we studied TACE, which is a metalloprotease relevant to a variety of diseases, and report here a unique structural feature of the protein. Our results offer new insights that may aid in the design of novel inhibitory anti-TACE antibody therapeutics that exhibit superb specificity.
Materials and methods
Construction and expression of chimeric TACE variants
DNA encoding full length TACE or ADAM10 (NCBI reference NP_003174.3 and NP_001101.1, respectively), recombinant soluble TACE ECD, and MEDI3622 (MedImmune, human IgG1/κ) were generated at MedImmune. DNA encoding chimeric TACE/ADAM10 variants were assembled using PCR by overlap extension, and cloned into the mammalian expression vector pcDNA3.1 (Invitrogen). Chinese hamster ovary (CHO) cells were transiently transfected with DNA constructs using Lipofectamine® LTX reagent with PLUS™ reagent (Invitrogen) according to the manufacturer's instructions. Transfected cells were harvested for flow cytometry characterization 24 h post-transfection for full-length proteins with transmembrane domains. Supernatants were harvested for SPR characterization 6 d post-transfection for soluble proteins, namely the entire extracellular domain or the catalytic M-domain.
Flow cytometry binding of MEDI3622 to TACE variants
About 106 CHO cells were transfected and incubated with 1 μg/ml MEDI3622 in 50 µl phosphate-buffered saline (PBS) containing 1% BSA for 30 min on ice. Cells were washed 3 times with 200 µl ice-cold PBS, and incubated with 1 μg/ml of an anti-human IgG antibody conjugated to fluorescein isothiocyanate (FITC) (Invitrogen) in 50 µl PBS containing 1% BSA for 30 min on ice. Protein expression was monitored by incubating the cells with 2 µg/ml goat anti-TACE poly antibody (R&D Systems) followed by FITC-conjugated anti-goat IgG antibody (Invitrogen) or PE-conjugated anti-ADAM10 monoclonal antibody (BioLegend) in 50 µl PBS containing 1% BSA for 30 min on ice. Cells were then washed 3 times with 200 µl ice-cold PBS. All samples were analyzed using a LSRII flow cytometer and FlowJo (BD Biosciences).
SPR characterization of MEDI3622 to TACE variants
Binding of MEDI3622 to TACE variants was assessed using a ProteOn XPR36 instrument (Bio-Rad). Standard amine coupling was used to immobilize an anti-His polyclonal antibody (MedImmune) in 10 mM sodium acetate (pH 5.0) to the surface of a ProteOn GLC biosensor chip (Bio-Rad) at ∼5,000 resonance units (RU) for each channel. TACE variants with a 6×His tag in transfected cell supernatant were captured on the chip surface by the immobilized anti-His polyclonal antibody. Expression levels of the variants were normalized using appropriate dilutions to achieve comparable levels of ligand density. Un-transfected cell supernatant was also injected under the same conditions in a reference channel. Fifty nM MEDI3622 or anti-TACE poly (R&D Systems) in PBS (pH 7.4) with 0.005% (v/v) Tween-20 was then flowed over the captured surface for 150 sec at 90 μL/min with 200 sec dissociation time. The sensor surface was regenerated twice by injecting glycine buffer (10 mM, pH 1.5) at 100 μL/min for 30 sec. Data were processed with the ProteOn Manager 3.0.1 software.
Competition binding between MEDI3622 and TIMP-3 to TACE
Competition binding between MEDI3622 and TIMP-3 for binding to TACE was assessed using an Octet QK384 (ForteBio). MEDI3622 and TIMP-3 (MedImmune) were biotinylated through primary amine groups using sulfo-NHS-biotin (Thermo Scientific) following manufacture instructions. Biotinylated Medi3622 or TIMP-3 at 5 µg/ml in PBS containing 1% BSA was captured on Octet streptavidin-sensors (ForteBio) for 380 sec. Sensors were then incubated with 5 µg/ml TACE-ECD (MedImmune) in PBS containing 1% BSA or buffer for 450 sec, followed with the incubation of TIMP-3 (5 µg/ml) or MEDI3622 in PBS containing 1% BSA for 450 sec.
MEDI3622 structure modeling and protein docking
The structure of MEDI3622 variable domains was predicted using Discovery Studio 4.5 (Biovia). Default antibody structure modeling protocols were used to model the frameworks and CDRs. Briefly, a BLAST search was performed against the protein data bank (Berman HM, The Protein Data Bank. Nucleic Acids Res. 2000) to identify framework templates for VH, VL and VH/VL interface with the highest sequence homology to MEDI3322. Fifty models were then constructed through homology modeling using these 3 structural templates. The top-scored framework model with the lowest probability density function energy was further selected for modeling of the CDR loops. CDRs were built by homology modeling using CDR templates sharing the highest sequence identity compared with MEDI3622 CDRs. The top-ranked model was then inspected for clashes between atoms, in which case limited minimization was performed for side-chains using CHARMm. The quality of the MEDI3622 model was validated using Ramachandran plots of DS 4.5. Illustrations were prepared using PyMOL (Schrödinger).
ZDOCK26 in DS 4.5 was used to dock the human MEDI3622 model structure to the TACE M-domain. The coordinates of TACE M-domain were prepared for docking using PDB ID number 3E8R16 and the protein preparation tool in Discovery studio 4.5. CHARMm force field27 was applied throughout the simulation. Rigid-body docking was performed at a 6° angular step size and clustered for the top 2000 poses. The clusters with the highest density of poses were further refined using RDOCK. Top poses with low RDOCK28 energies were analyzed for consistency with the experimental epitope mapping results.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Study supported by MedImmune, Gaithersburg, MD, 20878, USA.
References
- 1.Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med 2008; 29:258-89; PMID:18762209; http://dx.doi.org/ 10.1016/j.mam.2008.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, et al.. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997; 385:729-33; PMID:9034190; http://dx.doi.org/ 10.1038/385729a0 [DOI] [PubMed] [Google Scholar]
- 3.Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, et al.. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 1997; 385:733-6; PMID:9034191; http://dx.doi.org/ 10.1038/385733a0 [DOI] [PubMed] [Google Scholar]
- 4.Sunnarborg SW, Hinkle CL, Stevenson M, Russell WE, Raska CS, Peschon JJ, Castner BJ, Gerhart MJ, Paxton RJ, Black RA, et al.. Tumor necrosis factor-alpha converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J Biol Chem 2002; 277:12838-45; PMID:11823465; http://dx.doi.org/ 10.1074/jbc.M112050200 [DOI] [PubMed] [Google Scholar]
- 5.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol 2004; 164:769-79; PMID:14993236; http://dx.doi.org/ 10.1083/jcb.200307137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murumkar PR, Giridhar R, Yadav MR. Novel methods and strategies in the discovery of TACE inhibitors. Exp Opin Drug Discovery 2013; 8:157-81; PMID:23231541; http://dx.doi.org/ 10.1517/17460441.2013.744745 [DOI] [PubMed] [Google Scholar]
- 7.DasGupta S, Murumkar PR, Giridhar R, Yadav MR. Current perspective of TACE inhibitors: a review. Bioorg Med Chem 2009; 17:444-59; PMID:19095454; http://dx.doi.org/ 10.1016/j.bmc.2008.11.067 [DOI] [PubMed] [Google Scholar]
- 8.Maskos K, Fernandez-Catalan C, Huber R, Bourenkov GP, Bartunik H, Ellestad GA, Reddy P, Wolfson MF, Rauch CT, Castner BJ, et al.. Crystal structure of the catalytic domain of human tumor necrosis factor-alpha-converting enzyme. Proc Natl Acad Sci U S A 1998; 95:3408-12; PMID:9520379; http://dx.doi.org/ 10.1073/pnas.95.7.3408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerhardt S, Hassall G, Hawtin P, McCall E, Flavell L, Minshull C, Hargreaves D, Ting A, Pauptit RA, Parker AE, et al.. Crystal structures of human ADAMTS-1 reveal a conserved catalytic domain and a disintegrin-like domain with a fold homologous to cysteine-rich domains. J Mol Biol 2007; 373:891-902; PMID:17897672; http://dx.doi.org/ 10.1016/j.jmb.2007.07.047 [DOI] [PubMed] [Google Scholar]
- 10.Mosyak L, Georgiadis K, Shane T, Svenson K, Hebert T, McDonagh T, Mackie S, Olland S, Lin L, Zhong X, et al.. Crystal structures of the two major aggrecan degrading enzymes, ADAMTS4 and ADAMTS5. Protein Sci 2008; 17:16-21; PMID:18042673; http://dx.doi.org/ 10.1110/ps.073287008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shieh HS, Mathis KJ, Williams JM, Hills RL, Wiese JF, Benson TE, Kiefer JR, Marino MH, Carroll JN, Leone JW, et al.. High resolution crystal structure of the catalytic domain of ADAMTS-5 (aggrecanase-2). J Biol Chem 2008; 283:1501-7; PMID:17991750; http://dx.doi.org/ 10.1074/jbc.M705879200 [DOI] [PubMed] [Google Scholar]
- 12.Li NG, Shi ZH, Tang YP, Wei L, Lian Y, Duan JA. Discovery of selective small molecular TACE inhibitors for the treatment of rheumatoid arthritis. Curr Med Chem 2012; 19:2924-56; PMID:22519396; http://dx.doi.org/ 10.2174/092986712800672120 [DOI] [PubMed] [Google Scholar]
- 13.Stawikowska R, Cudic M, Giulianotti M, Houghten RA, Fields GB, Minond D. Activity of ADAM17 (a disintegrin and metalloprotease 17) is regulated by its noncatalytic domains and secondary structure of its substrates. J Biol Chem 2013; 288:22871-9; PMID:23779109; http://dx.doi.org/ 10.1074/jbc.M113.462267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rios-Doria J, Sabol D, Chesebrough J, Stewart D, Xu L, Tammali R, Cheng L, Du Q, Schifferli K, Rothstein R, et al.. A monoclonal antibody to ADAM17 inhibits tumor growth by inhibiting EGFR and Non-EGFR-mediated pathways. Mol Cancer Ther 2015; 14:1637-49; PMID:25948294; http://dx.doi.org/ 10.1158/1535-7163.MCT-14-1040 [DOI] [PubMed] [Google Scholar]
- 15.Martin AC, Thornton JM. Structural families in loops of homologous proteins: automatic classification, modelling and application to antibodies. J Mol Biol 1996; 263:800-15; PMID:8947577; http://dx.doi.org/ 10.1006/jmbi.1996.0617 [DOI] [PubMed] [Google Scholar]
- 16.Mazzola RD Jr., Zhu Z, Sinning L, McKittrick B, Lavey B, Spitler J, Kozlowski J, Neng-Yang S, Zhou G, Guo Z, et al.. Discovery of novel hydroxamates as highly potent tumor necrosis factor-alpha converting enzyme inhibitors. Part II: optimization of the S3′ pocket. Bioorg Med Chem Lett 2008; 18:5809-14; PMID:18835710; http://dx.doi.org/ 10.1016/j.bmcl.2008.09.045 [DOI] [PubMed] [Google Scholar]
- 17.DasGupta S, Murumkar PR, Giridhar R, Yadav MR. Studies on novel 2-imidazolidinones and tetrahydropyrimidin-2(1H)-ones as potential TACE inhibitors: design, synthesis, molecular modeling, and preliminary biological evaluation. Bioorg Med Chem 2009; 17:3604-17; PMID:19394232; http://dx.doi.org/ 10.1016/j.bmc.2009.04.003 [DOI] [PubMed] [Google Scholar]
- 18.Wisniewska M, Goettig P, Maskos K, Belouski E, Winters D, Hecht R, Black R, Bode W. Structural determinants of the ADAM inhibition by TIMP-3: crystal structure of the TACE-N-TIMP-3 complex. J Mol Biol 2008; 381:1307-19; PMID:18638486; http://dx.doi.org/ 10.1016/j.jmb.2008.06.088 [DOI] [PubMed] [Google Scholar]
- 19.Yu W, Tong L, Kim SH, Wong MK, Chen L, Yang DY, Shankar BB, Lavey BJ, Zhou G, Kosinski A, et al.. Biaryl substituted hydantoin compounds as TACE inhibitors. Bioorg Med Chem Lett 2010; 20:5286-9; PMID:20663669; http://dx.doi.org/ 10.1016/j.bmcl.2010.06.134 [DOI] [PubMed] [Google Scholar]
- 20.Dai C, Li D, Popovici-Muller J, Zhao L, Girijavallabhan VM, Rosner KE, Lavey BJ, Rizvi R, Shankar BB, Wong MK, et al.. 2-(2-Aminothiazol-4-yl)pyrrolidine-based tartrate diamides as potent, selective and orally bioavailable TACE inhibitors. Bioorg Med Chem Lett 2011; 21:3172-6; PMID:21458257; http://dx.doi.org/ 10.1016/j.bmcl.2011.01.002 [DOI] [PubMed] [Google Scholar]
- 21.Tape CJ, Willems SH, Dombernowsky SL, Stanley PL, Fogarasi M, Ouwehand W, McCafferty J, Murphy G. Cross-domain inhibition of TACE ectodomain. Proc Natl Acad Sci U S A 2011; 108:5578-83; PMID:21415364; http://dx.doi.org/ 10.1073/pnas.1017067108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwok HF, Botkjaer KA, Tape CJ, Huang Y, McCafferty J, Murphy G. Development of a ‘mouse and human cross-reactive’ affinity-matured exosite inhibitory human antibody specific to TACE (ADAM17) for cancer immunotherapy. Protein Eng Des Sel 2014; 27:179-90; PMID:24769623; http://dx.doi.org/16239146 10.1093/protein/gzu010 [DOI] [PubMed] [Google Scholar]
- 23.Janes PW, Saha N, Barton WA, Kolev MV, Wimmer-Kleikamp SH, Nievergall E, Blobel CP, Himanen JP, Lackmann M, Nikolov DB. Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell 2005; 123:291-304; PMID:16239146; http://dx.doi.org/ 10.1016/j.cell.2005.08.014 [DOI] [PubMed] [Google Scholar]
- 24.Althoff K, Reddy P, Voltz N, Rose-John S, Mullberg J. Shedding of interleukin-6 receptor and tumor necrosis factor alpha. Contribution of the stalk sequence to the cleavage pattern of transmembrane proteins. Eur J Biochem 2000; 267:2624-31; PMID:10785383; http://dx.doi.org/ 10.1046/j.1432-1327.2000.01278.x [DOI] [PubMed] [Google Scholar]
- 25.Hinkle CL, Sunnarborg SW, Loiselle D, Parker CE, Stevenson M, Russell WE, Lee DC. Selective roles for tumor necrosis factor alpha-converting enzyme/ADAM17 in the shedding of the epidermal growth factor receptor ligand family: the juxtamembrane stalk determines cleavage efficiency. J Biol Chem 2004; 279:24179-88; PMID:15066986; http://dx.doi.org/ 10.1074/jbc.M312141200 [DOI] [PubMed] [Google Scholar]
- 26.Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins 2003; 52:80-7; PMID:12784371; http://dx.doi.org/ 10.1002/prot.10389 [DOI] [PubMed] [Google Scholar]
- 27.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. Charmm - a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 1983; 4:187-217; http://dx.doi.org/ 10.1002/jcc.540040211 [DOI] [Google Scholar]
- 28.Li L, Chen R, Weng Z. RDOCK: refinement of rigid-body protein docking predictions. Proteins 2003; 53:693-707; PMID:14579360; http://dx.doi.org/ 10.1002/prot.10460 [DOI] [PubMed] [Google Scholar]
- 29.Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol 1987; 196:901-17; PMID:3681981; http://dx.doi.org/ 10.1016/0022-2836(87)90412-8 [DOI] [PubMed] [Google Scholar]
- 30.Shieh HS, Tomasselli AG, Mathis KJ, Schnute ME, Woodard SS, Caspers N, Williams JM, Kiefer JR, Munie G, Wittwer A, et al.. Structure analysis reveals the flexibility of the ADAMTS-5 active site. Protein Sci 2011; 20:735-44; PMID:21370305; http://dx.doi.org/ 10.1002/pro.606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Camodeca C, Nuti E, Tepshi L, Boero S, Tuccinardi T, Stura EA, Poggi A, Zocchi MR, Rossello A. Discovery of a new selective inhibitor of A Disintegrin And Metalloprotease 10 (ADAM-10) able to reduce the shedding of NKG2D ligands in Hodgkin's lymphoma cell models. Eur J Med Chem 2016; 111:193-201; PMID:26871660; http://dx.doi.org/ 10.1016/j.ejmech.2016.01.053 [DOI] [PubMed] [Google Scholar]
- 32.Liu H, Shim AH, He X. Structural characterization of the ectodomain of a disintegrin and metalloproteinase-22 (ADAM22), a neural adhesion receptor instead of metalloproteinase: insights on ADAM function. J Biol Chem 2009; 284:29077-86; PMID:19692335; http://dx.doi.org/ 10.1074/jbc.M109.014258 [DOI] [PMC free article] [PubMed] [Google Scholar]