

ABSTRACT
Concentration-dependent reversible self-association (RSA) of monoclonal antibodies (mAbs) poses a challenge to their pharmaceutical development as viable candidates for subcutaneous delivery. While the role of the antigen-binding fragment (Fab) in initiating RSA is well-established, little evidence supports the involvement of the crystallizable fragment (Fc). In this report, a variety of biophysical tools, including hydrogen exchange mass spectrometry, are used to elucidate the protein interface of such non-covalent protein-protein interactions. Using dynamic and static light scattering combined with viscosity measurements, we find that an IgG1 mAb (mAb-J) undergoes RSA primarily through electrostatic interactions and forms a monomer-dimer-tetramer equilibrium. We provide the first direct experimental mapping of the interface formed between the Fab and Fc domains of an antibody at high protein concentrations. Charge distribution heterogeneity between the positively charged interface spanning complementarity-determining regions CDR3H and CDR2L in the Fab and a negatively charged region in CH3/Fc domain mediates the RSA of mAb-J. When arginine and NaCl are added, they disrupt RSA of mAb-J and decrease the solution viscosity. Fab-Fc domain interactions between mAb monomers may promote the formation of large transient antibody complexes that ultimately cause increases in solution viscosity. Our findings illustrate how limited specific arrangements of amino-acid residues can cause mAbs to undergo RSA at high protein concentrations and how conserved regions in the Fc portion of the antibody can also play an important role in initiating weak and transient protein-protein interactions.
KEYWORDS: Hydrogen exchange, monoclonal antibody, protein aggregation, protein-protein interactions, reversible self-association
Abbreviations
- RSA
reversible self-association
- CG-MALS
composition-gradient multi-angle light scattering
- HX-MS
hydrogen exchange mass spectrometry
- CDR
complementarity-determining regions
Introduction
Protein-protein interactions play a critical role in many biological and biochemical processes. Like many biological processes however, protein-protein interactions can have negative as well as positive effects.1-4 In vivo, cellular proteins are usually present in a very crowded, highly concentrated environment.5 At such high protein concentrations, due to increases in molecular crowding and decreases in intermolecular distances between molecules, the extent of specific and non-specific protein-protein interactions driven by exposed charged and apolar regions on the protein surface increase.6-8 Independent of whether a protein is in vivo or in vitro (e.g., a purified protein drug candidate stored in a vial), molecular crowding causes protein solutions to deviate from ideality, thereby affecting macromolecular interactions and potentially protein conformation.9 Identification of the interfaces that mediate protein-protein interactions can open new avenues for drug targeting and discovery, and guide protein engineers in the development of macromolecule candidates that are more stable and easier to administer.
Monoclonal antibodies (mAbs) comprise a major class of biotherapeutics used for the treatment of many chronic conditions.10 The subcutaneous delivery route enables patients to self-administer 1-2 mL of injectable volume per dose. To deliver the amount of drug needed, which is often tens to hundreds of milligrams, in this volume, the mAbs must be formulated at very high protein concentrations.2,10 Due to decreases in intermolecular distances between protein molecules at high protein concentrations, attractive protein-protein interactions may overcome repulsive interactions, thereby favoring the formation of reversibly associating intermolecular protein complexes.11,12 Formation of large protein complexes increases the solution shear modulus, which can result in dramatic increases in viscosity at higher concentration,13,14 leading to formulation and manufacturing-related challenges. In addition, antibody clusters may act as seeds for the formation of irreversible aggregates at high protein concentration.15 Moreover, high shear stress during pumping of viscous solutions of self-associated proteins has, in some cases, been shown to also contribute to the formation of irreversible protein aggregates.16 Such irreversible aggregates can decrease protein activity and stability, and may elicit adverse immunogenic reactions in patients.17-19 In addition, parenteral administration of highly viscous liquids requires thicker gauge needles that may cause more painful injections.2
Previous work has shown that reversible self-association (RSA) between different IgG1 mAbs can result from different binding interfaces, despite high sequence similarity between the mAbs.20-22 Studies of enzymatic fragmentation of IgG1 monomers into Fab and Fc domains,21,23,24 site-specific mutations in complementarity-determining region (CDR),25 and coarse-grained simulations of such protein interactions26,27 suggest that intermolecular reversible interactions between mAb molecules can be initiated by either Fab-Fab or Fab-Fc associations and to a lesser extent through Fc-Fc interactions. Despite providing experimental insights into how solution conditions modulate the rate and extent of reversible protein-protein interactions, and which major regions of the mAb might be involved in such phenomena, these studies offer an incomplete and low-resolution picture of mAb reversible self-association. A more complete understanding of the specific molecular mechanisms of reversible protein-protein interactions requires site-specific information about the surfaces that mediate such associations.
Hydrogen exchange (HX) is a robust bioanalytical tool used to study protein dynamics and protein-protein interactions.28-33 The rate of hydrogen exchange depends on the higher order structure of the protein: backbone amides that are fully solvated (lacking hydrogen bonding) undergo rapid HX while amides located in structurally protected or strongly hydrogen-bonded regions exchange significantly more slowly.34-36 Thus, HX measurements can be used to map protein interfaces of intermolecular protein-protein interactions because the formation of intermolecular contacts directly affects the solvation and hydrogen bond strengths at the protein interface.33 Mass spectrometry coupled to HX (HX-MS), extends the HX technique to complex, multi-domain macromolecules like mAbs.37-43 Recently, we described a novel HX-MS method to map protein interfaces formed between mAbs undergoing reversible protein-protein interactions directly at up to 60 g/L.39
Here, we applied this technique to investigate the molecular mechanism by which an IgG1 mAb (mAb-J) undergoes reversible self-association, and further probed this mechanism by a variety of other biophysical techniques. We also mapped the interface of the reversible, concentration-dependent intermolecular interactions between mAb-J monomers using hydrogen exchange mass spectrometry (HX-MS). The results of this study augment our knowledge of how proteins interact with each other at high protein concentrations under crowded environments.
Results
Increases in mAb hydrodynamic diameter suggests reversible self-association
In an initial set of experiments done to better understand the possible mechanism(s) of mAb-J RSA, the effects of solution properties and additives on the magnitude of RSA at relatively low protein concentration (1-10 g/L) were examined. All results are described relative to mAb-J in a control solution (20 mM citrate-phosphate, 30 mM NaCl, pH 6.0). Fig. 1 shows the effects of charged co-solutes and sugars and changes in solution pH on the average hydrodynamic diameter of mAb-J, as determined by dynamic light scattering (DLS). The average hydrodynamic diameter of a full length, monomeric IgG1 mAb molecule is typically 9-12 nm.44,45 At 10 g/L, the hydrodynamic diameter of mAb-J in the control solution was 17.5 nm, significantly higher than a typical monomeric IgG1 mAb. The large hydrodynamic diameter suggests that mAb-J undergoes self-association even at the relatively low concentration of 10 g/L at low ionic strength. The average hydrodynamic diameter of mAb-J in solution decreased, from 17.5 nm at low ionic strength to 12.8 nm, in the presence of an additional 100 mM NaCl. This same hydrodynamic diameter vs. ionic strength trend also was evident in the presence of either sucrose or trehalose (Fig. 1A). The average hydrodynamic diameter of mAb-J decreased, from 17.5 nm to 10.8 nm, in the presence of 100 mM arginine. In contrast, the addition of sucrose and trehalose caused increases in the average hydrodynamic diameter, from 17.5 nm to 18.8 and to 19.7 nm, respectively (Fig. 1A). Raising the pH from 5.0 to 7.0 also resulted in an increased hydrodynamic diameter, from 13.0 nm to 18.8 nm (Fig. 1B). Taken together, these data suggest that the extent of mAb-J RSA, even at 10 g/L, decreases as the ionic strength increases. MAb-J RSA increases, however, in higher pH solutions or upon addition of sugars. Since the experimental limit for DLS is around 10 g/L protein concentration, which limits the investigation of association at higher concentrations, we then used dynamic viscosity to investigate protein-protein interactions at higher mAb-J concentrations.
Figure 1.
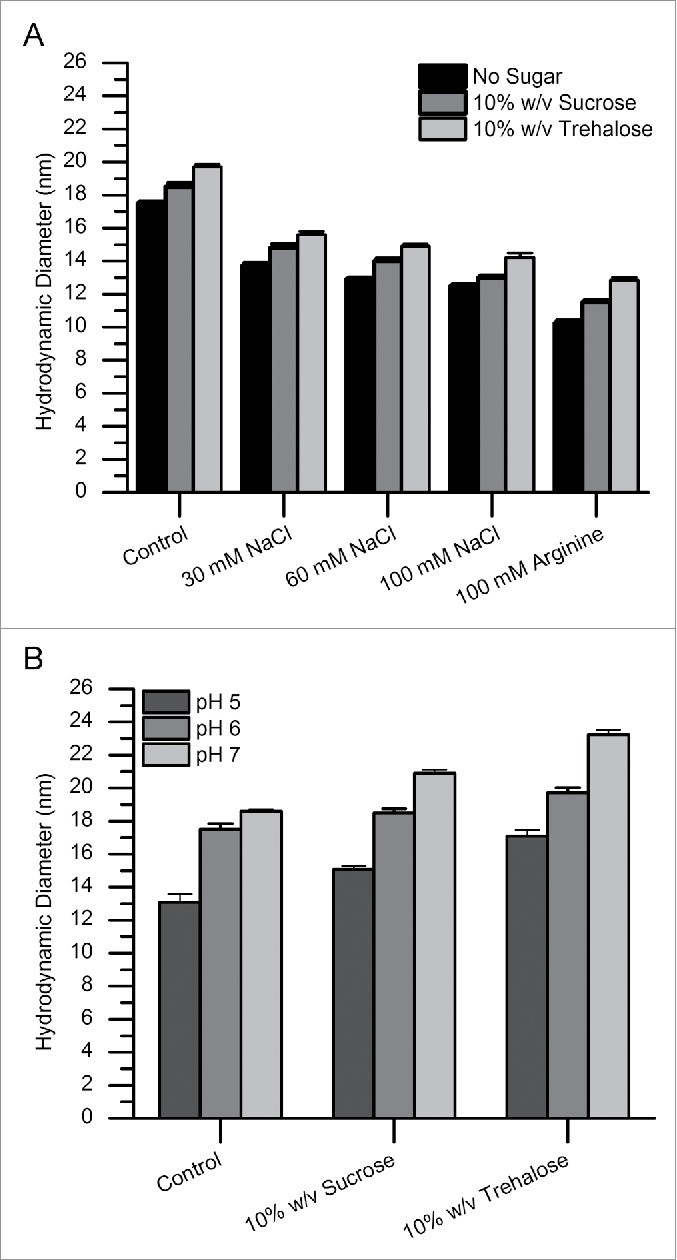
Effects of additives and pH on the hydrodynamic diameter of mAb-J as measured by dynamic light scattering. (A) Addition of incremental amounts of NaCl and 100 mM arginine to the control buffer and (B) addition of sugars and the effect of pH on the hydrodynamic diameter of mAb-J. All measurements were taken at 25°C. For panel A, mAb-J was prepared at 10 g/L in control solution (20 mM citrate-phosphate buffer containing 30 mM NaCl at pH 6.0) containing either additional NaCl (30, 60 and 100 mM) or 100 mM arginine. For panel B, additional 10% (w/v) sucrose and trehalose were added to the control solution with the pH adjusted to (5.0, 6.0 and 7.0). The error bars represent one standard deviation from 3 independent DLS measurements.
Effects of solutes on the dynamic viscosity of mAb-J solutions
Highly associated protein will usually cause a dramatic increase in solution viscosity at high protein concentrations.20,21 Trends in mAb-J solution viscosity with increased protein concentration under various solution conditions are shown in Fig. 2. The solution viscosity of mAb-J in the control buffer increased exponentially with protein concentration, an effect that can be attributed to RSA between mAb monomers. Addition of NaCl or arginine to the control solution at concentrations up to 100 mM weakened the effect. Consistent with DLS results, the effect of arginine on the viscosity of mAb-J was stronger than NaCl. The effect of pH and sugars can be seen by comparing the solution viscosity of mAb-J at 60 g/L. As the pH of the control buffer was increased from 5.0 to 7.0, the viscosity of 60 g/L mAb-J increased (Fig. 2D). When either trehalose or sucrose was added, the viscosity of the mAb-J solution increased, with the effect of trehalose stronger than sucrose. The solution viscosity of buffer in the absence of protein only increased slightly upon addition of sugars. Taken together, the viscosity results provide further evidence supporting the presence of concentration-dependent transient protein-protein interactions between mAb monomers. An increase in the ionic strength of the solution or addition of arginine decreased solution viscosity, while an increase in pH from 5.0 to 7.0 caused the dynamic viscosity of mAb-J solution to increase. These observations suggest that mAb-J intermolecular interactions are primarily driven by attractive electrostatic interactions.
Figure 2.

Concentration-dependent effects of additives and pH on the dynamic viscosity of mAb-J solutions. Effects of (A) NaCl and (B) arginine on solution viscosity as a function of mAb-J concentration. Effects of (C) 10% (w/v) sucrose and trehalose and (D) pH on solution viscosity of mAb-J solutions at 60 g/L protein concentration. All measurements of solution viscosity were taken at 25°C. In panels A, B and C, all samples of mAb-J were prepared in the control solution containing additional amounts of NaCl, arginine, or sugars. In panel D, mAb-J samples were prepared in the control with pH adjusted to (5.0, 6.0 and 7.0). The error bars represent one standard deviation from a set of 3 independent measurements in A and B the error bars are smaller than the symbols.
Reversible self-association of mAb-J involves a monomer-dimer-tetramer equilibrium
The nature of mAb-J intermolecular interactions was further studied using composition-gradient multi-angle light scattering (CG-MALS). Static light scattering quantifies the excess Rayleigh ratio (R), the fractional amount of incident light that is scattered by the macromolecule per unit volume of the solution. Intermolecular interactions affect the magnitude of the Rayleigh ratio. If the relationship between R, as a function of the scattering angle (θ), and protein concentration is known, then the apparent molecular weight, size, and extent of self-association of the molecule can be determined using values of the osmotic second virial coefficient (A2) combined with stoichiometry estimates based various association models (as described in detail in the Experimental Section).
The mole fractions of monomeric, dimeric, and tetrameric forms of mAb-J under the experimental conditions were determined (Fig. 3) based on fitting the static light scattering data to a monomer, dimer, tetramer association equilibrium model. First, for mAb-J at 20 g/L in control buffer containing 10% w/v trehalose, the solution had equal mole fractions of monomeric mAb-J (0.48) and tetrameric mAb-J (0.48) (Fig. 3A). This observation suggests extensive protein-protein interactions, supported by an osmotic second virial coefficient (A2) value of −9.6 × 10−5 mol mL g−2. In the presence of an additional 100 mM NaCl in the control buffer, the mole fraction of monomeric mAb-J did not decrease as steeply, reaching a value of 0.63 at 20 g/L. Interestingly, the mole fraction of dimer was 0.27, whereas tetramer was only 0.1 under these conditions (Fig. 3B). The value of A2 for mAb-J in the presence of additional 100 mM NaCl became positive, 2 × 10−5 mol mL g−2, in contrast to the negative value under low ionic strength solution conditions. The positive A2 value indicates disruption of protein-protein interactions. In the presence of 100 mM arginine, mAb-J was completely monomeric even at 20 g/L, suggesting complete disruption of attractive intermolecular interaction between mAb-J monomers over the concentration range tested (Fig. 3C). Disruption of protein-protein interactions by arginine is further supported by a large, positive value of A2 (9 × 10−5 mol mL g−2) for mAb-J in the presence of arginine.
Figure 3.

Effects of additives on the reversible self-association of mAb-J monomers based on analysis of static light scattering showing the mole fractions of monomeric (black squares), dimeric (red circles), and tetrameric (blue triangles) mAb-J species over a protein concentration range of 0.2-20 g/L in (A) control solution, (B) control solution + NaCl, and (C) control solution + arginine. In Panel C, the mole fractions of both dimer and tetramer are superimposed at zero. Samples of mAb-J were prepared in the control solution containing 20 mM citrate-phosphate buffer, 30 mM NaCl, 10% (w/v) trehalose at pH 6.0 with or without additional 100 mM NaCl or arginine. Panel A, B and C show mean of triplicate measurements at 25°C and error bars represent one standard deviation from the calculated mean.
Hydrogen exchange mass spectrometry reveals association between the Fc and Fab
To map the interfaces responsible for protein-protein interactions, the non-associating and associating protein states must be compared. We chose to compare the protein concentrations 5 g/L (∼90% monomeric) and 60 g/L (less than 50% monomeric) by HX-MS analysis. To conduct hydrogen exchange at high protein concentration, we followed an approach we recently developed,39 which involves reconstituting lyophilized mAb-J with D2O-containing buffer rather than diluting the solution with D2O. A combination of size-exclusion chromatography (SEC), circular dichroism (CD), and viscosity measurements confirms the structural integrity of mAb-J samples after lyophilization, as described in the Supporting Information (Fig. S1, and Table S3). To maintain the high concentration of mAb-J during hydrogen exchange, the lyophilized samples of mAb-J were reconstituted with pure D2O to yield a final solution composition of 20 mM citrate phosphate buffer (pH 6) containing 10% w/v trehalose and 30 mM NaCl (i.e., control buffer + 10% w/v trehalose in D2O). To eliminate artifacts that might potentially arise from altered chemical exchange kinetics, the composition of the buffer was held constant while only the mAb-J concentration was changed. Following selected intervals of hydrogen exchange, the reaction was quenched by lowering the pH to 2.5, and the sample was then digested with pepsin, and analyzed by LC-MS.
No significant differences were observed in the measured HX between low and high protein concentration for almost 95% of the peptic peptides of mAb-J (see Fig. S3). Deuterium uptake plots for some representative peptides in this category are shown in Fig. 4A. There were certain peptides, however, that exhibited significant protection against hydrogen exchange at 60 g/L mAb-J. Fig. 4B shows representative deuterium uptake plots for some of the peptides that became significantly protected. Deuterium uptake plots for all of the peptides from mAb-J are shown in Fig. S3 and a list of all the peptides and their sequential numbering is given in Table S4. A global representation of all of the changes in hydrogen exchange between mAb-J at 5 and 60 g/L is shown in Fig. 5. Relative mass difference or the differences in deuterium uptake between mAb-J peptides from 60 and 5 g/L samples (Eq.4) are plotted on the vertical axis and ordinal peptide number on the horizontal axis of the plot with:
| (Eq. 1) |
These plots show the location of protected () and de-protected () regions in mAb-J.
Figure 4.

Representative deuterium uptake plots for 8 peptide segments of mAb-J. Black squares and red circles represent HX-MS experiments at low (5 g/L) and high (60 g/L) protein concentrations, respectively. The data in panel A are representative of peptides that showed no significant differences in hydrogen exchange kinetics between the 2 concentrations of mAb-J. Peptide segments shown in panel B are representative of peptide segments that showed significant protection (slowed HX) at high protein concentration. The domain location and peptide index number are shown in parenthesis at the top of each plot. Error bars represent one standard deviation from 3 independent HX measurements. (For deuterium uptake plots of all peptide segments of mAb-J, refer to Fig. S3).
Figure 5.
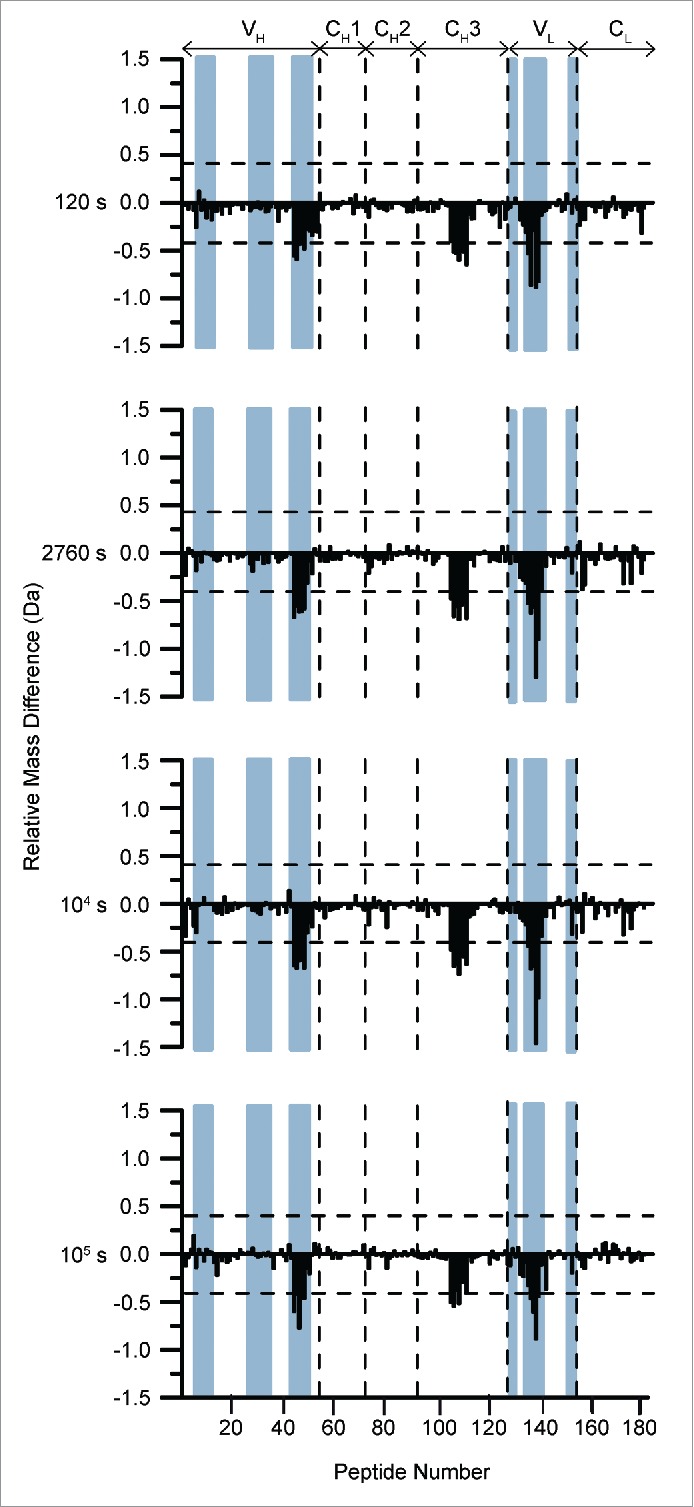
Differential deuterium uptake by 182 peptides at 4 different hydrogen exchange times comparing high (60 g/L) and low (5 g/L) concentrations of mAb-J at pH 6.0. Peptides are numbered in order from N terminus of the heavy chain to the C terminus of the light chain on the horizontal axis of the plots (see Table S4 for exact locations). The vertical axis is difference in HX between high vs. low protein concentrations, . Positive bars show peptide segments with faster HX at high protein concentration and negative bars indicate peptide segments with slower HX at high protein concentration. The dashed horizontal bars at ± 0.40 Da are the 99% confidence intervals. Vertical dashed lines separate boundaries of different domains of mAb-J (domains are listed at the top of the plot). The locations of the CDR segments are shaded in blue. Some peptides span more than one domain thus the boundaries are approximate. An average of 3 independent HX measurements was used to calculate each mass difference.
Comparison of the level of deuteration revealed various regions within mAb-J that became significantly protected at 60 g/L, conditions where mAb-J was substantially self-associated. The protected regions are located in the variable heavy chain (VH), variable light chain (VL), and constant domain of the heavy chain (CH3) of the antibody. In the heavy chain of the antibody, these regions cover HC 92-116 (peptide numbers 42 to 47) located in the third complementarity-determining region of the heavy chain (CDR3H) and HC 381-408 (peptide numbers 105 to 111) that surrounds a region in the CH3 domain. In addition, in the light chain, a segment that became significantly protected (LC 39-76), peptide numbers 135 to 140, spans the second CDR of the light chain (CDR2L) located in the VL domain of the antibody. None of the regions in mAb-J became significantly more flexible (higher HX) at high protein concentration.
Thus, significant decreases in hydrogen exchange (i.e., increased protection against deuterium uptake) were observed upon RSA of mAb-J in 2 of the 6 CDR regions (i.e., the CDR2 region of the light and CDR3 region of the heavy chain) and in the CH3 domain of mAb-J. Fig. 6 shows the protected regions mapped onto a homology model of mAb-J (for details about the homology model, see the Materials and Methods section). The segments that became significantly protected at high protein concentration are highlighted in yellow (Fig. 6). A surface representation of the Fab and Fc of mAb-J is also shown in Fig. 6 where protected segments, presumably the primary protein interface of RSA, are colored in yellow as in the ribbon representation. In the surface representation, the negatively-charged residues that became protected are colored red, the positively charged residues that became protected are blue, and surface exposed hydrophobic residues are colored in green. It should be noted that while the level of confidence in the location and surface exposure of residues in the highly-conserved CH3 domain is high, the exact location and surface exposure information of the VH and VL domain residues may be less reliable.
Figure 6.

Locations of significant protection at high protein concentration mapped onto a homology model of mAb-J. The protected regions are colored yellow. A zoomed-in surface representation of Fab and Fc domains of mAb-J is presented in the inset panels to the right. Surface exposed, positively and negatively charged residues located in protected segments during RSA are highlighted in blue and red, respectively, and solvent-exposed hydrophobic residues are colored in green. Segments with no significant difference in HX between high and low protein concentrations of mAb-J are gray; regions with no mass spectrometry data are white.
To confirm that the changes in hydrogen exchange resulted from specific interactions between mAb-J monomers rather than non-specific interactions arising from non-ideality at high protein concentrations, we compared a mAb-J control at 60 g/L to mAb-J samples at 60 g/L containing control buffer and either 100 mM arginine or 100 mM NaCl (Fig. S4). As described above, arginine and sodium chloride both reduced the extent of self-association as indicated both by decreased hydrodynamic diameter (Fig. 1A) and reduced solution viscosity (Fig. 2), although the effect of arginine was much stronger. The regions of mAb-J that became significantly protected under control solution conditions were the same regions that became protected at high protein concentration when compared to low concentration non-associating control (Fig. 5), confirming the validity of the analysis. Stronger protection in the control mAb-J sample when compared to that with 100 mM arginine reflects arginine's more potent action at disrupting mAb-J RSA in comparison to 100 mM NaCl. Regions in the heavy chain, HC 87-93 (peptide number 41) and LC 35-38 (peptide number 132) became significantly more rigid upon addition of arginine at 60 g/L (see Fig. S4A). These regions are at the N-terminal of the RSA interface present in VH and VL domains of the antibody. A decrease in local flexibility upon addition of arginine could be attributed to allosteric effects of the arginine-inhibited self-association.46
We also compared hydrogen exchange by mAb-J at 60 g/L in control buffers containing either 100 mM NaCl or 100 mM arginine to a sample of mAb-J at 5 g/L in control buffer without any additional charged solutes. Difference plots comparing HX between these samples are shown in Fig. S5. None of the 182 peptides analyzed in this experiment became either protected or de-protected. These observations suggest that a solution of mAb-J containing additional 100 mM NaCl or 100 mM arginine at 60 g/L protein concentration behaves similarly to a dilute solution of mAb-J without any additional charged solutes, indicating the absence of significant specific intermolecular interactions.
Discussion
MAbs may form intermolecular protein-protein interaction networks at high protein concentration.20,21 These large, associated protein complexes typically cause a dramatic increase in solution viscosity that can introduce a number of challenges to their pharmaceutical use. This work has 2 primary goals: 1) to determine the macroscopic nature of protein-protein interactions between mAb-J monomers at high protein concentration; and 2) to further delineate the underlying molecular mechanisms that promote RSA by mapping the protein interface of interaction at high protein concentration.
To investigate the nature of reversible non-covalent protein-protein interactions, their extent was measured indirectly by measuring the hydrodynamic diameter and dynamic viscosity of mAb-J solutions at varying pH values, with and without charged and uncharged solutes. The hydrodynamic diameter and the viscosity of mAb-J decreased as ionic strength increased. Based on fitting static light scattering measurements to various association models, we found that a monomer-dimer-tetramer equilibrium provided the best fit for mAb-J self-association. A sharp decline in monomer mole fraction with increasing protein concentration and a negative A2 value at low ionic strength was observed. On the contrary, a slower decrease in the monomer mole fraction and positive A2 values in the presence of additional NaCl or arginine was observed. A decrease in the propensity of mAb-J to form protein-protein complexes with an increase in ionic strength can be attributed to electrostatic attractive interactions being the dominant contributor governing protein-protein interactions between mAb-J monomers.47-49 Based on Derjaguin-Landau-Verwey-Overbeek theory, as ionic strength is increased, the Debye screening length (the thickness of the electrical double layer surrounding the protein molecule) shortens and the effective charge on the mAb will decrease due to electrostatic charge screening.50 Under such conditions, the strength of both repulsive and attractive electrostatic interactions decreases, thereby causing disruption of intermolecular protein interactions between mAb-J molecules.
The predicted isoelectric point for mAb-J is ∼7.3. Increasing the pH from 5.0 to 7.0 promoted the extent of protein-protein interactions between mAb-J monomers. This observation contradicts predictions based on the electroviscous effect that describes the viscosity of dilute colloidal solutions as directly proportional to the electrostatic charge on the molecule.51 According to the electroviscous effect, solution viscosity should decrease as the pH is moved toward the pI of the protein where the overall net charge on the molecule approaches its minimum.52-54 However, we observed the opposite. Yadav et al.47 also observed an increase in the extent of mAb1 association near the pI of the antibody. In another report, Yadav et al.48 showed that the mutual diffusion coefficient for an antibody decreased as the pH value approached the pI of the protein. This observation was attributed to an increase in the extent of specific protein-protein interactions between mAb-2 molcules near the pI. At a pH value below or above the pI, the overall net charge on a protein will be either positive or negative, respectively. Away from the pI, the electrostatic repulsive interactions between protein molecules dominate and contributions by attractive dipolar interactions are weakest. At the pI, although the overall net charge on the protein reaches its minimum, thus decreasing global intermolecular repulsions, large numbers of charged amino acids can still be present at the surface. These charged side chains, if present in a specific distribution, can cause formation of localized charged patches on the protein surface. Charged patches on the protein surface can lead to spatial reorientation of protein molecules, which might initiate favorable dipole-dipole and charge-dipole interactions between protein molecules.47 At high protein concentrations the intermolecular distances between protein molecules decrease to a few Angstroms. Around the pI, where intermolecular repulsive interactions reach a minimum, short-ranged and non-specific attractive interactions between surface-exposed hydrophobic residues, hydrogen bonding, and van der Waals interactions can also become significant contributors to intermolecular protein-protein interactions. These attractive non-covalent interactions could cause the monomers to form protein-protein associated complexes that further cause a dramatic increase in solution viscosity at high protein concentrations.
The addition of sugars and polyols has been shown to affect protein structure and protein-protein interactions.55,56 Our results (Figs. 1B and 2D) show both sucrose and trehalose enhanced the extent of protein-protein interactions as reflected by increased hydrodynamic diameter and solution viscosity of mAb-J. Sugars are preferentially excluded from the protein surface, causing water molecules to populate around protein domains. Accumulation of water at the surface produces an unfavorable increase in protein chemical potential.57,58 To counter such thermodynamically unfavorable interactions between apolar side-chains and water molecules, the protein structure responds by minimizing the exposed surface area, leading to lower preferential exclusion. Thus, sugars are often added to protein solutions to increase protein conformational stability. At high protein concentrations, however, where the intermolecular distances between protein monomers are relatively shorter, preferential exclusion can also lead to formation of reversible (or potentially irreversible) protein-protein interactions because the interactions decrease solvent exposure, thereby lowering the extent of unfavorable preferential exclusion. While stabilizing, added sugars can also contribute to the formation of protein-protein complexes that cause an enhanced effect on the exponential increases in solution viscosity at high protein concentrations, as seen here.
To map the protein interfaces of mAb-J self-association at high protein concentration, we used HX-MS to compare associated and non-associated mAb-J. Based on our HX-MS results (see Fig. 5), we conclude that both Fab and Fc regions are involved in formation of the primary interface for intermolecular protein-protein interactions between mAb-J monomers. This is supported by control HX experiments comparing 60 g/L mAb-J with and without viscosity-lowering additives (Figs. S4 and S5) where the same regions in mAb-J in control buffer became significantly protected. Among all the charged side-chains in the region spanning CDR2L in the VL domain, LC 39-76 (YQQLPGTAPKLLIYDNFNRPSGVPDRFSGSKSGTSASL) D53, R57, R64 and K69 are solvent exposed in the homology model. Only one lysine (K102) was solvent exposed in the VH domain interface of mAb-J, which spans the CDR3H, HC 92-116 (AVYYCATVMGKWIKGGYDYWGRGTL). Taken together, solvent-exposed residues give the Fab region interface a net surface charge of +3 at pH 6.0 (Fig. 6B). On the contrary, in the region of the Fc domain of the antibody that became significantly protected, LC 381-408 (IAVEWESNGQPENNYKTTPPVLDSDGSF), all the charged residue side-chains are either fully or partially solvent exposed, giving the interface a net surface charge of −4 at pH 6.0 (Fig. 6C).
As mentioned previously, confidence in the homology model is lowest in the variable regions; however, CDR loops are primarily unstructured, dynamic, and solvent exposed,59 thus it is reasonable to expect that the lysine and arginine side-chains will be accessible. The interface on the surfaces of the VL and VH domains of the antibody forms a patch with a net positive charge and the interface on the surface of the Fc region has a net negative charge. The presence of charge anisotropy between the protein interfaces of the Fab and Fc regions is consistent with the biophysical characterization of mAb-J RSA that suggests protein association is initiated by long-range electrostatic attractive interactions.
The involvement of the CDR in the Fab region in intermolecular protein-protein interactions has been well documented.21,22,24,25,39 Few reports, however, have implicated the Fc region in mediating mAb RSA.23, 26 Nishi et al. used low resolution biophysical tools to show the involvement of the Fc region in mediating reversible Fc-Fc interactions between mAb monomers. Chaudhri et al. used coarse-grained molecular dynamic simulations to probe the nature of site-site interactions between mAb monomers. Significant intermolecular Fab-Fc interactions were present in one of the 2 antibodies that they simulated. Ido et al.,60 using frequency modulation atomic force microscopy (FM-AFM), showed that mAbs self-assemble into rosette-shaped hexamers through lateral Fc-Fc interactions between monomers. Based on our HX measurements, mAb-J monomers could undergo association through Fab-Fab, Fc-Fc or Fab-Fc interactions. However, due to the presence of opposite charges on Fab and Fc domain interfaces of mAb-J, Fab-Fc mediated protein-protein interactions seem most likely.
To the best of our knowledge, this report provides the first direct interfacial mapping of mAb RSA at high protein concentration with protein interfaces in both Fab and Fc region of the antibody. The protein sequence of the mAb-J Fc interface is conserved between all antibodies of IgG1 subclass. Thus, the negatively-charged patch would be present in all IgG1s. In contrast, the corresponding interface in the Fab region spans the CDR2L and CDR3H regions of this specific antibody. Amino acid sequences of the hypervariable regions or the CDR regions differ greatly between different mAbs. A special case of specific orientation and distribution of charged, aliphatic and aromatic amino acids in the Fab region interface of mAb-J might contribute significantly to initiating favorable interactions between Fab and Fc domain interfaces of mAb-J.
Another observation of interest was the more potent action of arginine compared to NaCl on decreasing the hydrodynamic size of mAb-J complexes and reducing the viscosity of the mAb-J solutions (Figs. 1A and 2A, B). Analysis of the static light scattering data showed that arginine completely disrupts the interactions between mAb-J monomers, and that its effect is stronger than the effect of NaCl, which still allows formation of dimers and tetramers of mAb-J (Fig. 3). These results suggest that, although the interactions between mAb-J monomers are dominated by electrostatic attraction, there may also be additional secondary contributions by other short range non-covalent interactions, such as van der Waals interactions, hydrogen bonding, π-π stacking interactions, and hydrophobic interactions. These observations collectively suggest that arginine's effect is not limited to only electrostatic charge shielding and disruption of charge-charge attractive interactions.
Arginine's potent effect on protein-protein interactions has been previously reported by other researchers.61-63 In addition to its charge-charge interactions with the ionizable side-chains of amino-acids, arginine has also been shown to interact favorably with the apolar and aromatic amino acids.61 Kita et al. and Arakawa et al.64,65 suggested that arginine penetrates the protein solvation shell to interact with specific amino acid side chains in the protein. Shukla et al. performed molecular dynamics simulations that suggested formation of favorable interactions between arginine co-solute and aromatic and charged side-chains present on the protein surface.63 Formation of favorable cation-π interactions between the guanidinium side chain of arginine and aromatic side chains of tryptophan, tyrosine, and phenylalanine and formation of salt bridges with charged side chains of amino acids on the protein surface help to explain the more potent action of arginine in suppression of protein-protein interactions.66
In addition to possessing surface-exposed oppositely charged amino acids at protein interfaces in the Fab and Fc regions of the mAb-J, the patches also contain surface-exposed hydrophobic residues. The interaction interfaces in the VL and VH domain together contain a tyrosine, a phenylalanine, a valine, and an isoleucine residue that are fully or partially surface-exposed in the homology model. The protein interface in the Fc domain contains a tyrosine, a tryptophan, and a valine residue that are surface exposed. These surface-exposed aliphatic and aromatic residues at protein interfaces might be involved in mediating short-range non-covalent van der Waals and hydrophobic interactions. The effect of arginine on short-range non-covalent interactions in associating mAb systems is a topic currently being further evaluated in our laboratories.
Our proposed hypothetical model for mAb-J RSA is shown in Fig. 7. Because mAb-J associates through Fab-Fc interactions at high protein concentrations, depending upon the kinetics of association, the associated species might be tetramers or the association could potentially extend to the formation of either linear fibrous complexes or rosette-shaped protein complexes. Higher-order associated complexes formed at high protein concentrations would increase the shear modulus of the mAb solutions, causing a dramatic increase in solution viscosity.
Figure 7.

A model of mAb-J reversible self-association illustrating 2 possible morphologies that an associated mAb network could form at high protein concentrations resulting in elevated solution viscosity. On the left, the Fab domain of the antibody is shown in orange and the Fc is green. On the right homology model of mAb-J with protein interfaces of RSA colored in yellow are arranged to show contact points between mAb-J monomers and possible arrangement of associated complexes.
In conclusion, our experiments show that mAb monomers can form reversibly-associated protein complexes mediated through Fab-Fc interactions. We also demonstrated that, although mAb-J intermolecular interactions are primarily driven by electrostatic attractive interactions, other short-ranged non-covalent forces may also play a role in mediating complex formation at high protein concentrations. Our results contribute to a better understanding of the nature of mAb RSA at high protein concentrations. This study further supports the notion that HX-MS can substantially aid protein engineering and candidate selection efforts directed toward the development of improved mAb therapeutics with superior physiochemical properties.67
Materials and methods
Sample preparation
A purified IgG1 mAb, mAb-J, at a concentration of 150 g/L was obtained from MedImmune LLC, Gaithersburg, MD. The stock solution of mAb-J was dialyzed against the “control buffer” (20 mM citrate-phosphate buffer at selected pH values, containing 30 mM NaCl) with or without additional NaCl, arginine hydrochloride, or sugars (sucrose and trehalose) at 4°C using 3500 kDa molecular-weight cutoff membranes (Slide-A-Lyzer, Thermo Scientific, Rockford, IL) for 24 hours. For static light scattering and lyophilization, additional 10% w/v trehalose was added to the samples. Subsequently, after dialysis stock solutions of mAb-J were diluted using corresponding buffer solutions.
Dynamic light scattering
Samples of mAb-J at 10 g/L prepared in control (pH 5.0-7.0) with or without additional NaCl, arginine hydrochloride or sugars (sucrose and trehalose) were centrifuged at 12,000 × g for 5 minutes before analysis. DLS was measured in triplicate using the DynaPro Plate Reader (Wyatt Technology, Santa Barbara, CA). Scattered light was analyzed using a backscatter detector fixed at an angle of 173˚. Fifteen runs of 5 second acquisitions were collected and averaged to determine the hydrodynamic diameter for each sample.
Viscosity measurements
Samples of mAb-J, ranging from 5 to 60 g/L were prepared by diluting the dialyzed stock with corresponding buffer solutions. Solution dynamic viscosity was measured at 25°C with an m-VROC viscometer (Rheosence, San Ramon, CA) at a rate of 100 μL/min with a shear rate of 1420 1 s−1. Triplicate viscosity measurements were recorded over a duration of 100 seconds.
Composition-gradient multi-angle light scattering
Dialyzed stock of mAb-J at 150 g/L in control (containing 10% w/v trehalose) with or without additional 100 mM NaCl and arginine was diluted with corresponding buffer solutions to prepare samples of mAb-J at 2 and 20 g/L. Thereafter, these samples were filtered through 0.22 μm Millex-GV syringe filter units (EMD Millipore, Billerica, MA). A fully automated CG-MALS instrument with a dual syringe-pump Calypso sample preparation and delivery unit (Wyatt Technology, Santa Barbara, CA) was used to measure light scattering at room temperature. A Dawn Heleos II light scattering instrument (Wyatt Technology, Santa Barbara, CA), equipped with a 661 nm laser and an Optilab Rex refractive index detector (Wyatt Technology, Santa Barbara, CA), was used to measure both light scattering and protein concentration. Filtered HPLC grade toluene (Fisher Scientific, Fair Lawn, New Jersey) was used to calibrate voltage and light scattering intensities. Rayleigh ratio light scattering intensities were obtained over a protein concentration range of 0.2–20 g/L. The light scattering and concentration data were fit to a set of association models using Calypso software (Wyatt Technology, Santa Barbara, CA).
To obtain a stoichiometric analysis of RSA of mAb-J under different solution conditions, light scattering data were fit to various association models using equation 2, a virial expansion for non-ideal solutions containing associating components;
| (Eq. 2) |
Rθ is the excess Rayleigh ratio, M the molecular weight, i is the stoichiometry of the associated species (e.g., i = 2 for a dimer), Ci are the concentrations of the individual species, and Ctot is the total concentration. A2 is the osmotic second virial coefficient, left unconstrained during data fitting. A2, provides useful insights into intermolecular interactions between protein molecules. A negative value of A2 indicates that the overall interactions between protein molecules are attractive, while a positive value indicates that the overall interactions are repulsive. K in equation 2 is the optical constant described by equation 3
| (Eq. 3) |
with n0 as the refractive index of the solvent (1.33), NA is Avogadro's number (mol−1), is the refractive index increment of the protein/solvent pair (0.185 mL/g),68,69 and λ0 is the wavelength of the incident light in vacuum. Following an iterative procedure, A2 and Ci values were optimized to achieve the best fit (based on χ2) between the light scattering data and various association models (see Table S1 for the χ2 values). A monomer-dimer-tetramer association equilibrium model best fit the light scattering data for mAb-J (see Table S2 for the A2 values).
Lyophilization of mAb-J samples
Stock solutions of mAb-J at 150 g/L, dialyzed against the control buffer (containing 10% w/v trehalose), were diluted to 5 and 60 g/L using control buffer, and dispensed into 3 mL FIOLAX® clear Type 1 glass vials (Schott North America, Elmsford, NY) with a fill volume of 500 μL. The vials were partially stoppered by 2-leg, 13 mm siliconized rubber stoppers (Wheaton Industries Inc., Millville, NJ). The samples were then lyophilized using a LyoStar II lyophilizer (SP Scientific, Warminster, PA) using an optimized lyophilization cycle described previously.39
Circular dichroism
Lyophilized mAb-J samples at both 5 and 60 g/L were reconstituted with D2O to a concentration of 0.3 g/L. CD experiments were carried out with a Chirascan Plus Circular Dichroism Spectrometer (Applied Photophysics Ltd., Leatherhead, UK) equipped with a Peltier temperature controller and a 4-position cuvette holder. Far-ultraviolet (UV) CD spectra of non-lyophilized control and freeze-dried mAb-J samples (0.3 g/L) were collected from 200 nm to 260 nm using 0.1 cm path length quartz cuvettes. CD scans were collected at 10°C using a sampling time of 1 second and a bandwidth of 1 nm. Non-lyophilized mAb-J samples were prepared in H2O-based control buffer. Ellipticity values obtained from the instrument were then converted to molar ellipticity by dividing ellipticity by protein concentration (M) and cuvette path length (m).
Size-exclusion chromatography
Freeze-dried mAb-J samples at both 5 and 60 g/L were reconstituted to 0.5 g/L using D2O and then centrifuged at 14000 × g for 5 minutes to remove any insoluble aggregates prior to SEC analysis. A Shimadzu high performance liquid-chromatography system equipped with a photodiode array detector capable of recording UV absorbance spectra from 200 to 400 nm was used. A 7.8 mm × 30 cm Tosoh TSK-Gel BioAssist G3SWxL (TOSOH Biosciences, King of Prussia, PA) and a corresponding guard column were preconditioned with 0.2 M sodium phosphate, pH 6.8, and then calibrated using gel-filtration molecular weight standards (Bio-Rad, Hercules, CA). mAb-J species were separated at 0.7 mL min−1 based on their size. A dual wavelength quantification method described previously70 was used to quantify the amounts of various species of mAb-J in solution. Non-lyophilized samples were prepared in H2O-based control buffer. To calculate the amount of insoluble aggregates in reconstituted mAb-J samples, the total area of all the species (soluble aggregates, monomer, and fragment) in the chromatogram was calculated. The difference between the total peak areas of the sample and the control was defined as total insoluble aggregates.
Deuteration of arginine
To remove exchangeable 1H from arginine, arginine hydrochloride was dissolved in D2O at appropriate concentrations and then dried at 30 ˚C for 24 hours in a Vacufuge™ vacuum concentrator (Eppendorf, Hauppauge, NY). The process was repeated 3 times. After the third drying cycle, the deuterated arginine powder was reconstituted with D2O-based 5 mM citrate-phosphate buffer (pH 6.0). The final pH was adjusted with deuterium chloride or deuterium oxide to be within 0.02 pH unit of the desired pH. The pH values was recorded without correction for the deuterium isotope effect.71
Hydrogen-exchange mass spectrometry
Hydrogen exchange was initiated by adding 500 μL of D2O to vials of lyophilized mAb-J as described in more detail previously.39 Labeling was thermostated to 25°C on an Echotherm chilling/heating plate (Torrey Pines Scientific, Inc. Carlsbad, CA). Hydrogen exchange was quenched after 120, 2760, 104 and 105 seconds by adding 20 μL of the exchange reaction mixture to 180 μL of quench buffer containing 0.5 M tris(2-carboxyethyl)phosphine hydrochloride, 4 M guanidine hydrochloride, and 0.2 M sodium phosphate at pH 2.5 pre-equilibrated at 1°C. An H/DX PAL robot (LEAP Technologies, Carrboro, NC) was used for sample handling and injection. Subsequent to the quench step, samples were loaded into the sample loop of the refrigerated column compartment containing 3 valves connected to an Agilent 1260 infinity series LC (Agilent Technologies, Santa Clara, CA), an immobilized pepsin column, a peptide desalting trap, and a C18 column. The level of deuteration in each peptide was measured using an Agilent 6530 quadrupole-time of flight mass spectrometer (Agilent Technologies, Santa Clara, CA), equipped with a standard electrospray ionization source operated in positive mode. A complete description of the hydrogen exchange methodology was previously reported.39
Hydrogen exchange mass spectrometry (HX-MS) with viscosity-decreasing solutes
A D2O-based 5 mM citrate-phosphate buffer (pH 6.0) was used to make reconstitution solutions containing 100 mM of deuterated arginine and 100 mM NaCl, respectively. Lyophilized mAb-J samples at 5 g/L were reconstituted using 500 μL of D2O-based 5 mM citrate-phosphate buffer (pH 6.0). Half of the lyophilized mAb-J samples at 60 g/L were reconstituted using 5 mM citrate-phosphate buffer (pH 6.0) containing 100 mM deuterated arginine while the other half was reconstituted using the same buffer containing 100 mM NaCl instead of 100 mM arginine, yielding a final solution composition of 25 mM citrate phosphate buffer (pH 6.0) containing 30 mM sodium chloride, 10 % (w/v) trehalose with or without 100 mM arginine or 100 mM NaCl. The hydrogen exchange reaction was quenched after 2760 seconds. A full description of the hydrogen exchange process is described elsewhere.39
HX-MS data processing and analysis
mAb-J peptic peptides were identified using accurate mass (± 10 ppm) and tandem MS with collision-induced dissociation on a quadrupole-time of flight mass spectrometer. A total of 182 peptides covering 92% of the primary sequence of mAb-J were identified and used for analysis. HDExaminer (Sierra Analytics, Modesto, CA) was used for initial processing of the HX-MS data. Deuterium uptake plots with average deuterium uptake values and standard deviations from triplicate hydrogen exchange runs for each peptide were generated using an R script, written in-house. A 99% confidence interval of ±0.40 Da for the differences in our dataset was calculated using a procedure we described previously.39 HX-MS results were mapped onto a homology model of mAb-J.
mAb-J homology model construction
An in silico Padlan structure was used as a template to construct the homology model of mAb-J, using Modeler version 9.12.72,73 The Fab domain homology model was constructed from the structure of the human antibody molecule Kol.74 The homology model of the Fc domain was derived from PDBI:3AVE, which has the same amino acid sequence as mAb-J. Both the structures were superimposed over the in silico template to produce a homology model of the whole antibody.75 The atomic coordinates of the residues in the hinge were obtained from the in silico hypothetical structure assembled by Padlan.73
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Dr. Cavan Kalonia for his assistance with composition-gradient multi-angle light scattering data acquisition and analysis. This study was supported by a research grant from MedImmune LLC, Gaithersburg, MD and an equipment loan from Agilent Technologies, Inc., Santa Clara, CA.
Funding
The present study was funded by MedImmune Inc. JA and YH are graduate students and CRM, DBV and DDW are professors at The University of Kansas. RE, HAS, SMB are employees of MedImmune.
References
- 1.Jeong H, Mason SP, Barabási A-L, Oltvai ZN. Lethality and centrality in protein networks. Nature 2001; 411:41-2; PMID:11333967; http://dx.doi.org/ 10.1038/35075138 [DOI] [PubMed] [Google Scholar]
- 2.Shire SJ, Shahrokh Z, Liu J. Challenges in the development of high protein concentration formulations. J Pharm Sci 2004; 93:1390-402; PMID:15124199; http://dx.doi.org/ 10.1002/jps.20079 [DOI] [PubMed] [Google Scholar]
- 3.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med 2004; 10 Suppl:S10-7; PMID:15272267; http://dx.doi.org/ 10.1038/nm1066 [DOI] [PubMed] [Google Scholar]
- 4.Ballatore C, Lee VM-Y, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci 2007; 8:663-72; PMID:17684513; http://dx.doi.org/ 10.1038/nrn2194 [DOI] [PubMed] [Google Scholar]
- 5.Zimmerman SB, Trach SO. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J Mol Bio 1991; 222:599-620; http://dx.doi.org/ 10.1016/0022-2836(91)90499-V [DOI] [PubMed] [Google Scholar]
- 6.van den Berg B, Ellis RJ, Dobson CM. Effects of macromolecular crowding on protein folding and aggregation. EMBO J 1999; 18:6927-33; PMID:10601015; http://dx.doi.org/ 10.1093/emboj/18.24.6927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simpanya MF, Ansari RR, Suh KI, Leverenz VR, Giblin FJ. Aggregation of lens crystallins in an in vivo hyperbaric oxygen guinea pig model of nuclear cataract: dynamic light-scattering and HPLC analysis. Invest Ophthalmol Vis Sci 2005; 46:4641-51; PMID:16303961; http://dx.doi.org/ 10.1167/iovs.05-0843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connolly BD, Petry C, Yadav S, Demeule B, Ciaccio N, Moore JM, Shire SJ, Gokarn YR. Weak interactions govern the viscosity of concentrated antibody solutions: high-throughput analysis using the diffusion interaction parameter. Biophys J 2012; 103:69-78; PMID:22828333; http://dx.doi.org/ 10.1016/j.bpj.2012.04.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci 2001; 26:597-604; PMID:11590012; http://dx.doi.org/ 10.1016/S0968-0004(01)01938-7 [DOI] [PubMed] [Google Scholar]
- 10.Eisenstein M. Something new under the skin. Nat Biotech 2011; 29:107-9; http://dx.doi.org/ 10.1038/nbt.1768 [DOI] [PubMed] [Google Scholar]
- 11.Krieger IM, Eguiluz M. The second electroviscous effect in polymer latices. Trans Soc Rheol (1957-1977) 1976; 20:29-45; http://dx.doi.org/ 10.1122/1.549428 [DOI] [Google Scholar]
- 12.van der Vorst B, van den Ende D, Aelmans NJJ, Mellema J. Shear viscosity of an ordering latex suspension. Phys Rev E 1997; 56:3119-26; http://dx.doi.org/ 10.1103/PhysRevE.56.3119 [DOI] [Google Scholar]
- 13.Graessley WW. The entanglement concept in polymer rheology The Entanglement Concept in Polymer Rheology. Berlin, Heidelberg: Springer Berlin Heidelberg, 1974:1-179. [Google Scholar]
- 14.Buscall R, Goodwin JW, Hawkins MW, Ottewill RH. Viscoelastic properties of concentrated latices. Part 1—Methods of examination. Journal of the Chemical Society, Faraday Transactions 1: Physical Chemistry in Condensed Phases 1982; 78:2873-87; http://dx.doi.org/ 10.1039/f19827802873 [DOI] [Google Scholar]
- 15.Cromwell ME, Hilario E, Jacobson F. Protein aggregation and bioprocessing. AAPS J 2006; 8:E572-E9; PMID:17025275; http://dx.doi.org/ 10.1208/aapsj080366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas C, Nienow A, Dunnill P. Action of shear on enzymes: studies with alcohol dehydrogenase. Biotechnol Bioeng 1979; 21:2263-78; PMID:42450; http://dx.doi.org/ 10.1002/bit.260211208 [DOI] [PubMed] [Google Scholar]
- 17.Braun A, Kwee L, Labow MA, Alsenz J. Protein aggregates seem to play a key role among the parameters influencing the antigenicity of interferon alpha (IFN-α) in normal and transgenic mice. Pharm Res 1997; 14:1472-8; PMID:9358564; http://dx.doi.org/ 10.1023/A:1012193326789 [DOI] [PubMed] [Google Scholar]
- 18.Hermeling S, Schellekens H, Maas C, Gebbink MF, Crommelin DJ, Jiskoot W. Antibody response to aggregated human interferon alpha2b in wild‐type and transgenic immune tolerant mice depends on type and level of aggregation. J Pharm Sci 2006; 95:1084-96; PMID:16552750; http://dx.doi.org/ 10.1002/jps.20599 [DOI] [PubMed] [Google Scholar]
- 19.Joubert MK, Hokom M, Eakin C, Zhou L, Deshpande M, Baker MP, Goletz TJ, Kerwin BA, Chirmule N, Narhi LO. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J Biol Chem 2012; 287:25266-79; PMID:22584577; http://dx.doi.org/ 10.1074/jbc.M111.330902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J, Nguyen MD, Andya JD, Shire SJ. Reversible self‐association increases the viscosity of a concentrated monoclonal antibody in aqueous solution. J Pharm Sci 2005; 94:1928-40; PMID:16052543; http://dx.doi.org/ 10.1002/jps.20347 [DOI] [PubMed] [Google Scholar]
- 21.Kanai S, Liu J, Patapoff TW, Shire SJ. Reversible self‐association of a concentrated monoclonal antibody solution mediated by Fab–Fab interaction that impacts solution viscosity. J Pharm Sci 2008; 97:4219-27; PMID:18240303; http://dx.doi.org/ 10.1002/jps.21322 [DOI] [PubMed] [Google Scholar]
- 22.Bethea D, Wu S-J, Luo J, Hyun L, Lacy ER, Teplyakov A, Jacobs SA, O'Neil KT, Gilliland GL, Feng Y. Mechanisms of self-association of a human monoclonal antibody CNTO607. Protein Eng Des Sel 2012; 25:531-8; PMID:22915597; http://dx.doi.org/ 10.1093/protein/gzs047 [DOI] [PubMed] [Google Scholar]
- 23.Nishi H, Miyajima M, Wakiyama N, Kubota K, Hasegawa J, Uchiyama S, Fukui K. Fc domain mediated self-association of an IgG1 monoclonal antibody under a low ionic strength condition. J Biosci Bioeng 2011; 112:326-32; PMID:21783411; http://dx.doi.org/ 10.1016/j.jbiosc.2011.06.017 [DOI] [PubMed] [Google Scholar]
- 24.Esfandiary R, Parupudi A, Casas‐Finet J, Gadre D, Sathish H. Mechanism of Reversible Self‐Association of a Monoclonal Antibody: Role of Electrostatic and Hydrophobic Interactions. J Pharm Sci 2015; 104:577-86; PMID:25407315; http://dx.doi.org/ 10.1002/jps.24237 [DOI] [PubMed] [Google Scholar]
- 25.Yadav S, Sreedhara A, Kanai S, Liu J, Lien S, Lowman H, Kalonia DS, Shire SJ. Establishing a link between amino acid sequences and self-associating and viscoelastic behavior of two closely related monoclonal antibodies. Pharm res 2011; 28:1750-64; PMID:21626060; http://dx.doi.org/ 10.1007/s11095-011-0410-0 [DOI] [PubMed] [Google Scholar]
- 26.Chaudhri A, Zarraga IE, Kamerzell TJ, Brandt JP, Patapoff TW, Shire SJ, Voth GA. Coarse-grained modeling of the self-association of therapeutic monoclonal antibodies. J Phys Chem B 2012; 116:8045-57; PMID:22694284; http://dx.doi.org/ 10.1021/jp301140u [DOI] [PubMed] [Google Scholar]
- 27.Chaudhri A, Zarraga IE, Yadav S, Patapoff TW, Shire SJ, Voth GA. The role of amino acid sequence in the self-association of therapeutic monoclonal antibodies: insights from coarse-grained modeling. J Phys Chem B 2013; 117:1269-79; PMID:23316912; http://dx.doi.org/ 10.1021/jp3108396 [DOI] [PubMed] [Google Scholar]
- 28.Hvidt A, Linderstrøm-Lang K. Exchange of hydrogen atoms in insulin with deuterium atoms in aqueous solutions. Biochim Biophys Acta 1954; 14:574-5; PMID:13198919; http://dx.doi.org/ 10.1016/0006-3002(54)90241-3 [DOI] [PubMed] [Google Scholar]
- 29.Hvidt A, Linderstrøm-Lang K. The kinetics of the deuterium exchange of insulin with D 2 O. An amendment. Biochim Biophys Acta 1955; 16:168-9; PMID:14363248; http://dx.doi.org/ 10.1016/0006-3002(55)90200-6 [DOI] [PubMed] [Google Scholar]
- 30.Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys 1983; 16:521-655; PMID:6204354; http://dx.doi.org/ 10.1017/S0033583500005217 [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci 1993; 2:522-31; PMID:8390883; http://dx.doi.org/ 10.1002/pro.5560020404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bai Y, Sosnick TR, Mayne L, Englander SW. Protein folding intermediates: native-state hydrogen exchange. Science 1995; 269:192-7; PMID:7618079; http://dx.doi.org/ 10.1126/science.7618079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev 2006; 25:158-70; PMID:16208684; http://dx.doi.org/ 10.1002/mas.20064 [DOI] [PubMed] [Google Scholar]
- 34.Hvidt A, Nielsen SO. Hydrogen exchange in proteins. Adv Protein Chem 1966; 21:287-386; PMID:5333290; http://dx.doi.org/ 10.1016/S0065-3233(08)60129-1 [DOI] [PubMed] [Google Scholar]
- 35.Skinner JJ, Lim WK, Bédard S, Black BE, Englander SW. Protein dynamics viewed by hydrogen exchange. Protein Sci 2012; 21:996-1005; PMID:22544544; http://dx.doi.org/ 10.1002/pro.2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skinner JJ, Lim WK, Bédard S, Black BE, Englander SW. Protein hydrogen exchange: Testing current models. Protein Sci 2012; 21:987-95; PMID:22544567; http://dx.doi.org/ 10.1002/pro.2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Houde D, Peng Y, Berkowitz SA, Engen JR. Post-translational modifications differentially affect IgG1 conformation and receptor binding. Mol Cell Proteomics 2010; 9:1716-28; PMID:20103567; http://dx.doi.org/ 10.1074/mcp.M900540-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Majumdar R, Manikwar P, Hickey JM, Samra HS, Sathish HA, Bishop SM, Middaugh CR, Volkin DB, Weis DD. Effects of salts from the Hofmeister series on the conformational stability, aggregation propensity, and local flexibility of an IgG1 monoclonal antibody. Biochemistry 2013; 52:3376-89; PMID:23594236; http://dx.doi.org/ 10.1021/bi400232p [DOI] [PubMed] [Google Scholar]
- 39.Arora J, Hickey JM, Majumdar R, Esfandiary R, Bishop SM, Samra HS, Middaugh CR, Weis DD, Volkin DB. Hydrogen exchange mass spectrometry reveals protein interfaces and distant dynamic coupling effects during the reversible self-association of an IgG1 monoclonal antibody. mAbs 2015; 7:525-39; PMID:25875351; http://dx.doi.org/ 10.1080/19420862.2015.1029217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jensen PF, Larraillet V, Schlothauer T, Kettenberger H, Hilger M, Rand KD. Investigating the interaction between the neonatal Fc receptor and monoclonal antibody variants by hydrogen/deuterium exchange mass spectrometry. Mol Cell Proteomics 2015; 14:148-61; PMID:25378534; http://dx.doi.org/ 10.1074/mcp.M114.042044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walters BT, Jensen PF, Larraillet V, Lin K, Patapoff T, Schlothauer T, Rand KD, Zhang J. Conformational Destabilization of Immunoglobulin G Increases the Low pH Binding Affinity with the Neonatal Fc Receptor. J Biol Chem 2016; 291:1817-25; PMID:26627822; http://dx.doi.org/ 10.1074/jbc.M115.691568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang J, Richardson J, Du Z, Zhang Z. Effect of Fc-Glycan Structure on the Conformational Stability of IgG Revealed by Hydrogen/Deuterium Exchange and Limited Proteolysis. Biochemistry 2016; 55:860-8; PMID:26812426; http://dx.doi.org/ 10.1021/acs.biochem.5b01323 [DOI] [PubMed] [Google Scholar]
- 43.Houde D, Nazari ZE, Bou-Assaf GM, Weiskopf AS, Rand KD. Conformational Analysis of Proteins in Highly Concentrated Solutions by Dialysis-Coupled Hydrogen/Deuterium Exchange Mass Spectrometry. J Am Soc Mass Spectrom 2016; 27:669-76; PMID:26860088; http://dx.doi.org/ 10.1007/s13361-015-1331-7 [DOI] [PubMed] [Google Scholar]
- 44.Armstrong J, Wenby R, Meiselman H, Fisher T. The hydrodynamic radii of macromolecules and their effect on red blood cell aggregation. Biophys J 2004; 87:4259-70; PMID:15361408; http://dx.doi.org/ 10.1529/biophysj.104.047746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Esfandiary R, Hayes DB, Parupudi A, Casas‐Finet J, Bai S, Samra HS, Shah AU, Sathish HA. A systematic multitechnique approach for detection and characterization of reversible self‐association during formulation development of therapeutic antibodies. J Pharm Sci 2013; 102:3089-99; PMID:23794522; http://dx.doi.org/ 10.1002/jps.23654 [DOI] [PubMed] [Google Scholar]
- 46.Fischer G, Rossmann M, Hyvönen M. Alternative modulation of protein–protein interactions by small molecules. Curr Opin Biotechnol 2015; 35:78-85; PMID:25935873; http://dx.doi.org/ 10.1016/j.copbio.2015.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yadav S, Liu J, Shire SJ, Kalonia DS. Specific interactions in high concentration antibody solutions resulting in high viscosity. J Pharm Sci 2010; 99:1152-68; PMID:19705420; http://dx.doi.org/ 10.1002/jps.21898 [DOI] [PubMed] [Google Scholar]
- 48.Yadav S, Shire SJ, Kalonia DS. Viscosity behavior of high‐concentration monoclonal antibody solutions: Correlation with interaction parameter and electroviscous effects. J Pharm Sci 2012; 101:998-1011; PMID:22113861; http://dx.doi.org/ 10.1002/jps.22831 [DOI] [PubMed] [Google Scholar]
- 49.Yadav S, Laue TM, Kalonia DS, Singh SN, Shire SJ. The influence of charge distribution on self-association and viscosity behavior of monoclonal antibody solutions. Mol Pharm 2012; 9:791-802; PMID:22352470; http://dx.doi.org/ 10.1021/mp200566k [DOI] [PubMed] [Google Scholar]
- 50.Zhang J. Protein-protein interactions in salt solutions. INTECH Open Access Publisher, 2012; ISBN 978-953-51-0397-4 [Google Scholar]
- 51.Smoluchowski Mv. Theoretische Bemerkungen über die Viskosität der Kolloide. Kolloid Z 1916; 18:190-5; http://dx.doi.org/ 10.1007/BF01433350 [DOI] [Google Scholar]
- 52.Buzzell JG, Tanford C. The effect of charge and ionic strength on the viscosity of ribonuclease. J Phys Chem 1956; 60:1204-7; http://dx.doi.org/ 10.1021/j150543a014 [DOI] [Google Scholar]
- 53.Komatsubara M, Suzuki K, Nakajima H, Wada Y. Electroviscous effect of lysozyme in aqueous solutions. Biopolymers 1973; 12:1741-6; PMID:4739048; http://dx.doi.org/ 10.1002/bip.1973.360120804 [DOI] [PubMed] [Google Scholar]
- 54.Bull HB. The electroviscous effect in egg albumin solutions. Trans Faraday Soc 1940; 35:80-4; http://dx.doi.org/ 10.1039/tf9403500080 [DOI] [Google Scholar]
- 55.Arakawa T, Timasheff SN. Stabilization of protein structure by sugars. Biochemistry 1982; 21:6536-44; PMID:7150574; http://dx.doi.org/ 10.1021/bi00268a033 [DOI] [PubMed] [Google Scholar]
- 56.Chi EY, Krishnan S, Randolph TW, Carpenter JF. Physical stability of proteins in aqueous solution: mechanism and driving forces in nonnative protein aggregation. Pharm Res 2003; 20:1325-36; PMID:14567625; http://dx.doi.org/ 10.1023/A:1025771421906 [DOI] [PubMed] [Google Scholar]
- 57.Lee JC, Timasheff SN. The stabilization of proteins by sucrose. J Biol Chem 1981; 256:7193-201; PMID:7251592 [PubMed] [Google Scholar]
- 58.Kendrick BS, Chang BS, Arakawa T, Peterson B, Randolph TW, Manning MC, Carpenter JF. Preferential exclusion of sucrose from recombinant interleukin-1 receptor antagonist: role in restricted conformational mobility and compaction of native state. Proc Natl Acad Sci 1997; 94:11917-22; PMID:9342337; http://dx.doi.org/ 10.1073/pnas.94.22.11917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chothia C, Gelfand I, Kister A. Structural determinants in the sequences of immunoglobulin variable domain. J Mol Biol 1998; 278:457-79; PMID:9571064; http://dx.doi.org/ 10.1006/jmbi.1998.1653 [DOI] [PubMed] [Google Scholar]
- 60.Ido S, Kimiya H, Kobayashi K, Kominami H, Matsushige K, Yamada H. Immunoactive two-dimensional self-assembly of monoclonal antibodies in aqueous solution revealed by atomic force microscopy. Nat Mater 2014; 13:264-70; PMID:24441879; http://dx.doi.org/ 10.1038/nmat3847 [DOI] [PubMed] [Google Scholar]
- 61.Arakawa T, Ejima D, Tsumoto K, Obeyama N, Tanaka Y, Kita Y, Timasheff SN. Suppression of protein interactions by arginine: A proposed mechanism of the arginine effects. Biophys Chem 2007; 127:1-8; PMID:17257734; http://dx.doi.org/ 10.1016/j.bpc.2006.12.007 [DOI] [PubMed] [Google Scholar]
- 62.Baynes BM, Wang DI, Trout BL. Role of arginine in the stabilization of proteins against aggregation. Biochemistry 2005; 44:4919-25; PMID:15779919; http://dx.doi.org/ 10.1021/bi047528r [DOI] [PubMed] [Google Scholar]
- 63.Shukla D, Trout BL. Interaction of arginine with proteins and the mechanism by which it inhibits aggregation. J Phys Chem B 2010; 114:13426-38; PMID:20925358; http://dx.doi.org/ 10.1021/jp108399g [DOI] [PubMed] [Google Scholar]
- 64.Kita Y, Arakawa T, Lin T-Y, Timasheff SN. Contribution of the surface free energy perturbation to protein-solvent interactions. Biochemistry 1994; 33:15178-89; PMID:7999778; http://dx.doi.org/ 10.1021/bi00254a029 [DOI] [PubMed] [Google Scholar]
- 65.Arakawa T, Timasheff S. The mechanism of action of Na glutamate, lysine HCl, and piperazine-N, N'-bis (2-ethanesulfonic acid) in the stabilization of tubulin and microtubule formation. J Biol Chem 1984; 259:4979-86; PMID:6325414 [PubMed] [Google Scholar]
- 66.Gallivan JP, Dougherty DA. Cation-π interactions in structural biology. Proc Natl Acad Sci 1999; 96:9459-64; PMID:10449714; http://dx.doi.org/ 10.1073/pnas.96.17.9459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Geoghegan JC, Fleming R, Damschroder M, Bishop SM, Sathish HA, Esfandiary R. Mitigation of reversible self-association and viscosity in a human IgG1 monoclonal antibody by rational, structure-guided Fv engineering. mAbs 2016:00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Holde KE, Johnson WC, Ho PS. Principles of physical biochemistry. 2006. [Google Scholar]
- 69.Barer R, Joseph S. Refractometry of living cells. J Cell Sci 1955; 3:423-47. [Google Scholar]
- 70.Bond MD, Panek ME, Zhang Z, Wang D, Mehndiratta P, Zhao H, Gunton K, Ni A, Nedved ML, Burman S et al. Evaluation of a dual‐wavelength size exclusion HPLC method with improved sensitivity to detect protein aggregates and its use to better characterize degradation pathways of an IgG1 monoclonal antibody. J Pharm Sci 2010; 99:2582-97; PMID:20039394; http://dx.doi.org/ 10.1002/jps.22034 [DOI] [PubMed] [Google Scholar]
- 71.Glasoe PK, Long F. Use of glass electrodes to measure acidities in deuterium oxide. J Phys Chem 1960; 64:188-90; http://dx.doi.org/ 10.1021/j100830a521 [DOI] [Google Scholar]
- 72.Sali A, Blundell T. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol 1993; 234:779-815; PMID:8254673; http://dx.doi.org/ 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- 73.Padlan EA. Anatomy of the antibody molecule. Mol Immunol 1994; 31:169-217; PMID:8114766; http://dx.doi.org/ 10.1016/0161-5890(94)90001-9 [DOI] [PubMed] [Google Scholar]
- 74.Colman PM, Deisenhofer J, Huber R, Palm W. Structure of the human antibody molecule kol (immunoglobulin G1): An electron density map at 5 Å resolution. J Mol Biol 1976; 100:257-78; PMID:1255713; http://dx.doi.org/ 10.1016/S0022-2836(76)80062-9 [DOI] [PubMed] [Google Scholar]
- 75.Matsumiya S, Yamaguchi Y, Saito J-i, Nagano M, Sasakawa H, Otaki S, Satoh M, Shitara K, Kato K. Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J Mol Biol 2007; 368:767-79; PMID:17368483; http://dx.doi.org/ 10.1016/j.jmb.2007.02.034 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.