

Summary
Cumulative evidence indicates a role for the complement system in both pathology and recovery after ischemic stroke. Here, we review the current understanding of the dual role of complement in post-stroke injury and recovery, and discuss the challenges of anti-complement therapies. Most complement directed therapeutics currently under investigation or development systemically inhibit the complement system, but since complement is important for immune surveillance and is involved in various homeostatic activities, there are potential risks associated with systemic inhibition. Depending on the target within the complement pathway, other concerns are high concentrations of inhibitor required, low efficacy and poor bioavailability. To overcome these limitations, approaches to target complement inhibitors to specific sites have been investigated. Here we discuss targeting strategies, with a focus on strategies developed in our lab, to specifically localize complement inhibition to sites of tissue injury and complement activation, and in particular to the post-ischemic brain. We discuss various injury site-specific targeted complement inhibitors as potential therapeutic agents for the treatment of ischemic stroke treatment, as well as their use as investigate tools for probing complement-dependent pathophysiological processes.
Keywords: Complement system, stroke, tissue targeting, stroke therapeutics, inflammation
Introduction
Stroke is among the leading causes of death worldwide and is a major cause of disability (1–3). Although there has been a remarkable improvement in stroke prevention and management of high-risk patients (4), treatment options for ischemic stroke patients remain extremely limited and unsatisfactory. The only currently available acute treatment option for stroke is thrombolytic therapy using tissue-plasminogen activator (t-PA), which has a very short window of efficacy (up to 3 hrs. post-stroke) and carries the risk of fatal intracranial hemorrhage. As a consequence, less than 10% of stroke patients receive t-PA therapy (5–7). Beyond an acute treatment window, efforts to improve outcomes of stroke patients have been focused on rehabilitation therapy that has shown mild improvements in select subsets of chronic stroke patients (8). The main challenge in developing novel stroke treatments is designing a therapeutic with an extended window of efficacy, and that can safely reduce the extent of injury after stroke without impairing recovery mechanisms.
At the pathophysiological level, stroke or cerebral ischemia and reperfusion injury (IRI) involves a complex dynamic of multi-phasic events. These include acute ischemia-induced cell death within the ischemic core, inflammatory responses that exacerbate damage and result in secondary injury, and neuroregenerative responses that underlie functional recovery. The inflammatory response not only contributes to the propagation of secondary injury that radiates from the ischemic core to the penumbra, but also provides a major inhibitory drive to recovery and reparatory mechanisms (9, 10). The complement system has been recognized as a central mediator in the activation and pathological amplification of inflammation after IRI. Data from studies on human stroke patients have shown that there is significant activation and deposition of complement in the postmortem ischemic brain (11, 12). In addition, serum complement proteins and activation products in stroke patients have been shown to correlate with severity of stroke and subsequent functional disability (13–18). Complement has also been extensively studied in animal models of experimental stroke, in which an important role for complement in the pathology of acute and chronic stroke has been demonstrated (reviewed in (19)). On the other hand, studies in animal models have shown that complement is also involved in reparatory and neuroregenerative processes after stroke, as well as other homeostatic functions across multiple organ systems. Thus, these dual functions of complement present a challenge to the clinical development of anti-complement therapeutics that have adequate efficacy and safety profiles (20, 21). For these reasons, it is important to understand the roles of the different complement pathways and activation products in the pathogenesis of stroke, as targeting a particular complement pathway or activation product may allow for a therapeutic that dissects the dueling roles of complement. In addition, targeting complement inhibition to the site of cerebral injury is likely to improve the efficacy and safety profile of an anti-complement therapeutic.
We review the role of complement in post-stroke pathophysiological events and discuss the design, application and challenges of site-targeted complement inhibitors for the treatment of stroke.
Overview of complement cascade and regulatory proteins
The complement system consists of secreted and membrane-bound proteins. The complement activation cascade can be triggered via one of three different pathways; the classical, lectin or alternative pathway (Fig. 1). The classical pathway is usually initiated by the binding of C1q to Ab Fc domains or by direct deposition of C1q on apoptotic cells and debris. The lectin pathway is initiated by the binding of mannose binding lectin (MBL) or ficolins to certain carbohydrates on microbial surfaces, as well as to glycosylation patterns on IgM. The alternative pathway can be spontaneously activated, but also serves as an amplification loop for the other pathways, and may account for up to 80% of complement activation. All three complement pathways converge at the formation of the C3 convertase that cleaves C3 into C3a and C3b, with subsequent protein cleavage and assembly steps that culminate with the formation of the cytolytic membrane attack complex (MAC) (Fig. 1). In addition to the MAC, other biologically active proteins and products of complement activation products that function via receptor engagement are bound C1q and MBL, deposited C3 activation products (C3b, iC3b, C3d), and the soluble C3a and C5a anaphylatoxins. Of note, more recently an extrinsic protease pathway of activation has been described, which includes plasmin and thrombin mediated activation of complement by direct cleavage of C3 and C5 proteins (22, 23), findings that may have implications in the context of stroke and thrombolytic therapy.
Figure 1. Overview of the complement system and complement regulators.
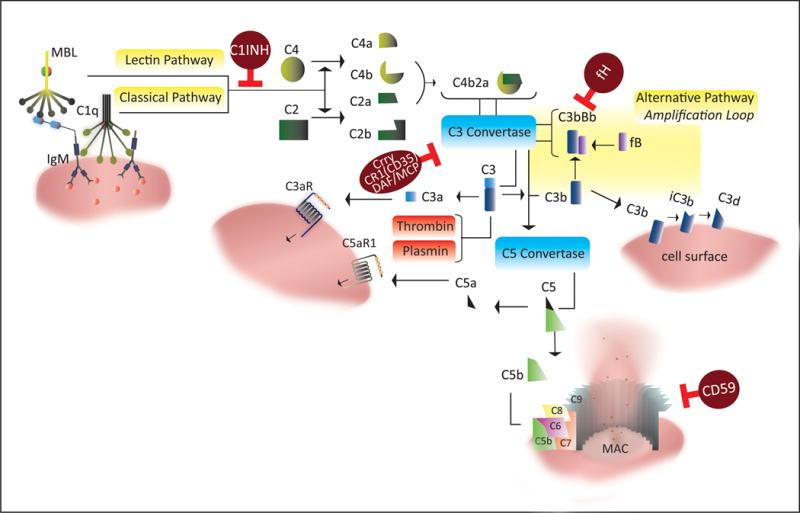
In addition to extrinsic pathways that may contribute to complement activation through serum proteases (plasmin and thrombin), complement is activated by one of three pathways: 1. The classical pathway, via C1q binding to Fc-domains of antigen-bound antibodies or directly to apoptotic or stressed cells, 2. The lectin pathway, via binding of MBL to sugar moieties on pathogenic surfaces and glycosylated proteins, and 3. The alternative pathway, via spontaneous hydrolysis of serum complement C3 (“tick-over”) with the involvement of factors B and D. Both classical and lectin pathway form a C3 convertase by cleaving C4 and C2 to form C4b2a. The alternative pathway C3 convertase is formed by association of C3b with factor B and subsequent cleavage of factor B by factor D forming C3bBb. The initial products of C3 cleavage are C3b and C3a. Deposited C3b can further amplify C3 cleavage by recruitment of the alternative pathway, and additionally serves as an opsonin that is subsequently processed into iC3b and C3d. C3b can also associate with C3 convertase (C4b2a or C3bBb), forming a C5 convertase that cleaves C5 into C5b and C5a. C3a and C5a are termed anaphylatoxins and signal through G-protein coupled receptors. C5b deposits on cell surfaces and recruits downstream complement proteins C6-C9 leading to the formation of membrane attack complex (MAC). Several complement regulators that function at different points in the complement pathway serve as checkpoints that prevent uncontrolled complement activation (brown circles in the figure).
Complement activation on host cells is controlled by various proteins; decay accelerating factor (DAF) and membrane cofactor protein (MCP), and complement receptor 1 (CR1/CD35) that control C3 activation, and CD59 that inhibits formation of the MAC. In mice, MCP expression is restricted and DAF and Crry, the latter being a structural and functional analogue of DAF and MCP, represent the primary membrane bound inhibitors of C3 activation. Factor H (fH) and C4-binding protein are soluble inhibitors of the alternative and classical pathway, respectively (Reviewed in (24)).
Complement activation and function in the post-ischemic brain
Triggers for complement activation after stroke
Stroke or cerebral IRI is associated with depletion of cellular energy resources, release of reactive oxygen species, activation of apoptotic and necrotic cell death mechanisms, and excitotoxic insult due to glutamate excess and ionic imbalances (19). These pathological processes can lead to the activation of complement via different mechanisms (Fig. 2). Metabolic stress to central nervous system (CNS) parenchyma and endothelial cells is associated with the exposure of normally hidden antigens, often referred to as danger associated molecular patterns or neoepitopes (25). This phenomenon is a general paradigm and occurs in other tissues following IRI, although the specificity and pattern of expression of post-ischemic neoepitopes may be different (26–28). Post-ischemic neoepitopes are recognized by circulating natural self-reactive IgM antibodies that in turn can bind C1q and MBL, via Fc-domain or agalactosyl (G0) glycosylation pattern, respectively (25, 29). Although with lower affinity, C1q and MBL can also directly deposit on apoptotic cells and debris leading to complement activation (30, 31). The classical pathway can also be activated by C-reactive protein binding to stressed cells, and the lectin pathway can also be activated by ficolins deposited on stressed or apoptotic surfaces (32, 33).
Figure 2. Triggers and consequences of complement activation after stroke.
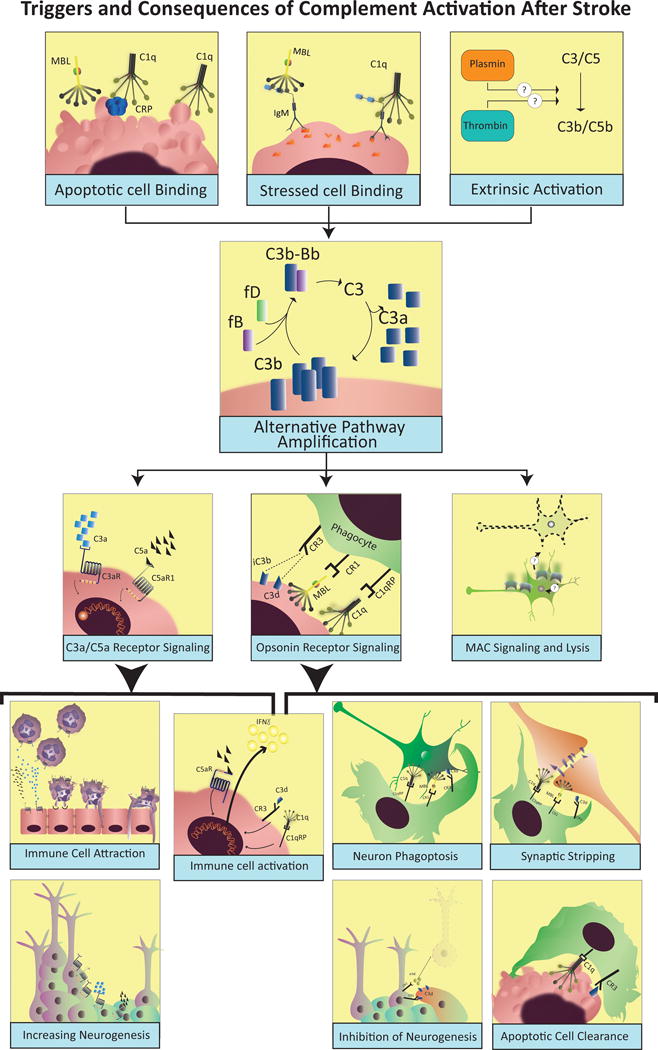
Following stroke or cerebral ischemia-reperfusion injury, complement can be activated by the direct binding of C1q and MBL to stressed cells, or via natural antibodies that recognize danger-associated molecular patterns (DAMPs). Reperfusion and activation of serum proteases like thrombin and plasmin can also provide an extrinsic mechanism to activate complement after stroke. Following initial activation, complement activity is amplified by the alternative pathway leading to deposition of opsonins, MAC and release of anaphylatoxins, all of which are involved in injury and recovery after stroke. Signaling through C3aR and C5aR1 has been implicated in promoting expression of adhesion molecules on endothelial surfaces, and to directly stimulate activation and migration of neutrophils and monocytes/microglia. Complement anaphylatoxins have also been shown to play a role in promoting neuronal stem cell proliferation and migration after stroke. C3d and other complement opsonins can also serve as targets and activators for phagocytic cells, but can also contribute to exacerbated loss of function after stroke by promoting synaptic and neuronal uptake by microglia and other phagocytic cells. Through a similar mechanism, complement opsonins can also beneficially assist with the clearance of apoptotic and dead cells, the resolution of inflammation, and synaptic remodeling after stroke. Finally, although several studies have shown MAC deposition in the brain after stroke, the role of MAC in post-stroke injury and recovery is still not well understood.
Although there are multiple potential mechanisms of complement activation after stroke, we have shown a key role for natural self-reactive IgM in activating complement and propagating injury in murine ischemic stroke. Antibody (and T cell) deficient Rag1−/− mice were protected from cerebral complement activation and injury after transient middle cerebral artery occlusion (MCAO), but reconstitution of Rag1−/− mice with natural IgM mAbs that recognized specific post-ischemic neoepitopes reversed the phenotype and restored complement activation and injury (25). Since MBL deficient mice (34), but not C1q deficient mice (35) are protected from ischemic stroke, the data taken together indicate that complement activation and propagation of injury in murine ischemic stroke occurs via IgM mediated activation of the lectin pathway. Indeed, in human stroke, serum level of MBL was shown to be an independent risk factor of ischemic stroke (36), and genetically-defined deficiency of serum MBL results in smaller lesion sizes, and more favorable acute (3 days) and sub-acute outcomes (3 months) in stroke patients (37–40). Similarly, lower serum levels of ficolin-3 on patient admission, which is indicative of ficolin consumption, correlated with increased injury markers and worse functional outcome in stroke patients (41).
Although the lectin pathway appears to play an important role in complement activation after cerebral ischemia, we have shown an equally important role for the alternative pathway in the propagation of injury following murine ischemic stroke, since genetic or pharmacological inhibition of the alternative pathway also resulted in a significant reduction in complement activation and improved outcomes after stroke (42). These data indicate that alternative pathway amplification of the lectin pathway drives injury in murine ischemic stroke.
Complement dependent pathophysiology in the CNS
Complement activation results in the generation of three types of activation products, namely opsonins (C1q, MBL and C3b/iC3b/C3d), anaphylatoxins (C3a and C5a) and the MAC. These activation products have been variously implicated in both degenerative and regenerative mechanisms after brain injury ((19, 20), summarized in Fig. 2). Although much is still unknown about the role and contributions of these activation products in the context of ischemic stroke, studies across a spectrum of neurodegenerative diseases have provided insight on how complement contributes to CNS pathology.
Complement opsonins play a key role in promoting microglial-mediated phagocytosis of stressed, but living and potentially salvageable neurons, and also in the stripping of synapses during neurodegenerative processes (43, 44). Signaling through complement opsonin receptors not only stimulates phagocytosis, but also promotes the activation of microglia and macrophages through intracellular signaling pathways culminating in the release of interferon gamma and other cytokines (45). Signaling of C3d through complement receptor 2 (CR2) has also been shown to inhibit proliferation of neuronal progenitor cells (46). The complement anaphylatoxins C3a and C5a are potent activators of microglia, monocytes and granulocytes (47); they can further increase interferon gamma release (45), and can serve as chemoattractants by signaling through their cognate receptors, C3aR and C5aR1. Complement activation within the CNS can also induce the endothelial expression of P-selectin, facilitating immune cell recruitment (35, 48–50). The terminal activation product of complement, the MAC, can be cell-activating or cytolytic, depending on the level of deposition on the cell surface. However, a pathological role for the MAC has not been clearly defined, and at least in murine models, may be dependent on gender and severity of insult (42, 51, 52).
Source and regulation of CNS complement activity
Important for developing strategies to target complement activity is understanding the origin of complement within the CNS, and how complement activity within the CNS is regulated. The principle source of serum complement proteins is the liver, and they circulate in their inactive form prior to cleavage and activation by a triggering insult. Although serum complement is normally excluded from the CNS by the blood-brain-barrier (BBB), breaches in BBB integrity, as occurs for a period of several days after stroke, allows the influx of serum complement proteins into the CNS (53, 54). However, the CNS is capable of locally producing complement proteins, and expression is upregulated in many neurodegenerative diseases and during development (55–57). The role of CNS-derived complement versus serum-derived complement in post-stroke pathology, however, has not been investigated. As in other organs and tissues, cells within the CNS also express complement regulatory proteins, including microglia, astrocytes, oligodendrocytes and neurons (53, 55, 58). Interestingly, stressed and apoptotic neurons during neuronal pathology express reduced levels of complement regulatory proteins on their surface (59).
Role of complement in reparatory and regenerative responses after injury
Understanding the role of complement in promoting pathology after CNS injury, and specifically after stroke, is complicated by the fact that the same complement activation products that promote injury are also involved in homeostatic processes, as well as reparatory responses to injury (20). For example, the deposition of complement opsonins on apoptotic cells and debris represents a mechanism for the clearance of dead and dying cells and resolution of inflammation. C3a and C5a signaling can also be involved in promoting neurogenesis and neuroblast migration, as well as in providing neuronal protection from excitotoxic insult (60–62). At sub-lytic concentrations, the MAC can contribute to anti-apoptotic signaling in oligodendrocytes via PI3K dependent mechanism (63). Because of this duality of function, it is a challenge from a therapeutic perspective to fine-tune complement inhibition strategies and bisect the pathological versus protective roles of complement.
Injury site-specific targeting of complement modulation
General considerations of site-targeted and systemic complement inhibition
Despite a well-defined role for complement in the pathology of inflammatory diseases, including stroke, there are various potential concerns with the application of systemic complement inhibition (21, 64, 65). One major concern for therapeutic strategies that utilize a systemic approach to inhibit activation of certain complement proteins is poor bioavailability and low efficacy. For instance, we have previously shown that site-targeted forms of human DAF and CD59 (by means of linking the proteins to CR2, see below) are more than 20-fold more effective at inhibiting complement in-vitro than their untargeted soluble forms (64). A similarly targeted form of murine Crry, that targets the inhibitor to sites of complement activation, required a 10-fold lower dose than an untargeted soluble form of Crry to provide equivalent protection in a model of intestinal IRI (66). Depending on the target, a systemic approach would require very large doses of inhibitor; for example circulating levels of C3 are greater than 1 mg/ml. Additional concerns regarding the use of systemic complement inhibitors include high turnover of complement proteins, high concentration of some target complement proteins, such as C3 which is present at greater than 1 mg/ml in serum, the potential contribution of local complement production to pathology (particularly relevant for CNS injury and disease), and the increased risk of infectious complications (21, 64, 65). Risk of infection is a major concern for stroke patients, as they have increased vulnerability to infections that can significantly deteriorate outcome and affect recovery (67, 68). Systemic complement inhibition may also interfere with various homeostatic functions of complement such as the catabolism of immune complexes and apoptotic cells, tissue regeneration, lipid metabolism and angiogenesis (reviewed in (54)).
Beyond therapeutic applications, site-targeted inhibitors can also provide a toolbox for the dissection and exploration of the role of complement in the pathophysiological response after injury, as we have applied previously in the context of different models of IRI using a targeting moiety linked to different complement inhibitors (21). While transgenic mice lacking different complement proteins have provided important insights into the role of complement in injury and disease, complement inhibition allows investigation of mechanisms in a clinical setting. This is important since transient complement inhibition can, and indeed sometimes does, produce different outcomes when compared to a mouse deficient in the equivalent targeted complement protein. This is not surprising given that complement deficiency from birth can affect processes from synaptic maturation during development to various other ongoing homeostatic mechanisms.
A final consideration here, although complement inhibitor biologics (whether targeted or systemic) have limited ability to cross the BBB and may thus have limited application in several neurodegenerative diseases, this is of less concern for ischemic stroke. During the acute phase of ischemic stroke, both ischemia and reperfusion insults are associated with a breach of BBB integrity which lasts for several days after injury, both in human and experimental models (69). This will allow access of complement inhibitory proteins to the ischemic brain. The temporary disruption to the BBB also highlights a potential advantage of a site-targeted inhibitor in that once it has gained access, the inhibitor will have an increased half-life at the target site. Indeed, we have shown that a complement inhibitor that targets to the site of injury after ischemic stroke can be detected in the ipsilateral hemisphere 5 days after stroke (47) (Fig. 3). Even in the context of chronic stroke, site-targeted complement inhibitors may maintain their therapeutic utility given accumulating evidence that there is sustained oxidative stress and inflammation in the brain endothelium of chronic stroke patients, and that this is associated with deteriorated outcome and higher incidence of thrombotic events (70–72). Therefore, targeting of complement inhibitors to the inflamed endothelium may still provide therapeutic efficacy despite an intact BBB as is found in chronic stroke.
Figure 3. Brain localization of targeted versus untargeted fH to ischemic brain.
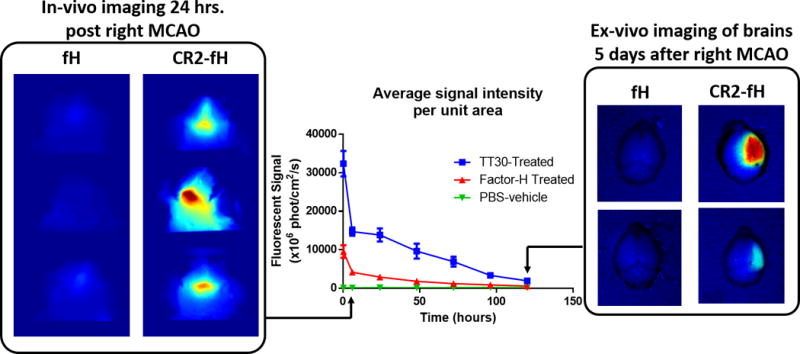
Adult male C57BL/6 mice were subjected to 60 min right MCAO followed by reperfusion, and fluorescently labeled fH or CR2-fH were administered 30 min after reperfusion. In-vivo fluorescence tomography was performed daily and the average signal per unit area in the head was calculated. Figure shows localization of CR2-fH in brains of mice after ischemic stroke with a calculated half-life of 48.5 hrs. The right panel shows ex-vivo imaging of isolated brains at 5 days after injury, revealing the presence of CR2-fH specifically in the ipsilateral hemisphere.
Targeting sites of complement activation
The fact that complement components circulate as inactive proteins in high concentrations and are selectively and pathologically activated and deposited at sites of injury, make deposited complement activation products an attractive target for the delivery of complement inhibitors (or indeed other therapeutics) to sites of inflammation and injury.
The approach we have taken is to target C3 activation products that are deposited at high density at sites of complement activation. C3 cleavage is the central event in the complement cascade, and C3 activation products are produced regardless of which complement pathway is engaged.
Following C3 cleavage into C3b and C3a, C3b irreversibly binds through amide or ester bonds to cell surface molecules, and is then cleaved into iC3b and subsequently C3dg and C3d (73). Whereas the half-life of C3b is short, the C3d breakdown product remains membrane-bound for an extended period. C3d is a ligand for CR2, a receptor expressed predominantly on B cells and dendritic cells, and we used the N-terminal C3d binding portion of CR2 as a targeting vehicle for complement inhibitory proteins or inhibitory domains thereof (64, 65). Specifically, we have prepared and characterized human CR2-DAF and CR2-CD59 (64), and for in vivo characterization, murine CR2-Crry (65), CR2-fH (66) and CR2-CD59 (74). The different inhibitors provide a toolbox for investigating the role and contribution of certain complement pathways and activation products in disease processes; Crry inhibits all complement pathways at the C3 activation step, fH inhibits only the alternative pathway, and CD59 inhibits generation of the terminal MAC, leaving the ability to produce earlier complement activation products intact. Importantly, we have shown that therapeutic doses of CR2-Crry do not interfere with systemic complement activity and do not increase susceptibility to infection, unlike C3 deficiency (65). Also of note, the CR2 targeting vehicle can have independent activity. The CR2 moiety can serve as an antagonist of CR3 receptor engagement on phagocytes, and thus inhibit the interaction between C3d and CR3, which is involved in phagocytic cell activation (45). In addition, the CR2 moiety of fusion proteins has been shown to modulate autoimmunity in a murine model of lupus, presumably by interfering with the engagement of C3d with CR2 expressed on B cells which has a costimulatory function (75).
Ruseva et al (76) recently reported a new, albeit similar, approach for complement activation site-specific targeting of CD59. Their construct consisted of CD59 linked to CRIg (complement receptor of Ig superfamily) via the hinge region of mouse IgG2a, and they demonstrated targeting of the construct to the brain following traumatic brain injury. The CRIg moiety targets to C3b and iC3b, and the CD59 moiety inhibits generation of the MAC, which provided protection from brain injury.
Other site-targeting strategies
Another approach that has been used to target complement inhibition to the brain after ischemic stroke is the decoration of soluble complement receptor 1 (sCR1) with sialyl-Lewis (sLex) carbohydrate. The sLex moieties on the sCR1sLex complex bind P and E-selectin, adhesion molecules that are upregulated on the endothelium at sites of inflammation. Characterization of sCR1sLex in a model of ischemic stroke is discussed below.
We recently developed a new targeting strategy for delivery of a complement inhibitor to post-ischemic tissue. Single chain Abs (scFv) derived from IgM mAbs recognizing post-ischemic neoepitopes (see above) were linked to complement inhibitors, and one such inhibitor, B4scFv-Crry, has been shown to specifically target to transplanted hearts and significantly reduce graft inflammation and injury. Furthermore, unlike complement deficiency, and similar to CR2-Crry, B4scFv-Crry did not affect immunity to infection (26).
The neoepitope recognized by B4 mAb and B4scFv is a modified form of annexin IV, and we have shown that this neoepitope, as well as a neoepitope recognized by an IgM mAb designated C2 which recognizes a subset of phospholipids (25), are expressed in the post-ischemic murine (25) and human (unpublished) brain. We are currently investigating B4 and C2 scFv targeting strategies of targeted complement inhibition in models of ischemic stroke. Neoepitope targeting has potential advantages over CR2 targeting approaches that include: i. A contribution of the scFv vehicle to therapeutic activity by inhibiting binding of pathogenic IgM and, as a consequence reduction in C1q and MBL binding, ii. Higher specificity for injured tissue since CR2 has several other non-injury associated ligands (21), iii. Lower immunosuppressive activity since unlike the CR2 target C3d, there will be an absence of neoepitope expression on pathogenic surfaces and, iv. Unlike CR2, a scFv that targets neoepitopes will not directly affect generation of its own ligand. Figure 4 outlines current approaches to target complement inhibition.
Figure 4.
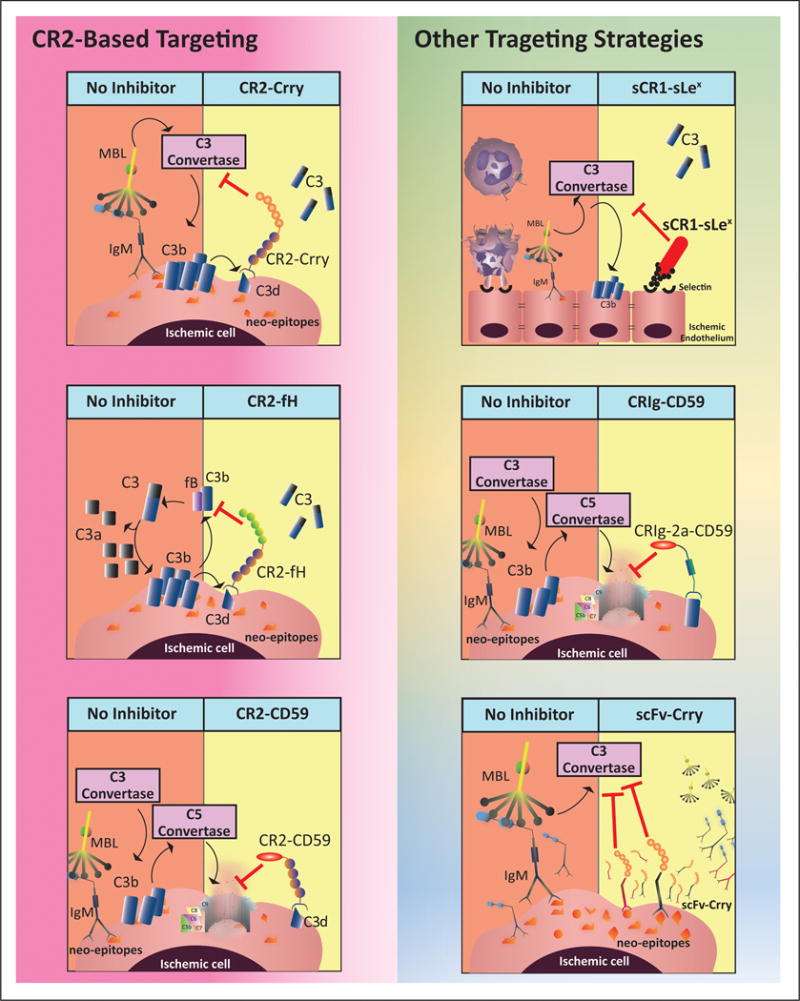
Mechanism of action of different site-targeted complement inhibitors in the context of ischemic stroke
Use of targeted C inhibitors in the context of stroke
Early studies using a site-targeted complement inhibitor in the context of cerebral IRI, characterized the protective effects of sCR1-sLex (see above) that binds to selectin-expressing cells and inhibits both neutrophil migration and complement activation (73). When administered prior to MCAO in mice, sCR1sLex specifically localized to the cerebral vasculature following ischemic stroke, and was significantly more effective at providing protection than its untargeted counterpart, sCR1 (77). However, sCR1sLex failed to provide neuroprotection following ischemic stroke in neonatal rats (78) and non-human primates (79), and to our knowledge has not been developed further.
The only other site-targeted approach of complement inhibition applied to the treatment of ischemic stroke is our CR2 targeting strategy, which is the focus of the remainder of this review. It was shown that a single dose of CR2-Crry administered 60 min after MCAO and 30 min of reperfusion, resulted in a similar level of protection as C3 deficiency at 24 hrs. post-stroke. CR2-Crry treatment was associated with a significant reduction in IgM and complement deposition in the brain, lower levels of P-selectin, decreased formation of microthrombi, and reduced neutrophil infiltration (48).
Subsequent studies demonstrated that CR2-fH, an alternative pathway specific inhibitor, similarly provided protection in the acute phase after stroke, as did deficiency in fB, an alternative pathway protein (42), implicating this pathway as an important mediator of stroke pathogenesis. In this study, we also investigated the effects of combined C1q/MBL deficiency and C6 deficiency on outcomes after stroke. C1q/MBL deficiency was highly protective, supporting previous studies indicating an important role for the lectin pathway (34, 38, 40, 80, 81), and consistent with IgM-mediated activation of complement (see above). C6 deficiency had no effect on acute outcome after ischemic stroke, indicating that the MAC is not involved in mediating injury in the model used (60 min MCAO, male C57BL/6 mice). Of note, however, deficiency of CD59a, an inhibitor of MAC formation, exacerbated injury in mice subjected to 30 min MCAO, indicating that the MAC may contribute to injury in less severe settings (52).
Given the evidence for a role of complement in reparatory and homeostatic mechanisms during stroke recovery, we further investigated how complement deficiency and targeted complement inhibition influenced sub-acute outcome measures (47). We showed that although C3 deficiency, CR2-Crry or CR2-fH treatment all had comparable protective effects in the acute phase after stroke (24 hrs.), only CR2-fH provided sustained neuroprotection into the subacute phase (7 days) (47). In fact, C3 deficient and CR2-Crry treated animals showed a rebound increase in infarct volume at 7 days compared to 24 hrs. Nevertheless, CR2-Crry was associated with a lower extent of cell death and microglial activation compared to C3 deficiency, a finding that suggests transient inhibition of complement by CR2-Crry induces an improved anti-inflammatory response compared to C3 deficiency. On the other hand, compared to C3 deficiency and CR2-Crry treatment, CR2-fH treatment was associated with higher SVZ neurogenesis, more pronounced neuronal migration, and enhanced cognitive outcomes at 7 days after stroke. Live animal fluorescence tomography demonstrated that CR2-fH targeted specifically to the ischemic brain with a tissue half-life of a little over 2 days (Fig 3).
To investigate a potential reason for this difference between CR2-Crry and CR2-fH treatment, we assayed for C5a in brain homogenates 3 days after stroke, since the anaphylatoxins have been implicated in promoting neurogenesis (60, 82). Although C5a levels were significantly reduced in CR2-fH treated mice compared to PBS treated controls, they were higher than in CR2-Crry treated mice, which had almost undetectable levels of C5a. These data are consistent with the alternative pathway amplification loop being a major contributor to post-stroke pathology, while inhibition of this pathway allows for residual complement activation via the lectin and/or classical pathway that generates anaphylatoxins for regenerative processes in the subacute phase. It is possible that low level deposition of complement opsonins generated in the subacute phase via the lectin/classical pathway may also play a role in recovery, for example via clearance of dead cells and debris or even synaptic remodeling.
Complement-dependent neuroprotective mechanisms are not well understood, but studies on cerebral IRI and neurodegenerative diseases have provided insight into potential mechanisms involved in the protective effects of complement inhibitors (summarized in Fig. 5). In the acute phase after stroke, inhibition of complement opsonization is associated with increased neuronal density and decreased cell death in the ischemic penumbra (47, 83, 84), a finding that can potentially be explained by the inhibition of neuronal apoptosis and phagocytic activity of CNS-resident and hematogenous phagocytes. In addition, inhibition of either opsonins or anaphylatoxins using targeted complement inhibitors or a C3aR antagonist is associated with a significant reduction in neutrophil and granulocyte infiltration to the ischemic penumbra, and a reduction in microgliosis and astrogliosis, in both the acute and sub-acute phases after ischemic stroke (42, 47, 77, 85). Collectively, these effects explain the robust acute reduction in functional deficit after stroke with complement inhibition. However, we have also investigated the effects of acute complement inhibition on subacute outcome measures, and found that acute inhibition with CR2-fH resulted in a significant reduction in inflammatory cytokine levels, increased expression of neuronal growth factors, and concurrent elevation of neuroblast adhesion molecules at 7 days after stroke (47). Both TNF-alpha and C3d have been shown to inhibit neurogenesis after brain injury (46), and taking the findings together, we hypothesize that the effect of complement inhibition on recovery mechanisms is primarily through inhibition of inflammation which releases a brake on recovery and growth-promoting processes (Fig. 5).
Figure 5. Pathophysiological responses after ischemic stroke and its modulation by targeted inhibition of complement amplification.
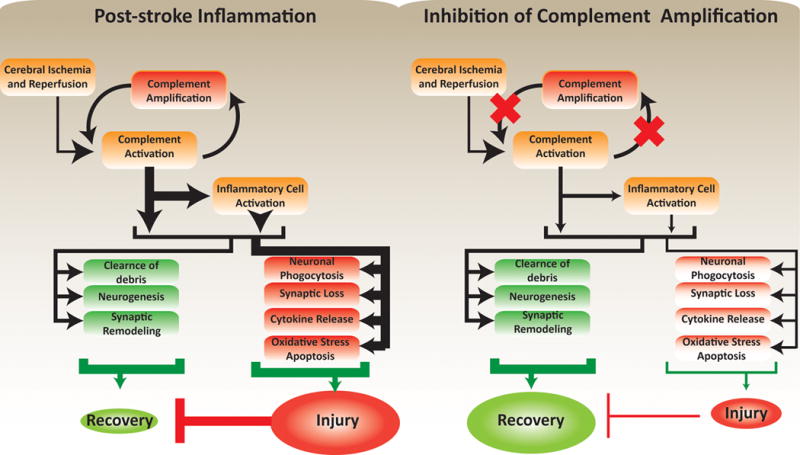
Inhibition of complement amplification reduces inflammation-associated neuropathology. The reduced injury lessens the inhibitory effects of injury on regeneration, leading to optimal recovery.
Concluding Remarks
Development and application of complement inhibitory strategies in the context of ischemic stroke has not only led to the characterization of novel therapeutic candidates with potentially better safety profiles than t-PA, but also has provided key insights into the pathophysiological role of complement in promoting injury and recovery after stroke. From a therapeutic standpoint, there is a requirement for additional and rigorous pre-clinical characterization in multiple models. Also, it will be necessary to investigate complement inhibition, and indeed any other neuroprotective strategy, in the context of t-PA therapy as a standard of care. At a minimum, it will be necessary to show that there is no contraindication for the combined use of complement inhibition and thrombolytic therapy. There are also many important, yet unanswered questions concerning the role of complement in post-stroke pathology. For example, how CNS-derived versus serum-derived complement contributes to injury, how each complement activation product is involved in injury versus repair mechanisms, whether and how complement is involved in neuronal circuit remodeling during recovery, and whether targeting the inflamed endothelium during chronic stroke after BBB repair will aid recovery.
Acknowledgments
This work was supported by grants from the NIH (1P20GM109040), the Department of Veterans Affairs (Merit Award 1I01RX001141 and 1BX001218), and an American Heart Association Predoctoral Fellowship to AA (15PRE25250009). We thank Zahraa Alawieh for assistance with illustrations.
Footnotes
The authors have no conflicts of interest
References
- 1.Mozaffarian Dariush, et al. Executive Summary: Heart Disease and Stroke Statistics—2016 Update A Report From the American Heart Association. Circulation. 2016;133.4:447–454. doi: 10.1161/CIR.0000000000000366. APA. [DOI] [PubMed] [Google Scholar]
- 2.Kochanek K, Murphy S, Xu J, Arias E. Mortality in the United States, 2013. Hyattsville, MD: National Center for Health Statistics; 2014. (NCHS data brief, no. 178). 2014. [PubMed] [Google Scholar]
- 3.Murray CJ, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2197–2223. doi: 10.1016/S0140-6736(12)61689-4. [DOI] [PubMed] [Google Scholar]
- 4.Towfighi A, Saver JL. Stroke declines from third to fourth leading cause of death in the United States: historical perspective and challenges ahead. Stroke. 2011;42:2351–2355. doi: 10.1161/STROKEAHA.111.621904. [DOI] [PubMed] [Google Scholar]
- 5.Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med. 1995;333:1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 6.Barber PA, Zhang J, Demchuk AM, Hill MD, Buchan AM. Why are stroke patients excluded from TPA therapy? An analysis of patient eligibility. Neurology. 2001;56:1015–1020. doi: 10.1212/wnl.56.8.1015. [DOI] [PubMed] [Google Scholar]
- 7.Heuschmann PU, et al. Frequency of thrombolytic therapy in patients with acute ischemic stroke and the risk of in-hospital mortality: the German Stroke Registers Study Group. Stroke. 2003;34:1106–1113. doi: 10.1161/01.STR.0000065198.80347.C5. [DOI] [PubMed] [Google Scholar]
- 8.Langhorne P, Coupar F, Pollock A. Motor recovery after stroke: a systematic review. Lancet Neurol. 2009;8:741–754. doi: 10.1016/S1474-4422(09)70150-4. [DOI] [PubMed] [Google Scholar]
- 9.Murphy TH, Corbett D. Plasticity during stroke recovery: from synapse to behaviour. Nature reviews Neuroscience. 2009;10:861–872. doi: 10.1038/nrn2735. [DOI] [PubMed] [Google Scholar]
- 10.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nature medicine. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindsberg PJ, et al. Complement activation in the central nervous system following blood-brain barrier damage in man. Ann Neurol. 1996;40:587–596. doi: 10.1002/ana.410400408. [DOI] [PubMed] [Google Scholar]
- 12.Pedersen ED, Loberg EM, Vege E, Daha MR, Maehlen J, Mollnes TE. In situ deposition of complement in human acute brain ischaemia. Scandinavian journal of immunology. 2009;69:555–562. doi: 10.1111/j.1365-3083.2009.02253.x. [DOI] [PubMed] [Google Scholar]
- 13.Stokowska A, et al. Cardioembolic and small vessel disease stroke show differences in associations between systemic C3 levels and outcome. PLoS One. 2013;8:e72133. doi: 10.1371/journal.pone.0072133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pedersen ED, Waje-Andreassen U, Vedeler CA, Aamodt G, Mollnes TE. Systemic complement activation following human acute ischaemic stroke. Clinical and experimental immunology. 2004;137:117–122. doi: 10.1111/j.1365-2249.2004.02489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Napoli M. Systemic complement activation in ischemic stroke. Stroke. 2001;32:1443–1448. doi: 10.1161/01.str.32.6.1443-a. [DOI] [PubMed] [Google Scholar]
- 16.Mehta N, et al. Platelet C4d is associated with acute ischemic stroke and stroke severity. Stroke. 2008;39:3236–3241. doi: 10.1161/STROKEAHA.108.514687. [DOI] [PubMed] [Google Scholar]
- 17.Zhang ZG, et al. Prognostic value of mannose-binding lectin: 90-day outcome in patients with acute ischemic stroke. Mol Neurobiol. 2015;51:230–239. doi: 10.1007/s12035-014-8682-0. [DOI] [PubMed] [Google Scholar]
- 18.Zhang B, Yang N, Gao C. Is plasma C3 and C4 levels useful in young cerebral ischemic stroke patients? Associations with prognosis at 3 months. Journal of thrombosis and thrombolysis. 2015;39:209–214. doi: 10.1007/s11239-014-1100-7. [DOI] [PubMed] [Google Scholar]
- 19.Alawieh A, Elvington A, Tomlinson S. Complement in the Homeostatic and Ischemic Brain. Frontiers in immunology. 2015;6:417. doi: 10.3389/fimmu.2015.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alawieh A, Narang A, Tomlinson S. Complementing regeneration. Oncotarget. 2015;6:21769–21770. doi: 10.18632/oncotarget.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holers VM, Rohrer B, Tomlinson S. CR2-mediated targeting of complement inhibitors: bench-to-bedside using a novel strategy for site-specific complement modulation. Advances in experimental medicine and biology. 2013;735:137–154. doi: 10.1007/978-1-4614-4118-2_9. [DOI] [PubMed] [Google Scholar]
- 22.Amara U, et al. Interaction between the coagulation and complement system. Advances in experimental medicine and biology. 2008;632:71–79. doi: 10.1007/978-0-387-78952-1_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huber-Lang M, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nature medicine. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 24.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nature reviews Immunology. 2009;9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 25.Elvington A, et al. Pathogenic natural antibodies propagate cerebral injury following ischemic stroke in mice. Journal of immunology. 2012;188:1460–1468. doi: 10.4049/jimmunol.1102132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Atkinson C, et al. Targeting pathogenic postischemic self-recognition by natural IgM to protect against posttransplantation cardiac reperfusion injury. Circulation. 2015;131:1171–1180. doi: 10.1161/CIRCULATIONAHA.114.010482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulik L, et al. Pathogenic natural antibodies recognizing annexin IV are required to develop intestinal ischemia-reperfusion injury. Journal of immunology. 2009;182:5363–5373. doi: 10.4049/jimmunol.0803980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weiser MR, et al. Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. The Journal of experimental medicine. 1996;183:2343–2348. doi: 10.1084/jem.183.5.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jordan JE, Montalto MC, Stahl GL. Inhibition of mannose-binding lectin reduces postischemic myocardial reperfusion injury. Circulation. 2001;104:1413–1418. doi: 10.1161/hc3601.095578. [DOI] [PubMed] [Google Scholar]
- 30.Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. Journal of neurochemistry. 2010;112:733–743. doi: 10.1111/j.1471-4159.2009.06494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tenner AJ, Robinson SL, Ezekowitz RA. Mannose binding protein (MBP) enhances mononuclear phagocyte function via a receptor that contains the 126,000 M(r) component of the C1q receptor. Immunity. 1995;3:485–493. doi: 10.1016/1074-7613(95)90177-9. [DOI] [PubMed] [Google Scholar]
- 32.McGrath FD, Brouwer MC, Arlaud GJ, Daha MR, Hack CE, Roos A. Evidence that complement protein C1q interacts with C-reactive protein through its globular head region. Journal of immunology. 2006;176:2950–2957. doi: 10.4049/jimmunol.176.5.2950. [DOI] [PubMed] [Google Scholar]
- 33.Kuraya M, Ming Z, Liu X, Matsushita M, Fujita T. Specific binding of L-ficolin and H-ficolin to apoptotic cells leads to complement activation. Immunobiology. 2005;209:689–697. doi: 10.1016/j.imbio.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Orsini F, et al. Targeting mannose-binding lectin confers long-lasting protection with a surprisingly wide therapeutic window in cerebral ischemia. Circulation. 2012;126:1484–1494. doi: 10.1161/CIRCULATIONAHA.112.103051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mocco J, et al. Complement component C3 mediates inflammatory injury following focal cerebral ischemia. Circulation research. 2006;99:209–217. doi: 10.1161/01.RES.0000232544.90675.42. [DOI] [PubMed] [Google Scholar]
- 36.Wang ZY, Sun ZR, Zhang LM. The relationship between serum mannose-binding lectin levels and acute ischemic stroke risk. Neurochemical research. 2014;39:248–253. doi: 10.1007/s11064-013-1214-x. [DOI] [PubMed] [Google Scholar]
- 37.Osthoff M, et al. Mannose-binding lectin deficiency is associated with smaller infarction size and favorable outcome in ischemic stroke patients. PLoS One. 2011;6:e21338. doi: 10.1371/journal.pone.0021338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morrison H, et al. The contribution of mannose binding lectin to reperfusion injury after ischemic stroke. Current neurovascular research. 2011;8:52–63. doi: 10.2174/156720211794520260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zangari R, et al. Early ficolin-1 is a sensitive prognostic marker for functional outcome in ischemic stroke. Journal of neuroinflammation. 2016;13:16. doi: 10.1186/s12974-016-0481-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cervera A, et al. Genetically-defined deficiency of mannose-binding lectin is associated with protection after experimental stroke in mice and outcome in human stroke. PLoS One. 2010;5:e8433. doi: 10.1371/journal.pone.0008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fust G, et al. Low ficolin-3 levels in early follow-up serum samples are associated with the severity and unfavorable outcome of acute ischemic stroke. Journal of neuroinflammation. 2011;8:185. doi: 10.1186/1742-2094-8-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elvington A, et al. The alternative complement pathway propagates inflammation and injury in murine ischemic stroke. Journal of immunology. 2012;189:4640–4647. doi: 10.4049/jimmunol.1201904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nature reviews Neuroscience. 2014;15:209–216. doi: 10.1038/nrn3710. [DOI] [PubMed] [Google Scholar]
- 44.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annual review of neuroscience. 2012;35:369–389. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 45.Hajishengallis G, Lambris JD. Crosstalk pathways between Toll-like receptors and the complement system. Trends in immunology. 2010;31:154–163. doi: 10.1016/j.it.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moriyama M, et al. Complement receptor 2 is expressed in neural progenitor cells and regulates adult hippocampal neurogenesis. J Neurosci. 2011;31:3981–3989. doi: 10.1523/JNEUROSCI.3617-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alawieh A, et al. Modulation of post-stroke degenerative and regenerative processes and subacute protection by site-targeted inhibition of the alternative pathway of complement. Journal of neuroinflammation. 2015;12:247. doi: 10.1186/s12974-015-0464-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Atkinson C, et al. Complement-dependent P-selectin expression and injury following ischemic stroke. Journal of immunology. 2006;177:7266–7274. doi: 10.4049/jimmunol.177.10.7266. [DOI] [PubMed] [Google Scholar]
- 49.Ducruet AF, et al. C3a receptor modulation of granulocyte infiltration after murine focal cerebral ischemia is reperfusion dependent. J Cereb Blood Flow Metab. 2008;28:1048–1058. doi: 10.1038/sj.jcbfm.9600608. [DOI] [PubMed] [Google Scholar]
- 50.Garrett MC, et al. Synergistic neuroprotective effects of C3a and C5a receptor blockade following intracerebral hemorrhage. Brain Res. 2009;1298:171–177. doi: 10.1016/j.brainres.2009.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pedersen ED, et al. Expression of complement regulators and receptors on human NT2-N neurons–effect of hypoxia and reoxygenation. Molecular immunology. 2007;44:2459–2468. doi: 10.1016/j.molimm.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 52.Harhausen D, et al. Membrane attack complex inhibitor CD59a protects against focal cerebral ischemia in mice. Journal of neuroinflammation. 2010;7:15. doi: 10.1186/1742-2094-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gasque P, Dean YD, McGreal EP, VanBeek J, Morgan BP. Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology. 2000;49:171–186. doi: 10.1016/s0162-3109(00)80302-1. [DOI] [PubMed] [Google Scholar]
- 54.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nature immunology. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gasque P, Fontaine M, Morgan BP. Complement expression in human brain. Biosynthesis of terminal pathway components and regulators in human glial cells and cell lines. Journal of immunology. 1995;154:4726–4733. [PubMed] [Google Scholar]
- 56.Levi-Strauss M, Mallat M. Primary cultures of murine astrocytes produce C3 and factor B, two components of the alternative pathway of complement activation. Journal of immunology. 1987;139:2361–2366. [PubMed] [Google Scholar]
- 57.Terai K, Walker DG, McGeer EG, McGeer PL. Neurons express proteins of the classical complement pathway in Alzheimer disease. Brain Res. 1997;769:385–390. doi: 10.1016/s0006-8993(97)00849-4. [DOI] [PubMed] [Google Scholar]
- 58.Gasque P, Morgan BP. Complement regulatory protein expression by a human oligodendrocyte cell line: cytokine regulation and comparison with astrocytes. Immunology. 1996;89:338–347. doi: 10.1046/j.1365-2567.1996.d01-756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cole DS, Hughes TR, Gasque P, Morgan BP. Complement regulator loss on apoptotic neuronal cells causes increased complement activation and promotes both phagocytosis and cell lysis. Molecular immunology. 2006;43:1953–1964. doi: 10.1016/j.molimm.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 60.Rahpeymai Y, et al. Complement: a novel factor in basal and ischemia-induced neurogenesis. The EMBO journal. 2006;25:1364–1374. doi: 10.1038/sj.emboj.7601004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Osaka H, Mukherjee P, Aisen PS, Pasinetti GM. Complement-derived anaphylatoxin C5a protects against glutamate-mediated neurotoxicity. Journal of cellular biochemistry. 1999;73:303–311. [PubMed] [Google Scholar]
- 62.van Beek J, et al. Complement anaphylatoxin C3a is selectively protective against NMDA-induced neuronal cell death. Neuroreport. 2001;12:289–293. doi: 10.1097/00001756-200102120-00022. [DOI] [PubMed] [Google Scholar]
- 63.Cudrici C, et al. C5b-9 terminal complex protects oligodendrocytes from apoptotic cell death by inhibiting caspase-8 processing and up-regulating FLIP. Journal of immunology. 2006;176:3173–3180. doi: 10.4049/jimmunol.176.5.3173. [DOI] [PubMed] [Google Scholar]
- 64.Song H, He C, Knaak C, Guthridge JM, Holers VM, Tomlinson S. Complement receptor 2-mediated targeting of complement inhibitors to sites of complement activation. The Journal of clinical investigation. 2003;111:1875–1885. doi: 10.1172/JCI17348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Atkinson C, et al. Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection. The Journal of clinical investigation. 2005;115:2444–2453. doi: 10.1172/JCI25208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang Y, Qiao F, Atkinson C, Holers VM, Tomlinson S. A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury. Journal of immunology. 2008;181:8068–8076. doi: 10.4049/jimmunol.181.11.8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vargas M, et al. Clinical consequences of infection in patients with acute stroke: is it prime time for further antibiotic trials? Stroke. 2006;37:461–465. doi: 10.1161/01.STR.0000199138.73365.b3. [DOI] [PubMed] [Google Scholar]
- 68.Kumar S, Selim MH, Caplan LR. Medical complications after stroke. Lancet Neurol. 2010;9:105–118. doi: 10.1016/S1474-4422(09)70266-2. [DOI] [PubMed] [Google Scholar]
- 69.Borlongan CV, Rodrigues AA, Jr, Oliveira MC. Breaking the barrier in stroke: what should we know? A mini-review. Current pharmaceutical design. 2012;18:3615–3623. doi: 10.2174/138161212802002670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Emsley HC, et al. An early and sustained peripheral inflammatory response in acute ischaemic stroke: relationships with infection and atherosclerosis. J Neuroimmunol. 2003;139:93–101. doi: 10.1016/s0165-5728(03)00134-6. [DOI] [PubMed] [Google Scholar]
- 71.Beamer NB, Coull BM, Clark WM, Briley DP, Wynn M, Sexton G. Persistent inflammatory response in stroke survivors. Neurology. 1998;50:1722–1728. doi: 10.1212/wnl.50.6.1722. [DOI] [PubMed] [Google Scholar]
- 72.Ford ES, Giles WH. Serum C-reactive protein and self-reported stroke: findings from the Third National Health and Nutrition Examination Survey. Arteriosclerosis, thrombosis, and vascular biology. 2000;20:1052–1056. doi: 10.1161/01.atv.20.4.1052. [DOI] [PubMed] [Google Scholar]
- 73.Law SK, Lichtenberg NA, Levine RP. Covalent binding and hemolytic activity of complement proteins. Proc Natl Acad Sci U S A. 1980;77:7194–7198. doi: 10.1073/pnas.77.12.7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marshall KM, He S, Zhong Z, Atkinson C, Tomlinson S. Dissecting the complement pathway in hepatic injury and regeneration with a novel protective strategy. The Journal of experimental medicine. 2014;211:1793–1805. doi: 10.1084/jem.20131902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sekine H, et al. The benefit of targeted and selective inhibition of the alternative complement pathway for modulating autoimmunity and renal disease in MRL/lpr mice. Arthritis and rheumatism. 2011;63:1076–1085. doi: 10.1002/art.30222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ruseva MM, Ramaglia V, Morgan BP, Harris CL. An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice. Proc Natl Acad Sci U S A. 2015;112:14319–14324. doi: 10.1073/pnas.1513698112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang J, et al. Neuronal protection in stroke by an sLe x-glycosylated complement inhibitory protein. Science. 1999;285:595–599. doi: 10.1126/science.285.5427.595. [DOI] [PubMed] [Google Scholar]
- 78.Imm MD, Feldhoff PW, Feldhoff RC, Lassiter HA. The administration of complement component C9 augments post-ischemic cerebral infarction volume in neonatal rats. Neurosci Lett. 2002;325:175–178. doi: 10.1016/s0304-3940(02)00271-9. [DOI] [PubMed] [Google Scholar]
- 79.Ducruet AF, et al. Pre-clinical evaluation of an sLe x-glycosylated complement inhibitory protein in a non-human primate model of reperfused stroke. Journal of medical primatology. 2007;36:375–380. doi: 10.1111/j.1600-0684.2007.00213.x. [DOI] [PubMed] [Google Scholar]
- 80.de la Rosa X, et al. Mannose-binding lectin promotes local microvascular thrombosis after transient brain ischemia in mice. Stroke. 2014;45:1453–1459. doi: 10.1161/STROKEAHA.113.004111. [DOI] [PubMed] [Google Scholar]
- 81.Ducruet AF, et al. The Neuroprotective Effect of Genetic Mannose-binding Lectin Deficiency is not Sustained in the Sub-acute Phase of Stroke. Translational stroke research. 2011;2:588–599. doi: 10.1007/s12975-011-0104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shinjyo N, Stahlberg A, Dragunow M, Pekny M, Pekna M. Complement-derived anaphylatoxin C3a regulates in vitro differentiation and migration of neural progenitor cells. Stem cells. 2009;27:2824–2832. doi: 10.1002/stem.225. [DOI] [PubMed] [Google Scholar]
- 83.De Simoni MG, Rossi E, Storini C, Pizzimenti S, Echart C, Bergamaschini L. The powerful neuroprotective action of C1-inhibitor on brain ischemia-reperfusion injury does not require C1q. The American journal of pathology. 2004;164:1857–1863. doi: 10.1016/S0002-9440(10)63744-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Storini C, et al. C1-inhibitor protects against brain ischemia-reperfusion injury via inhibition of cell recruitment and inflammation. Neurobiology of disease. 2005;19:10–17. doi: 10.1016/j.nbd.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 85.Costa C, et al. Role of complement component C5 in cerebral ischemia/reperfusion injury. Brain Res. 2006;1100:142–151. doi: 10.1016/j.brainres.2006.05.029. [DOI] [PubMed] [Google Scholar]