

Abstract
Aim: Iron accumulation in foam cells was previously shown to be involved in atherogenesis. However, the mechanism for iron accumulation was not clarified. Ceruloplasmin (Cp) is an important factor in cellular iron efflux and was found to be downregulated in atherosclerotic plaques in our previous study. The current study is to investigate the role of Cp in atherosclerosis.
Methods: We used RAW264.7 cells, a well-accepted cell model of atherosclerosis, which were treated with lipopolysaccharides (LPS), ferric ammonium citrate (FAC) or deferoxamine, and oxidized low density lipoprotein (ox-LDL) to detect the regulation of Cp and its influence in iron efflux and lipid accumulation using biochemical and histological assays.
Results: Our results showed that the Cp protein level increased after 200-µM FAC treatment in LPS-activated RAW264.7 cells. Ox-LDL treatment (50 µg/ml) moderately reduced both mRNA and protein levels and ferroxidase activity of Cp (p < 0.05). No significant difference was observed in the expression of ferritin and ferroportin, two important iron-related proteins for iron storage and efflux, respectively, after ox-LDL treatment. However, co-treatment with ox-LDL and FAC drastically reduced the expression of Cp. Accordingly, the ferroxidase activities simultaneously decreased, whereas the protein levels of Ft and Fpn1 significantly increased, indicating further iron accumulation. Moreover, co-treatment with FAC and ox-LDL enhanced the accumulation of cholesterol compared with ox-LDL-only treatment to trigger apoptosis.
Conclusion: Our findings suggest that physiological interaction of iron and lipid obstructs iron efflux and accelerates the lipid accumulation in macrophages during foam cell formation, which implicates the role of iron in the pathology of atherosclerosis.
Keywords: Atherosclerosis, Foam cell formation, Iron efflux, Ceruloplasmin
Introduction
Atherosclerosis is a systemic multifactorial disease characterized by lipid deposition and hardening of the vascular walls of the large- and medium-sized arteries. The recruitment of monocytes from circulation to the arterial wall is a noteworthy feature of atherogenesis1). A few decades ago, Sullivan proposed the iron hypothesis2), which suggests that iron deficiency in lipid-laden macrophages, namely foam cells, plays a protective role against atherosclerosis. In foam cells, overloaded iron catalyzes the generation of reactive oxygen species (ROS) and could therefore promote lipid oxidation and contribute to atherosclerotic plaque instability3). Ferroportin (Fpn1), encoded by SLC40A1, is the only known mammalian free iron exporter expressed by macrophages and intestinal epithelial cells4). Reducing intracellular free iron levels within macrophages by increasing the expression of Fpn1 may be a promising strategy to promote macrophage cholesterol efflux and steady the plaque.
Iron taken up by epithelial cells is either sequestered to iron storage in ferritin (in ferric form) or transported across the cell membrane (in ferrous form) by Fpn1 into plasma5). Fpn1 is systemically degraded through ubiquitination after binding to a hepatic hormone, hepcidin6). Clinical studies demonstrated that serum hepcidin was associated with the presence of atherosclerosis7–9); however, this is still controversial10). It was also reported that pharmacological suppression of hepcidin could increase lipid efflux and reduce foam cell formation in ApoE−/− mice11). However, a recent in vivo study in a mouse model showed that the specific mutation of Fpn1 in macrophages had no significant influence on atherogenesis12). Therefore, there may be other molecules involved in iron deposition in foam cells.
Fpn1 is not the only key protein in macrophage iron efflux. Ceruloplasmin (Cp) is another factor that is very important in this process. After being transported by Fpn1 to the outside of the cells, ferrous iron needs to be oxidized to ferric form by ferroxidases such as Cp and then released to bind transferrin (Tf), which is the primary carrier of soluble iron in plasma13). Absence of functional Cp (aceruloplasminemia in human or Cp knockout in mice) leads to iron accumulation in several tissues including the liver and brain14). The ferroxidase activity of Cp prevents ferrous iron-mediated production of ROS and thus Cp can have a potent antioxidant activity15). Cp is a positive acute-phase reactant that is involved in host defense and repair processes mediated by the immune system16). As a circulating antioxidant and oxygen free radical scavenger17), Cp appears to be an effective protective agent against tissue injuries generated by oxygen free radicals. More recently, we found that the expression of Cp is markedly reduced in atherosclerotic plaques18). Therefore, we propose that Cp may function to delay the progression of atherosclerosis.
The main purpose of this study was to investigate the influence of iron and lipid deposition on the expression of Cp in RAW264.7 cells and its role in foam cell formation. Our results support the notion that iron with lipid, and not alone, downregulates the protein levels of Cp in macrophage and further accelerates lipid-laden macrophage foam cell formation.
Materials and Methods
Cell Cultures
Cell culture growth medium, antibiotics (penicillin and streptomycin), and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific® (Waltham, MA, USA). The RAW264.7 cell line was purchased from the American Type Culture Collection® (ATCC®, Rockefeller, MD, USA). RAW 267.4 macrophage cells were maintained at 37°C in a 5% CO2 incubator in a basal medium, which consisted of RPMI 1640, 10% FBS, 10 mM HEPES (4-(2-Hydroxyethyl) piperazine-1-ethanesulfonic acid) Buffer, 2 mM L-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. Cells (1 × 105 cells/ml) were plated 1 day before experiment in 6-well plates (1 × 105 cells/ml) or 3-cm-diameter culture plates.
Cell Treatment
Ferric ammonium citrate (FAC), deferoxamine (DFO), lipopolysaccharides (LPS), and oxidized low density lipoprotein (ox-LDL) were purchased from Sigma-Aldrich® (St. Louis, MO, USA). Cell iron interference comprised incubating cells for 24 h in the presence of 200 µM FAC (in H2O) or in the presence of 100 µM DFO (in H2O) after 12-h stimulation of LPS (50 ng/ml in H2O). To format the foam cell, RAW264.7 cells were treated with ox-LDL (50 µg/ml) for 24 h19) after 12-h activation by LPS.
RNA Isolation and Real-Time Quantitative PCR Analyses
Total cellular RNA was isolated from RAW264.7 cells using Trizol (Invitrogen™, Carlsbad, CA, USA) and was reverse-transcribed to create cDNA. cDNA was quantified by quantitative polymerase chain reaction on a StepOne Plus system (Applied Biosystems®, Foster, CA, USA). Primers for Cp were designed spanning the intron-exon boundaries to amplify the corresponding mRNAs and minimize amplification of potentially contaminating genomic DNA. For Cp, the forward primer was 5′-GTAAACAAAGACAACGAGGAA T-3′ and the reverse primer was 5′-TATTTCATTCAGCCAGACTTAG-3′20). mRNA levels were normalized to those of GAPDH.
Western Blot Analysis
Western blot was performed as previously described21). Briefly, cells were harvested and completely homogenized using lysis buffer (Thermo Fisher Scientific®, Waltham, MA, USA) and were then placed on ice for 10 min. After centrifugation at 12,000 rpm for 10 min at 4°C, the supernatant was collected. Protein concentration was determined with a BCA kit (Beyotime Biotechnology®, Shanghai, China). Proteins (35 µg) were denatured at 95°C for 5 min in SDS and β-mercaptoethanol-containing sample buffer, separated on 15% gradient SDS-PAGE, and electro-blotted onto nitrocellulose membranes (Pall® Corporation, New York, NY, USA). The blots were incubated overnight at 4°C in 5% (w/v) skim milk with the primary antibodies for ferritin (Abcam®, Cambridge, MA, UK) (1:1000), Cp (GeneTex®, San Antonio, TX, USA) (1:1000), transferrin receptors (TfR) (Santa Cruz Biotechnology®, Inc., Santa Cruz, CA, USA) (1:200), hephaestin (Hp) (GeneTex®) (1:1000), CD36 (ProteintechTM Group, Inc., Chicago, IL, USA) (1:1000), ATP-binding cassette transporter A1 (ABCA1) (Signalway Antibody® LLC, College Park, MD, USA) (1:500), B-cell lymphoma-2 (Bcl2) (Santa Cruz Biotechnology®, Inc.) (1:1000), Bax (Santa Cruz Biotechnology®, Inc.) (1:1000), β-actin (Abcam®) (1:1000), or GAPDH (Bioworld Technology®, St. Louis Park, MN, USA) (1:10000). Following this, the blots were incubated with HRP-conjugated secondary antibodies (goat anti-rabbit). Blotted protein bands were visualized by enhanced chemiluminescence Western blot detection reagents (Thermo Fisher Scientific®). Relative changes in protein expression were estimated from the mean pixel density using Image J software, normalized to that of β-actin or GAPDH.
Iron Staining
Iron staining was performed to detect cellular iron content as previously described22). RAW264.7 cells were seeded in 24-mm glass-bottom coverslip microscopy dishes. Cells were fixed using 4% paraformaldehyde in PBS for 10 min. Following this, dishes were washed with PBS and then incubated in the dark with freshly made Prussian blue staining solution (2% K4[Fe(CN)6] and 2% HCl) for 1 h. After being washed with PBS again, the dishes were incubated in the dark with freshly made diaminobenzidine solution (30 mg DAB tablet, 40 ml 1 M Tris pH 7.5, 1 ml 3% H2O2) for 1 h. The dishes were then washed with 1 × PBS twice and mounted in glycerinum. Photographs were obtained under a light microscope (at 400 × magnification) by an investigator blinded to the treatment. Five photographs from each dish were obtained, and quantitation was performed using Image J software.
Cholesterol Content
The content of lipids including total cholesterol (TC) and free cholesterol (FC) of ox-LDL-treated RAW264.7 cells were measured using enzymatic assay kits according to manufacturer instructions (Abcam®). The conjugated cholesterol was calculated as cholesteryl ester (CE) using the following formula: CE = TC − FC.
Ferroxidase Enzyme Activity Assay
Cp catalyzes the transfer of electrons from Fe2+ through its T1–T3 Cu2 sites, resulting in a 4-electron reduction of O2 to 2H2O, forming Fe3+. Para-phenylenediamine (pPD) assay is a surrogate for Fe2+ oxidation and was used in the present study to measure the activity of Cp. As previously described23), pPD can be converted to a fused ring aromatic compound with the formula C18H18N6. It has an absorption maxima at ∼530 nm, which can be quantified by reading A530 in a spectrophotometer. Cells were lysed and proteins were harvested to perform the pPD assay.
Oil Red Stain
For oil red O staining, cells were seeded on 6-well plate and treated with FAC for 12 h, followed by ox-LDL for 24 h. After treatment, cells were fixed in 4% paraformaldehyde (10 min), washed with distilled water, and completely dried before applying 0.6% oil red O solution in 60:40 (v/v) isopropyl alcohols:H2O (room temperature, 30 min).
Flow Cytometric Analysis
RAW264.7 cells were treated with iron or/and ox-LDL as needed. Apoptotic cells were quantified by flow cytometric analysis (FACSCalibur®, Becton, Dickinson and Company, NJ, USA) using Annexin V-FITC and propidium iodide double staining using a detection kit (KeyGE N Biotech®, Nanjing, China) according to the manufacturer instructions.
Statistical Analysis
All statistical analyses were performed using Statistical Product and Service Solutions (SPSS) statistics for Windows (Release 21.0; SPSS®, Armonk, NY, USA). All data were subjected to one-way ANOVA. Differences between different samples were determined by the Fisher's LSD post-test. Statistical significance was inferred at p < 0.05. Quantitative analysis and comparison for band-intensity in western blot were based on the averages of four repeats.
Results
Treatment of FAC Upregulates the Expression of Cp in Activated RAW264.7 Cells
Previous studies have demonstrated that Cp was expressed in monocytic cells and can be induced by inflammation or iron loading24–26). Little data are available about the expression of Cp in macrophage-like RAW264.7 cells. Because RAW264.7 cells are well-known and well-accepted cell model to investigate atherosclerosis, we examined the iron effect on Cp expression in LPS-activated RAW264.7 cells. The results showed that iron content increased after FAC treatment, which was revealed by iron Perl's staining (Fig. 1A). Iron-related proteins were then determine d. The expression of ferritin (Ft) and Fpn1 were upregulated and TfR was downregulated by FAC treatment. On the contrary, the iron chelator DFO downregulated the expression of Ft and upregulated that of TfR (Fig. 1B), indicating that iron repletion reduces iron uptake, whereas iron depletion enhances iron uptake and diminishes the storage. LPS-alone treatment did not significantly change the iron status compared with the non-treated control. However, co-treatment with LPS and FAC made cells form more sharp spikes compared with FAC or LPS treatment only (Fig. 1A), suggesting that LPS-mediated activation of macrophages was further enhanced by iron for phagocytosis. Interestingly, the treatment with LPS and FAC could independently and/or synergistically induce the expression of Cp at the mRNA and protein levels and ferroxidase activity of Cp (Fig. 1C). This suggests that induced increase of Cp occurred in response to iron and/or LPS challenge for facilitating the release of iron from cells.
Fig. 1.
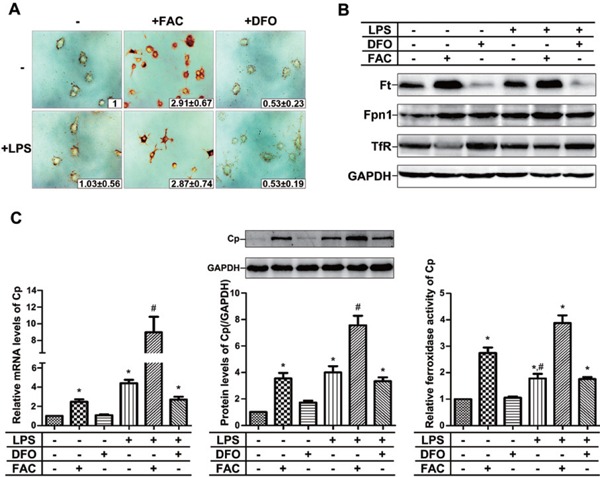
Both iron and LPS promote the expression and activities of ceruloplasmin
(A) Cellular iron Perl's stain. The relative values are shown (inset). (B) Western blot for iron related proteins, Ft, Fpn1, and TfR. (C) The expression and ferroxidase activity of Cp, revealed by real-time quantitative PCR, western blotting and ferroxidase enzyme activity assays. FAC: ferric ammonium citrate; DFO: deferoxamine; LPS: lipopolysaccharides; Ft: ferritin; Fpn1: Ferroportin 1; TfR: Transferrin receptor; Cp: ceruloplasmin; PCR: polymerase chain reaction. * p < 0.05 compared with PBS-treated group, # p < 0.05 compared with LPS-treated group.
Treatment of the Oxidized LDL Significantly Reduces the Expression of Cp in the Activated RAW264.7 Cells
Foam cells derived from macrophages are the main components in atherosclerotic plaques. We treated LPS-activated RAW264.7 cells with ox-LDL to mimic the formation of foam cells and determined the effect of ox-LDL on Cp expression. As shown in Fig. 2A, the cholesterol content, including TC, FC, and cholesterol ester, was significantly increased after the treatment of ox-LDL, indicating that RAW264.7 cells were transformed into foam cells. The expression of the iron-related proteins was then determined by western blotting. We found that the expression of Cp, but not Ft and Fpn1, was moderately and significantly reduced (down to 76.2 ± 10.3%) after the treatment of ox-LDL in LPS-activated RAW264.7 cells (Fig. 2B). Given the role of Cp in iron metabolism, this moderate reduction is likely not enough to change cellular iron status because of the unchanged protein levels of Ft and Fpn1.
Fig. 2.
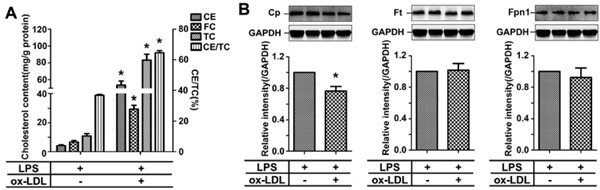
Uptake of the oxidized-LDL by activated RAW264.7 cells significantly reduces the expression of Cp
(A) The content of CE, FC, TC, and CE/TC(%) in LPS-activated RAW264.7 cells after the treatment of ox-LDL. (B) Representative western blots to show the effect of ox-LDL on Cp, Ft, and Fpn1 expression. ox-LDL: oxidized low density lipoprotein; TC: total cholesterol; FC: free cholesterol; CE: cholesteryl ester. * p < 0.05 compared with LPS-treated group.
Iron Overload Worsens Cp Reduction in Lipid-Laden Macrophages Leading to Obstructed Iron Efflux
Human atherosclerosis was always associated with micro-vessel hemorrhage and therefore, foam cells were often iron-overloading27). As the moderately reduced Cp expression had no significant influence on iron efflux in lipid-laden macrophages (so called foam cells) under the low and normal iron conditions (Fig. 2B), we then investigated Cp expression under high iron condition in foam cells. Strikingly, we found that not only the mRNA levels, but also protein levels of Cp dramatically decreased (Fig. 3A). This is opposite to that after treatment with FAC alone and is more severe than that after treatment with ox-LDL alone. Accordingly, the ferroxidase activities of Cp were reduced by co-treatment with iron and ox-LDL (Fig. 3A), suggesting that the iron efflux might be obstructed. As shown in Fig. 3B, the ferritin levels significantly increased in FAC + ox-LDL-treated group, compared with that in ox-LDL or FAC-treated group, indicating cellular iron accumulation. Although the protein levels of Fpn1 in FAC + ox-LDL and FAC groups were comparable (Fig. 3C), the remarkably reduced protein levels of Cp may have caused the higher ferritin levels in FAC + ox-LDL group than in FAC group.
Fig. 3.
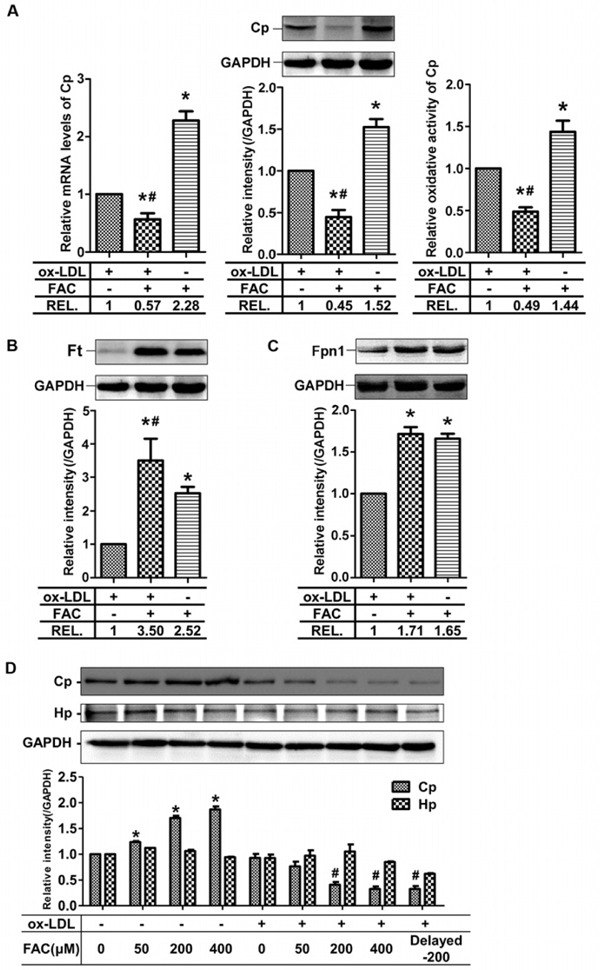
Physiological interaction of iron and ox-LDL accelerates the decrease of ceruloplasmin expression in LPS-treated RAW264.7 cells
(A) Co-effect of iron and ox-LDL on mRNA and protein levels and activity of Cp, (B) the protein levels of Ft, and (C) the protein levels of Fpn1. (D) Effects of ox-LDL on the expression of Cp in an iron dependent manner. Delayed-200 in the last lane means 2-h delayed treatment of FAC after ox-LDL addition. The treatment for the rest lanes is simultaneous addition of FAC and ox-LDL. Hp: hephaestin; REL: Relative intensity. * p < 0.05 vs. ox-LDL treated group, # p < 0.05 vs. FAC group in (A–C); * p < 0.05 vs. non-treated group, # p < 0.05 vs. ox-LDL-only group in (D).
Because of the positive correlation between FAC treatment and Cp expression and the negative correlation between FAC + ox-LDL treatment and Cp expression, we treated the cells with different concentration of iron with or without ox-LDL to see if the increase or decrease of Cp expression is iron-dependent. The results showed that Cp expression increased without ox-LDL and decreased with ox-LDL in an iron-dependent manner (Fig. 3D), which strongly supported the notion that ox-LDL completely reverses the effect of iron on Cp expression to make the cells accumulate more iron in the foam cells. We examined another ferroxidase, Hp, which showed constant expression in the presence of iron or iron + ox-LDL (Fig. 3D). To exclude the physical interaction of iron and ox-LDL, we treated RAW264.7 cells first with ox-LDL for 2 h then added iron. The expression of Cp is similar to that when iron and ox-LDL were added simultaneously (Fig. 3D, last lane).
Cp is found as a soluble isoform in plasma or as a membrane-associated isoform (GPI-Cp) or just as a cytosolic isoform in specific cell types. To see the cellular localization of Cp, immunofluorescent assay was performed. The results clearly showed the dotted and scattered Cp after FAC treatment, which was hardly visible in the control and FAC + ox-LDL groups (Fig. 4). Because we treated the cells with Triton X-100 to make the cells permeable in the immunofluorescent assay, more cytosolic Cp than membrane Cp was observed, which is in line with a previous report26). The cytosolic dotted Cp is supposed to be in lipid rafts as suggested26). Consistent with the above western blot results, co-treatment of iron with ox-LDL brought the level of Cp back to basic line (Fig. 4).
Fig. 4.
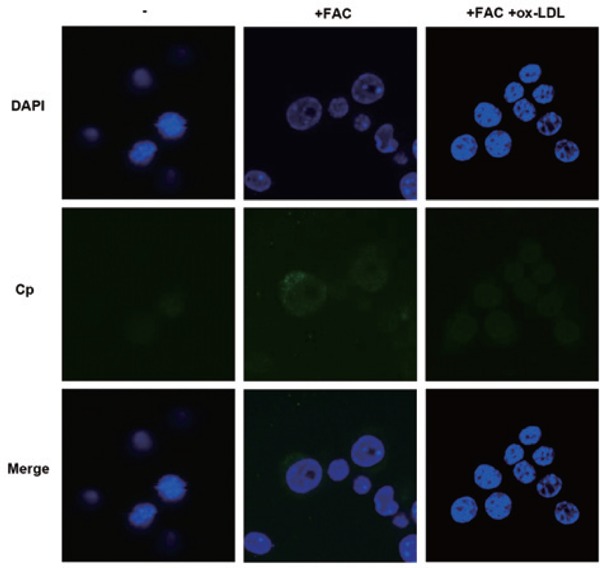
Cellular localization of Cp is determined in RAW264.7 cells
Immuno-fluorescence assays showed upregulated cellular expression of Cp by iron is reversed by the additon of ox-LDL in RAW264.7 cells. Most Cp is localized in the cytoplasm as shown by dots, which are suggested to be lipid rafts26). DAPI: 4′,6-diamidino-2-phenylindole.
Iron Overload Accelerates Ox-LDL-Mediated Foam Cell Formation in the Activated RAW264.7 Cells
To visualize the effect of iron overload on lipid content in ox-LDL-treated cells, oil-red staining was performed and cholesterol content was measured. The results showed that co-treatment of FAC and ox-LDL did not only make the macrophages take up more lipids (Fig. 5A and 5B), but also made the macrophages aggregated and deformed (Fig. 5B). This observation suggests that a strong physiological interaction between the overloaded iron and laden lipid brings about the damage of the foam cells.
Fig. 5.
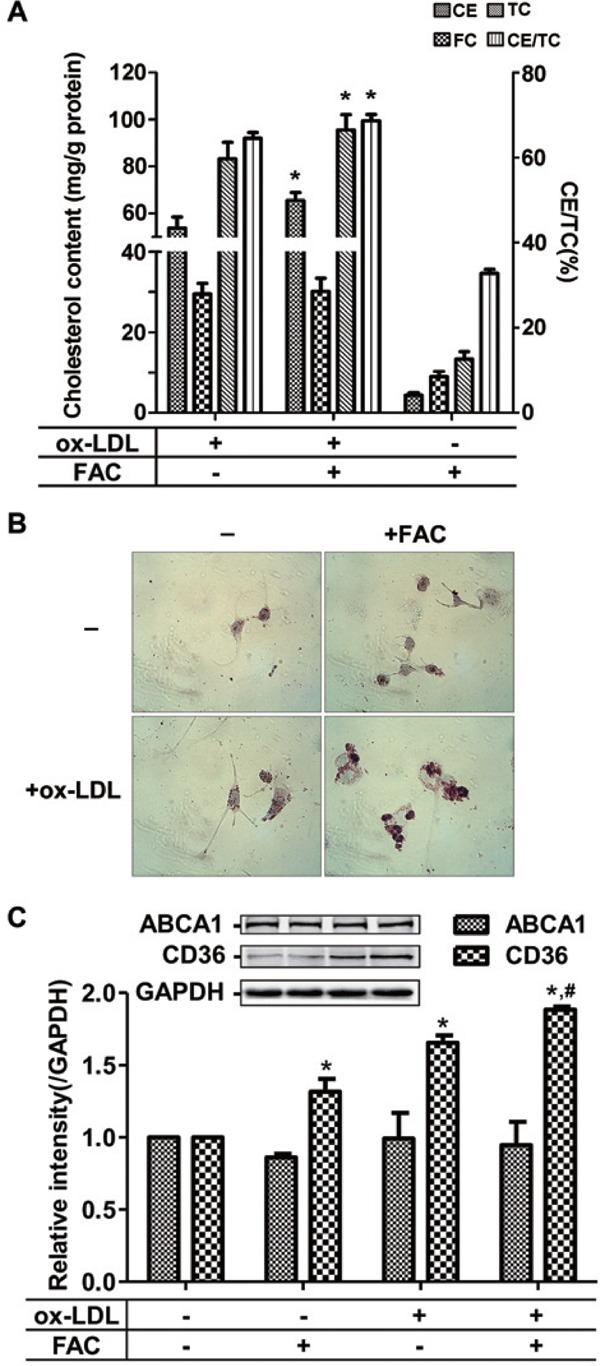
Iron promotes the lipid deposition in ox-LDL-treated RAW264.7 cells
(A) Iron enhancement of lipid uptake in RAW264.7 cells. (B) More lipid in the deformed RAW264.7 cells when co-treated with iron and ox-LDL, revealed by oil red stain. (C) A severe imbalance between lipid influx and efflux after the co-treatment of FAC with ox-LDL. CD36: a scavenger receptor of cholesterol influx; ABCA1 (ATP-binding cassette transporter A1): an important transporter for cholesterol efflux. *p < 0.05 vs. ox-LDL treated group in (A); *p < 0.05 vs. non-treated group, # p < 0.05 vs. ox-LDL group in (C).
To reveal the mechanism of how the presence of iron worsens lipid accumulation in macrophages, we detected the expression of CD36, a scavenger receptor of cholesterol influx, and ABCA1, an important transporter for cholesterol efflux after co-treatment of FAC with ox-LDL. The results showed that the expression of ABCA1 kept constant when the cells were treated with ox-LDL and/or iron, whereas ox-LDL alone induced the upregulation of CD36 (Fig. 5C), which level was elevated further when co-treatment with iron was performed. These data suggest that the stimulated lipid accumulation by ox-LDL or by ox-LDL plus iron likely results from the enhanced influx of lipid.
Iron Overload Stimulates the Detrimental Effects of Ox-LDL in the Activated RAW264.7 Cells
The aggregated and deformed RAW264.7 cells induced by co-treatment of FAC with ox-LDL suggested the occurrence of apoptosis. Therefore, we determined if iron stimulated the apoptosis of oxLDL-mediated foam cells using flow cytometry, directly counted the living cells, and conducted biochemical assays. The results showed an increased number of apoptotic cells after ox-LDL treatment (9.753 ± 0.510% vs. 2.730 ± 0.140% in control), whereas co-treatment of iron with ox-LDL pushed apoptosis up to 13.900 ± 1.434% (Fig. 6A and 6B). Consistently, the survival rate data supported the results (Fig. 6C). The ratio of Bcl2, an integral outer mitochondrial membrane protein that blocks apoptotic death, to Bax, a pro-apoptotic regulator, is often used to judge the early apoptotic event. The decreased ratio of Bcl2/Bax after co-treatment with iron and ox-LDL supported the elevated rate of apoptosis compared with ox-LDL treatment (Fig. 6D).
Fig. 6.
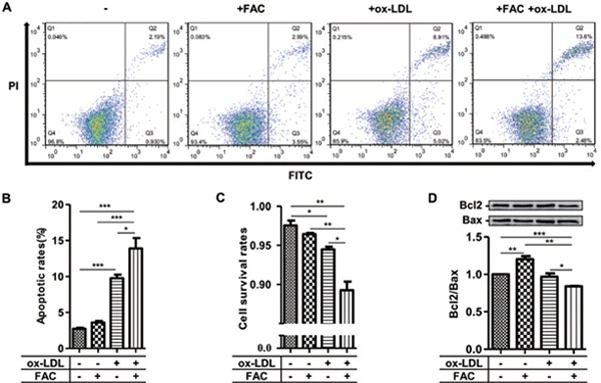
Physiological interaction of iron with ox-LDL increases apoptosis of RAW264.7 cells
(A–B) Flow cytometry assays demonstrated the enhancement of apoptotic rates of RAW264.7 by co-treatment of FAC with ox-LDL versus FAC or ox-LDL treatment alone. (C) Cell survival rate was significantly decreased after the co-treatment of FAC with ox-LDL versus FAC or ox-LDL treatment alone by counting the living cells. (D) Apoptotic index (Bcl2/Bax ratio) confirmed the occurrence of apoptosis. PI: propidium iodide; Bcl2 (B-cell lymphoma-2): an anti-apoptotic regulator; Bax: a proapoptotic regulator; *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
The association between iron overload and atherogenesis has been widely studied28, 29). However, iron hypothesis of cardiovascular diseases (CVD) is still controversial. The reason is probably that people focus on the association between CVD and systemic iron status. The parameters tested generally include serum ferritin, iron, and hepcidin30). CVD, in the case of atherosclerosis, may display a focal iron deposition or accumulation in atherosclerotic plaques rather than a systemic iron overload. Focal plaque expansion can be promoted when microvessels become thrombotic or rupture prone27). Apart from supplying plaques with leukocytes and lipoproteins, focal intraplaque micro-hemorrhages initiate erythrocyte phagocytosis, further leading to iron deposition and neovascularization. Therefore, iron will accumulate in the macrophages of plaques. Cp is a key protein in macrophage iron efflux. In the present study, Cp expression in RAW264.7 cells was closely linked with cell iron content, indicating that Cp is important in iron efflux in RAW264.7 cells. This is consistent with a previous study in bone marrow-derived macrophages (BMDM)26). Unexpectedly, foam cell-like formation of RAW264.7 cells after lipid loading moderately and significantly reduced Cp expression but did not lead to obstructed iron efflux (because of the constant ferritin and Fpn1 as shown in Fig. 2) when without FAC treatment. This phenomenon can be partly explained by the ubiquitous expression of the glycosylphosphatidylinositol-anchored (GPI)-Cp isoform in membrane and cytosol of macrophages26). The expression level of GPI-Cp is sufficient for iron homeostasis within the tested time frame. Strikingly, the protein levels and activities of Cp in lipid-laden cells in the presence of FAC were remarkably reduced and were accompanied by increased protein levels of Fpn1 and Ft. Iron efflux through Fpn1 from the macrophage is a passive-gradient mechanism. Therefore, the maintenance of iron gradient by removing iron from the site of iron efflux is important, especially in iron-overloaded cells. Cp has ferroxidase activity, which can convert ferrous iron to ferric form and accelerate the release of iron across the cellular membrane31). Reduction in both protein levels and activities of Cp attenuated the effects of increased Fpn132, 33) and resulted in the potent toxicity of iron, which concurred with the deformed cells in the presence of iron and lipid loading.
Atherosclerosis is a chronic inflammation and serum Cp level is an indicator reflecting the systemic inflammation. Previous clinical studies reported that serum Cp was positively associated with the arterial stiffness in subjects with type 2 diabetes mellitus34) and weakly with atherosclerosis35). The correlations suggest the role of iron in CVD, although it is still controversial. These studies largely focus on plasma Cp levels and the expression of Cp in macrophages in atherosclerosis plaques has not been clarified. Because of the poor blood supply in the area of atherosclerotic plaques, serum Cp very likely does not have a significant role in iron efflux of macrophages. On the contrary, this iron efflux might be more dependent on locally expressed GPI-Cp, as this current study suggests. On the other hand, macrophages expressing GPI-Cp may be sensitive to iron overloading26) and play a protective role in chronic inflammation36). When Cp level is low along with iron overloading, more oxidative damage will occur because of the Fen-ton reaction and this oxidation further stimulates CE accumulation37). Thus, Cp expression by macrophages might be a protective factor in atherosclerosis. We propose that the reduced Cp promoted by lipid loading, particularly further diminished by subsequent iron deposition, may trigger detrimental effects on foam cells in atherosclerotic plaques. Thus, our results support this notion that reduction of macrophage intracellular iron may be a promising avenue to increase macrophage reverse cholesterol transport38).
How ox-LDL and FAC interact after uptake to drastically reduce the expression of Cp is an interesting question to be addressed. Little is known about this phenomenon. The complexes formed by myeloperoxidase (MPO), Cp, and LDL have been reported39), and it is known that Cp inhibits peroxidase activity of MPO40). The complex formation might shield the ferroxidase activity of Cp, which could be one of the reasons for the reduced activity of Cp besides the low protein levels of Cp when co-treatment of iron with ox-LDL was performed in the present study. Cp mRNA level is known to be regulated by a cis-regulatory GAIT (IFN-γ activated inhibitor of translation complex) element and binding of the GAIT complex leads to translational silencing of Cp to some minimal basal level41). However, the effect of ox-LDL on GAIT and how iron enhances the effect of ox-LDL remain elusive.
In summary, the present study shows that Cp expression and activity were reduced in lipid-laden foam cells, which was especially further diminished by iron-overloading. This synergized reduction leads to obstructed iron efflux and enhanced cholesterol influx, possibly causing iron toxicity and oxidative stress to trigger apoptosis. The detailed mechanism on how Cp is regulated in the macrophages of atherosclerotic plaques by the interaction of ox-LDL with iron needs to be further elucidated. Nevertheless, this study opens new prospects for exploring the role of iron in atherosclerosis.
Acknowledgments
This work was supported by the National Nature Science foundation of China, Grant numbers: 81370387, 31071085, 31371060.
Conflicts of Interest
None.
References
- 1). Hansson GK: Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med, 2005; 352: 1685-1695 [DOI] [PubMed] [Google Scholar]
- 2). Sullivan JL: Macrophage iron, hepcidin, and atherosclerotic plaque stability. Exp Biol Med (Maywood), 2007; 232: 1014-1020 [DOI] [PubMed] [Google Scholar]
- 3). Ramakrishna G, Rooke TW, Cooper LT: Iron and peripheral arterial disease: revisiting the iron hypothesis in a different light. Vasc Med, 2003; 8: 203-210 [DOI] [PubMed] [Google Scholar]
- 4). Abboud S, Haile DJ: A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem, 2000; 275: 19906-19912 [DOI] [PubMed] [Google Scholar]
- 5). Weiss G: Genetic mechanisms and modifying factors in hereditary hemochromatosis. Nat Rev Gastroenterol Hepatol, 2010; 7: 50-58 [DOI] [PubMed] [Google Scholar]
- 6). Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J: Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science, 2004; 306: 2090-2093 [DOI] [PubMed] [Google Scholar]
- 7). Galesloot TE, Holewijn S, Kiemeney LALM, de Graaf J, Vermeulen SH, Swinkels DW: Serum Hepcidin Is Associated With Presence of Plaque in Postmenopausal Women of a General Population. Arterioscl Throm Vas, 2014; 34: 446-456 [DOI] [PubMed] [Google Scholar]
- 8). Abdel-Khalek MA, El-Barbary AM, Essa SAM, Ghobashi AS: Serum Hepcidin: A Direct Link Between Anemia of Inflammation and Coronary Artery Atherosclerosis in Patients with Rheumatoid Arthritis. J Rheumatol, 2011; 38: 2153-2159 [DOI] [PubMed] [Google Scholar]
- 9). Valenti L, Dongiovanni P, Motta BM, Swinkels DW, Bonara P, Rametta R, Burdick L, Frugoni C, Fracanzani AL, Fargion S: Serum Hepcidin and Macrophage Iron Correlate With MCP-1 Release and Vascular Damage in Patients With Metabolic Syndrome Alterations. Arterioscl Throm Vas, 2011; 31: 683-690 [DOI] [PubMed] [Google Scholar]
- 10). Aursulesei V, Cozma A, Krasniqi A: Iron hypothesis of cardiovascular disease: still controversial. Rev Med Chir Soc Med Nat Iasi, 2014; 118: 901-909 [PubMed] [Google Scholar]
- 11). Saeed O, Otsuka F, Polavarapu R, Karmali V, Weiss D, Davis T, Rostad B, Pachura K, Adams L, Elliott J, Taylor WR, Narula J, Kolodgie F, Virmani R, Hong CC, Finn AV: Pharmacological suppression of hepcidin increases macrophage cholesterol efflux and reduces foam cell formation and atherosclerosis. Arterioscler Thromb Vasc, 2012; 32: 299-307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12). Kautz L, Gabayan V, Wang X, Wu J, Onwuzurike J, Jung G, Qiao B, Lusis AJ, Ganz T, Nemeth E: Testing the iron hypothesis in a mouse model of atherosclerosis. Cell Rep, 2013; 5: 1436-1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13). Osaki S, Johnson DA, Frieden E: The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J Biol Chem, 1966; 241: 2746-2751 [PubMed] [Google Scholar]
- 14). Di Patti MC, Maio N, Rizzo G, De Francesco G, Persichini T, Colasanti M, Polticelli F, Musci G: Dominant mutants of ceruloplasmin impair the copper loading machinery in aceruloplasminemia. J Biol Chem, 2009; 284: 4545-4554 [DOI] [PubMed] [Google Scholar]
- 15). Cherukuri S, Tripoulas NA, Nurko S, Fox PL: Anemia and impaired stress-induced erythropoiesis in aceruloplasminemic mice. Blood Cells Mol Dis, 2004; 33: 346-355 [DOI] [PubMed] [Google Scholar]
- 16). Vassiliev V, Harris ZL, Zatta P: Ceruloplasmin in neurodegenerative diseases. Brain Res Brain Res Rev, 2005; 49: 633-640 [DOI] [PubMed] [Google Scholar]
- 17). Gutteridge JM, Winyard PG, Blake DR, Lunec J, Brailsford S, Halliwell B: The behaviour of caeruloplasmin in stored human extracellular fluids in relation to ferroxidase II activity, lipid peroxidation and phenanthroline-detectable copper. Biochem J, 1985; 230: 517-523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18). Ji J, Zhou Y, Hao S, Wang Q, Li K, Qiao T: Low expression of ferroxidases is implicated in the iron retention in human atherosclerotic plaques. Biochem Biophys Res Commun, 2015; 464: 1134-1138 [DOI] [PubMed] [Google Scholar]
- 19). Zhang Q, Ma AZ, Song ZY, Wang C, Fu XD: Nifedipine enhances cholesterol efflux in RAW264.7 macrophages. Cardiovasc Drugs Ther, 2013; 27: 425-431 [DOI] [PubMed] [Google Scholar]
- 20). Patel BN, Dunn RJ, David S: Alternative RNA splicing generates a glycosylphosphatidylinositol-anchored form of ceruloplasmin in mammalian brain. J Biol Chem, 2000; 275: 4305-4310 [DOI] [PubMed] [Google Scholar]
- 21). Yan H, Zhang D, Hao S, Li K, Hang CH: Role of Mitochondrial Calcium Uniporter in Early Brain Injury After Experimental Subarachnoid Hemorrhage. Mol Neurobiol, 2015; 52: 1637-1647 [DOI] [PubMed] [Google Scholar]
- 22). Yan H, Hao S, Sun X, Zhang D, Gao X, Yu Z, Li K, Hang CH: Blockage of mitochondrial calcium uniporter prevents iron accumulation in a model of experimental subarachnoid hemorrhage. Biochem Biophys Res Commun, 2015; 456: 835-840 [DOI] [PubMed] [Google Scholar]
- 23). Roeser HP, Lee GR, Nacht S, Cartwright GE: The role of ceruloplasmin in iron metabolism. J Clin Invest, 1970; 49: 2408-2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24). Persichini T, Maio N, di Patti MC, Rizzo G, Toscano S, Colasanti M, Musci G: Interleukin-1beta induces ceruloplasmin and ferroportin-1 gene expression via MAP kinases and C/EBPbeta, AP-1, and NF-kappaB activation. Neurosci Lett, 2010; 484: 133-138 [DOI] [PubMed] [Google Scholar]
- 25). Yang F, Naylor SL, Lum JB, Cutshaw S, McCombs JL, Naberhaus KH, McGill JR, Adrian GS, Moore CM, Barnett DR: Characterization, mapping, and expression of the human ceruloplasmin gene. Proc Natl Acad Sci USA, 1986; 83: 3257-3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26). Marques L, Auriac A, Willemetz A, Banha J, Silva B, Canonne-Hergaux F, Costa L: Immune cells and hepatocytes express glycosylphosphatidylinositol-anchored ceruloplasmin at their cell surface. Blood Cells Mol Dis, 2012; 48: 110-120 [DOI] [PubMed] [Google Scholar]
- 27). Kockx MM, Cromheeke KM, Knaapen MW, Bosmans JM, De Meyer GR, Herman AG, Bult H: Phagocytosis and macrophage activation associated with hemorrhagic microvessels in human atherosclerosis. Arterioscler Thromb Vasc, 2003; 23: 440-446 [DOI] [PubMed] [Google Scholar]
- 28). Zheng H, Cable RG, Spencer BR, Katz SD: Iron stores and vascular endothelial function in voluntary blood donors. Transfusion, 2005; 45: 23a-23a [DOI] [PubMed] [Google Scholar]
- 29). Engberink MF, Geleijnse JM, Durga J, Swinkels DW, de Kort WLAM, Schouten EG, Verhoef P: Blood donation, body iron status and carotid intima-media thickness. Atherosclerosis, 2008; 196: 856-862 [DOI] [PubMed] [Google Scholar]
- 30). Munoz-Bravo C, Gutierrez-Bedmar M, Gomez-Aracena J, Garcia-Rodriguez A, Navajas JF: Iron: protector or risk factor for cardiovascular disease? Still controversial. Nutrients, 2013; 5: 2384-2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31). Potdar AA, Sarkar J, Das NK, Ghosh P, Gratzl M, Fox PL, Saidel GM: Computational modeling and analysis of iron release from macrophages. PLoS Comput Biol, 2014; 10: e1003701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32). Kono S, Yoshida K, Tomosugi N, Terada T, Hamaya Y, Kanaoka S, Miyajima H: Biological effects of mutant ceruloplasmin on hepcidin-mediated internalization of ferroportin. Biochim Biophys Acta, 2010; 1802: 968-975 [DOI] [PubMed] [Google Scholar]
- 33). De Domenico I, Ward DM, Di Patti MCB, Jeong SY, David S, Musci G, Kaplan J: Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. Embo Journal, 2007; 26: 2823-2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34). Lee MJ, Jung CH, Hwang JY, Shin MS, Yu JH, Lee WJ, Park JY: Association Between Serum Ceruloplasmin Levels and Arterial Stiffness in Korean Men with Type 2 Diabetes Mellitus. Diabetes Technology & Therapeutics, 2012; 14: 1091-1097 [DOI] [PubMed] [Google Scholar]
- 35). Dadu RT, Dodge R, Nambi V, Virani SS, Hoogeveen RC, Smith NL, Chen FJ, Pankow JS, Guild C, Tang WHW, Boerwinkle E, Hazen SL, Ballantyne CM: Ceruloplasmin and Heart Failure in the Atherosclerosis Risk in Communities Study. Circulation-Heart Failure, 2013; 6: 936-943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36). Bakhautdin B, Febbraio M, Goksoy E, de la Motte CA, Gulen MF, Childers EP, Hazen SL, Li X, Fox PL: Protective role of macrophage-derived ceruloplasmin in inflammatory bowel disease. Gut, 2013; 62: 209-219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37). Kraml PJ, Klein RL, Huang Y, Nareika A, Lopes-Virella MF: Iron loading increases cholesterol accumulation and macrophage scavenger receptor I expression in THP-1 mononuclear phagocytes. Metabolism, 2005; 54: 453-459 [DOI] [PubMed] [Google Scholar]
- 38). Finn AV, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao X, Yazdani S, Otsuka F, Davis T, Habib A, Narula J, Kolodgie FD, Virmani R: Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol, 2012; 59: 166-177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39). Sokolov AV, Ageeva KV, Cherkalina OS, Pulina MO, Zakharova ET, Prozorovskii VN, Aksenov DV, Vasilyev VB, Panasenko OM: Identification and properties of complexes formed by myeloperoxidase with lipoproteins and ceruloplasmin. Chem Phys Lipids, 2010; 163: 347-355 [DOI] [PubMed] [Google Scholar]
- 40). Sokolov AV, Ageeva KV, Pulina MO, Cherkalina OS, Samygina VR, Vlasova II, Panasenko OM, Zakharova ET, Vasilyev VB: Ceruloplasmin and myeloperoxidase in complex affect the enzymatic properties of each other. Free Radic Res, 2008; 42: 989-998 [DOI] [PubMed] [Google Scholar]
- 41). Yao P, Potdar AA, Arif A, Ray PS, Mukhopadhyay R, Willard B, Xu YC, Yan J, Saidel GM, Fox PL: Coding Region Polyadenylation Generates a Truncated tRNA Synthetase that Counters Translation Repression. Cell, 2012; 149: 88-100 [DOI] [PMC free article] [PubMed] [Google Scholar]