

Abstract
Atherosclerosis is a complex process responsible for a major burden of cardiovascular morbidity and mortality. Macrophages and smooth muscle cells (SMCs) are abundant within atherosclerotic plaques. This review discusses the role of macrophages and SMCs in plaque progression and provides an overview of nanoparticle-based approaches and other current methods for local targeting of atherosclerotic plaques.
Keywords: atherosclerosis, plaque, macrophage, smooth muscle cell, nanoparticle, nanotechnology
Introduction
Atherosclerotic cardiovascular disease is the leading cause of mortality in the world, accounting for approximately one-third of all global deaths.1 Atherosclerosis is a complex process that contributes to the development of myocardial infarctions, chronic angina, cerebrovascular events, aortic disease, and peripheral arterial disease. The pathogenesis of atherosclerosis is multifaceted and includes endothelial dysfunction, a myriad of inflammatory and immunologic factors, oxidative stress, and plaque rupture. Smoking, dyslipidemia, hypertension, and diabetes are well known risk factors for the development of atherosclerosis and its subsequent complications. The role of inflammation and the immune system is well established as central to the development of atherosclerosis and to arterial remodeling.2
While 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (i.e., statins) have revolutionized the treatment of hyperlipidemia since their first clinical use in the 1980s, there is still an unmet need for novel therapies due to statin intolerance, inadequate reduction of low-density lipoprotein cholesterol (LDL-C), and the residual risk of cardiovascular (CV) events that persists despite statin use. For example, despite intensive lipid therapy with statins, the residual annual risk of CV events such as myocardial infarction or stroke remains at approximately 9%.3 While the pleiotropic effect of statin therapy is thought to include some anti-inflammatory properties, therapies directed to specifically target the inflammatory and immune pathways are lacking in the clinical arena.
Role of Macrophages and Smooth Muscle Cells in Atherosclerosis
Although the composition of atherosclerotic plaques is complex and may include lymphocytes, dendritic cells, mast cells, neutrophils, and endothelial cells, macrophages and smooth muscle cells are major determinants of plaque progression and plaque stability.
Macrophages in Atherosclerosis
Macrophages are phagocytic cells that ensure clearance of any foreign or unwanted cells and waste material. In early atherosclerosis, the endothelium is activated by oxidized LDL-C (oxLDL) retained in the arterial intima and secretes chemokines that attract circulating monocytes. The latter transmigrate into the intima and differentiate into macrophages.4 There are several phenotypically distinct populations of macrophages found in atherosclerotic lesions, including M1, M2, and Mox. M1 and Mox macrophages are inflammation-promoting cells enriched in progressing plaques, whereas M2 macrophages are inflammation-resolving cells found in regressing plaques.5
In the arterial intima, macrophages oxidize and uptake lipoproteins. LDL-C digestion in lysosomes releases free cholesterol that can either be effluxed from the cell or esterified to store in cytosol as lipid droplets.6,7 Because there is little negative feedback following lipoprotein uptake, macrophages ingest LDL-C until they become overloaded with lipids and esterified cholesterol.6 These enlarged “foamy” cells, with diminished capacity to migrate, accumulate in the intima and build up the atherosclerotic plaque.8
Excessive LDL-C uptake leads to disruption of cholesterol metabolism and accumulation of free cholesterol. The latter promotes activation of inflammatory signaling and ultimately results in apoptotic cell death.9 Since these apoptotic lipid-laden macrophages cannot be efficiently cleared, they undergo secondary necrosis, thereby releasing cellular components and lipids that form the lipid-rich necrotic core of the plaque.10 The core enlarges in size as atherosclerosis progresses. If the fibrous cap of the plaque ulcerates or ruptures, it can cause vessel occlusion or embolism with respective clinical symptoms.11 Furthermore, proinflammatory macrophages secrete proteolytic enzymes that degrade collagen, thus facilitating thinning and rupture of the fibrous cap.12
Recently, it was shown that the amount of macrophages within the plaque is not solely dependent on monocyte recruitment.13 Although macrophages do proliferate within the plaque, they have a relatively short life span there. In the murine model of atherosclerosis, macrophage turnover within the developed plaques is about 1 month.13 Monocyte recruitment occurs as an initial response to LDL-C accumulation in the intima, with activation of the endothelium and expression of endothelial adhesion molecules. Macrophage proliferation within the plaques drives further disease progression.
Vascular Smooth Muscle Cells in Atherosclerosis
Vascular smooth muscle cells (SMCs) are responsible for vessel contraction and relaxation to maintain blood pressure and blood flow distribution. In the vessel wall, these cells reside underneath the basal lamina, which is overlaid by the endothelium monolayer. It is known that vascular SMCs can undergo phenotype switching and participate in the development of atherosclerotic plaque.14 In addition, extensively proliferating SMCs play a pivotal role in restenosis that occurs after vascular procedures such as endarterectomy and percutaneous coronary intervention.15
Intact vascular SMCs have a very low proliferation rate and express proteins responsible for physical contraction of the cells. This phenotype is referred to as “contractile.” In response to retained lipoproteins and proinflammatory signals of endothelial cells and macrophages, these contractile, quiescent SMCs lose contraction-specific markers and migrate through the basal lamina. In the artery intima, SMCs proliferate and produce fibrous tissue, thereby forming the fibrous cap of the plaque.16 Moreover, these cells can produce proinflammatory cytokines and engulf lipoproteins. The molecular regulation of SMC phenotype switching involves transcriptional factors, microRNAs, and epigenetic modifications.14 Whether SMC phenotype switching is a physiologic defensive response to injury or a pathologic component of the disease process remains unclear. On one hand, the fibroproliferative response of SMCs seems to sequester the injury agent (e.g., oxLDL) by forming the cap of the plaque. On the other hand, this fibroproliferative response progresses above and beyond that required to repair a small area of injury.16 In addition, normal fibrotic response to injury is ensured by fibroblasts rather than SMCs, therefore SMCs are thought to produce pathologic fibrous tissue.
The ability of activated SMCs to engulf lipoproteins also makes these cells a major contributor to plaque development.17 Because they are devoid of SMC-specific markers and can express macrophage-specific markers, it is difficult to estimate what fraction of foam cells within the atherosclerotic lesion is SMC derived. Recent SMC in vivo fate-tracing studies proved that SMCs do indeed undergo clonal expansion, transdifferentiate into macrophage-like cells, and make up large areas of the plaques.18,19 The phenotype of these cells resembles that of Mox macrophages. Furthermore, some SMCs within the plaque were shown to express markers specific to other cell types, such as mesenchymal stem cells and myofibroblasts. This plasticity of SMCs likely leads to misidentification of these cells and underestimation of their importance in disease pathogenesis. For instance, the lineage-tracing study has determined that more than 80% of SMCs within brachiocephalic artery lesions in mice are phenotypically modulated and therefore undetectable by conventional techniques.19
Vascular SMCs are also involved in calcification of the vascular wall seen in advanced atherosclerosis. Calcifying SMCs produce matrix vesicles that contain a number of molecules essential to induce hydroxyapatite crystallization.20 The mechanism of replicative senescence is thought to induce calcifying SMCs through the activation of several transcription factors.21 Calcification decreases vessel elasticity and changes local hemodynamics and thus may aggravate systolic hypertension and endothelial dysfunction. Moreover, microcalcifications in the fibrous cap may be associated with plaque rupture.22 The role of SMCs and macrophages in atherogenesis is summarized in Figure 1, and major inflammatory pathways in which these cells are involved are depicted in Figure 2.
Figure 1.
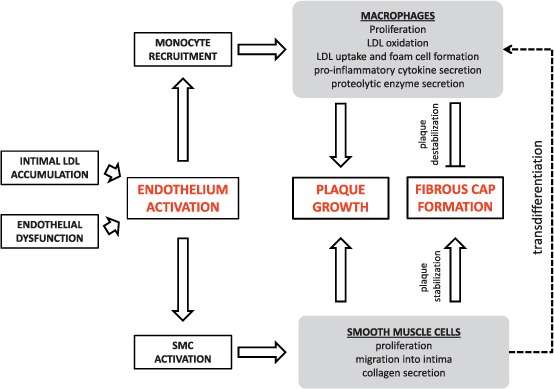
The role of macrophages and smooth muscle cells (SMCs) in atherogenesis.
Figure 2.
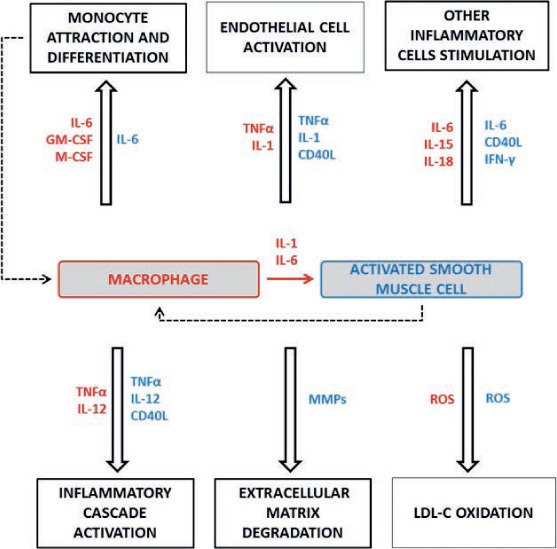
Macrophage- and smooth muscle cell (SMC)-related inflammatory pathways involved in atherosclerosis progression and plaque rapture. Bioactive molecules produced by macrophages are noted in red, the molecules produced by SMCs in blue. IL: interleukin, GM-CSF: granulocyte macrophage colony-stimulating factor, M-CSF: macrophage colony-stimulating factor, TNFα: tumor necrosis factor α, IFN-γ: interferon γ, CD40L: soluble CD40 ligand, MMP: matrix metalloproteinase, ROS: reactive oxygen species.
Current Strategies to Suppress Plaque Progression
Lowering blood cholesterol levels is a key component of the therapeutic strategy used to clinically treat atherosclerosis and its complications. Cholesterol lowering can be achieved in modest amounts by dietary modifications and by increasing physical activity. Pharmacological approaches have been dominated by statins, which inhibit cholesterol synthesis,23 while other agents such as ezetimibe, niacin, and fibrates are sometimes used in patients with poor tolerance or response to statins or who have predominantly non-LDL-C lipid abnormalities. Ezetimibe reduces cholesterol absorption in the small intestine24 and predominantly lowers LDL-C, whereas fibrates predominantly lower triglyceride levels and increase high-density lipoprotein cholesterol (HDL-C) levels. Niacin lowers LDL-C levels and increases HDL-C by mechanisms that are not clearly elucidated.25 Compared to statins, these agents have a lesser magnitude of clinical benefit.
Recently, the U.S. Food and Drug Administration approved new agents that are monoclonal antibodies targeting proprotein convertase subtilisin kexin 9 (PCSK9). These PCSK9 inhibitors lower LDL-C substantially—as much as 70% in patients intolerant to statins or 60% in patients on statins—by improving its uptake by the liver.26 These agents are currently being studied to determine their effect on CV event risk reduction, and they are approved for use in patients with familial hypercholesterolemia and for those with clinical atherosclerotic disease who require additional LDL-C lowering beyond the effect of statins.
Antihypertensive drugs targeting the renin-angiotensin-aldosterone system, such as angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers, have also proved to be beneficial in preventing clinical complications of atherosclerosis in high-risk patients.27 Angiotensin II suppresses the release of the atheroprotective molecule nitric oxide (NO) and induces NAD(P)H oxidase activity in endothelium, hence promoting oxidative stress and LDL peroxidation. Furthermore, angiotensin II is involved in activation of inflammatory signaling pathways, the recruitment of inflammatory cells into injured endothelium, and proliferation of vascular SMCs.28 Inhibiting the renin-angiotensin-aldosterone system, therefore, may slow inflammatory processes and atherosclerosis progression by increasing NO bioavailability and reducing oxidative stress. Since cigarette smoking also significantly promotes vascular oxidative stress and is highly associated with CV disease, the benefits of tobacco cessation should be stressed for all patients with atherosclerosis.29
Since atherosclerosis is a chronic inflammatory disease, a broad range of molecules targeting distinct inflammation-related signaling pathways are currently being evaluated as possible therapeutic agents. Macrophages and SMCs are prime targets for the development of novel agents since they are intricately involved in the inflammatory pathways of plaque development and progression. A number of recombinant antibodies developed to interfere with proinflammatory signaling molecules are now in trials.30 In addition, the inhibitors of leukotriene synthesis and those of the p38 MAPK pathway demonstrate promising results in clinical studies.31,32 Both leukotriene and p38 MAPK pathways transduce intracellular proinflammatory responses that are implicated in monocyte recruitment and activation and in SMC proliferation.
Of note, low doses of cyclooxygenase inhibitors such as aspirin not only affect platelets but can also prevent formation of thromboxane A2 by macrophages in atherosclerotic lesions. Thromboxane A2 can induce proliferation of SMCs and activate inflammatory and endothelial cells.30,33 Statins, in addition to lowering cholesterol, are also known to reduce levels of C-reactive protein (CRP), a biomarker of inflammation that independently predicts CV events.34 Because of this effect, statins may be considered for patients who do not have elevated cholesterol but, rather, have high CRP levels that put them at risk for CV events.
Inhibiting the cell cycle with antiproliferative agents, such as rapamycin or paclitaxel, is another promising therapeutic strategy for suppressing plaque growth. Rapamycin inhibits the mTORC1 protein complex that regulates cell growth, proliferation, motility, survival, protein synthesis, and transcription.35 Rapamycin affects SMC proliferation, macrophage and lipid accumulation within the plaque and suppresses intraplaque neoangiogenesis. There are many preclinical studies that show a significant reduction in plaque (up to 50%–80%) after systemic administration of rapamycin or its synthetic analog everolimus.36 Although chronic systemic administration of rapamycin in doses necessary to achieve such a dramatic effect seems to be toxic, local administration of rapamycin or everolimus through drug-eluting stents is successfully used to prevent restenosis.
In addition to mTOR inhibitors, another antiproliferative agent, paclitaxel, is used in drug-eluting stents to prevent restenosis. Paclitaxel promotes polymerization of tubulin, thereby inhibiting mitotic spindle formation.37 Other approaches to inhibit cell proliferation include local delivery of β or γ radiation to the stented artery and gene therapy with antisense oligodeoxynucleotides complimentary to cell cycle regulatory genes.38
Innovative Approaches for Local Targeting of Atherosclerotic Plaques
Systemic drug administration is a common and simple way to deliver a certain therapeutic molecule to its target (usually consumed orally or intravenously injected). Since the drug is introduced to the entire body, we are actually using more drug then we need, as opposed to the case in which the drug is “delivered” directly to the target site. In addition, when a drug is delivered systemically, it predisposes other non-targeted to potential side effects. To overcome this, scientists have been investigating ways in which to improve drug administration. A growing area of intense study is the use of nanoparticles, specialized drug carriers aimed at specific targets.39,40 Nanoparticles (NPs) have the ability to affect biological systems on the cellular and molecular level that cause an overall change in the tissue microenvironment. They also are designed to target a specific site and release drugs in a controlled fashion, thereby increasing the therapeutic index for molecules considered toxic.41 For example, multiple clinical trials are currently investigating immune-modulatory treatments for atherosclerosis.42 A major drawback of such treatment is the risks inherent in systemic immune-suppression. Encapsulating anti-inflammatory agents in NPs would mediate an anti-inflammatory effect but spare the off-target effects. A variety of NP systems have recently been developed, offering the potential to attack atherosclerotic plaques using different pathways. The following highlights some recent examples.
Given their role in the development of atherosclerotic plaque,43 many efforts are focused on targeting macrophages. Innovative approaches for macrophage targeting include the design of hybrid poly(lactic-co-glycolic acid)/HDL-C nanoparticles. These HDL-C mimetic NPs were shown to have HDL-C characteristics, including specific uptake by macrophages and accumulation in the plaque in Apo E-/- mouse models.44 In another study, HDL-C NPs were loaded with statin and shown to inhibit macrophage proliferation in plaques, thereby decreasing plaque.45 Another attempt to target macrophages was done using gold-coated iron oxide NPs conjugated with an anti-CD163 antibody, a membrane receptor expressed by macrophages and overexpressed in inflammatory sites.46 Macrophages use scavenger receptors, such as CD36, to incorporate oxidized LDL (oxLDL). Based on that principle, NPs decorated with CD36 ligand have demonstrated active targeting and efficient biological activity when encapsulated with an agent that reduces cholesterol accumulation and inflammatory response in macrophages.47 The use of lipid-latex NPs bearing phagocytic signals is a relatively new approach of targeting macrophages for atherosclerosis treatment. Although initial results are promising, additional investigation is required to optimize the specific active-targeting abilities of NPs.48 Since macrophages can play both anti- and pro-inflammatory roles, a biological signal that induces the anti-inflammatory pathway, as opposed to simply inhibiting the deleterious inflammatory activity, may prove to be beneficial in supporting the “self-healing” of the atherosclerotic vascular tissue.
Given that the formation of plaque is an ongoing inflammatory event, studies are increasingly exploring methods that target the inflammatory pathway. For example, we previously demonstrated the effect of leukolike vectors (LLV), a biomimetic platform composed of nanoporous silicon particles coated with leukocyte membrane, on inflamed endothelia.49 The LLV were shown to exert leukocyte properties, such as avoiding immune recognition and specifically interacting with inflamed endothelia. Other attempts to target inflammation include the design of glucocorticoid-encapsulated NPs that passively target plaque according to the Enhanced Permeability and Retention (EPR) effect50,51 and the targeting of collagen IV, which is exposed at the site of injury and inflammation. NPs targeting collagen IV were coated with a small peptide Ac-26 and specifically targeted plaque lesion in vivo to improve diseased vessels by increasing the protective collagen layer of the fibrous cap, reducing oxidative stress, and attenuating plaque necrosis.52 Other molecular targets currently being explored include the receptors for hyaluronic acid, stabilin-2, and CD44, which are overexpressed in atherosclerotic vessels.53
Major advances in both nanotechnology and understanding of atherosclerosis pathobiology have led to the design of new NPs for plaque treatment. As the field of NP biologics is being established, there is potential for the design of drug delivery systems that will target atherosclerotic processes. In theory, any of the pathways depicted in Figure 2 could be targeted locally with NPs. Recently, our group has proposed local delivery of an mTOR inhibitor rapamycin to suppress intraplaque proliferation of macrophages and SMCs. As a delivery system, we proposed using leukosomes - bioinspired lipid nanoparticles enriched with purified cell membranes isolated from circulating leukocytes. The latter display the broad spectrum of leukocyte membrane proteins on their surface, and therefore are not trapped by immune cells in the circulation and feature tropism towards inflamed vasculature.54 Although preliminary studies are promising, in vivo experiments on the animal model of atherosclerosis is still in progress.
Conclusion
The role of the immune system and inflammatory pathways in the development of atherosclerosis is well established. Macrophages, activated by oxLDL in the arterial intima, play a critical role in the early stages and subsequent progression of atherosclerosis. Smooth muscle cells also play a major role via mechanisms such as intimal migration and proliferation as well as their uptake of lipoproteins and subsequent proinflammatory state. Therapies directed at the traditional risk factors (hypercholesterolemia, hypertension, diabetes mellitus, and tobacco exposure) have had major clinical benefits, but there remains a high residual risk of CV events. There is a lack of therapies directly targeting macrophages and smooth muscle cells, largely due to the concern for systemic toxicities. The use of nano-technology to specifically target these pathways without causing global suppression of the immune system is a promising approach.
Key Points
Macrophages and smooth muscle cells (SMCs) are major contributors to plaque growth.
Macrophages and SMCs proliferate locally within the atherosclerotic plaque.
SMCs may transdifferentiate into macrophage-like cells.
Although macrophages and SMCs are natural targets for novel therapies, current approaches are limited.
Using nanotechnology to specifically target macrophage- and SMC-mediated inflammatory pathways without causing systemic toxicity is a promising strategy to suppress plaque progression.
Footnotes
Conflict of Interest Disclosure: The authors have completed and submitted the Methodist DeBakey Cardiovascular Journal Conflict of Interest Statement and none were reported.
Funding: This work was funded by an award from George and Angelina Kostas Research Center for Cardiovascular Nanomedicine.
References
- 1. World Health Organization [Internet]. Geneva, Switzerland: World Health Organization; c2016. Cardiovascular diseases: fact sheet N°317; 2015 Jan [cited 2016 May 8]. Available from: http://www.who.int/mediacentre/factsheets/fs317/en/. [Google Scholar]
- 2. Tellides G, Pober JS. Inflammatory and immune responses in the arterial media. Circ Res. 2015. January 16; 116( 2): 312– 22. [DOI] [PubMed] [Google Scholar]
- 3. Sampson UK, Fazio S, Linton MF.. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Curr Atheroscler Rep. 2012. February; 14( 1): 1– 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tabas I, Williams KJ, Borén J.. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007. October 16; 116( 16): 1832– 44. [DOI] [PubMed] [Google Scholar]
- 5. Chinetti-Gbaguidi G, Colin S, Staels B.. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015. January; 12( 1): 10– 7. [DOI] [PubMed] [Google Scholar]
- 6. Moore KJ, Sheedy FJ, Fisher EA.. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013. October; 13( 10): 709– 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McLaren JE, Michael DR, Ashlin TG, Ramji DP.. Cytokines, macrophage lipid metabolism and foam cells: implications for cardiovascular disease therapy. Prog Lipid Res. 2011. October; 50( 4): 331– 47. [DOI] [PubMed] [Google Scholar]
- 8. Shashkin P, Dragulev B, Ley K.. Macrophage differentiation to foam cells. Curr Pharm Des. 2005; 11( 23): 3061– 72. [DOI] [PubMed] [Google Scholar]
- 9. Feng B, Yao PM, Li Y, . et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003. September; 5( 9): 781– 92. [DOI] [PubMed] [Google Scholar]
- 10. Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005. November; 25( 11): 2255– 64. [DOI] [PubMed] [Google Scholar]
- 11. Lyaker MR, Tulman DB, Dimitrova GT, Pin RH, Papadimos TJ.. Arterial embolism. Int J Crit Illn Inj Sci. 2013. January; 3( 1): 77– 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Libby P. Collagenases and cracks in the plaque. J Clin Invest. 2013. August; 123( 8): 3201– 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robbins CS, Hilgendorf I, Weber GF, . et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013. September; 19( 9): 1166– 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012. July 15; 95( 2): 156– 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marx SO, Totary-Jain H, Marks AR.. Vascular smooth muscle cell proliferation in restenosis. Circ Cardiovasc Interv. 2011. February 1; 4( 1): 104– 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Frink RJ. Inflammatory atherosclerosis: characteristics of the injurious agent. Sacramento, CA: Heart Research Foundation; 2002. Chapter 2, The smooth muscle cell. The pivot in atherosclerosis 111 p. [Google Scholar]
- 17. Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA.. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014. April 15; 129( 15): 1551– 9. [DOI] [PubMed] [Google Scholar]
- 18. Feil S, Fehrenbacher B, Lukowski R, . et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. 2014. September 12; 115( 7): 662– 7. [DOI] [PubMed] [Google Scholar]
- 19. Shankman LS, Gomez D, Cherepanova OA, . et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015. June; 21( 6): 628– 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anderson HC. Matrix vesicles and calcification. Curr Rheumatol Rep. 2003. June; 5( 3): 222– 6. [DOI] [PubMed] [Google Scholar]
- 21. Nakano-Kurimoto R, Ikeda K, Uraoka M, . et al. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am J Physiol Heart Circ Physiol. 2009. November; 297( 5): H1673– 84. [DOI] [PubMed] [Google Scholar]
- 22. Kataoka Y, Puri R, Hammadah M, . et al. Spotty calcification and plaque vulnerability in vivo: frequency-domain optical coherence tomography analysis. Cardiovasc Diagn Ther. 2014. December; 4( 6): 460– 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gotto AM., Jr. The cardiology patient page. Statins: powerful drugs for lowering cholesterol: advice for patients. Circulation. 2002. April 2; 105( 13): 1514– 6. [DOI] [PubMed] [Google Scholar]
- 24. Hayek S, Canepa Escaro F, Sattar A, . et al. Effect of ezetimibe on major atherosclerotic disease events and all-cause mortality. Am J Cardiol. 2013. February 15; 111( 4): 532– 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ruparelia N, Digby JE, Choudhury RP.. Effects of niacin on atherosclerosis and vascular function. Curr Opin Cardiol. 2011. January; 26( 1): 66– 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Everett BM, Smith RJ, Hiatt WR.. Reducing LDL with PCSK9 Inhibitors--The Clinical Benefit of Lipid Drugs. N Engl J Med. 2015. October 22; 373( 17): 1588– 91. [DOI] [PubMed] [Google Scholar]
- 27. Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G.. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000. January 20; 342( 3): 145– 53. [DOI] [PubMed] [Google Scholar]
- 28. Mason RP. Optimal therapeutic strategy for treating patients with hypertension and atherosclerosis: focus on olmesartan medoxomil. Vasc Health Risk Manag. 2011; 7: 405– 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004. May 19; 43( 10): 1731– 7. [DOI] [PubMed] [Google Scholar]
- 30. Bäck M, Hansson GK. Anti-inflammatory therapies for atherosclerosis. Nat Rev Cardiol. 2015. April; 12( 4): 199– 211. [DOI] [PubMed] [Google Scholar]
- 31. Tardif JC, L'allier P L, Ibrahim R, . et al. Treatment with 5-lipoxygenase inhibitor VIA-2291 (Atreleuton) in patients with recent acute coronary syndrome. Circ Cardiovasc Imaging. 2010. May; 3( 3): 298– 307. [DOI] [PubMed] [Google Scholar]
- 32. Fisk M, Gajendragadkar PR, Mäki-Petäjä KM, Wilkinson IB, Cheriyan J.. Therapeutic potential of p38 MAP kinase inhibition in the management of cardiovascular disease. Am J Cardiovasc Drugs. 2014. June; 14( 3): 155– 65. [DOI] [PubMed] [Google Scholar]
- 33. Petri MH, Tellier C, Michiels C, Ellertsen I, Dogné JM, Bäck M.. Effects of the dual TP receptor antagonist and thromboxane synthase inhibitor EV-077 on human endothelial and vascular smooth muscle cells. Biochem Biophys Res Commun. 2013. November 15; 441( 2): 393– 8. [DOI] [PubMed] [Google Scholar]
- 34. Ridker PM. The JUPITER trial: results, controversies, and implications for prevention. Circ Cardiovasc Qual Outcomes. 2009. May; 2( 3): 279– 85. [DOI] [PubMed] [Google Scholar]
- 35. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012. April 13; 149( 2): 274– 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martinet W, De Loof H, De Meyer GR.. mTOR inhibition: a promising strategy for stabilization of atherosclerotic plaques. Atherosclerosis. 2014. April; 233( 2): 601– 7. [DOI] [PubMed] [Google Scholar]
- 37. Axel DI, Kunert W, Göggelmann C, . et al. Paclitaxel inhibits arterial smooth muscle cell proliferation and migration in vitro and in vivo using local drug delivery. Circulation. 1997. July 15; 96( 2): 636– 45. [DOI] [PubMed] [Google Scholar]
- 38. Dzau VJ, Braun-Dullaeus RC, Sedding DG.. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med. 2002. November; 8( 11): 1249– 56. [DOI] [PubMed] [Google Scholar]
- 39. Suri SS, Fenniri H, Singh B.. Nanotechnology-based drug delivery systems. J Occup Med Toxicol. 2007. December 1; 2: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Godin B, Hu Y, La Francesca S, Ferrari M.. Cardiovascular nanomedicine: challenges and opportunities. : Homeister JW, Willis MS, . Molecular & translational vascular medicine. New York: Springer Science & Business Media; 2012. p 249– 281. [Google Scholar]
- 41. Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004. March 19; 303( 5665): 1818– 22. [DOI] [PubMed] [Google Scholar]
- 42. Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013. January 11; 339( 6116): 166– 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011. April 29; 145( 3): 341– 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sanchez-Gaytan BL, Fay F, Lobatto ME, . et al. HDL-mimetic PLGA nanoparticle to target atherosclerosis plaque macrophages. Bioconjug Chem. 2015. March 18; 26( 3): 443– 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tang J, Lobatto ME, Hassing L, . et al. Inhibiting macrophage proliferation suppresses atherosclerotic plaque inflammation. Sci Adv. 2015. April; 1( 3): e1400223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tarin C, Carril M, Martin-Ventura JL, . et al. Targeted gold-coated iron oxide nanoparticles for CD163 detection in atherosclerosis by MRI. Sci Rep. 2015. November 30; 5: 17135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang J, Nie S, Martinez-Zaguilan R, Sennoune SR, Wang S.. Formulation, characteristics and antiatherogenic bioactivities of CD36-targeted epigallocatechin gallate (EGCG)-loaded nanoparticles. J Nutr Biochem. 2016. April; 30: 14– 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bagalkot V, Badgeley MA, Kampfrath T, Deiuliis JA, Rajagopalan S, Maiseyeu A.. Hybrid nanoparticles improve targeting to inflammatory macrophages through phagocytic signals. J Control Release. 2015. November 10; 217: 243– 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Parodi A, Quattrocchi N, van de Ven AL, . et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013. January; 8( 1): 61– 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fang J, Nakamura H, Maeda H.. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011. March 18; 63( 3): 136– 51. [DOI] [PubMed] [Google Scholar]
- 51. Lobatto ME, Fayad ZA, Silvera S, . et al. Multimodal clinical imaging to longitudinally assess a nanomedical anti-inflammatory treatment in experimental atherosclerosis. Mol Pharm. 2010. December 6; 7( 6): 2020– 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fredman G, Kamaly N, Spolitu S, . et al. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci Transl Med. 2015. February 18; 7( 275): 275ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee GY, Kim JH, Choi KY, . et al. Hyaluronic acid nanoparticles for active targeting atherosclerosis. Biomaterials. 2015; 53: 341– 8. [DOI] [PubMed] [Google Scholar]
- 54. Molinaro R, Corbo C, Martinez JO, . et al. Biomimetic proteolipid vesicles for targeting inflamed tissues. Nat Mater. 2016. September; 15( 9); 1037– 46. [DOI] [PMC free article] [PubMed] [Google Scholar]