

Significance
The immune system plays an essential role in the pathophysiology of major diseases such as atherosclerosis, diabetes, and cancer, which has inspired the development of numerous small molecules to modulate immune cells, intending to create immunotherapies for these diseases. Tissue- and cell-specific delivery of these small molecules is the key to transform these compounds to safe, potent immunotherapies. In this study, we present an in vivo nanoparticle screen approach that involves designing and evaluating a library of nanoparticles with distinct immune cell targeting specificity. This study carries out a systematic in vivo immune cell screening to create effective nanoparticle-based immunotherapy for modulating the pathological immune cells in atherosclerosis.
Keywords: nanomedicine, drug delivery, immunotherapy, molecular imaging, atherosclerosis
Abstract
Immunological complexity in atherosclerosis warrants targeted treatment of specific inflammatory cells that aggravate the disease. With the initiation of large phase III trials investigating immunomodulatory drugs for atherosclerosis, cardiovascular disease treatment enters a new era. We here propose a radically different approach: implementing and evaluating in vivo a combinatorial library of nanoparticles with distinct physiochemical properties and differential immune cell specificities. The library’s nanoparticles are based on endogenous high-density lipoprotein, which can preferentially deliver therapeutic compounds to pathological macrophages in atherosclerosis. Using the apolipoprotein E-deficient (Apoe−/−) mouse model of atherosclerosis, we quantitatively evaluated the library’s immune cell specificity by combining immunological techniques and in vivo positron emission tomography imaging. Based on this screen, we formulated a liver X receptor agonist (GW3965) and abolished its liver toxicity while still preserving its therapeutic function. Screening the immune cell specificity of nanoparticles can be used to develop tailored therapies for atherosclerosis and other inflammatory diseases.
Research in the past decades has revealed the immune system’s central role in the pathophysiology of cancer (1), diabetes (2), and atherosclerosis (3, 4). Because macrophages drive pathological progression of these diseases, immunomodulatory small-molecule compounds modulating macrophage function are promising therapeutic candidates for treating these maladies. However, these compounds' lack of cellular specificity necessitates a strategy for targeted delivery to harmful immune cells without negatively affecting beneficial immune cells. Despite numerous studies of nanoparticle-based delivery, rational attempts to screen meticulously designed nanoparticles for immune cell specificity in vivo have never been reported.
We created a combinatorial library of hybrid lipoprotein-inspired nanoparticles with distinct physiochemical properties (particularly size and chemical composition) with differential immune cell specificity. We then chose atherosclerosis, a lipid-driven inflammatory process of the large arteries, as a model disease to evaluate our nanoparticle library. Atherosclerosis accounts for the majority of cardiovascular deaths worldwide (5), and macrophages are the major immune cells to drive the pathological inflammation and the progression of atherosclerotic plaques (6, 7). These plaques, which are present throughout the vasculature, have a complicated cellular composition (8). To improve the therapeutic index of small-molecule immunomodulatory compounds, plaque macrophage-specific delivery, with minimal delivery to nonpathological cells in healthy tissues, is essential.
We used the apolipoprotein E-deficient (Apoe−/−) mouse model of atherosclerosis to evaluate our nanoparticle library, because it accurately recapitulates some important immunological aspects of human atherosclerosis (9). Using a combination of optical methods, immunological techniques, and in vivo positron emission tomography (PET) imaging, we carefully selected candidate nanoparticles from the library for subsequent atherosclerosis drug delivery studies. As a proof of concept, we incorporated the liver X receptor agonist GW3965, a therapeutic compound that did not reach clinical application due to its serious adverse effects on the liver (10, 11), in two nanoparticles—one with favorable organ distribution and immune cell specificity and one without. In Apoe−/− mice with advanced disease, the favorable nanoparticle from the library screen was shown to abolish GW3965's liver toxicity while remaining effective on atherosclerotic plaque macrophages.
Results
Study Design.
We created a combinatorial library comprising 15 high-density lipoprotein-mimicking nanoparticles and two extensively studied nanoparticles, a PEGylated micellar and a long-circulating liposomal nanoparticle. We then combined in vitro assays and in vivo experiments in atherosclerotic Apoe−/− mice to study the nanoparticle library’s biological behavior by using near-infrared fluorescence imaging (NIRF), flow cytometry, immunofluorescence, and radiolabeling. Based on the results of this library screening, we formulated two GW3965-loaded HDL nanoparticles with distinctly different immune cell specificity and organ distribution. Finally, we quantitatively studied the pharmacokinetics, immune cell specificity, liver toxicity, and therapeutic effects of these drug-loaded nanoparticles in Apoe−/− mice with advanced atherosclerosis (Fig. 1A).
Fig. 1.

Study design and in vitro characterization of the nanoparticle library. (A) In the in vivo immune cell screen study (left), we first created a combinatorial nanoparticle library and then evaluated the library in atherosclerotic Apoe−/− mice by using blood half-life determination, NIRF, and flow cytometry. The library screening data lead to rational design of one GW3965-loaded nanoparticle with favorable characteristics and one without. In the second study, the two nanoparticles were radio- and fluorescence-labeled, and they were quantitatively and therapeutically evaluated by using PET-CT imaging, mRNA profiling, flow cytometry, and liver toxicity assays (right). (B) Representative high-magnification TEM images of negatively stained nanoparticles. Low-magnification images and a discussion of the different structures are presented in Fig. S1. (Scale bar, 50 nm.) (C) The size of nanoparticles as measured by dynamic laser scattering (DLS). (D) Cholesterol efflux capacity of the nanoparticles in primary macrophages normalized to natural human HDL (n = 6). Error bars are SDs. Color-coded bar at the bottom shows the relative rank of each nanoparticle, with the red indicating high cholesterol efflux efficiency and the blue indicating low efficiency.
HDL Nanoparticle Library.
Previous studies indicate that the size, phospholipid composition, ratio of phospholipid to apolipoprotein A-1 (APOA1), or the inclusion of payloads can affect HDL-mimicking nanoparticles’ in vivo performance (12–14). In our current study, we created a library containing HDL-mimicking nanoparticles that differ in size, shape, composition, and payload, all of which have been reported to affect nanoparticles’ in vivo targeting efficiency (15). To fine-tune nanoparticle size and morphology, we added either triglyceride or polymers [poly-lactic-co-glycolic acid (PLGA) or polylactic acid (PLA)] to the HDL core (Table S1); this allowed us to modulate nanoparticle size from about 10 nm (NP1, NP2, NP3, and NP9) or 30 nm (NP6, NP7, NP10, and NP11) to over 100 nm (NP12) (Fig. 1 B and C). We observed that the inclusion of a core component, namely triglycerides or polymers, results in a nanoparticle shape change from discoidal to spherical, as can be clearly appreciated when comparing NP5 to NP7 and NP15. In addition to size and shape, we also varied phospholipid composition (Table S1). Because oxidization greatly affects HDL function (16), we sought to test if this modification also changed the HDL-mimicking nanoparticles’ drug delivery capability. We oxidized the phospholipids and APOA1 of NP1 to produce NP2 (Fig. S1B). The nanoparticle sizes were measured by dynamic light scattering, and their morphologies were visualized by transmission electron microscopy (TEM) (Fig. 1 B and C). Micellar (17) and liposomal (18) nanoparticles are established lipid-based platforms and served as references in this study.
Table S1.
Chemical composition of the nanoparticle library
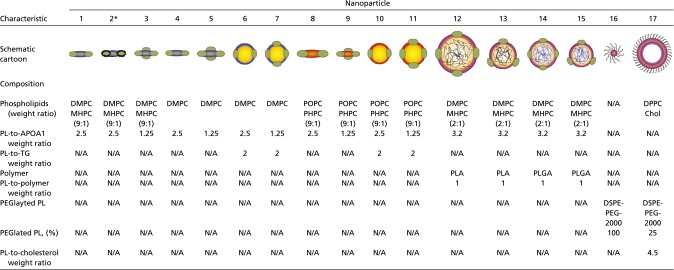 |
Chol, cholesterol; N/A, not available; PL, phospholipid; TG, triglyceride.
APOA1 of nanoparticle 2 were oxidized by a chemical process.
Fig. S1.

Chemical characterization of the nanoparticle library. (A) Low-magnification TEM images of the 17 nanoparticles, as well as the two therapeutic nanoparticles—Rx-HDL and Rx-PLGA-HDL. NP1-5 have a discoidal shape, as is typical for HDL nanoparticles reconstituted from phospholipids and APOA1. POPC-based nanoparticles require a higher amount of APOA1 to force disk formation, as can be observed when comparing NP8 and NP9. The inclusion of triglycerides as a core material results in nanoparticles with ∼20 nm (NP6, NP7, NP10, and NP11), whereas the inclusion of PLGA or PLA polymers allows fine-tuning of nanoparticle sizes, ranging from ∼40 nm for NP15 to over 100 nm for NP12. (Scale bar, 100 nm.) (B) Mass spectrometry of oxidized nanoparticle. An HDL-mimicking nanoparticle made mainly of DMPC and APOA1 (NP1) was oxidized to generate NP2, which was analyzed by mass spectrometry. Mass spectrometry shows the increase of APOA1 M.W. by about 146.7 on average, indicating the oxidization of APOA1 (Ox-APOA1).
HDL Nanoparticle Library’s Cholesterol Efflux Efficiency.
Natural HDL facilitates reverse cholesterol transport, the intrinsic mechanism that removes cholesterol from macrophages in atherosclerotic plaques and protects against the atherosclerosis (19, 20). Cholesterol efflux capacity indicates the nanoparticles’ biological similarity to native HDL. Therefore, we measured the 17 nanoparticles’ ability to induce cholesterol efflux from cholesterol-laden primary macrophages. We found 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)-based nanoparticles (NP8, NP9, NP10, and NP11) to be the most efficient in extracting cholesterol from macrophages; in contrast, polymer-core HDL nanoparticles produced the least cholesterol efflux (NP12, NP13, NP14, and NP15). Liposomal and micellar nanoparticles, without APOA1 on their surface to bind the cholesterol efflux receptors Abca1 and Abcg1, performed similarly to polymer-core HDL nanoparticles (Fig. 1D). These results demonstrate the essential role of APOA1, as well as the impact of phospholipid and core composition, in promoting cholesterol efflux. It has been proposed that APOA1 changes conformation when interacting with its specific receptors to extract cholesterol (19). The artificial polymeric cores of NP12, NP13, NP14, and NP15 might limit conformational flexibility of APOA1, leading to impaired cholesterol efflux. On the other hand, POPC-based HDL nanoparticles display less rigidity, are more similar to natural HDL, and therefore result in more efficient cholesterol efflux.
Nanoparticles’ Physiochemical Properties Affect Their in Vivo Behavior.
We i.v. injected the library’s 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR)-labeled nanoparticles into atherosclerotic Apoe−/− mice. We found that NP1 and NP10, with diameters between 7 nm and 30 nm, exhibited the longest blood half-lives of 5.0 h and 6.3 h, respectively. NP12 and NP14, with diameters larger than 70 nm, had the shortest blood half-lives of 0.71 h and 0.67 h, respectively (Fig. 2A). The difference between the longest and the shortest blood half-life was almost 10-fold (Table S2).
Fig. 2.

In vivo evaluation of the nanoparticle library. (A) Relative nanoparticle plasma concentration in Apoe−/− mice that were fed a 12-wk high-cholesterol diet. The values were derived from the near-infrared dye DiR incorporated in the nanoparticles. Heat map below the graphs ranks the blood half-lives, with the red indicating a long and the blue a short blood half-life (n = 5 per nanoparticle). The values of blood half-lives are provided in Table S2. (B) Representative near-infrared fluorescence images of nanoparticle accumulation in aorta, liver, and spleen. The heat map below the aorta images ranks the mean total fluorescent tissue signal, the heat map below the liver images ranks the total aorta-to-liver signal, and the heat map below the spleen images ranks the mean aorta-to-spleen accumulation (Ao-to-sp) ratio, with the red indicating a high and the blue a low ratio (n = 5 for each nanoparticle). Bar graphs are provided in Fig. S2. (C) Blood half-lives of four selected nanoparticles radiolabeled with 89Zr (n = 3 per nanoparticle) were 7.0 h for 89Zr-NP10, 5.7 h for 89Zr-NP14, 7.1 h for 89Zr-NP15, and 6.6 h for 89Zr-NP17. Blood radioactivity (percentage injected dose per gram of tissue) of all time points is normalized to that of the first time point—2 min after injection. (D) Representative autoradiography images of aortas, livers, and spleens 24 h after nanoparticle injection. Dashed windows indicate the aortic root and arch area analyzed in E. (E) Nanoparticle accumulation in aortic roots and arches as measured by percentage of injected dose (%ID) (n = 3 per nanoparticle). (F) Relative accumulation of nanoparticles between aortas and liver or spleen. Arbitrary units (A.U.) were defined by the aortic accumulation (percentage injected dose per gram of tissue) divided by the hepatic or splenic accumulation (percentage injected dose per gram of tissue). Error bars are SDs. Statistics was calculated with nonparametric two-tailed Student’s t test. N.S., not statistically significant; *P < 0.05; **P < 0.01.
Table S2.
Blood half-lives of nanoparticle library
| Characteristic | Nanoparticle | ||||||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | |
| Blood half-life, h | 5.01 | 2.99 | 2.49 | 2.53 | 3.47 | 0.97 | 0.92 | 1.73 | 1.70 | 6.26 | 3.99 | 0.71 | 5.00 | 0.67 | 0.98 | 3.24 | 0.78 |
Blood half-life values were determined from five mice per nanoparticle.
Using NIRF, we investigated nanoparticle accumulation in the heart, aorta, lung, liver, spleen, kidney, brain, and muscle 24 h after i.v. administration (Fig. 2B and Fig. S2A). Among all nanoparticles, liver accumulation was generally the highest, followed by spleen, kidney, and lung accumulation (Fig. S2B). Because the aorta is the primary target tissue whereas the liver and spleen are clearance organs, we determined accumulation in the aorta relative to these two organs (21). Our measurements showed a 3.4-fold difference between the highest and lowest aorta-to-liver accumulation ratios (NP5 vs. NP12, P < 0.01, Fig. S2D), and a 4.7-fold difference between the aorta-to-spleen accumulation ratios (NP5 vs. NP12, P < 0.01, Fig. S2E). For the nanoparticles’ relative performance, see Fig. 2B. Distribution of DiR-labeled nanoparticle in certain tissues is difficult to quantify in vivo due to the limited penetrating depth of light and varying absorbance rates among tissues (22). Radiolabeled nanoparticles’ biodistribution and pharmacokinetics can be quantitatively measured. From our initial optical imaging screen, we selected NP10, NP14, NP15, and NP17 to be labeled with Zr through the hydrophobic chelator deferoxamine (DFO)-C34, which serves as a surrogate for hydrophobic payloads. In line with the optical imaging results, we observed that the smaller nanoparticles (NP10 and NP15) had longer blood half-lives than the larger ones (NP14 and NP17, Fig. 2C). We also observed that the HDL-based nanoparticles (NP10, NP14, and NP15), particularly NP10, accumulated more efficiently in atherosclerotic plaques than the liposomal nanoparticle (Fig. 2 D and E). Overall, NP10 displayed the most favorable performance among the four selected nanoparticles (Fig. 2 C–F).
Fig. S2.

Representative and quantitative NIRF data. (A) Representative NIRF images of key organs. Quantification of total signal of (B) all organs and (C) aortas. (D) The aortic-to-liver accumulation ratio is calculated by dividing the total signal of aorta with that of liver. (E) The aorta-to-spleen accumulation ratio is calculated by dividing the total signal of aroma with that of spleen. For B−E, n = 5 for per nanoparticle, and the error bars are SEM.
Distinct Immune Cell Targeting Patterns Within the Nanoparticle Library.
Macrophages and monocytes are the key immune cells that drive atherosclerosis progression (4). In Apoe−/− atherosclerotic mice, these cells mainly reside in atherosclerotic plaques, spleen, blood, and bone marrow (4). Using a robust flow cytometry procedure adapted from previous studies (23, 24), we were able to identify macrophages, monocytes, and nonmyeloid immune cells (Lin+) in the aortas; macrophages, Ly-6Chi monocytes, dendritic cells (DCs), and neutrophils in the spleens; and Ly-6Chi monocytes, Ly-6Clo monocytes, DCs, and neutrophils in the blood (Fig. 3A).
Fig. 3.

Nanoparticle immune cell specificity. (A) The flow cytometry gating procedures to identify relevant immune cells in aorta, spleen, and blood. Black histograms on the right show representative signal distribution of different immune cells in the mice injected with nanoparticles compared with the cells from control animals injected with PBS (gray histogram on the left in each graph). (B) Quantification of mean fluorescence intensity (MFI) of each immune cell type in different tissues; n = 5 for each nanoparticle, and error bars are SEMs. (C) Heat map ranks targeting efficiency in key immune cells (aortic macrophages, spleen macrophages, and blood Ly-6Chi monocytes), with red indicating a high and blue indicating a low MFI in the first three rows. The last row shows the aortic-to-splenic macrophage MFI ratio (Ao-to-sp MΦ), and its quantitative values are provided in Fig. S3A.
In the aortas, all HDL-mimicking nanoparticles efficiently targeted macrophages and monocytes. The difference between the highest and lowest nanoparticle accumulations was 5.7-fold in macrophages (NP3 vs. NP17, P < 0.01) and 2.7-fold in monocytes (NP3 vs. NP7, P < 0.001; Fig. 3B). In the spleens, all nanoparticles had the highest accumulation in macrophages, which do the bulk of nanoparticle clearance, with a 3.8-fold difference between the highest and lowest accumulation levels (NP12 vs. NP17, P < 0.01, Fig. 3B). In the blood, DCs and Ly-6Chi monocytes displayed the highest nanoparticle association, with a 3.8-fold difference between the highest and lowest association levels (NP10 vs. NP5, P < 0.01) in DCs and a 3.79-fold difference in Ly-6Chi monocytes (NP3 vs. NP14, P < 0.0001, Fig. 3B). In addition, the nanoparticles were far less effective in targeting Ly-6Clo monocytes, the patrolling monocytes in the blood, compared with Ly-6Chi monocytes (Fig. 3B).
Although aortic macrophages are the main target of immunomodulatory nanoparticles, splenic macrophages, which clear nanoparticles from the blood and reduce their bioavailability to aortic macrophages (25), need to be avoided. We evaluated the ratios of nanoparticle accumulation in aortic macrophages versus splenic macrophages, and we found a 3.8-fold difference between the highest and lowest aortic-to-splenic ratios (NP16 vs. NP12, P < 0.0001, Fig. S3A). This differential targeting specificity to atherosclerotic macrophages was confirmed by immunofluorescence in the aortic roots (Fig. S3B). Altogether, the flow cytometry data reveal that the distinct physiochemical nanoparticle properties within the library lead to drastically different immune cell targeting patterns (Fig. 3C).
Fig. S3.

Macrophage targeting efficiency of the nanoparticle library. (A) Aortic-to-splenic (Ao-to-sp) macrophage MFI (MΦ) ratio is calculated by dividing the MFI of aortic macrophages by that of splenic macrophages (n = 5 per nanoparticle). Error bars are SEM. (B) Representative immunostaining images of aortic roots from the animals injected with the nanoparticles. CD68 was used to identify macrophages in the frozen section. Nanoparticles were identified by detecting their fluorescent label DiR.
GW3965-Loaded Nanoparticle Development.
Liver X receptor (LXR) agonists promote cholesterol efflux from macrophages in atherosclerotic plaques, and they have been proposed as novel immunomodulatory drugs for the disease (26). However, most experimental LXR agonists fail clinical translation or early-stage clinical trials due to poor safety profiles. For example, GW3965, an effective LXR agonist promoting cholesterol efflux from atherosclerotic macrophages (27, 28), did not reach the clinical phase, because of its liver toxicity in hamsters and monkeys (10), as well as in human hepatocytes (11).
In our nanoparticle library studies, NP10 was found to have high cholesterol efflux promotion efficiency, a long blood half-life, high relative aorta-to-liver accumulation, and a high relative aortic-to-splenic macrophage association ratio (Fig. S4A). These features make NP10 a promising candidate for avoiding GW3965’s liver toxicity and enhancing its efficacy on atherosclerotic plaque macrophages. Therefore, we replaced NP10’s hydrophobic triglyceride cargo with hydrophobic GW3965 and created a GW3965-loaded nanoparticle (Rx-HDL) that was morphologically similar but not identical, due to the different nanoparticle composition, to NP10 in size (∼30 nm), phospholipid composition (POPC-dominant), and morphology (Fig. S4B). Further, we identified NP14 as an unfavorable nanoparticle for GW3965 delivery due to the nanoparticle’s poor cholesterol efflux efficiency, short blood half-life, and low relative aorta-to-liver accumulation (Fig. S4A). By loading GW3965 into the PLGA matrix of NP14, we created a PLGA-core GW3965-loaded nanoparticle (Rx-PLGA-HDL) with similar size, phospholipid composition, and morphology to NP14 (Fig. S4B). Notably, the size and cholesterol efflux capability of the two drug-loaded nanoparticles were drastically different (Fig. S4 B and C).
Fig. S4.

In vivo evaluation of drug-loaded nanoparticles. (A) The summary of the performance of the nanoparticle library in the immunological screening study, with the red showing the favorable and the blue showing the unfavorable performance according to the specific limitations of GW3965. High-rank nanoparticles show high cholesterol efflux capacity, long blood half-life, high relative aorta-to-liver (Ao-to-li) accumulation ration, and high relative aortic-to-splenic macrophage MFI ratio. NP10 was found to have the highest overall rank. (B) Representative TEM images of negatively stained drug-loaded nanoparticles. Overview of the images can be found in Fig. S1A. (Scale bar, 50 nm.) (C) Relative cholesterol efflux capacity of drug-loaded nanoparticles (n = 6). Nonparametric Student’s t test was used for statistics. (D) Radioactive HPLC eluting profiles of drug-loaded nanoparticles. The black profile is the UV absorbance, which indicates the nanoparticle itself. The blue and red profiles are the 89Zr radioactive signal. A 2.5-min delay between UV and radioactive profiles is added on purpose to better present the profiles. (E) Extensive biodistribution of radio-labeled and GW3965-loaded nanoparticles in relevant organs (n = 5 per nanoparticle). (F) Representative flow cytometry histograms of relevant immune cells in different tissues. All error bars are SEM.
Quantitative Evaluation of GW3965-Loaded Nanoparticles.
Nanoparticles that are 89Zr-labeled can be quantitatively characterized by in vivo PET imaging as well as by ex vivo radioactivity counting (29). To accurately understand the in vivo performance of the two nanoparticles, we loaded the hydrophobic 89Zr-DFO-C34 into Rx-HDL and Rx-PLGA-HDL (Fig. 4A), and radioactive high-performance chromatography showed both Rx-HDL and Rx-PLGA-HDL to be efficiently radiolabeled (Fig. S4D).
Fig. 4.

In vivo quantitative evaluation of GW3965-loaded nanoparticles. (A) Schematic depictions of small Rx-HDL plaque macrophage-targeting and large polymer-hybrid Rx-PLGA-HDL nanoparticles. The two nanoparticles were either radiolabeled with 89Zr or labeled with the near-infrared fluorescent dye DiR. (B) Blood half-lives were determined in three mice per nanoparticle. (C) Representative PET images of mice that received either the small or large nanoparticle at 0.5 h and 24 h after i.v. administration. The 3D-rendered images are provided as Movies S1–S4 (n = 5 per nanoparticle). (D) Quantification of radioactivity in the heart, liver, and spleen. (E) Representative autoradiographic images of key organs. Full biodistribution of all organs is provided in Fig. S4E. (F) Representative histograms of selected immune cell targeting specificity in the aorta, spleen, and blood. (G) MFI quantification of relevant immune cells in the tissues (n = 4 per nanoparticle). Error bars are SEM. Statistics was calculated with nonparametric two-tailed Student’s t test. *P < 0.05; **P < 0.01.
In Apoe−/− atherosclerotic mice, Rx-HDL circulated in the blood much longer (weighted t1/2 = 10.5 h, n = 3) than Rx-PLGA-HDL (weighted t1/2 = 5.0 h, n = 3, Fig. 4B) or its precursor NP10 (t1/2 = 7.0 h, Fig. 2C), demonstrating its favorable features. We then used PET-computed tomography hybrid imaging (PET-CT) to measure the nanoparticle dynamic accumulation in the cardiac blood pool, the liver, and the spleen 30 min and 24 h after i.v. administration (n = 5 per nanoparticle, Fig. 4C; also see Movies S1–S4). After 30 min, Rx-HDL had higher accumulation in the cardiac blood pool (29.9 maximum percentage injected dose per gram of tissue (Max %ID/g) vs. 24.5 Max %ID/g, P < 0.05) but lower accumulation in the liver than Rx-PLGA-HDL (21.7 Max %ID/g vs. 29.7 Max %ID/g, P < 0.05). After 24 h, Rx-HDL liver accumulation was still 36% lower than Rx-PLGA-HDL (26.5 Max %ID/g vs. 41.7 Max %ID/g, P < 0.05, Fig. 4 C and D). Autoradiography revealed that both nanoparticles displayed patchy aorta accumulation, in accordance with the heterogeneous distribution of atherosclerotic plaques in this tissue (30) (Fig. 4E). Additionally, autoradiography confirmed the highest nanoparticle accumulation to be in the liver and spleen (Fig. 4E), a result that was corroborated by an extensive biodistribution analysis (Fig. S4E).
Having labeled both nanoparticles with DiR (Fig. 4A), we used our flow cytometry protocol (Fig. 3A) to quantify immune cell targeting specificity (Fig. S4F). Rx-HDL predominantly targeted macrophages in the aortas, and its accumulation was twofold higher than Rx-PLGA-HDL (P < 0.01). In the spleen, Rx-HDL had 33% less accumulation in splenic macrophages than Rx-PLGA-HDL (P < 0.05). In the blood, Rx-HDL targeted Ly-6Chi monocytes 2.4-fold more efficiently than Rx-PLGA-HDL (P < 0.01, Fig. 4 F and G). Collectively, these data show that, compared with Rx-PLGA-HDL, Rx-HDL has a longer blood half-life, lower accumulation in the liver, higher accumulation in atherosclerotic plaque macrophages, and lower accumulation in splenic macrophages.
Nanoparticle Abolishes Liver Toxicity and Preserves Efficacy of GW3965.
To test if Rx-HDL’s optimal in vivo performance reduced GW3965s liver toxicity, we gave four i.v. injections (one injection every 2 d, at a dose of 10 mg/kg GW3865) of Rx-HDL, its vehicle control (HDL), Rx-PLGA-HDL, its vehicle control (PLGA-HDL), and PBS to Apoe−/− atherosclerotic mice (n = 12 per group).
In the liver, Rx-PLGA-HDL increased the expression of two of the three major GW3965-related toxicity genes, and Rx-HDL increased the expression of one gene (Fig. 5A). Importantly, we measured hepatic triglyceride and cholesterol levels, which are the major biomarkers of GW3965-induced hepatic steatosis (10). The Rx-PLGA-HDL group had 35.4% more hepatic triglyceride than its control vehicle PLGA-HDL group (P = 0.014) and 21% more than the PBS group (P = 0.12). For hepatic cholesterol, the Rx-PLGA-HDL group had 31% higher levels than the PLGA-HDL group (P = 0.018) and 17% higher levels than the PBS group (P = 0.014). These results suggest that the high liver accumulation of Rx-PLGA-HDL caused severe liver toxicity (Fig. 5 B and C). On the other hand, the Rx-HDL group had 26.5% lower hepatic triglyceride levels than its vehicle HDL control group (P = 0.06) and 22.7% lower levels than the PBS group (P = 0.076). The Rx-HDL group also had 20% lower hepatic cholesterol levels than the HDL group (P = 0.06) and 33.3% lower levels than the PBS group (P = 0.00043). A recent study suggested that high HDL levels are associated with a lower degree of steatosis, which might explain the reduced hepatic triglyceride and cholesterol levels in mice treated with vehicle HDL nanoparticles (31). Furthermore, GW3965 has been reported to increase HDL levels in mice (32), likely explaining the additional hepatic benefits in Rx-HDL-treated mice. Most importantly, compared with the Rx-PLGA-HDL group, the Rx-HDL group had 36.1% lower hepatic triglyceride (P = 0.00083) and 43.0% lower hepatic cholesterol levels (P < 0.0001). These results demonstrate that the two GW3965-loaded nanoparticles’ distinct liver accumulations resulted in differential liver toxicity profiles (Fig. 5 A–C).
Fig. 5.

Evaluation of the toxicity and efficacy of GW3965 (Rx)-loaded nanoparticles. In A−E, Apoe−/− mice received four i.v. administrations of nanoparticles or PBS on every other day (n = 12 per group). (A) The mRNA expression levels of three GW3965 target genes in liver homogenates. (B) Triglyceride and (C) cholesterol levels in the liver, the primary organ suffering from the toxic effects of GW3965. (D) The mRNA expression levels of five GW3965 target genes in aortic macrophages. (E) The mRNA expression levels of 19 inflammation-related genes in aortic macrophages. All gene expression was normalized to housekeeping gene Hprt1. The bar graph presentation of the heat maps is in Fig. S5 (n = 12 per group). In F−H, Apoe−/− mice (n = 10 in PBS; n = 9 in Rx-HDL) received 12 i.v. injections in 6 wk. Lipid levels of aortic cells were analyzed by flow cytometry. Cellular lipid levels were calculated by multiplying the mean fluorescence intensity of a cell type by the number of the cells per aorta (AU = MFI × number of cells). The total lipid levels of aortic (F) macrophages, (G) monocytes, or (H) nonimmune CD45− cells per aorta in the mice are presented here. Gating procedure is provided in Fig. S6 A and B. Error bars in all graphs are SEM. Statistics was calculated with nonparametric two-tailed Student’s t test.
To measure the treatments’ efficacies on aortic macrophages, we isolated the cells from aortic roots by laser capture microdissection and measured their mRNA expression levels by quantitative real-time PCR (qPCR). We found that Rx-HDL increased expression of five GW3965 target genes compared with the vehicle HDL control, whereas Rx-PLGA-HDL increased only four genes compared with the vehicle PLGA-HDL control (Fig. 5D). Furthermore, Rx-HDL produced elevated expression of the five target genes compared with Rx-PLGA-HDL (Fig. S5 A and B). These results suggest successful GW3965 delivery to macrophages in atherosclerotic plaques. We did not observe clear effects of either nanoparticle on genes related to macrophage inflammation, as the number of genes with increased expression was almost equal to the number with decreased expression (Fig. 5E and Fig. S5 A and B). Similarly, the macrophage levels in aortic roots were the same among all groups (Fig. S5 D and E).
Fig. S5.

Efficacy of drug-loaded nanoparticles on aortic macrophages. (A) Quantification of mRNA expression of the selected genes in atherosclerotic plaque macrophages. The relative expression of a gene is calculated by following these formulas: relative gene expression of gene X of Rx-HDL group = (expression of gene X in the Rx-HDL group/expression of gene X in the HDL group); relative gene expression of gene X of Rx-PLGA-HDL group = (expression of gene X in the Rx-PLGA-HDL group/expression of gene X in the PLGA-HDL group). (B) Relative gene expression normalized to average expression of both Rx-PLGA-HDL and Rx-HDL groups by following a formula: gene X expression in one group = (expression of gene X in the given group/average expression of gene X in both Rx-PLGA-HDL and Rx-HDL groups). (C) Relative gene expression normalized to PBS group by following the following formula: gene X expression of a given group = (expression of gene X in the given group/expression of gene X in the PBS group). (D) Representative CD68 immunostaining of aortic roots. CD68 was used as the macrophage marker in this experiment. (Left) The original image and (Right) the mask generated by a MATLAB procedure. The dark pixels in the mask were calculated and used to calculate the CD68 positive area in the original immunostaining image. (E) Quantification of macrophage levels in all treatment group. n = 12 per group (A–C and E). Nonparametric Student’s t test was used to calculate statistics, and all error bars are SEM.
Based on the favorable properties of Rx-HDL, we further evaluated its therapeutic effects in a 6-wk, long-term treatment regimen focusing on cellular lipid levels in aortas. Working from a BODIPY (4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene)-based flow cytometry protocol (33), we developed a procedure to quantify the cellular lipid levels of macrophages, monocytes, and CD45 negative nonimmune cells (CD45−) in the aortas (Fig. S6). Importantly, we found that the 6-wk treatment of Rx-HDL (10 mg/kg GW3965, two i.v. injections per week) resulted in 28% less total lipid in aortic macrophages (P = 0.224; Fig. 5F). Further, the treatment reduced total lipid levels in monocytes by 43.0% (P = 0.011, Fig. 5G) and in all CD45− nonimmune cells by 40.0% (P = 0.021, Fig. 5H). Furthermore, no therapeutic effects were observed when the compound was given orally with the same 6-wk treatment regimen (10 mg/kg GW3965, two gavages per week) compared with the placebo-treated group (Fig. S6 C–H). Consistent with the liver toxicity results after the short-term Rx-HDL treatment, blood cholesterol and triglyceride levels were similar to those in mice receiving PBS treatment (Fig. S7 A and B). In addition, the long-term treatment of Rx-HDL did not cause any observable toxicity in the liver, kidney, heart, and blood cells (Fig. S7 C–L). Taken together, the results show that long-term Rx-HDL treatment produces significant therapeutic benefits without causing toxicity in major organs, most notably the liver, whereas long-term oral treatment at the same dose did not produce any therapeutic benefits.
Fig. S6.

Cellular lipid measurement flow cytometry protocol and the efficacy evaluation of 6-wk treatment. Aortic cells from 1-y-old C57BL/6 wild-type (WT) mice (n = 2) and Apoe−/− mice (n = 2) with 10 mo of high-cholesterol diet were stained with BODIPY. Intracellular BODIPY signal was measured by a flow cytometry procedure used in the previous experiments (Fig. 3A). (A) Representative gating procedure shows the identification of CD45 negative (CD45−) nonimmune cells, macrophages, and monocytes in the aortas. (B) Representative BODIPY signal levels in (Left) nonimmune cells, (Middle) macrophages, and (Right) monocytes are shown in the histogram graphs. Gray histograms represent the non-BODIPY-stained cells; green histograms represent cellular BODIPY levels of the cells from wild-type mice; red histograms (Ctrl) represent cellular BODIPY levels of the cells from Apoe−/− mice. Cells from Apoe−/− mice show much higher BODIPY levels than those from wild-type mice, due to their higher cellular lipid levels than those in wild-type mice. (C−E) Apoe−/− mice received oral treatment of either GW3965 (Oral Rx, n = 6) or PBS (Oral PBS, n = 5) twice per week for 6 wk. Cellular lipid levels were calculated by multiplying the mean fluorescence intensity of a cell type with the number of the cells per aorta (AU = MFI X number of cells). Cellular lipids levels of the two groups were compared in aortic macrophages (C), aortic monocytes (D), and aortic nonimmune cells (E). (F−H) Two batches of female Apoe−/− mice with the same age and length of high-cholesterol diet were given either i.v. treatments (I.V. PBS or Rx-HDL; Fig. 5 F–H) or oral treatments (Oral Rx or Oral PBS). Due to the batch and measurement difference, the cellular lipid levels of aortic cell populations treated with either Rx-HDL or Oral Rx were normalized to those from mice treated with either i.v. PBS or oral PBS, respectively. Cellular lipid levels from the two batches of treated mice were compared with their corresponding control-treated mice in aortic macrophages (F), aortic monocytes (G), and aortic nonimmune cells (H). Error bars in all graphs are SEM. Statistics was calculated with nonparametric two-tailed Student’s t test.
Fig. S7.

A long-term Rx-HDL treatment does not cause toxic effects. Mice were treated with i.v. PBS (I.V. PBS, n = 10), i.v. Rx-HDL (Rx-HDL, n = 9), oral GW3965 (Oral Rx, n = 6), or oral PBS (Oral PBS, n = 5) two times per week for 6 wk, and key toxicity markers were measured. (A) Blood cholesterol, (B) triglyceride, and (C) glucose levels are metabolism markers. (D) Gamma-glutamyl transaminase, (E) alanine transaminase, and (F) aspartate transaminase are liver damage markers. (G) Creatine kinase is a cardiac toxicity marker. (H) Blood urea nitrogen is a kidney damage marker. The (I) red blood cell counts, (J) hemoglobin levels, (K) white blood cell counts, and (L) percentage of monocytes in blood white cells are shown here, too. No statistical significance was found in any measurements above. Nonparametric two-tailed Student’s t test was used to calculate statistics between treatment and PBS control, and all error bars are SD.
In summary, we developed a rational library screen strategy to identify nanoparticles with favorable immune cell specificity and biodistribution in an atherosclerosis mouse model. On the basis of this nanoparticle screen, we optimized GW3965 delivery to plaque macrophages and, while preserving its efficacy on atherosclerotic plaques (Fig. 5 D–H), abolished GW3965 liver toxicity (Fig. 5 A–C and Fig. S7), a well-known adverse effect of LXR agonists.
Discussion
By fine-tuning the components and synthesis procedures, we created a combinatorial library of 17 nanoparticles with distinct composition, size, and morphology. These distinct physiochemical properties resulted in an approximate sixfold difference in promoting cholesterol efflux from macrophages, 10-fold difference among blood half-lives, 3.4-fold difference in relative aorta-to-liver accumulation, and 3.8-fold difference in relative aortic-to-splenic macrophage accumulation. From this library screening, we identified the favorable lipid composition (POPC-dominant), pharmacokinetics (long blood half-life), size (around 30 nm), and morphology (spherical) to achieve optimal plaque macrophage-specific drug delivery. We hypothesize that the combination of long blood half-life and small size allow efficient and prolonged atherosclerotic plaque penetration and subsequent macrophage accumulation. A favorable nanoparticle lipid composition and morphology increases stability and promotes delivery of the encapsulated small molecules to the targeted cells, as suggested by our recent study (34). In a proof-of-concept application, we used these guidelines to identify two nanoparticles from the library as the most and least favorable nanoparticles for delivering the liver-toxic compound GW3965. Although the unfavorable nanoparticle Rx-PLGA-HDL caused severe GW3965-induced liver toxicity, the favorable nanoparticle Rx-HDL did not cause observable liver toxicity in treated animals but did preserve the compound’s therapeutic efficacy on the atherosclerotic plaques.
Despite the clinical introduction of antibody immunotherapies for atherosclerosis (35), such biological drugs can only modulate a limited number of extracellular targets, such as PCSK9 (36, 37) or receptors on cell surface (38). Intracellular entities present many more immunological targets that can be effectively controlled by immunomodulatory small molecules. Most experimental small molecules for atherosclerosis, however, failed clinical trials due to their unfavorable toxicity profiles, generally caused by high accumulation in nontargeted tissues or in nontargeted cells within the targeted tissues (39). To convert these immunomodulatory small molecules into precision medicines for atherosclerosis, organ-specific and cell-specific delivery is highly desirable.
Our approach allows the creation and immunological screening of a combinatorial nanoparticle library with distinct organ biodistribution and immune cell targeting specificities. By using PET imaging, NIRF, and flow cytometry to generate an extensive database detailing the in vivo performance of the library nanoparticles, we were able to rationally design a strategy that avoids the specific limitations of an immunomodulatory compound. Through this process, we successfully converted a liver-toxic immunomodulatory compound into a precision nanomedicine for atherosclerotic plaque macrophage treatment.
A polymer-core nanoparticle (NP13) in the library has similarly optimal performance to the best-performing NP10 (Fig. S4A). Polymer-based nanoparticles have been formulated with chemical compounds (40), peptides (41), and nucleic acids (42). This variety suggests that NP13 may be able to deliver a wide range of therapeutic molecules. In addition, a few nanoparticles (NP3, NP9, and NP10) in the library show high targeting specificity to Ly-6Chi monocytes and DCs, which are attractive targets in certain types of cancer (43), asthma (44), and diabetes (45). It would be interesting to apply the same library screening strategy to develop nanoparticle-based specific drug delivery to these immune cells in relevant diseases.
This nanoparticle library screening strategy in immune cells allowed us to improve the therapeutic index of an immunomodulatory molecule that causes hepatic toxicity and has failed clinical translation. Our precision nanomedicine strategy is radically different from the current clinical therapeutics as well as those in experimental phases. Moreover, the approach’s potential to deliver various compounds preferentially to other immune cells would expand its application to numerous immunologically implicated diseases, such as myocardial infarction, diabetes, and cancer.
Materials and Methods
Synthesis of Library Nanoparticles.
The compositions of all NP synthesis materials are listed in Table S1. The synthesis procedure for NP1 through NP11 was similar to a previous method (12). Briefly, phospholipids, DiR, and triglyceride were dissolved in a chloroform/methanol solvent, dried to form thin film, and then hydrated with human APOA1 solution. The homogenized solution was sonicated with a tip sonicator, and the aggregates and free lipids were removed by passing through a series of filters. The synthesis procedure for NP12 through NP15 was adapted from a previous microfluidics-based method (14). Briefly, a solution containing 0.79 mL of a PLGA or PLA solution in acetonitrile (100 mg/mL), 1.58 mL of a 3:1 molar ratio of 1,2-dimyristoyl-sn-glycero-3-phosphocholine/1-myristoyl-2-hydroxy-sn-glycero- phosphocholine (DMPC/MHPC) in ethanol (5 mg/mL), 0.36 mg of DiR, 7.9 mL of ethanol, and 14.5 mL of acetonitrile was prepared. The aforementioned solution was injected in the middle channel of the microfluidic device at a rate of 2 mL/min, and a solution of APOA1 (0.01 mg/mL in PBS) was injected in the outer channels at a rate of 10 mL/min. The product was collected, washed with PBS, and concentrated using tangential filtration [100,000-Da molecular mass cutoff (MMCO)] to remove acetonitrile, ethanol, and lipid-free APOA1. For micelle NP16, 2 mL of chloroform solution containing 211 mg of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DSPE-PEG2000) and 0.4 mg of DIR (0.5% mol) was slowly dripped into 10 mL of PBS solution heated at 85 °C under vigorous stirring. After dripping, the solution was kept at 85 °C until all of the chloroform was totally evaporated. The micelle solution was washed and concentrated in PBS using Millipore centrifugal filter (50,0000 Da MMCO). For liposome NP17, a lipid film was first prepared by evaporating a chloroform solution containing 35.4 mg of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 11.1 mg of DSPE-PEG2000, 10.24 mg of cholesterol, and 0.4 mg of DiR (61.1%, 2%, 33.4%, 0.5% in molar percentage). The residue of chloroform was removed by blowing nitrogen gas. The resulting film was hydrated with 10 mL PBS, vortexed, and subsequently sonicated for 25 min. After centrifugation at 18 × g for 10 min to remove aggregates, the liposome solution was washed and concentrated in PBS using Millipore centrifugal filter (100,000 Da MMCO). To oxidize NP1, a PBS solution containing NP1 was stirred at 37 °C in the presence of EDTA, α-tocopherol, and 2,2’-Azo-bis(2-amidinopropane) dihydrochloride for 20 h. The resulting oxidized nanoparticle (NP2) was washed thoroughly with PBS.
Synthesis of GW3965-Loaded Nanoparticles.
The synthesis procedure for Rx-HDL was similar to that used for NP10. In addition to phospholipids [POPC and 1-palmitoyl-2-hexadecyl-sn-glycero-3-phosphocholine (PHPC), 3:1 by weight] and DiR, GW3965 was added to make up 26% by weight of the total starting materials. For Rx-PLGA-HDL, a solution containing 1 mL of GW3965 in DMSO (50 mg/mL), 2 mL of PLGA (100 mg/mL), 2 mL of lipids (10 mg/mL), 53 mL of acetonitrile, and 22 mL of ethanol was prepared. The organic solution was injected in the middle channel of the microfluidic device at a rate of 2 mL/min, while a solution of APOA1 (0.01 mg/mL in 1× PBS) was injected in the outer channels at a rate of 10 mL/min. The product was collected, washed with PBS, and concentrated to ∼2 mg/mL using tangential and centrifugal filtration (100,000 Da MMCO and 300,000 Da MMCO, respectively). After synthesis, GW9365 was extracted from the final nanoparticles by acetonitrile, and the compound amount was measured by HPLC. The incorporation rate of Rx-HDL was above 90%, and that for Rx-PLGA-HDL was about 50%. The GW3965 injecting dose was adjusted on the basis of the measured GW3965 concentration in the nanoparticle solution.
Animals and Treatment Procedure.
All procedures and experiments were approved by the Institutional Animal Care and Use Committee of Icahn School of Medicine at Mount Sinai. About 300 female Apoe−/− mice (B6.129P2Apoetm1Unc/J) were purchased from Jackson Laboratories and then fed a high-fat diet (Harlan Teklad TD.88137, 42% calories from fat) for 16 wk. These mice developed advanced atherosclerosis in their aortic roots and aortas after 10 wk of high-fat diet and mimicked advanced disease in humans after 16 wk of the diet. Female C57BL/6 mice were fed on regular chow. All nanoparticles were i.v. injected into the lateral tail veins. Care was taken to ensure that each injection was less than 150 μL in volume. For the in vivo characterization study of the 17 nanoparticles, Rx-HDL, and Rx-PLGA-HDL, one i.v. injection was performed. No obvious toxicity or side effects were observed after the injections, except for NP12. After NP12 injection, mice had slow movement, and one out of five died within 24 h after the injection. For short-term toxicity study of Rx-HDL and Rx-PLGA-HDL, four injections in 8 d were given using a GW3965 dose of 10 mg/kg per injection. For the long-term treatment study, the mice received two i.v. injections of Rx-HDL (n = 12) per week for 6 wk at the dose of 10 mg/kg GW3965 per injection or equal volume of PBS (n = 12). A separate cohort of female Apoe−/− mice with the same length of high-fat diet received 6 wk of oral GW3965 (n = 6, 10 mg/kg GW3965, two times per week) or PBS (n = 5). No abnormal activities or deaths occurred during the treatment regimen.
Micro-PET/CT Imaging.
Apoe−/− atherosclerotic mice were injected with either 89Zr-labeled Rx-HDL or Rx-PLGA-HDL at about 200 μCi per mouse (n = 5). At 30 min and 24 h after the injection, the mice were imaged on an Inveon PET/CT scanner (Siemens Healthcare Global) under isoflurane-induced (Baxter Healthcare) anesthesia. Whole-body static PET scans recorded a minimum of 50 million coincident events in ∼15 min. The energy and coincidence timing windows were 350 keV to 700 keV and 6 ns, respectively. The image data were normalized to correct for nonuniform PET response, dead-time count losses, positron branching ratio, and physical decay to the time of injection, but no attenuation, scatter, or partial volume averaging correction was applied. The counting rates in the reconstructed images were converted to activity concentrations (percentage injected dose per gram of tissue) via a system calibration factor derived from imaging a mouse-sized water-equivalent phantom containing 89Zr. Images were analyzed using ASIPro VMTM software (Concorde Microsystems). Activity concentration was quantified by averaging the maximal values of at least 10 regions of interest (ROIs) drawn on consecutive slices of the chosen organs.
Liver mRNA Expression Measurement.
Total RNA was obtained from snap-frozen liver tissue and homogenized in TRIzol reagent (Ambion). The homogenate was spun down to pellet tissue, and the aqueous TRIzol supernatant was collected and processed using the Direct-Zol. RNA miniprep kit (Zymo Research Corporation) was used for RNA purification. The RNA was then reverse-transcribed using the Verso cDNA kit (Thermo Scientific) and diluted using RNase/DNase-free water. Quantitative real-time PCR was performed with Taqman Gene Expression Master Mix (Applied Biosystems), and with Taqman primer/probe mixes for Abca1, Abcg1, or Srebp1c. Gene expression was normalized to 18S ribosomal RNA (rRNA) expression.
Aortic Macrophage mRNA Expression Measurement.
The quality of RNA extracts from atherosclerotic plaques was measured by Agilent 2100 bioanalyzer (Agilent Technologies). High-quality [RNA integrity number (RIN) > 7] RNA samples were amplified with a WT-Ovation Pico RNA amplification system (NuGen). The cDNA from the amplification reactions were used to run a microfluidics-based mRNA profiling chip (BioMark; Fluidigm), which had high reproducibility. Hprt1 was used as the housekeeping gene. The following 24 genes were measured: Abca1, Abcg1, Srebp1c, Nr1h2, Hr1h3, Ccl2, Ccr2, Icam1, Ifng, Il12a, Il1a, Il1b, Il6, Nos2, Mmp2, Mmp9, Tnfa, Vcam1, Ccr7, Cd206, Foxp3, Il10, Mrc2, and Tgif1. Twelve biological repeats were included for each gene.
Cellular Lipid Measurement with Flow Cytometry.
Single cells were prepared from aortas from either Apoe−/− mice or wild-type C57BL/6 mice and then stained with antibody mixtures as described in SI Materials and Methods, Flow Cytometry. On the basis of a previous procedure (33), the stained cells were washed with PBS two times at room temperature and then incubated with 250 μL PBS containing 0.5 μg/mL BODIPY (D-3922; Molecular Probes) for 15 min. The cells were washed with flow cytometry buffer two times and analyzed in a Becton Dickinson LSRII flow cytometer.
Statistics.
Data are presented as mean ± SE of mean (SEM) unless otherwise noted. Two-tailed Student’s t test was used to calculate statistical significance. GraphPad Prism 5.0 for PC (GraphPad Software Inc.) was used for statistical analysis. P < 0.05 was regarded as significant; * denotes P value < 0.05, and ** denotes P value < 0.01 in all figures in the paper.
More description of materials and methods is included in Supporting Information.
SI Materials and Methods
Radiochemistry.
The 89Zr was produced at Memorial Sloan Kettering Cancer Center on an Ebco TR19/9 variable-beam energy cyclotron (Ebco Industries Inc.) via the 89Y(p,n)89Zr reaction and purified with a method previously reported (46). Radioactivity was measured with a Capintec CRC-15R dose Calibrator (Capintec).
Synthesis of C34-DFO.
The 2-Hexadecyl-octanoic acid (30 mg, 59 μmol), (Benzotriazol-1-yloxy) Tris(dimethylamino)phosphonium hexafluorophosphate (29 mg, 66 μmol), and N,N-diisopropylethylamine (DIEA, 10 μL) were dissolved in anhydrous dichloromethane (2 mL) and stirred at 40 °C for 10 min under nitrogen atmosphere. Next, a solution of deferoxamine mesylate (30 mg, 46 μmol) and DIEA (10 μL) in anhydrous DMSO (0.7 mL) was added, and the resulting mixture was stirred for 4 h at 40 °C under nitrogen. The cloudy suspension was allowed to cool to room temperature, and dichloromethane was removed under reduced pressure. Hydrochloric acid (0.1 M, 1 mL) was added, and the mixture was stirred for 10 min at room temperature. The solid was filtered and washed with 0.1 M HCl (3 × 1 mL), DMSO (3 × 1 mL), water (3 × 1 mL), and, finally, dichloromethane (3 × 1 mL) and dried to yield a white solid (31 mg, 64%). Mass cytometry analysis generated the following data: MS-ES+: 1,074, (M+Na)+; MS-ES−: 1,050 (M-H)−, 1,086 (M+Cl)−. Proton NMR analysis provided the subsequent values: 1H-NMR (CDCl3/CD3OD), δ in parts per million: 0.55 (triplet, 6H); 0.92 [broad signal (br), 60H]; 1.06 (br, 6H); 1.27 multiplet (m), 6H]; 1.32 (m, 6H); 1.76 (m, 1H); 1.80 singlet (s), 3H]; 2.15 (t, 4H); 2.46 (t, 4H); 2.87 (m, 6H); 3.29 (m, 6H). All chemicals were purchased from Sigma-Aldrich.
Radiolabeling of Library Nanoparticles and GW3965-Loaded Nanoparticles.
Nanoparticles were labeled using a slightly modified version of the synthesis procedure for regular screening nanoparticles or GW3965-loaded nanoparticles. Briefly, DiR of the library nanoparticles or 0.5% of GW3965 payload was replaced with C34-DFO while synthesizing radiolabeled nanoparticles. After synthesis and purification, the C34-DFO-labeled nanoparticles were incubated with 89Zr-oxalate in PBS (pH 7.1∼7.4) at 37 °C for 2 h at an activity-to-APOA1 ratio of ∼1 mCi/mg (or ∼1 mCi/10 mg lipids for NP17). The radiolabeled nanoparticles were purified by spin filtration by using 10,000 Da MMCO filter tubes. Radiochemical yields were in excess of 80%, and radiochemical purities, as determined by size exclusion chromatography, were greater than 95% for all nanoparticles.
Blood Half-Life Determination.
In the library study, all nanoparticles were injected at a fixed DiR dose of 1 mg/kg. The theoretical 0 time point was calculated by assuming that a mouse’s blood accounts for 6% of its body weight and the density of mouse blood was 1.06 g/mL. The mice were bled 1 h, 6 h, and 24 h after the injection. The DiR concentration was determined by comparing DiR fluorescence signal to a DiR standard curve. DiR signal was measured in an IVIS200 system (PerkinElmer) at a 750-nm excitation wavelength and an 820-nm emission wavelength. Blood-half was calculated by finding the x coordinate from the curve’s intersection with a horizontal line at 50% signal of time 0. Five mice were used for each nanoparticle. Regarding the radiolabeled nanoparticles, animals were injected with ∼20 μCi to ∼30 μCi of the corresponding nanoparticles, and blood samples (∼10 μL each) were collected 2 min, 30 min, 1 h, 2 h, 4 h, 8 h, and 24 h after the i.v. injection. The blood was weighted, and its radioactivity was determined using a gamma counter (PerkinElmer). Blood half-life was calculated via two-phase decay. Three mice were used for each nanoparticle.
Autoradiography and Biodistribution.
Upon sacrifice, animals were perfused with 20 mL of PBS through cardiac puncture. Lung, liver, aorta, spleen, kidney, femur muscle, and heart were collected, and their radiotracer distribution was determined by placing the tissues in a film cassette against a phosphorimaging plate (BASMSM-2325, Fujifilm) for either 14 h (mouse aortas) or 4 h (all other organs). Phosphorimaging plates were read in a Typhoon 7000IP plate reader (GE Healthcare). Biodistribution in aorta, heart, liver, spleen, blood, lung, skin, brain, pancreas, stomach, small intestine, large intestine, kidney, muscle, and bone was determined by first weighing the tissues and then measuring their radioactivity in a gamma counter (PerkinElmer). The relative activity per tissue is presented as percentage of injected dose per gram of tissue.
Nanoparticles Characterization.
The synthesized nanoparticles’ size was measured by DLS. To measure the DiR concentration in nanoparticles, we first extracted DiR from the nanoparticles with acetonitrile, and the amount of DiR was determined by measuring the signature absorbance of DiR at 750 nm. Nanoparticle morphology was determined by transition emission microscopy. Briefly, the original PBS solvent was replaced with an ammonium acetate buffer and then mixed with 2% (wt/vol) sodium phosphotungstate (pH = 7.4) to negatively stain the nanoparticles. The solution was then added to a TEM grid and imaged with a Hitachi H7650 system linked to a Scientific Instruments and Applications digital camera controlled by Maxim CCD software. To measure nanoparticle APOA1 concentration, APOA1 was separated from the other nanoparticle components by acetonitrile precipitation. The pellets’ protein component was then measured with a standard BCA assay (Thermo Scientific).
HPLC and Radio-HPLC.
HPLC was performed on a Shimadzu HPLC system equipped with two LC-10AT pumps and an SPD-M10AVP photodiode array detector. Radio-HPLC was performed using a Lablogic Scan-RAM Radio-TLC/HPLC detector. Size exclusion chromatography was performed on a Superdex 10/300 column (GE Healthcare Life Sciences) using PBS as an eluent at a flow rate of 1 mL/min. GW3865 detection was performed on a C18 column (Shimadzu) with an isocratic flow of water and acetonitrile at a flow rate of 1 mL/min.
Cholesterol Efflux Assay.
Bone marrow cells were flushed from the tibia and femurs of 6- to 8-wk-old C57BL/6 mice and differentiated into macrophages by incubation in DMEM media supplemented with 10% FBS, 1% P/S, and 15% L929-conditioned media for 7 d. Bone marrow-derived macrophages were subsequently incubated with media containing 0.5 μCi/mL [3H]-cholesterol and acLDL (50 μg/mL) for 30 h. Cells were washed twice with PBS and equilibrated overnight in media containing 2 mg/mL fatty acid-free albumin. Efflux to APOA1 (50 μg/mL) or various nanoparticles (50 μg/mL normalized to APOA1 concentration) was carried out for 8 h. Cell media were removed, and the cells were lysed in 0.1 M NaOH solution. The [3H]-cholesterol contents of media and cell lysates were measured by liquid scintillation counting. Efflux is expressed as a percentage of 3H-cholesterol in medium by using the following formula: [3H-cholesterol in medium/(3H-cholesterol in medium + 3H-cholesterol in cells)] × 100%.
NIRF.
Twenty-four hours after i.v. injection of DiR-containing nanoparticles, the mice were perfused with 20 mL of PBS, and brain, lung, heart, aorta, spleen, liver, kidney, and femur muscle were collected. Their total DiR fluorescent signal was measured in an IVIS200 at a 750-nm excitation wavelength and an 820-nm emission wavelength. The radiant efficiency was calculated using LiveImaing (PerkinElmer). For each nanoparticle, five mice were used.
Flow Cytometry.
We used a protocol similar to one previously reported (12). Briefly, at 24 h after i.v. injection of DiR-loaded nanoparticles, blood was collected and the animals were perfused. Afterward, aortas and spleens were collected, diced, and digested with a mixture of enzymes, including liberase TH (Roche), hyaluronidase (Sigma-Aldrich), and DNase (Sigma-Aldrich), in a 37 °C oven for 1 h. A single-cell suspension was made by removing tissue aggregates, extracellular matrix, and cell debris from the solution. Red blood cells were removed from the blood sample by lysis buffer. DiR was detected on the APC-Cy7 channel. To identify macrophages, monocytes, DCs, neutrophils, and other immune cells, a lineage of antibodies (Lin) recognizing CD90 (clone 53.2.1), B220 (clone RA3-6B2), CD49b (clone DX5), NK1.1 (clone PK136), Ly-6G (clone 1A8), and Ter-119 (clone TER-119) and antibodies recognizing Ly-6C (clone AL21), F4/80 (BM8), and CD11c (N418) were used. Antibodies were purchased from eBioscience, BD Biosciences, and Biolegend. Due to the large number of samples, two to three nanoparticles were measured per batch, each of which included 12 to 17 mice (10 to 15 from tested nanoparticles, 1 for reference DiR-nanoparticles, and 1 noninjected control to set up compensation). The APC-Cy7 channel of the flow cytometer was calibrated using APC-Cy7 calibration beads (Cat# ECFP-F7; Spherotech). Signal variation among batches was corrected by normalizing to the beads’ signal. All samples were measured on an LSRII (BD Biosciences) flow cytometer. Results were analyzed with FlowJo (Ashland), and statistics were calculated with Prism (GraphPad).
Hepatic Triglyceride and Cholesterol Measurement.
Lipids were extracted from the livers, which were snap-frozen in −80 °C immediately after sacrifice. Portions of liver tissue (generally <100 mg) were weighed, suspended in 2× weight volume of PBS, and homogenized. To analyze protein content, 10 μL of the total homogenate underwent a standard Lowry assay (BioRad); 50 μL of the total homogenate was suspended in 3 mL of isopropanol and incubated overnight at 4 °C while rocking to extract lipids. The solvent/lipid mixture was then centrifuged at 1,000 × g for 10 min, and supernatant was collected in glass tubes and dried under a stream of nitrogen. Following complete evaporation of the extraction isopropanol, lipids were stored at −20 °C until analysis. For quantification, dried lipids were resuspended in 1 mL of isopropanol, and analysis of triglycerides and total cholesterol was performed on diluted lipid solutions using standard colorimetric assays (Wako) as per manufacturer’s protocol. The cholesterol and triglyceride concentrations were normalized to the measured protein concentration per sample.
Immunostaining and Laser Capture Microdissection.
As previously described (2), 6-μm-thick frozen sections were made from the aortic roots of 60 mice. Six evenly distributed sections were stained with CD68 (clone MCA1957; Serotec). The stained slides were digitally scanned using a slide scanner (3DHistech Panoramic Scanner). When CD68-positive area was quantified, the positively stained area was calculated using a MATLAB procedure we had developed previously (2). A total of 360 sections were analyzed. For laser capture microdissection (LCM), 36 aortic roots sections per animal were used to extract atherosclerotic macrophages using Leica LDM6500. All LCM reagents were maintained, and procedures were done under RNase-free conditions. The sections were fixed in 70% ethanol for 1 min, washed in H2O, stained with Mayer’s hematoxylin (VWR Scientific) for 1 min, washed in H2O, incubated in PBS (to develop blue color) for 15 s, washed in H2O, partially dehydrated in 70% followed by 95% ethanol, stained in eosin Y (VWR Scientific) for 5 s, washed in 95% ethanol, and then completely dehydrated in 100% ethanol (30 s), xylene (30 s), and xylene (5 min). The sections were then air-dried for 10 min. Macrophages were identified under a microscope and verified by anti-CD68 staining on the guiding slides. Isolated macrophages were immediately lysed, and their RNA were extracted and stored in a −80 °C freezer. For LCM, 2,160 sections were used.
Supplementary Material
Acknowledgments
We thank the flow cytometry core facility, the preclinical imaging center of Translational and Molecular Imaging Institute, and the quantitative qPCR facility of Icahn School of Medicine at Mount Sinai. We thank the small animal imaging core facility, the genomic core facility, the molecular cytology core facility, and the radiochemistry and molecular imaging probe core of Memorial Sloan Kettering Cancer Center. We thank Dr. Kaley Joyes for editing the manuscript. This work was supported by National Institute of Health Grants R01 HL118440, R01HL125703, and R01 CA155432 (to W.J.M.M.), R01 EB009638 (to Z.A.F.), K25 EB016673 (to T.R.), R01HL123433 (to K.J.M.), and P30 CA008748-50 (to Dr. Jason S. Lewis), as well as a Harold S. Geneen Charitable Trust Award (to Z.A.F.), European Framework Program 7 grant (FP7-Health 309820: Nano-Athero) and the Dutch network for Nanotechnology NanoNext NL (A.A. and G.S.), and a Netherlands Organisation for Scientific Research Vidi (to W.J.M.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. L.Z. is a Guest Editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1609629113/-/DCSupplemental.
References
- 1.Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: Impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 2.Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol. 2010;10(7):501–513. doi: 10.1038/nri2787. [DOI] [PubMed] [Google Scholar]
- 3.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: A dynamic balance. Nat Rev Immunol. 2013;13(10):709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339(6116):161–166. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 6.Heidt T, et al. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20(7):754–758. doi: 10.1038/nm.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robbins CS, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19(9):1166–1172. doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol. 2009;27:165–197. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dutta P, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487(7407):325–329. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Groot PH, et al. Synthetic LXR agonists increase LDL in CETP species. J Lipid Res. 2005;46(10):2182–2191. doi: 10.1194/jlr.M500116-JLR200. [DOI] [PubMed] [Google Scholar]
- 11.Kotokorpi P, et al. Physiological differences between human and rat primary hepatocytes in response to liver X receptor activation by 3-[3-[N-(2-chloro-3-trifluoromethylbenzyl)-(2,2-diphenylethyl)amino]propyloxy]phenylacetic acid hydrochloride (GW3965) Mol Pharmacol. 2007;72(4):947–955. doi: 10.1124/mol.107.037358. [DOI] [PubMed] [Google Scholar]
- 12.Tang J, et al. Inhibiting macrophage proliferation suppresses atherosclerotic plaque inflammation. Sci Adv. 2015;1(3):e1400223. doi: 10.1126/sciadv.1400223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duivenvoorden R, et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun. 2014;5:3065. doi: 10.1038/ncomms4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanchez-Gaytan BL, et al. HDL-mimetic PLGA nanoparticle to target atherosclerosis plaque macrophages. Bioconjug Chem. 2015;26(3):443–451. doi: 10.1021/bc500517k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cormode DP, et al. HDL as a contrast agent for medical imaging. Clin Lipidol. 2009;4(4):493–500. doi: 10.2217/clp.09.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagano Y, Arai H, Kita T. High density lipoprotein loses its effect to stimulate efflux of cholesterol from foam cells after oxidative modification. Proc Natl Acad Sci USA. 1991;88(15):6457–6461. doi: 10.1073/pnas.88.15.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsumura Y. Poly (amino acid) micelle nanocarriers in preclinical and clinical studies. Adv Drug Deliv Rev. 2008;60(8):899–914. doi: 10.1016/j.addr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 18.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 19.Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N. HDL, ABC transporters, and cholesterol efflux: Implications for the treatment of atherosclerosis. Cell Metab. 2008;7(5):365–375. doi: 10.1016/j.cmet.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15(2):104–116. doi: 10.1038/nri3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schiener M, et al. Nanomedicine-based strategies for treatment of atherosclerosis. Trends Mol Med. 2014;20(5):271–281. doi: 10.1016/j.molmed.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Weissleder R, Ntziachristos V. Shedding light onto live molecular targets. Nat Med. 2003;9(1):123–128. doi: 10.1038/nm0103-123. [DOI] [PubMed] [Google Scholar]
- 23.Galkina E, et al. CXCR6 promotes atherosclerosis by supporting T-cell homing, interferon-gamma production, and macrophage accumulation in the aortic wall. Circulation. 2007;116(16):1801–1811. doi: 10.1161/CIRCULATIONAHA.106.678474. [DOI] [PubMed] [Google Scholar]
- 24.Galkina E, et al. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J Exp Med. 2006;203(5):1273–1282. doi: 10.1084/jem.20052205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weissleder R, Nahrendorf M, Pittet MJ. Imaging macrophages with nanoparticles. Nat Mater. 2014;13(2):125–138. doi: 10.1038/nmat3780. [DOI] [PubMed] [Google Scholar]
- 26.Hong C, Tontonoz P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat Rev Drug Discov. 2014;13(6):433–444. doi: 10.1038/nrd4280. [DOI] [PubMed] [Google Scholar]
- 27.Joseph SB, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci USA. 2002;99(11):7604–7609. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naik SU, et al. Pharmacological activation of liver X receptors promotes reverse cholesterol transport in vivo. Circulation. 2006;113(1):90–97. doi: 10.1161/CIRCULATIONAHA.105.560177. [DOI] [PubMed] [Google Scholar]
- 29.Deri MA, Zeglis BM, Francesconi LC, Lewis JS. PET imaging with ⁸⁹Zr: From radiochemistry to the clinic. Nucl Med Biol. 2013;40(1):3–14. doi: 10.1016/j.nucmedbio.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258(5081):468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 31.Wu KT, et al. Nonalcoholic fatty liver disease severity is associated with the ratios of total cholesterol and triglycerides to high-density lipoprotein cholesterol. J Clin Lipidol. 2016;10(2):420–425. doi: 10.1016/j.jacl.2015.12.026. [DOI] [PubMed] [Google Scholar]
- 32.Brunham LR, et al. Tissue-specific induction of intestinal ABCA1 expression with a liver X receptor agonist raises plasma HDL cholesterol levels. Circ Res. 2006;99(7):672–674. doi: 10.1161/01.RES.0000244014.19589.8e. [DOI] [PubMed] [Google Scholar]
- 33.Herber DL, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 2010;16(8):880–886. doi: 10.1038/nm.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Y, et al. Augmenting drug-carrier compatibility improves tumour nanotherapy efficacy. Nat Commun. 2016;7:11221. doi: 10.1038/ncomms11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mullard A. PCSK9 inhibitors are go. Nat Rev Drug Discov. 2015;14(9):593. doi: 10.1038/nrd4730. [DOI] [PubMed] [Google Scholar]
- 36.Sabatine MS, et al. Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500–1509. doi: 10.1056/NEJMoa1500858. [DOI] [PubMed] [Google Scholar]
- 37.Robinson JG, et al. ODYSSEY LONG TERM Investigators Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1489–1499. doi: 10.1056/NEJMoa1501031. [DOI] [PubMed] [Google Scholar]
- 38.Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br J Pharmacol. 2009;157(2):220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cook D, et al. Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework. Nat Rev Drug Discov. 2014;13(6):419–431. doi: 10.1038/nrd4309. [DOI] [PubMed] [Google Scholar]
- 40.Mieszawska AJ, et al. Synthesis of polymer-lipid nanoparticles for image-guided delivery of dual modality therapy. Bioconjug Chem. 2013;24(9):1429–1434. doi: 10.1021/bc400166j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fredman G, et al. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci Transl Med. 2015;7(275):275ra20. doi: 10.1126/scitranslmed.aaa1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choleris E, et al. Microparticle-based delivery of oxytocin receptor antisense DNA in the medial amygdala blocks social recognition in female mice. Proc Natl Acad Sci USA. 2007;104(11):4670–4675. doi: 10.1073/pnas.0700670104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hammad H, Lambrecht BN. Dendritic cells and epithelial cells: Linking innate and adaptive immunity in asthma. Nat Rev Immunol. 2008;8(3):193–204. doi: 10.1038/nri2275. [DOI] [PubMed] [Google Scholar]
- 45.Mukherjee G, Dilorenzo TP. The immunotherapeutic potential of dendritic cells in type 1 diabetes. Clin Exp Immunol. 2010;161(2):197–207. doi: 10.1111/j.1365-2249.2010.04157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pérez-Medina C, et al. PET imaging of tumor-associated macrophages with 89Zr-labeled high-density lipoprotein nanoparticles. J Nucl Med. 2015;56(8):1272–1277. doi: 10.2967/jnumed.115.158956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.