

Abstract
Stanniocalcin-1 (STC-1) is a multifunctional glycoprotein with anti-oxidant and anti-inflammatory properties. Ischemic myocardial necrosis generates ‘danger’ signals that perpetuate detrimental inflammatory reactions often involving monocyte recruitment and their subsequent differentiation into pro-inflammatory macrophages. Therefore, we evaluated the effects of recombinant STC-1 (rSTC-1) on monocyte phenotype and in a mouse model of myocardial infarction. Using an established protocol to differentiate human monocytes into macrophages, we demonstrated that rSTC-1 did not alter morphology of the differentiated cells, toll-like receptor (TLR) 4 expression, or expression of the myeloid cell marker CD11b. However rSTC-1 treatment prior to differentiation attenuated the rise in expression of CD14, a TLR4 co-receptor and pathogen sensor that propagates innate immune responses, and suppressed levels of inflammatory cytokines produced by the differentiated cells in response to the TLR4/CD14 ligand lipopolysaccharide (LPS). Moreover, rSTC-1 treatment reduced CD14 expression in monocytes stimulated with endogenous danger signals. Interestingly, the effects of rSTC-1 on CD14 expression were not reproduced by a superoxide dismutase mimetic. In mice with induced myocardial infarcts, intravenous administration of rSTC-1 decreased CD14 expression in the heart as well as levels of TNFα, CXCL2, IL-1β and myeloperoxidase (MPO). It also suppressed formation of scar tissue while enhancing cardiac function. The data suggests that one of the beneficial effects of STC-1 might be attributed to suppression of CD14 on recruited monocytes/macrophages that limits their inflammatory response. STC-1 may be a promising therapy to protect the heart and other tissues from ischemic injury.
Introduction
Stanniocalcin-1 (STC-1) is a conserved glycoprotein with an unusual history both in terms of its evolution and as a subject for biological research (1-3). STC-1, formerly called hypocalcin, was named after it was discovered to be secreted by the corpuscles of Stannius, small endocrine glands that were found in bony fish and that were initially assumed to be adrenal glands because of their location on the ventral surface of the kidneys. However, surgical removal of the glands caused toxic hypercalcemia and led to the finding that STC-1 is a critical regulator of plasma calcium and phosphate levels in the fishes. This role has since been replaced in mammals by parathyroid hormone, calcitonin, and 1, 25-dihydroxyvitamin D (3). Instead, STC-1 in mammals is produced by a variety of tissues and has been proposed to regulate a variety of physiological and cellular functions in a paracrine/intracrine fashion. A major intracellular target for STC-1 is mitochondria, the organelle also targeted by other intracrines such as angiotensin II, transforming growth factor-β, growth hormone, atrial natriuretic peptide, and Wnt 13 (2). Binding sites for STC-1 have been identified on cell membrane fractions and on mitochondria (4), but the major effects of STC-1 have been difficult to define. Some observations demonstrated that STC-1 reversibly inhibits transmembrane calcium currents through L-type channels in cardiomyocytes (5) and decreases cytokine-induced intracellular calcium signals in macrophages (6) suggesting that it has a continuing but diminished role in calcium metabolism. Other observations emphasized that STC-1 increases expression of mitochondrial uncoupling proteins (UCPs) that dissipate the proton gradient and thereby effectively uncouple oxidation from phosphorylation (7, 8). The increase in UCPs reduces the surge of reactive oxygen species (ROS) that is often seen with tissue injury and appears to explain, at least in part, the anti-inflammatory and anti-apoptotic properties of STC-1 in mouse models of sepsis (9), glomerulonephritis (10), and retinal degeneration (11). The decrease in ROS production also appears to explain some of the effects of STC-1 in altering macrophage function (12), and protecting cardiac myocytes from angiotensin II-mediated injury (13). However, the role of STC-1 on macrophage differentiation and in ischemic myocardial injury has not been studied.
Myocardial infarction is a leading cause of morbidity and mortality worldwide despite advances in therapy (14, 15). Myocardial tissue injury induces the release of endogenous inflammatory mediators or ‘danger’ signals including cytokines, ROS, and numerous intracellular factors that are normally inaccessible to the immune system but that are liberated from necrotic cells (15). Toll-like receptors (TLRs) are among the cellular receptors that sense many danger signals released by necrotic cells and provide a key molecular link between tissue injury and inflammatory response (14). A growing body of evidence suggests that inflammation after ischemic cardiac injury, especially when persistent, can exacerbate pathological remodeling of the heart and promote heart failure; therefore, strategies to regulate inflammatory pathways will improve prognosis after myocardial infarction (15, 16).
Accordingly in the present report, we tested the hypotheses that STC-1 regulates macrophage phenotype and response to inflammatory mediators, reduces inflammation in the post-infarcted heart, and protects the heart from damage associated with ischemic injury.
Materials and Methods
Cell Culture
The human monocyte cell line U937 was purchased from American Type Culture Collection (ATCC, Rockville, MD). The cells were cultured in a humidified atmosphere at 37°C and 5% CO2 in macrophage medium consisting of RPMI-1640 (ATCC) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Atlanta Biologicals, Flowery Branch, GA), 100 units/ml penicillin and 100 μg/ml streptomycin (Gibco, Grand Island, NY).
Monocyte Differentiation Assay
To induce macrophage differentiation of U937 monocytes, the cells were suspended at 500,000 cells/ml in petri dishes (VWR International, Radnor, PA) and stimulated for 48 hours with 100 ng/ml of phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich, St. Louis, MO). In separate experiments, U937 cells were stimulated concurrently with 5 μg/ml recombinant human high mobility group box1 protein (HMGB1, R&D systems, Minneapolis, MN) and recombinant human tumor necrosis factor alpha (TNFα), interleukin 1 beta (IL-1β), and IL-6 (10 ng/ml each, R& D Systems). Differentiation assays were performed in the presence or absence of 1.0 μg/ml of human rSTC-1 (BioVendor Research and Diagnostic Products, Asheville, NC or Czech Republic), 1 mM of the superoxide donor paraquat (Sigma-Aldrich), and/or 100 μM of the cell-permeable superoxide dismutase mimetic MnTBAP (Sigma-Aldrich). After 48 hours, images of the cells were captured on a Nikon Eclipse Ti-S inverted microscope using a Ds-Fi1 camera (Nikon, Melville, NY) and managed with NiS Elements AR 3.0 software (Nikon). The differentiated macrophages were subsequently harvested, counted, and processed for real-time reverse transcription polymerase chain reaction (RT-PCR) or flow cytometry analysis.
In Vitro Inflammatory Assay
U937 monocytes stimulated for 48 hours with PMA (in the presence or absence of rSTC-1) were seeded at 600,000 cells/ml into 24-well culture plates (Corning Incorporated, Corning, NY). After a 3 hour recovery period, 50 ng/ml of lipopolysaccharide (LPS, Sigma-Aldrich) was added to the cultures. In some experiments cultures of differentiated macrophages were treated with 1.0 μg/ml rSTC-1 just prior to LPS stimulation. Five hours later, media conditioned (CM) by unstimulated or LPS-stimulated macrophages were collected. Cells and debris were removed from the CM by centrifugation at 500 × g for 5-10 minutes. Several aliquots were prepared from the CM and stored at -80°C until they were used for ELISA.
Ischemic Cardiac Injury Model
The experimental protocols were approved by the Institutional Animal Care and Use Committee of Texas A&M University Health Science Center and in accordance with guidelines set forth by the National Institutes of Health. Male immunodeficient NOD/SCID mice (NOD.CB17-Prkdcscid/J, Jackson Laboratory, Bar Harbor, Maine), 7–8 weeks of age and 25-30 grams of body weight, were used for this study to reduce the immune reaction to human protein. The mice were kept on a 12 hour light-dark cycle and were provided sterile food and water ad libitum. To induce ischemic cardiac injury, mice were mechanically ventilated and maintained under anesthesia with isoflurane (1.5%) during the course of the procedure. The thoracic cavity was opened via an incision made at the left 5th intercostal space to visualize the left anterior descending (LAD) coronary artery. An 8-0 prolene suture (Ethicon, Somerville, NJ) was positioned around the LAD distal to the first diagonal branch and permanently tied. After myocardial infarction was confirmed by ventricular blanching, the chest was closed. rSTC-1 (2.0 mg/kg body weight) in 0.9% sodium chloride (Sigma-Aldrich) or equal volume of saline was injected intravenously 1 hour and 24 hours after ligation. To alleviate post-operative discomfort, buprenorphine (0.1 mg/kg) was administered subcutaneously twice daily for up to 5 days. For endpoint assays, the mice were euthanized by intraperitoneal injection of ketamine (80 mg/kg) and xylazine (8 mg/kg). Blood was collected via the facial vein, or via cardiac puncture for endpoint measurements, and processed to evaluate levels of serum cardiac troponin I (cTnI) by ELISA (Life Diagnostics Incorporated, West Chester, PA). Levels of inflammatory mediators in the heart were determined from cardiac tissue lysates prepared 48 hours after ligation. Cardiac function was evaluated on day 21 and mice were euthanized to determine infarct size.
Evaluation of Cardiac Function
Cardiac function was determined by transthoracic echocardiography 21 days after LLDCA using a 30 or 40 MHz transducer supported by a Vevo 2100 ultrasound instrument (VisualSonics Inc., Toronto, Canada). The procedure was performed on mice under isoflurane anesthesia. Heart rate was maintained at 320-400 beats per minute. Left ventricular ejection fraction (LVEF) and other parameters of cardiac function were evaluated from the parasternal long axis position (Supplemental Videos) by tracing end-systolic and end-diastolic endocardial boundaries using B-mode recordings. The average of two separate recordings containing 3 consecutive cardiac cycles were used per animal to obtain the results.
Infarct Size Measurement
Measurements of infarct size were performed on hearts obtained 21 days after surgery. Hearts were perfused with 3-5 ml of saline, excised from the chest, and fixed in 10-15 ml of 10% formalin (Sigma-Aldrich) to be processed for staining. Paraffin-embedded heart samples were cut into 5 μm sections from the apex to the base. Every 20th section was stained with Masson Trichrome. Quantitative assays for infarct size were performed as described by Takagawa and colleagues (17). Images of every fifth stained section covering both ventricles (total of 10 sections per heart) were acquired using a Photometrics Coolsnap HQ2 camera mounted on a Nikon Eclipse Ti-E inverted microscope (Nikon). NiS Elements AR 3.0 software (Nikon) was used to measure midline infarct length of heart.
Real-Time RT-PCR
Total RNA was isolated from U937 cells or homogenized heart tissue using RNeasy Mini Kit (Qiagen, Valencia, CA) with DNase I (RNase-Free DNase Set, Qiagen) digestion step. The isolated RNA was quantified with nanodrop spectrophotometer (ThermoFisher Scientific, Rockford, IL) and converted to complementary DNA (cDNA) with High-Capacity cDNA RT Kit (Applied Biosystems, Technologies, Grand Island, NY). Real-time RT-PCR was performed in triplicate for expression of human glyceraldehyde 3-phosphate dehydrogenase (GAPDH), CD11b (ITGAM), CD14, TLR2, and TLR4, as well as mouse Tnf, Ucp2, and Ucp3 using TaqMan® Gene Expression Assays (Applied Biosystems, Technologies) and TaqMan® Fast Master Mix (Life Technologies). Total of 20 ng of cDNA was used for each 20 μl reaction. Thermal cycling was performed with 7900HT System (Applied Biosystems, Technologies) by incubating the reactions at 95° C for 20 seconds followed by 40 cycles of 95° C for 1 second and 60° C for 20 seconds. Data were analyzed with Sequence Detection Software V2.3 (Applied Biosystems, Technologies) and relative quantitation (RQ) was calculated with comparative critical threshold (Ct) method using RQ Manager V1.2 (Applied Biosystems, Life Technologies).
Enzyme-linked immunosorbent assay (ELISA) for markers of inflammation
Levels of the inflammatory mediators TNFα and chemokine C-X-C motif ligand 2 (CXCL2) were determined in cell-free U937 CM using commercially available ELISA Kits (TNFα was from R&D Systems and CXCL2 from Abnova, Taipei City, Taiwan). To determine levels of inflammation in the heart, ventricular tissue was minced with surgical scissors, transferred to tissue extraction reagent I (Life Technologies) containing 1x of Halt protease inhibitor cocktail (ThermoFisher Scientific), and homogenized using a PowerGen model 125 tissue grinder (ThermoFisher Scientific). The lysates were centrifuged at 1,000 × g for 10 minutes 4°C. The protein rich supernatant was transferred to new tubes and centrifuged at 12,000 × g for 10 minutes. Several aliquots were prepared from the supernatants and stored at -80°C. Protein concentration was determined with micro BCA protein assay kit (ThermoFisher Scientific) per manufacturer's instructions. Levels of mouse myeloperoxidase (MPO), CD14, CXCL2, and IL-1β were measured using commercially available ELISA Kits (CD14 and IL-1β were form R&D Systems, CXCL2 from Abnova, and MPO from Hycult Biotech, Plymouth Meeting, PA). The optical density was determined on a plate reader (FLUOstarOmega; BMG Labtech) at an absorbance of 450 nm with wavelength correction at 540 nm. Where appropriate, samples were diluted prior to running the assay so that the optical density detected for the sample fit within the standard curve of the ELISA kit.
Flow Cytometry Analysis
Cell surface expression of CD11b and CD14 was determined by flow cytometry. U937 cells were suspended at 2,000 cells/μl in 100 μl of cold PBS (Gibco) containing 2% FBS and then incubated for 20 min with 20 μl of human Fc receptor binding inhibitor (Affymetrix eBioscience, San Diego, CA). Without washing by centrifugation, cell suspensions were labeled with 0.2 μg of fluorescein-conjugated antibodies to CD11b (Clone Bear1, Beckman Coulter, Brea, CA) or CD14 (Clone RMO52, Beckman Coulter) for 20 min at room temperature. Isotype matched antibodies were used as controls. After 2 washes in PBS, cells were again suspended in PBS containing 2% FBS and analyzed on an FC500 flow cytometer (Beckman Coulter). A minimum of 10,000 events was examined from the viable cell population. Data were analyzed using CXP Software (Beckman Coulter).
Statistical Analyses
Unpaired two-tailed Student's t-test was used to compare data sets consisting of two treatment groups. One-way ANOVA was used to calculate levels of significance between multiple groups. Statistical analysis was performed with Graphpad Prism 5 software.
Results
Recombinant STC-1 modulates the inflammatory response of differentiated human monocytes in vitro
To evaluate the effects of rSTC-1 treatment on monocyte differentiation and inflammatory response, we modified an established protocol that uses the protein kinase C (PKC) activator PMA to promote differentiation of U937 human monocytes into macrophages (18). Here, U937 cells were stimulated with 100 ng/ml of PMA for 48 hours in the presence or absence of 1 μg/ml rSTC-1. The differentiated U937 cells were harvested, re-plated, and activated with 50 ng/ml of LPS. Five hours later, CM was collected (Fig. 1A) and used to measure production of inflammatory cytokines/chemokines. As shown, PMA-differentiated U937 cells secreted high levels of TNFα and CXCL2 in response to LPS (Figs. 1B-C). Addition of rSTC-1 to the cultures prior to PMA treatment significantly reduced the secretion of these factors (Figs. 1B-C). We then examined the effects of rSTC-1 on inflammatory response of newly differentiated macrophages by first treating U937 cells with PMA for 48 hours to induce differentiation, and then with rSTC-1 immediately prior to LPS stimulation (Fig. 1D). Secretion of TNFα and CXCL2 in response to LPS was unchanged by treatment with rSTC-1 when the protein was applied to the cultures after the cells were differentiated (Figs. 1E-F). Similarly, the anti-inflammatory effects of rSTC-1 on differentiated mouse macrophages was negligible (data not shown). Together, the results showed that rSTC-1 treatment, during monocyte differentiation but not after, suppressed macrophage response to LPS, suggesting that STC-1 alters expression of cell-surface receptors that transmit signals from inflammatory stimuli.
Figure 1. Recombinant stanniocalcin-1 (rSTC-1) treatment, prior to monocyte differentiation but not after, suppressed the inflammatory response of macrophages to danger signals.
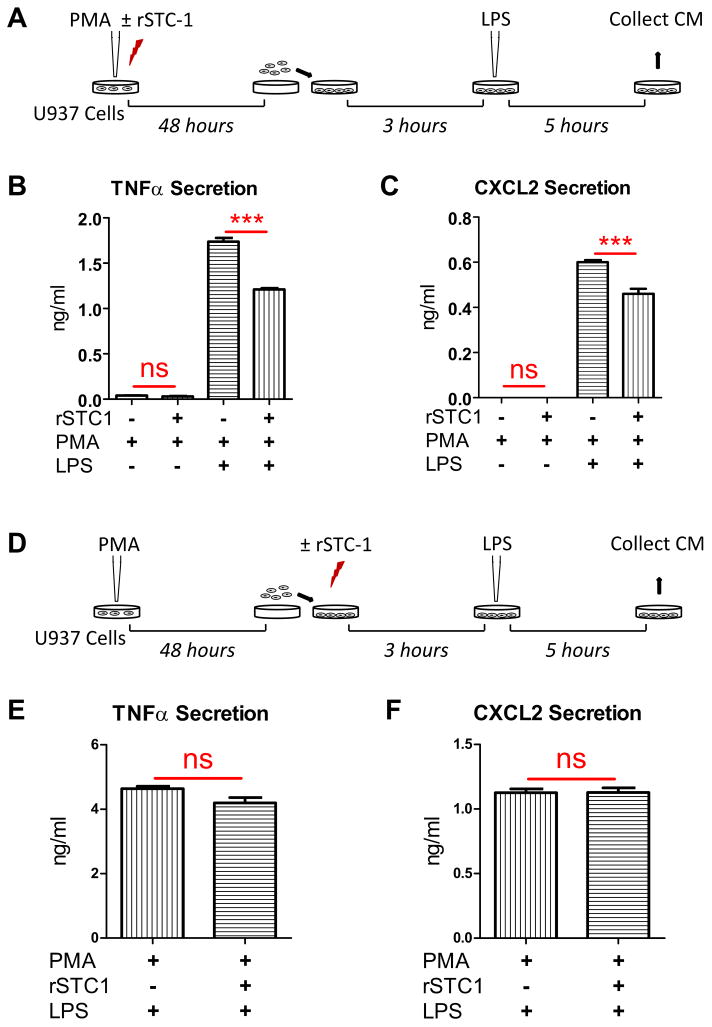
(A) Schematic illustrating the strategy used to evaluate the effects of rSTC-1 pre-treatment on macrophage response to lipopolysaccharide (LPS). Human U937 monocytes were induced to differentiate into macrophages by treatment with 100 ng/ml of phorbol 12-myristate 13-acetate (PMA) in the presence or absence of 1 μg/ml of rSTC-1. After 48 hours, differentiated macrophages were harvested, plated, and incubated for another 3 hours. Then, macrophages were stimulated with 50 ng/ml of LPS. After 5 hours, media conditioned (CM) by unstimulated or LPS-stimulated macrophages were collected and used to evaluate changes in levels of secreted TNFα (B) and CXCL2 (C) by ELISA. (D) Schematic illustrating the strategy used to evaluate the effects of rSTC-1 on the response of differentiated macrophages to LPS. U937 monocytes were plated in macrophage medium supplemented with 100 ng/ml of PMA for 48 hours. Differentiated macrophages were harvested, re-plated, and incubated with or without 1 μg/ml of rSTC-1. After 3 hours, macrophages were stimulated with LPS for an additional 5 hours. The CM was collected, clarified by centrifugation and used to determine levels of TNFα (E) and CXCL2 (F) by ELISA. Values are presented as mean + SEM (n=3). Statistical significance was determined using ANOVA (B-C) or Student's t-test (E-F) (not significant, ns p≥ 0.05; ***p< 0.001).
Expression of cell surface markers/receptors on differentiating monocytes are regulated by STC-1
To explore potential mechanism(s) driving the anti-inflammatory effects of rSTC-1 observed here, we evaluated changes in the phenotype of U937 cells in response to PMA and rSTC-1. Specifically, we measured changes in cell-surface markers associated with monocyte maturation and their response to LPS and other danger signals. In these experiments, U937 cells were stimulated with PMA for 48 hours in the presence or absence of rSTC-1 and then analyzed by microscopy, flow cytometry, and real-time RT-PCR (Fig. 2A). In culture, undifferentiated U937 monocytes propagate in suspension as single cells that ultimately form adherent colonies in the presence of PMA (Fig. 2B). Analysis by microscopy revealed that cells treated simultaneously with PMA and rSTC-1 shared a similar morphology to cells incubated with PMA alone (Fig. 2B). We next assayed for changes in expression of monocyte/macrophage surface markers by flow cytometry. We observed a dramatic increase in cell surface expression of CD11b and CD14 on PMA-differentiated macrophages (Fig. 2C). Interestingly, rSTC-1 treatment caused a marked reduction in cell surface expression of CD14 without changing CD11b levels (Fig. 2C). Since CD14 is a well-known co-receptor for TLR2 and TLR4 (19), we examined mRNA levels of these TLRs in addition to CD11b and CD14 by real-time RT-PCR. As expected, the expression of CD11b, CD14, TLR2, and TLR4 was increased in U937 cells differentiated with PMA (Figs. 2D-G); however, we detected a notable decline in CD14 message levels after treatment with PMA and rSTC-1 (Fig. 2E) but did not detect significant effects of rSTC-1 on amount of CD11b and TLR4 mRNA (Figs. 2D, F). In contrast, the expression of TLR2 was significantly increased in U937 cells treated with rSTC-1 (Fig. 2G).
Figure 2. Expression of CD14 was reduced in differentiating monocyte/macrophages by treatment with recombinant stanniocalcin-1 (rSTC-1).
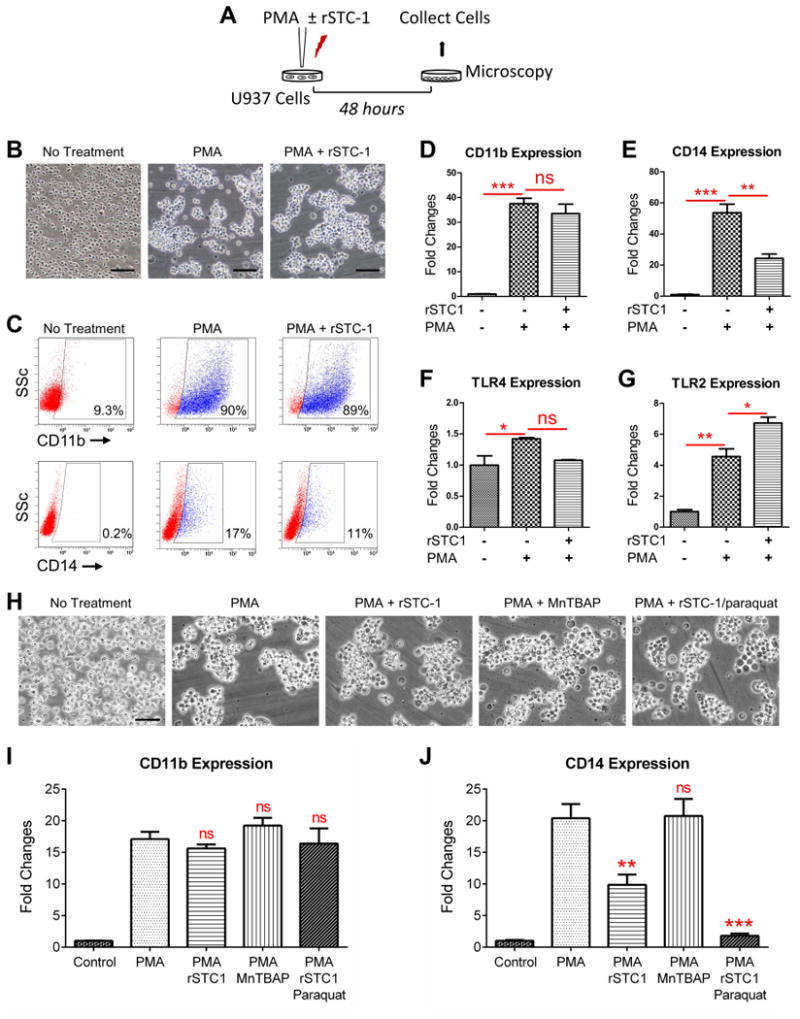
Human U937 monocytes were incubated in macrophage medium supplemented with 100 ng/ml of phorbol 12-myristate 13-acetate (PMA) in the presence or absence of 1.0 μg/ml of rSTC-1. After 48 hours, morphology of differentiated macrophages was evaluated by light microscopy. Then, macrophages were harvested and processed for real-time RT-PCR or flow cytometry analysis. (A) Schematic showing the workflow. (B) Representative images of undifferentiated U937 monocytes (No Treatment), PMA-differentiated U937 macrophages (PMA), and U937 cells stimulated with PMA and rSTC-1 (PMA+ rSTC-1). Scale bar, 100 μm. (C) Cell surface expression of CD11b and CD14 was assessed by flow cytometry. Real-time RT-PCR for CD11b (D), CD14 (E), TLR4 (F), and TLR2 (G) in undifferentiated U937 monocytes and cells stimulated with PMA for 48 hours (with and without rSTC-1 treatment). (H) Representative images of U937 cells treated for 48 hours with PMA in the presence and absence of rSTC-1, MnTBAP, and paraquat. Scale bar, 50 μm. Real-time RT-PCR for CD11b (I) and CD14 (J) in U937 cells stimulated for 48 hours with the compounds described in panel H. Values are expressed as mean + SEM (n=3-5). Data was analyzed using ANOVA (not significant, ns p≥ 0.05, *p< 0.05, **p< 0.01, ***p< 0.001).
Since the beneficial effects of STC-1 have been mostly attributed to its anti-oxidant properties, we determined if STC-1 suppressed CD14 by modifying levels of ROS, particularly superoxide. To this end, we employed a superoxide generator (paraquat) and a cell-permeable superoxide dismutase mimetic (MnTBAP) in our assays (20). As shown, treatment with MnTBAP and paraquat did not appear to prominently alter the effects of PMA on U937 cell aggregation (Fig. 2H) or expression of CD11b (Fig. 2I). MnTBAP unexpectedly did not decrease CD14 levels in PMA-stimulated monocytes in contrast to rSTC-1 (Fig. 2J). Moreover, paraquat did not reverse the inhibitory effects of rSTC-1, but instead decreased CD14 expression to a greater extent (Fig. 2J). The observed effects with paraquat were likely not due to its potential cyto-toxicity given that expression of CD11b was maintained (Fig. 2I). Moreover, U937 cells have been previously reported to be highly resistant to the toxic effects of paraquat (21). These data suggest that rSTC-1 suppresses monocyte/macrophage response to danger signals by blunting the upregulation of the TLR co-receptor CD14 in differentiating macrophages independent of its ability to reduce ROS.
Recombinant STC-1 suppresses the rise in CD14 expression induced by DAMPS and inflammatory cytokines
To further explore the role of STC-1 on monocyte-to-macrophage differentiation, we evaluated the effects of rSTC-1 on expression of CD11b and CD14 in U937 monocytes in response to numerous inflammatory stimuli classically associated with ischemic tissue injury. As shown, U937 cells were stimulated with a pro-inflammatory cocktail consisting of 5 μg/ml human HMGB1 and 10 ng/ml each of recombinant human TNFα, IL-1β, and IL-6 (Figs. 3A). After 48 hours, differentiated macrophages were lysed to quantify mRNA levels of CD11b and CD14 by real-time RT-PCR. The expression of CD11b was increased significantly after the cells were stimulated with the inflammatory cocktail in the presence and absence of rSTC-1 (Fig. 3B). In contrast, we observed a dramatic reduction in U937 cell expression of CD14 in the presence of rSTC-1 (Fig. 3C) similar to that observed previously with PMA treated cells. The data indicates that STC-1 could suppress inflammatory response of newly differentiated macrophages in the ischemic tissue microenvironment by inhibiting CD14 expression.
Figure 3. Recombinant stanniocalcin-1 (rSTC-1) reduced CD14 expression by monocyte/macrophages stimulated with danger signals.
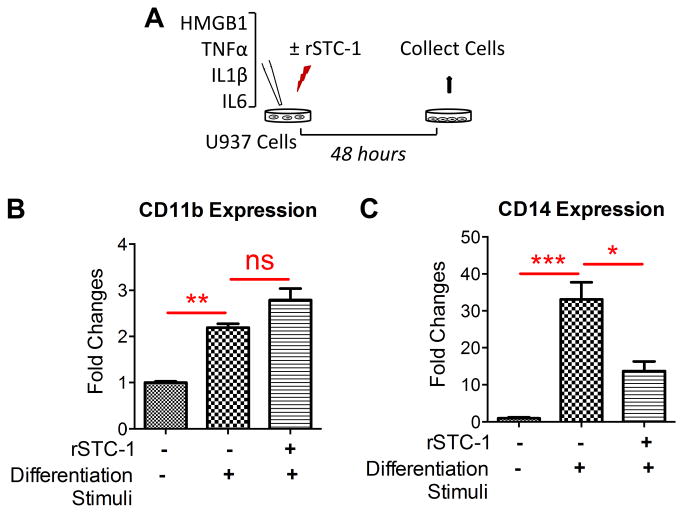
Human U937 monocytes were stimulated with 5 μg/ml human high mobility group box1 (HMGB1) and 10 ng/ml each of recombinant human tumor necrosis factor alpha (TNFa), interleukin-1 beta (IL-1β), and IL-6 in the presence or absence of 1.0 μg/ml rSTC-1 treatment. After 48 hours, cells were lysed for real-time RT-PCR. (A) Schematic showing the workflow. The fold changes of mRNA levels for CD11b (B) and CD14 (C) were measured in unstimulated U937 monocytes, stimulated U937 monocytes, and stimulated cells treated simultaneously with rSTC-1. Values are expressed as mean + SEM (n=3). Data were analyzed with ANOVA (not significant, ns p≥ 0.05, *p< 0.05, **p< 0.01, ***p< 0.001).
CD14 expression and inflammatory response in the ischemic heart were diminished by intravenous administration of rSTC-1
Based on our observations demonstrating the inhibitory effects of rSTC-1 on macrophage CD14 expression and response of the cells to TLR agonists, we hypothesized that rSTC-1 treatment reduces inflammation following myocardial infarction and protects the ischemic heart. For these experiments, NOD/SCID mice were chosen as a model to permit use of the same human STC-1 that we employed in our in vitro assays and prior studies (11), while limiting an immune response against the protein. A myocardial infarct was generated in the mice by permanent LLDCA (Fig 4A), a model which has been reported to impose sufficient myocardial deterioration and create the experimental window necessary to assess cardiac functional improvements and anti-inflammatory therapies (22). STC-1 was administered intravenously 1 hour and 24 hours after ligation (Fig 4A) at a dose (2.0 mg/kg) that effectively increased renal UCP2 expression in preliminary studies (Fig. 4B). Given that increased UCP2 has been shown to protect the kidneys from tissue damage following ligation of the renal artery and STC-1 treatment (23), we subsequently elected to use this dose of rSTC-1 for the remainder of our in vivo experiments. Reproducibility of myocardial infarction after LLDCA was assessed by measuring amount of cTnI released from the necrotic heart into the serum (24) as a function of time (Fig. 4C) and following rSTC-1 therapy (Fig. 4D). The data showed that cTnI levels peaked in the serum 24 hours after coronary ligation as expected (Fig 4C). Treatment with rSTC-1 did not alter peak cTnI levels (Fig. 4D), suggesting the level of infarction was uniform between groups.
Figure 4. Intravenous administration of recombinant stanniocalcin-1 (rSTC-1) reduced the expression of CD14 in cardiac tissue and attenuated inflammation following myocardial infarction.
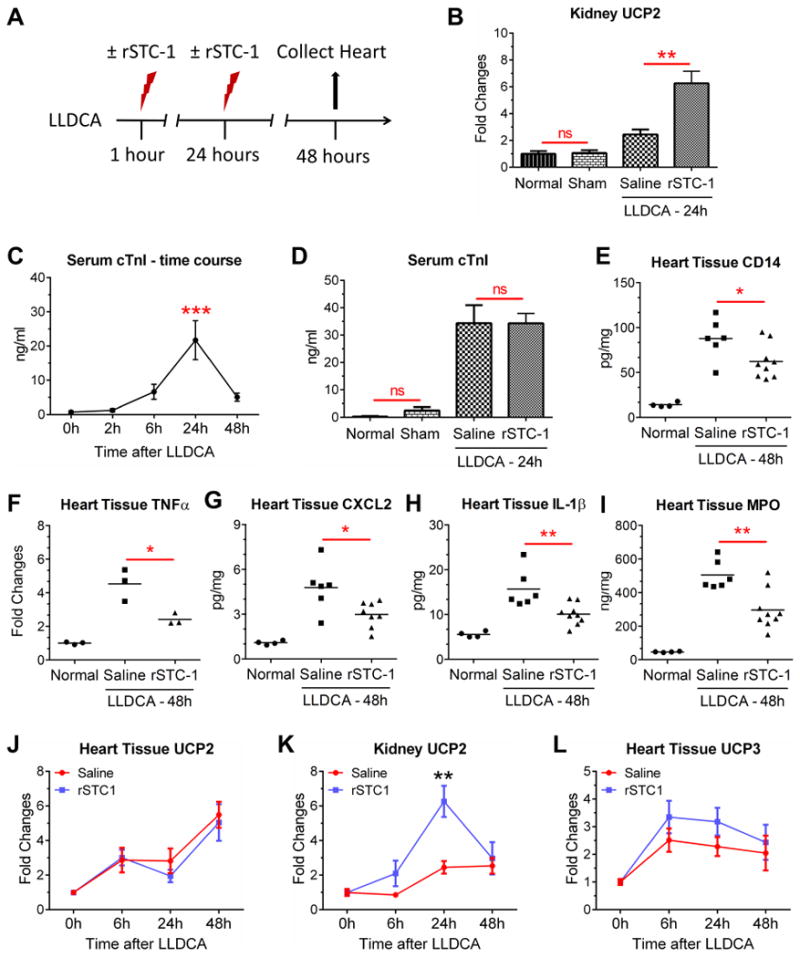
NOD/SCID mice were subjected to ischemic cardiac injury by permanent ligation of the left descending coronary artery (LLDCA). At 1 hour and 24 hours after ligation, 2.0 mg/kg of rSTC-1 or equal volume of 0.9% sodium chloride (saline) was administered intravenously. Mice were euthanized up to 48 hours after LLDCA to collect heart tissue and assess inflammatory response. Blood was collected to measure serum levels of cardiac troponin (cTnI). (A) Diagram showing the workflow of the cardiac injury model. (B) Changes in expression of kidney UCP2 were determined by real-time RT-PCR. (C) Time-dependent change in serum level of cTnI was determined by ELISA. (D) Assessment of serum cTnI, 24 hours after permanent ligation and rSTC-1 treatment. Intravenous rSTC-1 suppressed levels of (E) CD14, inflammatory cytokines TNFα (F), CXCL2 (G), and IL-1β (H), and (I) myeloperoxidase (MPO) in heart tissue lysate prepared 48 hours after LLDCA. Mean value is illustrated by the black lines. Real-time RT-PCR of UCP2 in cardiac (J) and kidney (K) tissue, and (L) UCP3 in cardiac tissue. Statistical significance was determined using Student's t-test when 2 groups were compared, or ANOVA for analysis of more than 2 groups (not significant, ns p> 0.05, * p< 0.05, ** p< 0.01).
To study the effects of rSTC-1 on inflammation, mice were euthanized 48 hours after surgery and the heart tissues were processed (Fig. 4A) for RT-PCR and ELISA. The amount of CD14 protein was increased in heart lysates following infarction. In a manner similar to our in vitro data using U937 cells, administration of rSTC-1 significantly reduced levels of CD14 (Fig. 4E) in the infarcted heart, as well as levels of other inflammatory mediators including TNFα (Fig. 4F), CXCL2 (Fig. 4G), and IL-1β (Fig. 4H). Moreover rSTC-1 treatment reduced cardiac MPO (Fig. 4I); a bio-marker of tissue injury that is produced mainly by activated neutrophils and macrophages, and that has powerful pro-oxidative and pro-inflammatory properties (15, 25). Collectively, these data suggest that STC-1 could be used therapeutically to suppress inflammation in the heart following myocardial infarction and protect cardiomyocytes from injury directly caused by MPO. STC-1 treatment, however, did not alter levels of UCP2 (Fig. 4J) in the infarcted heart as it did in the kidneys (Fig. 4K), and only moderately increased UCP3 expression (Fig. 4L), suggesting that the anti-inflammatory effects of STC-1 transpired through atypical mechanisms and regulators, at least in part.
Intravenous administration of rSTC-1 improved heart function and reduced scar formation in ischemic cardiac injury
To examine the therapeutic effects of rSTC-1, myocardial infarction was induced in NOD/SCID mice followed by 2 injections of rSTC-1 (2.0 mg/kg) as described previously. Changes in heart function were determined with echocardiography (echo) on anesthetized mice 21 days after LLDCA (Fig. 5A). All measurements were assessed in B-mode from the parasternal long axis position (Supplemental Videos). The results showed a substantial decrease in LVEF following coronary artery ligation that was improved significantly by treatment with rSTC-1 (Fig. 5B). STC-1 treatment also improved fractional shortening (FS) as well as end-systolic dimensions (Table 1), collectively suggesting that rSTC-1 treatment enhanced contractility and systolic function. After cardiac function was evaluated, the hearts were removed from the chest and infarct size measured from paraffin embedded sections stained with Masson Trichrome (Figs. 5C,D). We observed that rSTC-1 decreased the length of the ventricular infarct region (Figs. 5C,D). Thus, STC-1 treatment results in reduced cardiac scarring and functional improvements of the ischemic heart.
Figure 5. Recombinant stanniocalcin-1 (rSTC-1) administered intravenously improved heart function and reduced infarct size.
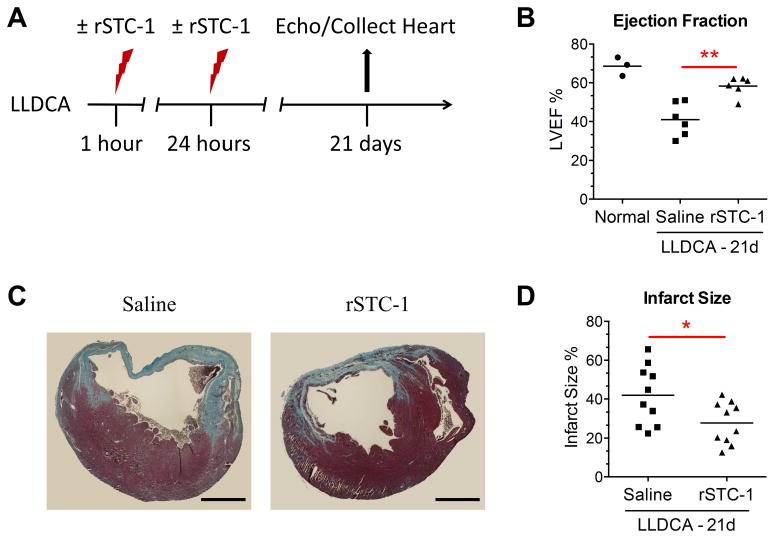
NOD/SCID mice were subjected to ischemic cardiac injury by permanent ligation of the left descending coronary artery (LLDCA). At 1 hour and 24 hours after ligation, 2.0 mg/kg of rSTC-1 or equal volume of 0.9% sodium chloride (saline) was administered intravenously. Twenty one days after ligation, the mice were anesthetized and their left ventricular ejection fraction (LVEF) was evaluated. Then, mice were euthanized to collect heart tissue and assess infarct size. (A) Diagram showing the workflow of the cardiac injury model. (B) Each data point represents the average LVEF of 2 independent recordings with 3 consecutive cardiac cycles (n=3-6). (C) Representative images showing fibrosis in heart sections stained with Masson Trichrome (Scale bar, 1 mm.). (D) Infarct size was measured at the ventricular midline of each section and expressed as the percent length of the fibrotic region relative to the ventricular midline length. Each data point represents the average of ten infarct size measurements per heart (n=10-12). Data was analyzed using Student's t-test (*p< 0.05, **p< 0.01).
Table 1. Assessment of cardiac function by echocardiography.
Mice were subjected to myocardial infarction by ligation of the left descending coronary artery (LLDCA). Saline or rSTC-1 were administered intravenously 1 h and 24 h following ligation, and cardiac function was assessed on day 21. Data are expressed as mean ± SEM. The p values shown are the result of a Student t-test comparing saline versus rSTC-1 treated mice. Abbreviations: diastolic (d), systolic (s), cardiac output (CO), fractional shortening (FS), stroke volume (SV), volume (V).
| MEASUREMENT | BASELINE VALUES | LLDCA (21 DAYS) + TREATMENT | P VALUE | |
|---|---|---|---|---|
| Saline | rSTC-1 | |||
| Area; d (mm2) | 14.6 ± 0.07 | 19.2 ± 1.12 | 16.1 ± 1.48 | P = 0.1261 |
| Area; s (mm2) | 6.5 ± 0.34 | 13.5 ± 1.28 | 9.4 ± 1.26 | P = 0.0466 |
| CO (ml/min) | 8.1 ± 0.28 | 7.9 ± 0.40 | 8.2 ± 0.79 | P = 0.8064 |
| FS (%) | 29.1 ± 3.37 | 7.1 ± 1.81 | 15.3 ± 2.74 | P = 0.0312 |
| SV (μl) | 21.3 ± 0.50 | 20.9 ± 1.24 | 21.8 ± 2.47 | P = 0.7748 |
| V; d (μl) | 30.3 ± 1.21 | 48.2 ± 3.71 | 38.2 ± 6.42 | P = 0.2082 |
| V; s (μl) | 9.0 ± 0.74 | 27.2 ± 3.61 | 16.4 ± 4.15 | P = 0.0789 |
Discussion
Macrophages and their precursor -monocytes- are among important immune effector cells that instruct inflammatory processes to defend against pathogens and promote wound healing (26). Upon tissue injury or infection, monocytes are recruited to foci of inflammation and, in response to a variety of signals emanating from the micro-environment, differentiate into functional tissue macrophages (15, 26). Tight control of these processes is essential to prevent excessive inflammation and collateral tissue destruction.
The glycoprotein STC-1 originally identified as a calcium regulatory hormone in fish (1, 3) has emerged, for multiple reasons, as a candidate to modulate monocyte behavior and inflammatory response. First, STC-1 expression is widely distributed and responsive to numerous stress-inducing factors including hypoxia (27, 28), oxidative stress, and inflammation (6, 9). We reported previously that STC-1 expression is upregulated in mesenchymal stem cells (MSCs) by signals from dying cells (29-31) and in response to caspase activation and inflammatory cytokines (31). Second, STC-1 is reported to blunt the rise in levels of mitochondrial ROS generated in stimulated macrophages (8) and decrease activation of the NLRP3 inflammasome (12), a cytosolic multiprotein complex present in myeloid cells that promotes cytokine maturation (32, 33). While mechanisms for the pleiotropic actions of STC-1 have not been entirely defined, most studies emphasize that the antioxidant property of the protein involves induction of UCPs and dissipation of the mitochondrial proton gradient (8, 13, 34).
In exploring the effects of STC-1 here, we discovered that STC-1 can potentially regulate inflammation by controlling the phenotype of differentiating monocytes and, therefore, their response to inflammatory mediators. Specifically, we observed that STC-1 suppresses the PKC-dependent rise in monocyte/macrophage expression of CD14, a myeloid cell co-receptor that detects indicators of ‘danger’ originating from pathogens or damaged tissue, and delivers these products to specific pro-inflammatory TLRs. It was intriguing here that this noteworthy effect on CD14 expression was not reproduced by the superoxide dismutase mimetic MnTBAP or reversed by paraquat, a superoxide generator, suggesting that the decrease in CD14 was not driven by the anti-ROS properties of STC-1. In subsequent experiments, we observed similar suppressive effects of STC-1 on CD14 expression in monocytes stimulated with a variety of cytokines and danger signals classically associated with tissue injury. Our immune system is designed to combat the threat of both infection and injury by responding to a particular set of molecular cues or patterns that are normally undetectable or absent. TLRs play a major role in this process by distinguishing the conserved motifs of pathogens termed pathogen associated molecular patterns (PAMPs), and the endogenous factors released upon sterile tissue injury collectively referred to as damage associated molecular patterns (DAMPs). There is considerable evidence that both PAMPs and DAMPs share many TLRs on macrophages and perpetuate inflammatory cytokine production (33). There is also considerable evidence indicating that DAMP-mediated TLR signaling is implicated in the pathobiology of numerous inflammatory and autoimmune diseases (35-37). For this reason, TLRs and factors associated with TLR signaling have become attractive therapeutic targets (14, 33).
One of these factors, CD14, can be highly expressed by monocytes/macrophages and is heavily implicated in the behavior of these cells. It is best known as a pattern recognition receptor of the innate immune system that directly interacts with bacterial endotoxin (LPS) to induce intracellular pro-inflammatory signaling cascades through TLR4. However, CD14 also recognizes peptidoglycans and products of apoptotic/necrotic cells such as the transcription factor HMGB1 (38). The role of CD14 in disease progression has become apparent with studies revealing that CD14 deficient mice are protected against LPS-induced cardiac inflammation (39), ischemic tissue injury (40), and septic shock (41). Moreover, several studies have demonstrated that TLR4 inhibition with antibodies or knockout strategies is associated with cardiac benefits following infarction that include attenuation of myocardial inflammation and reduction in infarct size (42-44). In some contexts, however, activation of particular TLRs such as TLR2 can exert cardioprotective effects (45). Specifically, TLR2 agonists have been shown to attenuate neutrophil infiltration into the infarcted heart, suppress infarct size, and enhance cardiac ejection fraction; results that were abrogated in TLR2-deficient mice (46). Here we report that in addition to CD14, TLR4 expression was decreased by STC1 treatment, albeit only moderately, while TLR2 expression was significantly enhanced, indicating that STC-1 can fine-tune the macrophage response to specific DAMPs/PAMPs. Interestingly, this combination of outcomes on CD14/TLR2/TLR4 expression recapitulates that previously observed with anti-inflammatory corticosteroid treatment (47) and could account for the broad therapeutic benefits of STC-1 in numerous injury models. Exactly how STC-1 differentially regulates CD14 and various TLRs is still not entirely clear but could be accredited to its ability to shift the balance of competing mitogen-activated protein kinase (MAPK) pathways such as ERK1/2 and p38. For instance, STC-1 can suppress ERK1/2 signaling (20, 48) and, therefore, decrease PMA-induced TLR4/CD14 expression which is dependent on ERK1/2 (49), while perhaps enhancing p38 activity which is important for TLR2 expression under some circumstances (50). Alternatively, the conserved calcium-regulatory properties of STC-1 might be implicated as CD14 expression is sensitive to calcium chelation and calcium channel antagonists (51).
Nonetheless, together these findings here support our initial observation that STC-1 effectively reduced cytokine production by monocytes/macrophages stimulated with LPS but only when the protein was applied concurrently with PMA which counteracted acquisition of a CD14 positive phenotype. These findings also explain, at least in part, our observations that STC-1 treatment suppressed inflammation in the heart and improved cardiac function after ischemic injury by reducing levels of cardiac CD14. Similar to the sequence of events that accompany ischemic injury in many tissues, myocardial ischemia and the resulting formation of necrotic cells triggers an intense inflammatory reaction that is important for monocyte recruitment and tissue repair but when lingering can become disadvantageous for tissue function (14-16). Interestingly, various cytokines such as IL-1β have been reported to extend the life of inflammatory cells (15). Moreover, it has been shown that both DAMP/TLR and IL-1 signaling can exacerbate post-infarction cardiac inflammation and lead to symptoms of heart failure (14, 15). Here we showed that STC-1 can potentially protect the heart by reducing levels of IL-1β, suggesting that it could be employed in therapeutic regimens for treatment of patients suffering from ischemic cardiac injury. Previous studies support this concept having demonstrated that STC-1 inhibits ischemic injury to the kidney (23) and brain (27). The improvements we noted in heart function with STC-1 treatment were also preceded by efficient reduction in levels of MPO, an enzyme found in neutrophils that catalyzes production of hypochlorous acid and other highly reactive moieties with important microbicidal properties (52). However, MPO can be toxic to cells and detrimental to the remodeling process after myocardial infarction therefore, reduction in levels of MPO can prevent deterioration of cardiac tissue. Our data suggest that STC-1 treatment might reduce cardiac MPO through downregulation of CD14 in the heart and subsequently decrease macrophage response to DAMPs that signal via CD14. Alternatively, reduction of MPO in the infarcted heart could result from the ability for STC-1 to suppress levels of CXCL2, a potent neutrophil chemoattractant produced by mast cells and macrophages (53); or prevent extravasation of neutrophils by decreasing permeability of vascular endothelial cells (54).
Regardless, STC-1 is an intriguing protein that exerts multiple beneficial effects in damaged mammalian tissue. Yet, the signaling pathways that drive STC-1 action have not been clearly defined and specific receptors that bind STC-1 have not been determined. Moreover, knowledge of the distribution of STC-1 following intravenous administration is lacking. As well as our understanding of how STC-1 might specifically regulate monocyte phenotype in vivo. While future studies are being designed to explore these concepts, it is important to note that CD14 expression is less restricted in mice. Instead, mouse CD11b monocytes are typically subcategorized by their expression of other factors such as Ly6C and CX3CR1 (55). Subsequently, our assessment of CD14 and inflammatory cytokines in the infarcted mouse heart was from a global perspective. While this limitation does not downplay the significance of our findings, it does stress the importance of executing future studies that evaluate the therapeutic efficacy of STC-1 in a system that closely mimics the human condition with respect to monocyte-specific features, such as a porcine model (56).
It is also important to note that we employed a permanent coronary artery ligation model (i.e. without reperfusion) devoid of the burst in ROS associated with reperfusion injury, suggesting that STC-1 might also exert therapeutic benefits in vivo independent of its familiar role as an anti-oxidant. This proposal was encouraged by our initial results that revealed an ROS scavenger, MnTBAP, did not suppress the PKC-dependent rise in CD14 expression and, therefore, did not mimic STC-1. Moreover, the superoxide generator paraquat did not reverse the effects of STC-1 on CD14 expression. The finding that myocardial UCP2/UCP3 was not significantly altered by intravenous rSTC-1 treatment also further supported our hypothesis. Nonetheless, it is still likely that STC-1 protects the ischemic heart from injury through mechanisms that involve its power to reduce ROS and regulate intracellular calcium, as well as other properties. It is also likely that with systemic administration, STC-1 might not suppress cardiac inflammation by directly interacting with cells in the heart, but instead mitigate inflammatory response by regulating the phenotype of splenic monocytes that are actively recruited to the infarcted heart (57). This hypothesis is further supported by previous observations that TLR inhibition prevents systemic inflammation following myocardial infarction (42-44, 58-60). In the end, this study has made a significant advancement in our understanding of the therapeutic arsenal employed by STC-1.
In conclusion, as shown in Figure 6, our data demonstrate that STC-1 regulates monocyte phenotype in response to a variety of differentiation stimuli and, in turn, suppresses levels of inflammatory factors produced by activated monocytes/macrophages. Moreover, we demonstrate that STC-1 treatment effectively attenuates inflammation in the ischemic heart and improves cardiac function. Our observations provide a novel mechanism for STC-1 action that could be important for tissue repair by preventing excessive inflammatory response without disrupting the tissue healing properties of macrophages.
Figure 6. Summary of recombinant stanniocalcin-1 (rSTC-1) effects on monocyte/macrophage differentiation and function.
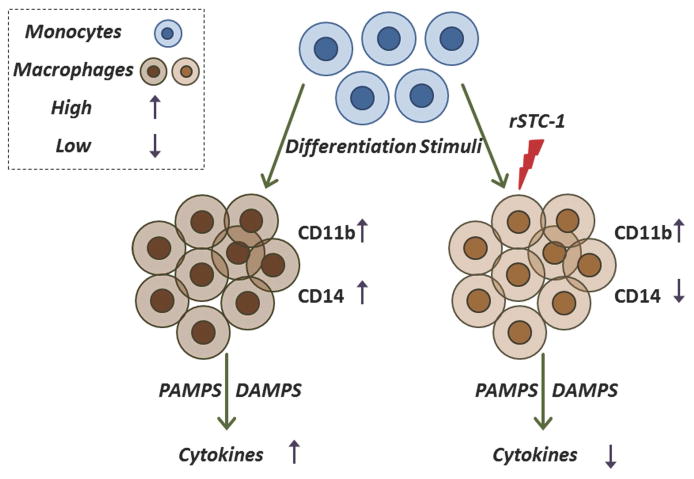
Tissue injury induces release of differentiation stimuli [such as pathogen-associated molecular pattern molecules (PAMPs), damage-associated molecular pattern molecules (DAMPs), cytokines, and chemokines] that promote monocyte differentiation and recruitment to the site of injury. Differentiation stimuli cause an increase in expression of surface molecules such as CD11b and CD14 in monocyte/macrophage and enhance secretion of inflammatory cytokines and chemokines (for example TNFα, IL-1β, and CXCL2) in these cells. Administration of rSTC-1 during the differentiation process reduces expression of CD14 by monocytes/macrophages, which subsequently decreases their response to inflammatory stimuli including DAMPS and PAMPS.
Supplementary Material
Supplemental Video 1. Representative video of a normal mouse heart (NOD/SCID) acquired from the parasternal long axis position by cardiac echo. The animals were anesthetized for the procedure as described in the methods section.
Supplemental Video 2. Representative video of a NOD/SCID mouse heart 21 days after permanent coronary ligation and saline treatment. Images were acquired from the parasternal long axis position by cardiac echo. The animals were anesthetized for the procedure as described in the methods section.
Supplemental Video 3. Representative video of a NOD/SCID mouse heart 21 days after permanent coronary ligation and rSTC-1 treatment. Images were acquired from the parasternal long axis position by cardiac echo. The animals were anesthetized for the procedure as described in the methods section.
Brief Commentary.
Background: The manuscript evaluates the effects of the anti-inflammatory protein stanniocalcin-1 (STC-1) on monocyte/macrophage differentiation and in a mouse model of myocardial infarction.
Translational Significance: We demonstrate that STC-1 treatment reduces levels of the pro-inflammatory TLR co-receptor CD14 on differentiating monocytes, and subsequently their response to destructive inflammatory stimuli. Moreover, we show that STC-1 treatment reduces inflammation and scarring in the infarcted heart, and improves cardiac function. Thus, we propose that STC-1 therapy may protect the heart and other tissues from ischemic injury by limiting harmful responses of monocytes/macrophages, which are recruited to sites of tissue injury, to inflammatory signals from damaged tissues.
Acknowledgments
This study was supported by NIH grant P40RR17447, NIH- National Heart, Lung, and Blood Institute grant 5RO1HL073755-06, and CPRIT award RP150637. We gratefully thank Monica Claypool for her assistance with animal surgeries and Dr. David Dostal for his guidance in analyzing cardiac ejection fraction.
Abbreviations
- ANOVA
Analysis of variance
- ATCC
American Type Culture Collection
- cDNA
Complementary deoxyribonucleic acid
- CM
Conditioned medium
- CO
Cardiac output
- Ct
Critical threshold
- CXCL2
Chemokine (C-X-C motif) ligand 2
- D
Diastolic
- DAMP
Damage associated molecular pattern
- DNase
Deoxyribonuclease
- ELISA
Enzyme-linked immunosorbent assay
- FBS
Fetal bovine serum
- FS
Fractional shortening
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- HMGB1
High mobility group box 1 protein
- IL
Interleukin
- IV
Intravenous
- LAD
Left anterior descending
- LLDCA
Ligation of left descending coronary artery
- LPS
Lipopolysaccharide
- LVEF
Left ventricle ejection fraction
- MPO
Myeloperoxidase
- mRNA
Messenger ribonucleic acid
- MSC
Mesenchymal stem cells
- NIH
National Institutes of Health
- NOD/SCID
Non-obese diabetic/severe combined immune deficiency
- PAMP
Pathogen associated molecular pattern
- PBS
Phosphate buffered saline
- PCR
Polymerase chain reaction
- PKC
Protein kinase C
- PMA
Phorbol 12-myristate 13-acetate
- RNA
Ribonucleic acid
- RPMI
Roswell Park Memorial Institute
- RQ
Relative quantity
- rSTC-1
recombinant human STC-1
- RT
Reverse transcription
- RT-PCR
Reverse transcription-polymerase chain reaction
- S
Systolic
- STC-1
Stanniocalcin-1
- SV
Stoke volume
- TLR
Toll-like receptor
- TNF
Tumor necrosis factor
- UCPs
Uncoupling proteins
Footnotes
All authors are in agreement with the content of the manuscript and have read the journal's authorship agreement as well as the policy on disclosure of potential conflicts of interest.
Disclosure: D.J.P is chair of the scientific advisory committee of a biotech (Temple Therapeutics LLC). The other authors declare they have no competing interests, or other interests that might be perceived to influence the results and discussion reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sheikh-Hamad D. Mammalian stanniocalcin-1 activates mitochondrial antioxidant pathways: new paradigms for regulation of macrophages and endothelium. Am J Physiol Renal Physiol. 2010;298(2):F248–54. doi: 10.1152/ajprenal.00260.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Re RN, Cook JL. The mitochondrial component of intracrine action. Am J Physiol Heart Circ Physiol. 2010;299(3):H577–83. doi: 10.1152/ajpheart.00421.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeung BH, Law AY, Wong CK. Evolution and roles of stanniocalcin. Mol Cell Endocrinol. 2012;349(2):272–280. doi: 10.1016/j.mce.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 4.Sazonova O, James KA, McCudden CR, Segal D, Talebian A, Wagner GF. Stanniocalcin-1 secretion and receptor regulation in kidney cells. Am J Physiol Renal Physiol. 2008;294(4):F788–94. doi: 10.1152/ajprenal.00553.2007. [DOI] [PubMed] [Google Scholar]
- 5.Sheikh-Hamad D, Bick R, Wu GY, et al. Stanniocalcin-1 is a naturally occurring L-channel inhibitor in cardiomyocytes: relevance to human heart failure. Am J Physiol Heart Circ Physiol. 2003;285(1):H442–8. doi: 10.1152/ajpheart.01071.2002. [DOI] [PubMed] [Google Scholar]
- 6.Kanellis J, Bick R, Garcia G, et al. Stanniocalcin-1, an inhibitor of macrophage chemotaxis and chemokinesis. Am J Physiol Renal Physiol. 2004;286(2):F356–62. doi: 10.1152/ajprenal.00138.2003. [DOI] [PubMed] [Google Scholar]
- 7.Ellard J, McCudden C, Tanega C, et al. The respiratory effects of stanniocalcin-1 (STC-1) on intact mitochondria and cells: STC-1 uncouples oxidative phosphorylation and its actions are modulated by nucleotide triphosphates. Mol Cell Endocrinol. 2007;264(1-2):90–101. doi: 10.1016/j.mce.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Huang L, Abdelrahim M, et al. Stanniocalcin-1 suppresses superoxide generation in macrophages through induction of mitochondrial UCP2. J Leukoc Biol. 2009;86(4):981–988. doi: 10.1189/jlb.0708454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang SE, Wu CP, Wu SY, et al. Stanniocalcin-1 ameliorates lipopolysaccharide-induced pulmonary oxidative stress, inflammation, and apoptosis in mice. Free Radic Biol Med. 2014;71C:321–331. doi: 10.1016/j.freeradbiomed.2014.03.034. [DOI] [PubMed] [Google Scholar]
- 10.Huang L, Garcia G, Lou Y, et al. Anti-inflammatory and renal protective actions of stanniocalcin-1 in a model of anti-glomerular basement membrane glomerulonephritis. Am J Pathol. 2009;174(4):1368–1378. doi: 10.2353/ajpath.2009.080476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roddy GW, Rosa RH, Jr, Oh JY, et al. Stanniocalcin-1 rescued photoreceptor degeneration in two rat models of inherited retinal degeneration. Mol Ther. 2012;20(4):788–797. doi: 10.1038/mt.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oh JY, Ko JH, Lee HJ, et al. Mesenchymal stem/stromal cells inhibit the NLRP3 inflammasome by decreasing mitochondrial reactive oxygen species. Stem Cells. 2014;32(6):1553–1563. doi: 10.1002/stem.1608. [DOI] [PubMed] [Google Scholar]
- 13.Liu D, Huang L, Wang Y, et al. Human stanniocalcin-1 suppresses angiotensin II-induced superoxide generation in cardiomyocytes through UCP3-mediated anti-oxidant pathway. PLoS One. 2012;7(5):e36994. doi: 10.1371/journal.pone.0036994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arslan F, de Kleijn D, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nature reviews Cardiology. 2011;8(5):292–300. doi: 10.1038/nrcardio.2011.38. [DOI] [PubMed] [Google Scholar]
- 15.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110(1):159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kempf T, Zarbock A, Vestweber D, Wollert K. Anti-inflammatory mechanisms and therapeutic opportunities in myocardial infarct healing. Journal of molecular medicine. 2012;90(4):361–9. doi: 10.1007/s00109-011-0847-y. [DOI] [PubMed] [Google Scholar]
- 17.Takagawa J, Zhang Y, Wong ML, et al. Myocardial infarct size measurement in the mouse chronic infarction model: comparison of area- and length-based approaches. J Appl Physiol (1985) 2007;102(6):2104–2111. doi: 10.1152/japplphysiol.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ylostalo J, Bartosh T, Coble K, Prockop D. Human mesenchymal stem/stromal cells cultured as spheroids are self-activated to produce prostaglandin E2 that directs stimulated macrophages into an anti-inflammatory phenotype. Stem Cells. 2012;30(10):2283–2296. doi: 10.1002/stem.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Gioia M, Zanoni I. Toll-like receptor co-receptors as master regulators of the immune response. Mol Immunol. 2014 doi: 10.1016/j.molimm.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 20.Huang L, Zhang L, Ju H, et al. Stanniocalcin-1 inhibits thrombin-induced signaling and protects from bleomycin-induced lung injury. Sci Rep. 2015;5:18117–18117. doi: 10.1038/srep18117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takeyama N, Tanaka T, Yabuki T, Nakatani T. The involvement of p53 in paraquat-induced apoptosis in human lung epithelial-like cells. Int J Toxicol. 2004;23(1):33–40. doi: 10.1080/10915810490265432. [DOI] [PubMed] [Google Scholar]
- 22.van Zuylen V, den Haan M, Roelofs H, Fibbe W, Schalij M, Atsma D. Myocardial infarction models in NOD/Scid mice for cell therapy research: permanent ischemia vs ischemia-reperfusion. Springerplus. 2015;4:336–336. doi: 10.1186/s40064-015-1128-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan J, Huang L, Belousova T, et al. Stanniocalcin-1 inhibits renal ischemia/reperfusion injury via an AMP-activated protein kinase-dependent pathway. J Am Soc Nephrol. 2015;26(2):364–378. doi: 10.1681/ASN.2013070703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santulli G, Xie W, Reiken S, Marks A. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A. 2015;112(36):11389–11394. doi: 10.1073/pnas.1513047112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loria V, Dato I, Graziani F, Biasucci LM. Myeloperoxidase: a new biomarker of inflammation in ischemic heart disease and acute coronary syndromes. Mediators Inflamm. 2008;2008:135625. doi: 10.1155/2008/135625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wynn T, Chawla A, Pollard J. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–55. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Westberg JA, Serlachius M, Lankila P, Penkowa M, Hidalgo J, Andersson LC. Hypoxic preconditioning induces neuroprotective stanniocalcin-1 in brain via IL-6 signaling. Stroke. 2007;38(3):1025–1030. doi: 10.1161/01.STR.0000258113.67252.fa. [DOI] [PubMed] [Google Scholar]
- 28.Westberg J, Serlachius M, Lankila P, Andersson L. Hypoxic preconditioning induces elevated expression of stanniocalcin-1 in the heart. American journal of physiology Heart and circulatory physiology. 2007;293(3):H1766–71. doi: 10.1152/ajpheart.00017.2007. [DOI] [PubMed] [Google Scholar]
- 29.Block GJ, Ohkouchi S, Fung F, et al. Multipotent stromal cells are activated to reduce apoptosis in part by upregulation and secretion of stanniocalcin-1. Stem Cells. 2009;27(3):670–681. doi: 10.1002/stem.20080742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartosh TJ, Wang Z, Rosales AA, Dimitrijevich SD, Roque RS. 3D-model of adult cardiac stem cells promotes cardiac differentiation and resistance to oxidative stress. J Cell Biochem. 2008;105(2):612–623. doi: 10.1002/jcb.21862. [DOI] [PubMed] [Google Scholar]
- 31.Bartosh TJ, Ylostalo JH, Bazhanov N, Kuhlman J, Prockop DJ. Dynamic compaction of human mesenchymal stem/precursor cells (MSC) into spheres self-activates caspase-dependent IL1 signaling to enhance secretion of modulators of inflammation and immunity (PGE2, TSG6 and STC1) Stem Cells. 2013 doi: 10.1002/stem.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–32. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 33.Chen G, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nature reviews Immunology. 2010;10(12):826–37. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohkouchi S, Block GJ, Katsha AM, et al. Mesenchymal stromal cells protect cancer cells from ROS-induced apoptosis and enhance the Warburg effect by secreting STC1. Mol Ther. 2012;20(2):417–423. doi: 10.1038/mt.2011.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Magna M, Pisetsky D. The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Molecular medicine. 2014;20:138–46. doi: 10.2119/molmed.2013.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith S, Jefferies C. Role of DNA/RNA sensors and contribution to autoimmunity. Cytokine Growth Factor Rev. 2014 doi: 10.1016/j.cytogfr.2014.07.019. [DOI] [PubMed] [Google Scholar]
- 37.Thwaites R, Chamberlain G, Sacre S. Emerging role of endosomal toll-like receptors in rheumatoid arthritis. Frontiers in immunology. 2014;5:1. doi: 10.3389/fimmu.2014.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim S, Kim SY, Pribis JP, et al. Signaling of high mobility group box 1 (HMGB1) through toll-like receptor 4 in macrophages requires CD14. Mol Med. 2013;19:88–98. doi: 10.2119/molmed.2012.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knuefermann P, Nemoto S, Misra A, et al. CD14-deficient mice are protected against lipopolysaccharide-induced cardiac inflammation and left ventricular dysfunction. Circulation. 2002;106(20):2608–15. doi: 10.1161/01.cir.0000038110.69369.4c. [DOI] [PubMed] [Google Scholar]
- 40.Kaczorowski D, Nakao A, Vallabhaneni R, et al. Mechanisms of Toll-like receptor 4 (TLR4)-mediated inflammation after cold ischemia/reperfusion in the heart. Transplantation. 2009;87(10):1455–63. doi: 10.1097/TP.0b013e3181a36e5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haziot A, Ferrero E, Köntgen F, et al. Resistance to endotoxin shock and reduced dissemination of gram-negative bacteria in CD14-deficient mice. Immunity. 1996;4(4):407–14. doi: 10.1016/s1074-7613(00)80254-x. [DOI] [PubMed] [Google Scholar]
- 42.Chong AJ, Shimamoto A, Hampton CR, et al. Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J Thorac Cardiovasc Surg. 2004;128(2):170–179. doi: 10.1016/j.jtcvs.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 43.Shimamoto A, Chong AJ, Yada M, et al. Inhibition of Toll-like receptor 4 with eritoran attenuates myocardial ischemia-reperfusion injury. Circulation. 2006;114(1 Suppl):I270–4. doi: 10.1161/CIRCULATIONAHA.105.000901. [DOI] [PubMed] [Google Scholar]
- 44.Timmers L, Sluijter JPG, van Keulen JK, et al. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res. 2008;102(2):257–64. doi: 10.1161/CIRCRESAHA.107.158220. [DOI] [PubMed] [Google Scholar]
- 45.Vilahur G, Badimon L. Ischemia/reperfusion activates myocardial innate immune response: the key role of the toll-like receptor. Front Physiol. 2014;5:496. doi: 10.3389/fphys.2014.00496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ha T, Hu Y, Liu L, et al. TLR2 ligands induce cardioprotection against ischaemia/reperfusion injury through a PI3K/Akt-dependent mechanism. Cardiovasc Res. 2010;87(4):694–703. doi: 10.1093/cvr/cvq116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oeff M, Seltmann H, Hiroi N, et al. Differential regulation of Toll-like receptor and CD14 pathways by retinoids and corticosteroids in human sebocytes. Dermatology (Basel) 2006;213(3):266–266. doi: 10.1159/000095056. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen A, Chang ACM, Reddel RR. Stanniocalcin-1 acts in a negative feedback loop in the prosurvival ERK1/2 signaling pathway during oxidative stress. Oncogene. 2009;28(18):1982–1992. doi: 10.1038/onc.2009.65. [DOI] [PubMed] [Google Scholar]
- 49.Traore K, Zirkin B, Thimmulappa R, Biswal S, Trush M. Upregulation of TLR1, TLR2, TLR4, and IRAK-2 Expression During ML-1 Cell Differentiation to Macrophages: Role in the Potentiation of Cellular Responses to LPS and LTA. ISRN Oncol. 2012;2012:641246–641246. doi: 10.5402/2012/641246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oshikawa K, Sugiyama Y. Regulation of toll-like receptor 2 and 4 gene expression in murine alveolar macrophages. Exp Lung Res. 2003;29(6):401–412. doi: 10.1080/01902140303756. [DOI] [PubMed] [Google Scholar]
- 51.Pedrinaci S, Ruiz Cabello F, Gomez O, Collado A, Garrido F. Protein kinase C-mediated regulation of the expression of CD14 and CD11/CD18 in U937 cells. Int J Cancer. 1990;45(2):294–298. doi: 10.1002/ijc.2910450215. [DOI] [PubMed] [Google Scholar]
- 52.Rosen H, Crowley JR, Heinecke JW. Human neutrophils use the myeloperoxidase-hydrogen peroxide-chloride system to chlorinate but not nitrate bacterial proteins during phagocytosis. J Biol Chem. 2002;277(34):30463–30468. doi: 10.1074/jbc.M202331200. [DOI] [PubMed] [Google Scholar]
- 53.De Filippo K, Dudeck A, Hasenberg M, et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. 2013;121(24):4930–7. doi: 10.1182/blood-2013-02-486217. [DOI] [PubMed] [Google Scholar]
- 54.Chen C, Jamaluddin MS, Yan S, Sheikh-Hamad D, Yao Q. Human stanniocalcin-1 blocks TNF-alpha-induced monolayer permeability in human coronary artery endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28(5):906–912. doi: 10.1161/ATVBAHA.108.163667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi C, Pamer E. Monocyte recruitment during infection and inflammation. Nature reviews Immunology. 2011;11(11):762–74. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fairbairn L, Kapetanovic R, Beraldi D, et al. Comparative analysis of monocyte subsets in the pig. J Immunol. 2013;190(12):6389–6396. doi: 10.4049/jimmunol.1300365. [DOI] [PubMed] [Google Scholar]
- 57.Swirski FK, Nahrendorf M, Etzrodt M, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325(5940):612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sakata Y, Dong JW, Vallejo JG, et al. Toll-like receptor 2 modulates left ventricular function following ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;292(1):H503–9. doi: 10.1152/ajpheart.00642.2006. [DOI] [PubMed] [Google Scholar]
- 59.Favre J, Musette P, Douin-Echinard V, et al. Toll-like receptors 2-deficient mice are protected against postischemic coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2007;27(5):1064–1071. doi: 10.1161/ATVBAHA.107.140723. [DOI] [PubMed] [Google Scholar]
- 60.Arslan F, Smeets MB, O'Neill LA, et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation. 2010;121(1):80–90. doi: 10.1161/CIRCULATIONAHA.109.880187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Video 1. Representative video of a normal mouse heart (NOD/SCID) acquired from the parasternal long axis position by cardiac echo. The animals were anesthetized for the procedure as described in the methods section.
Supplemental Video 2. Representative video of a NOD/SCID mouse heart 21 days after permanent coronary ligation and saline treatment. Images were acquired from the parasternal long axis position by cardiac echo. The animals were anesthetized for the procedure as described in the methods section.
Supplemental Video 3. Representative video of a NOD/SCID mouse heart 21 days after permanent coronary ligation and rSTC-1 treatment. Images were acquired from the parasternal long axis position by cardiac echo. The animals were anesthetized for the procedure as described in the methods section.