

Abstract
Neurodegeneration refers to a heterogeneous group of brain disorders that progressively evolve. It has been increasingly appreciated that many neurodegenerative conditions overlap at multiple levels and therefore traditional clinicopathological correlation approaches to better classify a disease have met with limited success. Neuronal network disintegration is fundamental to neurodegeneration, and concepts based around such a concept may better explain the overlap between their clinical and pathological phenotypes. In this Review, promoters of overlap in neurodegeneration incorporating behavioural, cognitive, metabolic, motor, and extrapyramidal presentations will be critically appraised. In addition, evidence that may support the existence of large-scale networks that might be contributing to phenotypic differentiation will be considered across a neurodegenerative spectrum. Disintegration of neuronal networks through different pathological processes, such as prion-like spread, may provide a better paradigm of disease and thereby facilitate the identification of novel therapies for neurodegeneration.
Keywords: COGNITION, DEMENTIA, MOTOR NEURON DISEASE, ALZHEIMER'S DISEASE, NEUROANATOMY
Introduction
Considerable debate exists regarding neurodegeneration, an umbrella term that incorporates a wide range of neurological disorders with heterogeneous clinical and pathological expressions affecting distinct subsets of neurons in specific functional anatomic systems showing a relentless progression.1 Traditionally, the study of neurodegenerative disorders has relied on a conventional clinicopathological approach, defining a particular clinical diagnoses (such as Alzheimer's disease), and attempting to match this to a pattern of pathology in the brain. However, it is increasingly recognised that there is considerable clinical and pathological overlap across neurodegenerative disorders (figure 1). Patients with frontotemporal dementia (FTD) may present with amyotrophic lateral sclerosis (ALS) and vice versa;2 patients with parkinisonian plus syndromes (including corticobasal degeneration (CBD) and progressive supranuclear palsy (PSP) phenotypes) may present with initial cognitive changes,3 while patients with FTD may develop prominent extrapyramidal symptoms resembling parkinsonian plus syndromes.4 Pathologically, there appears to be limited correlation linking specific syndromes such that both tau and Tar-DNA binding protein (TDP)-43 pathology can result in behavioural variant FTD (bvFTD),2 while corticobasal syndrome (CBS) may be associated with both CBD and Alzheimer's disease pathology.5 As such, current clinicopathological approaches fail to recognise the heterogeneity of these conditions.
Figure 1.
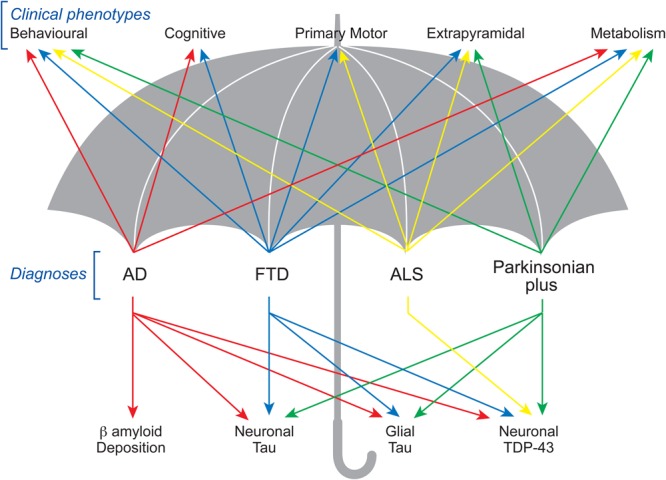
Clinical and pathological overlap in neurodegeneration: showing overlap at both a phenotypic and pathological level between multiple neurodegenerative conditions.
An alternative approach to the classification of neurodegenerative disorders invokes neural networks, defined as a series of interconnected neural nodes that determine physiological function.6 7 These networks occur at microscopic (neurons and synapses), macroscopic or structural (anatomical regions and fibres), and functional (physiological connections) levels.6 There is a wealth of literature describing specific networks based on functional and structural imaging modalities.8 More recently, the concept of pathological networks based on pathogenic proteins, known as ‘molecular nexopathies’,6 has been developed. It has been suggested that the susceptibility and phenotype will vary contingent on the involvement of specific neural networks.7 In this Review the overlap between neurological conditions in terms of clinical phenotype (behaviour, cognitive, primary motor, extrapyramidal, and metabolic) rather than syndromic diagnoses will be discussed, and the evidence suggesting widespread network involvement in neurodegeneration will be critically appraised. Network involvement in neurodegenerative language disorders will not be discussed, but extensive reviews are available.9 Evidence for network dysfunction at a genetic and pathological level, and how neurodegeneration may spread along these networks in a prion-like manner will be discussed in addition to transgenic mouse models that may play a role in mapping network involvement across neurodegenerative disorders.
Clinical phenotypes and neurodegenration
Behavioural phenotypes in neurodegeneration
Behavioural changes have been associated with a range of clinicopathological diagnoses, but are most characteristic of bvFTD, as reflected in current diagnostic criteria. Patients with bvFTD typically present with changes in behaviour ranging from apathy – with reduced motivation, inertia, and a lack of interest in previous hobbies – to disinhibition manifesting in impulsive and often socially inappropriate actions such as overspending, gambling, or sexually inappropriate remarks. Mental rigidity and stereotypical behaviour is also widely reported by caregivers, leading to alterations in food preferences and eating behaviour. Similar, prominent behavioural symptoms have been recorded in other neurodegenerative disorders, including ALS and a number of parkinsonian plus syndromes that display significant overlap with FTD.2 Patients with ALS may develop prominent behavioural features including apathy, loss of empathy, emotional lability, and less commonly gluttony, behavioural stereotypes and compulsions. Patients with ALS may occasionally develop neuropsychiatric and behavioural symptoms prior to the onset of classic motor symptoms.10 In a number of parkinsonian plus syndromes, such as PSP and CBS, there is increasing evidence for prominent behavioural change, including apathy,11 12 impaired social engagement,12 and impaired emotional expression.13 It has recently been suggested that behavioural changes may be present in up to 30% of patients with PSP.3 Patients with CBS and the language variants of FTD (semantic dementia) also develop difficulties with behaviour and emotion processing.14 Given the clinical overlap seen in behaviour and emotion processing in these disorders, it is likely that commonalities exist in the disruption or dysfunction of networks controlling these phenotypic behaviours.
Neural networks and behaviour
The last decade has witnessed a surge of evidence proposing dysfunction within large-scale neural networks; these relate to resting state and intrinsic connectivity functional MRI (fMRI), EEG and MEG studies that correlate with clinical presentations of abnormal behaviours in neurodegenerative diseases. Three major networks have been associated with aberrant behaviours in neurodegenerative diseases—the salience network (SN), the default mode network (DMN) and the executive-control network (ECN).
The SN is centred on the dorsal anterior cingulate cortex (dACC) and frontoinsular cortex with extensive links to both cortical and subcortical regions, including the superior temporal pole, the dorsolateral prefrontal cortex, the amygdala, thalamus, hypothalamus, and the substantia nigra/ventral tegmental area.15 In terms of behaviour, the SN is implicated in detecting, analysing, and integrating emotionally salient stimuli with respect to the internal environment.15 Within the network, the anterior insula is involved in salience detection, whereas the anterior cingulate functions as a modulator of behaviour. The anterior insula, together with the extended SN and input from the posterior insula, integrates internal sensory stimuli to produce a socially appropriate behavior, for example, wearing appropriate clothing for the season. Not only is the insula activated during internal sensory stimuli, it also shows activation to external emotionally salient stimuli such as love, compassion, empathy, disgust, anger, and happiness.16
While the SN is critical for self-awareness, self-control and emotion regulation, it also has a modulatory effect on the DMN – another large-scale network involved in behaviour and cognition (including autobiographical memory, prospection, and theory of mind) and intrinsically linked to the ECN.17 The DMN comprises a network of brain regions with core regions in the posterior cingulate cortex (PCC), lateral parietal cortex and medial prefrontal cortex (mPFC), and prominent connections to nodes in the medial temporal lobe (MTL) and angular gyrus. The DMN is reciprocally deactivated during attention-demanding tasks which activate the SN. In contrast, a number of specific non-stimulus driven cognitive functions activate the DMN; autobiographical memory, and episodic memory are associated with activation of nodes in the PCC, while theory of mind and moral reasoning are associated with activation of specific nodes in the mPFC.18 19 The reciprocity of DMN and SN activation in moral reasoning exemplifies the role of the anterior insula in modulating behaviour by switching between large-scale networks.20 As mentioned, the DMN is activated in healthy participants during moral reasoning tasks; however, during these tasks there is also directed functional connectivity to nodes within the SN. This functional connectivity between the DMN and SN is diminished when patients with FTD show poor moral reasoning.21 Finally, the ECN links the dorsolateral frontal and parietal neocortices as well as the dorsal caudate and anterior insula, and is primarily involved in manipulating and maintaining information in working memory, sustained attention, response selection to goal-directed behavior, and inhibition.15 22 Depending on the task, the ECN and the DMN show antagonistic responses. For example, when a participant is involved in an attention-demanding task in which self-referential and episodic memory is unnecessary, then the ECN is engaged and the DMN is disengaged,23 network changes that are facilitated by the SN and in particular, the anterior insula.
Common neural networks for the behavioural phenotype in neurodegeneration
As mentioned previously, many abnormal behaviours (including apathy and poor social interactions) are pervasive among an array of neurodegenerative conditions (including ALS, FTD, PSP, CBD and AD). Although these conditions show a variety of clinical phenotypes with distinctive neuroanatomical, neuroimaging, and genetic signatures, their shared behavioural deficits could be explained in terms of network disruption.
Mounting evidence exists for involvement of the SN in FTD, particularly in relation to bvFTD.24 These findings converge with results from structural imaging studies which demonstrate atrophy of key regions of the SN, including the anterior cingulate cortex, insula and subcortical structures (including the amygdala, striatum and thalamus), all regions which have been implicated in complex social and emotional processing.25 Furthermore, reduced SN connectivity correlates with disease severity.26 27 Considering the role of the SN and in particular, the anterior cingulate in terms of behaviour modulation, it is not surprising that patients with abnormalities in this network, and particularly patients with FTD, show profound disinhibited behaviour with loss of social graces and reduced empathy. In contrast, studies of DMN connectivity in FTD have shown conflicting results that perhaps can be reconciled in light of the top-down influence of the SN on the DMN, as discussed above.8 Theory of mind deficits are intrinsically linked to autobiographical memory and prospection through connectivity in the mPFC of the DMN in healthy participants.28 Deficits in ‘theory of mind (ToM)’ are often profound in FTD; these patients are unable to imagine the thoughts and experiences of others, and are often unsympathetic to issues which do not concern themselves and these, in turn, can cause conflict within personal relationships.29 30 Deficits in ToM have also been identified in patients with ALS and PSP, thus supporting the hypothesis that these conditions merge in terms of abnormalities in the DMN.31 32 In AD, the DMN consistently shows reduced connectivity, and this again corresponds to atrophy patterns in AD and has been shown to predict disease severity and progression.33 34 Similarly, the relative preservation or intensification of SN activation is in parallel with the preservation of manners and social decorum until late in AD, but has been shown to be associated with aberrant motor behaviour, anxiety, and irritability.35
In comparison, patients with ALS have shown inconsistent results in terms of network connectivity.24 36 37 A number of theories arise in terms of resolving the conflict between studies. The first enhanced connectivity may occur in ALS due to loss of cortical inhibition, and is in keeping with findings from one study that found a greater rate of disease progression in those with the most functional connectivity;38 39 the second that enhanced functional recruitment may occur in one region to compensate for loss of function in another, a compensatory mechanism that is early and may be lost as the network disintegrates and cognitive deficits increase.40 Finally, the heterogeneity of the ALS phenotype, particularly in terms of cognitive and behavioural features, may influence and be influenced by the pattern of network degeneration.
Further insights may be gained by exploring the complex interactions within and between large-scale neural networks in PSP and CBS, which show overlapping behavioural abnormalities and structural changes with FTD. The network degeneration theory has been confirmed in CBS,41 but as yet no published data exists regarding the nature of intrinsic connectivity abnormalities in these disorders in relation to behavioural changes.
Cognitive phenotype in neurodegeneration
Episodic memory dysfunction represents a hallmark feature of AD with patients typically experiencing difficulties in the encoding and retrieval of recently experienced events. Irrespective of the domain of testing, patients with AD display significant impairments on standard neuropsychological tests of visual and verbal recall, as well as gross impairments in the retrieval of personally relevant autobiographical memories from their past and adaptive forms of memory such as prospective memory. The MTL represent one of the earliest sites of pathology in AD, leading to the common assumption that episodic memory impairment reflects MTL dysfunction.42 It is evident, however, that regions beyond the MTL, such as the PCC/retrosplenial cortex, also play a crucial role in the origin of memory dysfunction in AD.43 It has been argued that disruption of functional connectivity between the hippocampus and posterior parietal regions represents a key modulator of episodic memory disruption in AD.44
While memory dysfunction is regarded as a phenotypic feature of AD, mounting evidence points to significant episodic memory disturbance in other neurodegenerative disorders, thus suggesting that the diagnostic utility of traditional neuropsychological memory tasks may be more limited. For example, a severe amnestic presentation remains an exclusion criterion for the diagnosis of bvFTD; yet a number of studies converge to reveal striking memory impairments equivalent to that of matched cases of AD.18 These deficits occur irrespective of the domain of testing of the standardised tests for verbal and visual, immediate and delayed, recall, autobiographical memory45 and prospective memory.46 Further impairments are evident for source memory, with bvFTD cases unable to correctly retrieve the spatial or temporal context of previously presented items.47 Given that the locus of atrophy in FTD typically affects frontoinsular cortices, it was initially assumed that episodic memory difficulties in bvFTD reflected prefrontal cortical degeneration. Recent studies unequivocally point to the involvement of MTL, notably the hippocampus, in driving episodic memory deficits in bvFTD.43 48 Accordingly, it is clear that significant overlap exists in the neural circuitry mediating episodic memory deficits in bvFTD and AD, with the hippocampus representing a critical nexus in this system.
Common neural networks for the cognitive phenotype in neurodegneration
The advent of sophisticated neuroimaging techniques has served to clarify the complexity of the neural circuitry which supports episodic memory in humans, confirming that engagement of a distributed network of cortical regions exhibiting strong connections with the MTL is essential for episodic memory retrieval.49 Key cortical regions in this network include frontal regions such as the medial prefrontal cortex as well as posterior parietal structures, including the retrosplenial/PCC and the angular gyrus.50 The hippocampus appears particularly well placed to support episodic memory retrieval given its dense projections with prefrontal51 and midline posterior cortical structures.44 In addition, subcortical structures, such as the thalamus and fornix, are becoming increasingly recognised for their role in supporting episodic retrieval, further underscoring the complexity of this system. Damage to any node of this large-scale network or alterations in the functional connectivity therein will result in compromised memory performance, as evident in the large-scale network dysfunction observed in the dementias.18
Primary motor/extrapyramidal phenotype and cognition in neurodegeneration
The archetypical overlap syndrome between loss of primary motor function and cognition is that of FTD and ALS. Symptoms of cognitive dysfunction and FTD can occur in ALS, and patients with FTD can develop limb weakness and bulbar dysfunction. There is also increasing awareness of an overlap in clinical phenotyping between extrapyramidal symptoms, including parkinsonism, bradykinesia, rigidity and gait abnormalities, and cognitive changes within the parkinsonian plus syndromes; in other words, it is often difficult to separate a diagnoses of FTD (behavioural and language variants) from CBS and PSP. Patients can often present with any of these movement disorders and go on to develop cognitive symptoms, most commonly in the form of behavioural change. Presentation may be with cognitive deficits and also involve other domains,4 including aphasia, apraxia of speech,52 limb apraxia, and visuoconstructive deficits.4 Further complicating the issue is that patients with different genetic causes of FTD, including the C9orf72 expansion, microtubule associated protein tau (MAPT) and progranulin (GRN) mutations, can develop features of parkinsonism, with both MAPT and progranulin mutations in particular being associated with CBS and PSP.
Common neural networks for the primary motor/extrapyramidal phenotype in neurodegneration
Structural and functional imaging studies have indicated that networks involved in the primary motor deficits of ALS functionally and anatomically involve regions associated with the motor cortices.53 On diffusion tensor imaging the corpus callosum has been found to be consistently affected in ALS; the hypothesis is that it is integral for the pathological spread in the brain.54 Voxel-based morphometry shows atrophy of motor cortices occurs in the primary motor phenotypes of both FTD and ALS, with additional atrophy also found in the left middle and inferior frontal gyri, the anterior portion of the superior frontal gyri, the superior temporal gyri, the temporal poles and the left posterior thalamus, and the non-motor regions known to be involved in pure bvFTD.55 56 This broader pattern of atrophy potentially explains the spectrum of motor and cognitive changes seen across FTD and ALS.
The extrapyramidal symptoms associated with parkinsonian plus syndromes differ depending on the clinical syndrome. Atrophy in PSP is localised to the midbrain, pons, thalamus and striatum; in CBS, bilateral atrophy of the premotor cortex, superior parietal lobules, and striatum occurs.57 It has been suggested that PSP participants with an extrapyramidal syndrome have more brainstem atrophy and less cortical atrophy than CBS participants.58 Studies examining network connections in PSP, using white matter tract diffusion tensor imaging and functional MRI, found disruption of network connectivity between the cerebellum, midbrain, thalamus, and premotor cortex.59 It has been suggested that PSP is associated with disrupted thalamocortical connectivity and the dentatorubrothalamic tract.60 An alternative study has suggested involvement of a network anchored by the dorsal midbrain and with connections including the brainstem, basal ganglia, diencephalic, cerebellar and cortical regions.61 A network such as this could potentially explain the mixed motor, behavioural and cognitive features observed in PSP. Further studies have suggested that white matter track degeneration differs between PSP, Parkinson's disease, multisystems atrophy, and CBS.62 63 Studies of overlap in networks responsible for the extrapyramidal symptoms in PSP, CBD and FTD have been limited, but such network overlap is likely given the strong clinical overlap between these conditions. Further research involving structural and functional imaging across these disorders would prove to be fruitful.
Metabolic phenotype in neurodegeneration
Recently it has been suggested that the overlap in neurodegenerative diseases may extend beyond motor and cognitive features, and involve more systemic bodily functions such as metabolism. Insulin resistance and type 2 diabetes appear to play an important role in modulating neurodegenerative diseases; an increased incidence of peripheral insulin resistance and diabetes is identified in a number of neurodegenerative conditions, including AD, Parkinson's disease, Huntington's disease ALS and FTD.64–68 How insulin promotes neurodegeneration remains unclear, but it may exert a peripheral and central influence. Proposed mechanisms include the promotion of oxidative stress, inflammation, vascular dysfunction, and impairment of neurogenesis and neuronal repair.69 Typically, insulin resistance and metabolic changes have been viewed as consequences of obesity. Mounting evidence indicates, however, that several neurodegenerative conditions – including AD, Huntington's disease and ALS – are associated with significant weight loss, often prior to diagnosis,70 implying that these metabolic changes may be part of the neurodegenerative process rather than a consequence of obesity. Further, metabolic change may underlie phenotypic changes in behavior, such as in bvFTD, where patients develop prominent changes in eating behaviour with hyperphagia and changes in food preference.71 This could have a subsequent effect on insulin and cholesterol levels.
Common neural networks for the metabolic phenotype in neurodegeneration
There is increasing evidence for networks controlling eating behavior, and neuroendocrine control and metabolism in conditions such as bvFTD,72 with involvement of reward networks, the hypothalamus and changes in neuroendocrine peptides.72 At a pathological and imaging level, other neurodegenerative conditions show changes in the neuroendocrine system, including ALS73 and Huntington's disease;68 these are conditions exhibiting hypothalamic atrophy. Further work is required to map the neural networks in neurodegeneration that influence behavioural change and metabolism directly.
Pathology and neurodegeneration
Does pathology correlate better to phenotypic networks than to clinical diagnostic syndromes?
Evidence for involvement of networks in neurodegeneration at a pathological level is growing.6 Recently, the concept of ‘molecular nexopathies’ proposes that pathological proteinopathies can target specific networks, and multiple functional networks can be targeted by these proteinopathies. This in turn leads to phenotypic variation and potential overlap between these disorders.6 This model is enticing especially given the hypothesis that proteinopathies may spread in a ‘prion’ like manner. Prions are defined as proteinaceous infectious particles consisting of misfolded prion protein that aggregate together and act as ‘seeds’ that result in a chain reaction causing further misfolding and aggregation, and these spread throughout the central nervous system. A prion hypothesis for disease spread has been hypothesised for a number of neurodegenerative conditions, including amyloid in AD,74 TDP-43 in FTD and ALS, Tau in Pick's disease, and PSP75 and synuclein in Parkinson's disease.74
In terms of the pathological networks corresponding to clinical phenotypic networks (ie, behaviour, cognitive, primary motor, extrapyramidal, and metabolic) this is an emerging area. To date, the examination of pathological networks has been based around the traditional clinical diagnostic syndromes (eg, Alzheimer's disease) rather than on the phenotypic networks highlighted above. The most developed pathological networks in terms of correlation to clinical phenotypic networks involve the β-amyloid and TDP-43 proteins, with tau an emerging area of research.
β-amyloid and the cognitive network
In AD it has been hypothesised that β-amyloid spreads in prion-like manner giving different pathological stages of β-amyloid deposition.74 β-amyloid first accumulates in the basal temporal and orbitofrontal neocortex, then spreads throughout the neocortex and then to the hippocampi, amygdala, diencephalon and basal ganglia. Then finally, in severe cases, this spreads to the mesencephalon, lower brainstem, and cerebellar cortex.74 76 How this pathological spread interacts with clinical phenotypic networks remains to be elucidated. There is emerging research examining this with β amyloid being found to preferentially deposit in the DMN network and higher order intrinsic connectivity networks, including the frontoparietal network, with areas showing decreased functional activity including both areas exhibiting β-amyloid deposition and atrophy.77 Frequently, however, in AD there is a disparity between the presence of pathology (eg, β-amyloid) and functional impairment.78 A recent study examined the functional impairment between variants of Alzheimer's disease (early onset, logopenic variant, and posterior cortical atrophy) as measured by 18F-labelled fluorodeoxyglucose positron emission tomography (FDG-PET), and β-amyloid deposition as measured by (11)C-labelled Pittsburgh compound B.78 It was found that the AD variants had degeneration in specific functional networks, while their pattern of β-amyloid deposition varied, often being diffuse.78 This suggests that the location of β-amyloid deposition may not fit the network patterns involved in the clinical variability seen between AD syndromes.78 Further work is required to understand the interaction between clinical phenotypic networks, and deposition of pathology along these networks and more diffusely.
TDP-43 and the primary motor and behavioural networks
In ALS it has being suggested that spread of TDP-43 pathology may occur in a recognised centrifugal pattern with four stages of spread.79 TDP-43 accumulation begins in the motor neocortex, progressing to the spinal cord and brainstem with involvement of frontal-parietal regions, and finally to the temporal lobes.79 Such a pattern of spread may potentially explain the eventual development of cognitive symptoms in ALS and the predominant clinical phenotype of motor signs. In bvFTD, TDP-43 pathology is suggested to develop with a frontooccipital gradient involving initial accumulation in frontal regions, then premotor, primary motor, parietal and occipital cortices.80 This pattern of pathological spread explains the initial prominent behavioural changes followed by involvement of other cognitive functions as the disease progresses.80
Tau Pathology and networks
Neuronal tau pathology networks have been described in AD with neuronal tau inclusions developing in the locus coeruleus, as well as in the transentorhinal and entorhinal cortices (stages I and II). This is followed by the presence of neuronal tau inclusions in the hippocampal formation and some parts of the neocortex (stages III and IV), followed by large parts of the neocortex (stages V and VI).74 How these stages relate to differing clinical phenotypic networks has not been examined, nor has the relationship between phenotypic networks and the glial tau pathologies of CBD and PSP.
How transmission occurs along pathological networks
There is growing evidence from animal models that transmission may occur in a prion-like fashion along neurons connected in networks. A recent study shows that injection of human brain extracts from patients suffering different tauopathies (including AD, Pick's disease, PSP, and CBD) resulted in the formation of tau inclusions in ALZ17 transgenic mice.81 Similar attempts were trialled several decades ago by Gajdusek and Gibbs in primates with limited success.82 Hypotheses of how cell-to-cell transmission occurs varies between diseases, with suggestions that include trans-synaptic transmission, transmission through membrane channels, and glial cell intrusions into neurons.83 84
In ALS it has been suggested that transmission between neuronal networks may occur via glia with which endocytose extracellular protein material;83 however, the oligodendrocytes along such connected pathways are unlikely to be involved since they do not accumulate TDP-43 protein.85 In PSP and CBD, with such obvious glial tau pathology, this may be a more dominant mechanism and the tau oligomers are considered as most responsible for the spread of pathology through the brain.86 It is potentially possible that susceptibility of specific cells to specific proteinaceous material may vary, and this along with the susceptibility of various functional networks may be responsible for the heterogeneity of clinical phenotypes as well as their overlap. Why some clinical phenotypic networks (eg, the behavioural network) may be susceptible to several proteins (eg, tau and TDP-43) while the primary motor network, as in ALS, seems more susceptible to only TDP-43 pathology remains elusive. It likely involves genetic, epigenetic and environmental interactions.
Do genetic abnormalities correlate better to phenotypic networks than to pathology and clinical diagnostic syndromes?
Genetic mutations involved in neurodegenerative diseases
A given clinical syndrome is often associated with several different genetic mutations and underlying pathologies. FTD is a good example. There are a wide range of causative genes for FTD, including C9orf72, MAPT and GRN, and other rarer causes including VCP, CHMP2B, FUS, TARBP, DCTN1 and SQSTM1. At a clinical level, C9orf72, MAPT and GRN are all most commonly associated with a bvFTD clinical presentation; however, even though they are associated with a similar clinical picture, C9orf72 and GRN are associated with TDP-43 pathology, while MAPT mutations are associated with underlying tau pathology. The clinical syndrome of pure ALS is also associated with a common clinical presentation, but with variable underlying genetic mutations and pathology. ALS is associated with mutations in SOD1, TARDBP, FUS and GGGGCC expansions in C9orf72. At a pathological level, most cases are associated with TDP-43 pathology;87 SOD-1 mutation cases, however, do not have TDP-43, but have neuronal SOD1 inclusions. CBD and PSP can be associated with MAPT mutations,88 and autosomal dominant AD has been associated with mutations in the Amyloid precursor Protein (APP) and presenilin 1 and 2, with late onset risk associated with allelic variation of apolipoprotein E. How this multitude of genetic mutations can be associated with differing pathologies, but overlapping clinical phenotype is not known; thus, the examination of genetic mutations in relation to network dysfunction may prove to be fruitful.
To date, despite a wealth of literature regarding the relationship between genetic mutations and pathological changes, and specific brain atrophy on imaging, there has been limited examination of the relationship between genetic mutations and either clinical phenotypic functional or pathological networks; however, this is an emerging area. In bvFTD, both C9orf72 mutation carriers and non-carriers showed convergent breakdown of large-scale networks despite different atrophy patterns, with behavioural symptoms correlating with diminished SN connectivity and heightened default node connectivity.89 This area of research will be likely to provide useful insights into the networks underlying neurodegeneration and clinical phenotypic heterogeneity, and will also provide useful insights into the molecular functioning of genes within networks and hence, in therapeutic trials. The mapping of networks in relation to genetic mutations also offers the prospect of being able to examine the network function of presymptomatic carriers and identify network involvement early on in the disease process. It has been found that GRN mutation carriers change decades before the onset of the disease in functional brain networks that begin in the parietal region and then spread to the anterior brain.90 The relationship between genes and neural networks will also provide an important link between animal models and these functional networks as many animal models are based on a specific genetic mutation.
Use of mouse models to map networks
Transgenic mice, and other animal models, have been instrumental to further our understanding of neurodegenerative diseases. While the first models used transgene-mediated expression of human amyloid precursor protein (APP) to model aspects of AD, other models followed to reflect tauopathies by transgenic expression of tau, ALS by expression of TDP-43, SOD1 or more recently through transgenic expansion of C9orf72 repeats. Neuronal network aberrations and hyperexcitability have been described for transgenic mouse models using EEG91 and imaging.92 Interestingly, network aberrations and hypersynchronous discharges and hyperexcitability91 are detectable in APP mice preceding notable β-amyloid deposition93, and cognitive deficits in APP transgenic mice are apparent at early stages. The early involvement of neuronal network abnormalities before marked pathology reflects findings in human functional imagining studies, and suggests that soluble APP products (eg, β-amyloid or β-amyloid oligomers) rather than β-amyloid plaques are involved in the generation of network impairments and likely cognitive deficits. The animal data also suggests that network aberrations and hyperexcitability are linked. Key techniques used in mouse models to analyse neuronal network function are electroencephalography (EEG) and computational methods to mine EEG data for network performance. Several recording types appear particularly relevant as their network features can be related to cognitive performance by computational means. Hippocampal recordings from the cornu ammonis (CA) region, for example, provide insight on performance and topology of pyramidal/interneuronal connections that are reflected in EEG γ and theta oscillations and their phase coupling.94 Spontaneous and induced hyperexcitability has also been observed in tau transgenic mouse models.95 However, detailed analysis of network topology by EEG or electrophysiology of single cells in situ in tau transgenic mice have not been performed. Network alterations in ALS mouse models using SOD1, TDP-43 overexpression, or C9orf72 repeat expansion have also not been addressed. Further research is required into how network aberrations and hyperexcitability are linked in such disease-relevant animal models.
In addition to functionally studying networks in animal models, the recent advances in optogenetics have now made manipulation of neuronal systems in vivo feasible.96 Combining functional network analysis by EEG, electrophysiology or imaging with induced changes in activity of specific neuronal ensembles using optogenetics could prove to be a valuable tool to address whether network aberrations can be manipulated or even reversed by specifically targeting subsets of neurons (eg, interneurons). Insights from these induced network outputs can then be correlated with cognitive performance. Mouse models, therefore, are a past and future tool to test these basic hypotheses on disease-relevant network mechanisms and to help drive better targeted therapies.
Future approaches
Based on the available literature, it is proposed that the clinical and pathological heterogeneity of neurodegenerative disorders remains fundamentally related to the functioning of underlying neural networks. Understanding these networks and how they are intrinsically involved will not only advance our clinical understanding of these conditions and their varying phenotypes, but also provide greater insight into their pathophysiology. The standard approach of examining one aspect of these conditions and mapping it to a particular brain region through neuroimaging and pathological examination has met with limited success, perhaps resulting in therapeutic failures. A potential way to overcome this problem would be to take ecologically valid tasks that show changes across multiple neurodegenerative conditions (eg, a task measuring behavioural change) and use multiple imaging techniques – both structural and functional – to map the brain networks in multiple conditions (figure 2) and understand their specific genetic susceptibilities. Using these networks, pathological involvement could be examined in addition to the potential for spread along these networks at the cellular and molecular levels. Insights into the processes and patterns of molecular spread of neurodegenerative conditions may also be gained by comparing animal models with relevant susceptible genetic mutations for the networks involved. Understanding a neural network of neurodegeneration and patterns of disease spread and progression via such networks seems likely to promote new treatment approaches.
Figure 2.

Neurodegenerative network map: proposed way forward for a collaborative approach based on phenotypic variation, imaging, pathology and genetics for investigating the neural networks and their contribution to the pathophysiological bases of neurodegenerative conditions. A better understanding of the neural networks involved is likely to translate into better targeted treatments based on these networks. DTI, diffusion tensor imaging; PET, positron emission tomography.
Footnotes
Contributors: RMA and MCK manuscript concept. All authors were involved in the drafting and editing of the manuscript.
Funding: This work was supported by funding to Forefront, a collaborative research group dedicated to the study of frontotemporal dementia and motor neurone disease, from the National Health and Medical Research Council of Australia (NHMRC) programme grant (#1037746 to GH, MK and JH) and the Australian Research Council Centre of Excellence in Cognition and its Disorders Memory Node (#CE110001021 JH) and other grants/sources (NHMRC project grant #1003139). RA is Royal Australasian College of Physicians PHD scholar and MND Research Australia PhD Scholar. MI is an ARC Discovery Early Career Researcher Award Fellow (#DE130100463). GH is a NHMRC Senior Principal Research Fellow (#1079679). LMI is a NHMRC Senior Research Fellow (#1003083). No funding source had a role in the writing of the manuscript.
Competing interests: None declared.
Patient consent: No.
Provenance and peer review: Commissioned; externally peer reviewed.
References
- 1.Przedborski S, Vila M, Jackson-Lewis V. Neurodegeneration: what is it and where are we? J Clin Invest 2003;111:3–10. 10.1172/JCI17522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002;59:1077–9. 10.1212/WNL.59.7.1077 [DOI] [PubMed] [Google Scholar]
- 3.Kobylecki C, Jones M, Thompson JC, et al. Cognitive-behavioural features of progressive supranuclear palsy syndrome overlap with frontotemporal dementia. J Neurol 2015;262:916–22. 10.1007/s00415-015-7657-z [DOI] [PubMed] [Google Scholar]
- 4.Kertesz A, McMonagle P, Jesso S. Extrapyramidal syndromes in frontotemporal degeneration. J Mol Neurosci 2011;45:336–42. 10.1007/s12031-011-9616-1 [DOI] [PubMed] [Google Scholar]
- 5.Boeve BF. The multiple phenotypes of corticobasal syndrome and corticobasal degeneration: implications for further study. J Mol Neurosci 2011;45:350–3. 10.1007/s12031-011-9624-1 [DOI] [PubMed] [Google Scholar]
- 6.Warren JD, Rohrer JD, Schott JM, et al. Molecular nexopathies: a new paradigm of neurodegenerative disease. Trends Neurosci 2013;36:561–9. 10.1016/j.tins.2013.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisen A, Turner MR. Does variation in neurodegenerative disease susceptibility and phenotype reflect cerebral differences at the network level? Amyotroph Lateral Scler Frontotemporal Degener 2013;14:487–93. 10.3109/21678421.2013.812660 [DOI] [PubMed] [Google Scholar]
- 8.Pievani M, de Haan W, Wu T, et al. Functional network disruption in the degenerative dementias. Lancet Neurol 2011;10:829–43. 10.1016/S1474-4422(11)70158-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mesulam MM, Rogalski EJ, Wieneke C, et al. Primary progressive aphasia and the evolving neurology of the language network. Nat Rev Neurol 2014;10:554–69. 10.1038/nrneurol.2014.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mioshi E, Caga J, Lillo P, et al. Neuropsychiatric changes precede classic motor symptoms in ALS and do not affect survival. Neurology 2014;82:149–55. 10.1212/WNL.0000000000000023 [DOI] [PubMed] [Google Scholar]
- 11.Borroni B, Alberici A, Agosti C, et al. Pattern of behavioral disturbances in corticobasal degeneration syndrome and progressive supranuclear palsy. Int Psychogeriatr 2009;21:463–8. 10.1017/S1041610209008862 [DOI] [PubMed] [Google Scholar]
- 12.Gerstenecker A, Duff K, Mast B, et al. Behavioral abnormalities in progressive supranuclear palsy. Psychiatry Res 2013;210:1205–10. 10.1016/j.psychres.2013.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh BC, Rowe JB, Calder AJ, et al. Emotion recognition in progressive supranuclear palsy. J Neurol Neurosurg Psychiatr 2009;80:1143–5. 10.1136/jnnp.2008.155846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumfor F, Sapey-Triomphe LA, Leyton CE, et al. Degradation of emotion processing ability in corticobasal syndrome and Alzheimer's disease. Brain 2014;137:3061–72. 10.1093/brain/awu246 [DOI] [PubMed] [Google Scholar]
- 15.Seeley WW, Menon V, Schatzberg AF, et al. Dissociable intrinsic connectivity networks for salience processing and executive control. J Neurosci. 2007;27:2349–56. 10.1523/JNEUROSCI.5587-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singer T, Critchley HD, Preuschoff K. A common role of insula in feelings, empathy and uncertainty. Trends Cogn Sci (Regul Ed) 2009;13:334–40. 10.1016/j.tics.2009.05.001 [DOI] [PubMed] [Google Scholar]
- 17.Bonnelle V, Ham TE, Leech R, et al. Salience network integrity predicts default mode network function after traumatic brain injury. Proc Natl Acad Sci U S A 2012;109:4690–5. 10.1073/pnas.1113455109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irish M, Piguet O, Hodges JR. Self-projection and the default network in frontotemporal dementia. Nat Rev Neurol 2012;8:152–61. 10.1038/nrneurol.2012.11 [DOI] [PubMed] [Google Scholar]
- 19.Greicius MD, Krasnow B, Reiss AL, et al. Functional connectivity in the resting brain: a network analysis of the default mode hypothesis. Proc Natl Acad Sci USA 2003;100:253–8. 10.1073/pnas.0135058100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menon V, Uddin LQ. Saliency, switching, attention and control: a network model of insula function. Brain Struct Funct 2010;214:655–67. 10.1007/s00429-010-0262-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiong W, Wilson SM, D'Esposito M, et al. The salience network causally influences default mode network activity during moral reasoning. Brain 2013;136(Pt 6):1929–41. 10.1093/brain/awt066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koechlin E, Summerfield C. An information theoretical approach to prefrontal executive function. Trends Cogn Sci (Regul Ed) 2007;11:229–35. 10.1016/j.tics.2007.04.005 [DOI] [PubMed] [Google Scholar]
- 23.Sridharan D, Levitin DJ, Menon V. A critical role for the right fronto-insular cortex in switching between central-executive and default-mode networks. Proc Natl Acad Sci U S A 2008;105:12569–74. 10.1073/pnas.0800005105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou J, Greicius MD, Gennatas ED, et al. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer's disease. Brain 2010;133(Pt 5):1352–67. 10.1093/brain/awq075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosen HJ, Gorno–Tempini ML, Goldman WP, et al. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology 2002;58:198–208. 10.1212/WNL.58.2.198 [DOI] [PubMed] [Google Scholar]
- 26.Day GS, Farb NA, Tang-Wai DF, et al. Salience network resting-state activity: prediction of frontotemporal dementia progression. JAMA Neurol 2013;70:1249–53. 10.1001/jamaneurol.2013.3258 [DOI] [PubMed] [Google Scholar]
- 27.Farb NAS, Grady CL, Strother S, et al. Abnormal network connectivity in frontotemporal dementia: evidence for prefrontal isolation. Cortex 2013;49:1856–73. 10.1016/j.cortex.2012.09.008 [DOI] [PubMed] [Google Scholar]
- 28.Spreng RN, Grady CL. Patterns of brain activity supporting autobiographical memory, prospection, and theory of mind, and their relationship to the default mode network. J Cogn Neurosci 2010;22:1112–23. 10.1162/jocn.2009.21282 [DOI] [PubMed] [Google Scholar]
- 29.Kipps CM, Hodges JR. Theory of mind in frontotemporal dementia. Soc Neurosci 2006;1:235–44. 10.1080/17470910600989847 [DOI] [PubMed] [Google Scholar]
- 30.Irish M, Hodges JR, Piguet O. Right anterior temporal lobe dysfunction underlies theory of mind impairments in semantic dementia. Brain 2014;137(Pt 4):1241–53. 10.1093/brain/awu003 [DOI] [PubMed] [Google Scholar]
- 31.Girardi A, Macpherson SE, Abrahams S. Deficits in emotional and social cognition in amyotrophic lateral sclerosis. Neuropsychology 2011;25:53–65. 10.1037/a0020357 [DOI] [PubMed] [Google Scholar]
- 32.Ghosh BC, Calder AJ, Peers PV, et al. Social cognitive deficits and their neural correlates in progressive supranuclear palsy. Brain 2012;135(Pt 7):2089–102. 10.1093/brain/aws128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petrella JR, Sheldon FC, Prince SE, et al. Default mode network connectivity in stable vs progressive mild cognitive impairment. Neurology 2011;76:511–17. 10.1212/WNL.0b013e31820af94e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brier MR, Thomas JB, Snyder AZ, et al. Loss of intranetwork and internetwork resting state functional connections with Alzheimer's disease progression. J Neurosci 2012;32:8890–9. 10.1523/JNEUROSCI.5698-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Balthazar MLF, Pereira FRS, Lopes TM, et al. Neuropsychiatric symptoms in Alzheimer's disease are related to functional connectivity alterations in the salience network. Hum Brain Mapp 2014;35:1237–46. 10.1002/hbm.22248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohammadi B, Kollewe K, Samii A, et al. Changes of resting state brain networks in amyotrophic lateral sclerosis. Exp Neurol 2009;217:147–53. 10.1016/j.expneurol.2009.01.025 [DOI] [PubMed] [Google Scholar]
- 37.Trojsi F, Esposito F, de Stefano M, et al. Functional overlap and divergence between ALS and bvFTD. Neurobiol Aging 2015;36:413–23. 10.1016/j.neurobiolaging.2014.06.025 [DOI] [PubMed] [Google Scholar]
- 38.Turner MR, Agosta F, Bede P, et al. Neuroimaging in amyotrophic lateral sclerosis. Biomark Med 2012;6:319–37. 10.2217/bmm.12.26 [DOI] [PubMed] [Google Scholar]
- 39.Turner MR, Kiernan MC. Does interneuronal dysfunction contribute to neurodegeneration in amyotrophic lateral sclerosis? Amyotroph Lateral Scler. 2012;13:245–50. 10.3109/17482968.2011.636050 [DOI] [PubMed] [Google Scholar]
- 40.Agosta F, Canu E, Valsasina P, et al. Divergent brain network connectivity in amyotrophic lateral sclerosis. Neurobiol Aging 2013;34:419–27. 10.1016/j.neurobiolaging.2012.04.015 [DOI] [PubMed] [Google Scholar]
- 41.Seeley WW, Crawford RK, Zhou J, et al. Neurodegenerative diseases target large-scale human brain networks. Neuron 2009;62:42–52. 10.1016/j.neuron.2009.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–49. 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- 43.Irish M, Piguet O, Hodges JR, et al. Common and unique gray matter correlates of episodic memory dysfunction in frontotemporal dementia and Alzheimer's disease. Hum Brain Mapp 2014;35:1422–35. 10.1002/hbm.22263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.La Joie R, Landeau B, Perrotin A, et al. Intrinsic connectivity identifies the hippocampus as a main crossroad between Alzheimer's and semantic dementia-targeted networks. Neuron 2014;81:1417–28. 10.1016/j.neuron.2014.01.026 [DOI] [PubMed] [Google Scholar]
- 45.Irish M, Hornberger M, Lah S, et al. Profiles of recent autobiographical memory retrieval in semantic dementia, behavioural-variant frontotemporal dementia, and Alzheimer's disease. Neuropsychologia 2011;49:2694–702. 10.1016/j.neuropsychologia.2011.05.017 [DOI] [PubMed] [Google Scholar]
- 46.Kamminga J, O'Callaghan C, Hodges JR, et al. Differential prospective memory profiles in frontotemporal dementia syndromes. J Alzheimers Dis 2014;38:669–79. 10.3233/JAD-131118 [DOI] [PubMed] [Google Scholar]
- 47.Irish M, Graham A, Graham KS, et al. Differential impairment of source memory in progressive versus non-progressive behavioral variant frontotemporal dementia. Arch Clin Neuropsychol 2012;27:338–47. 10.1093/arclin/acs033 [DOI] [PubMed] [Google Scholar]
- 48.Frisch S, Dukart J, Vogt B, et al. Dissociating memory networks in early Alzheimer's disease and frontotemporal lobar degeneration—a combined study of hypometabolism and atrophy. PLoS ONE 2013;8:e55251 10.1371/journal.pone.0055251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rugg MD, Vilberg KL. Brain networks underlying episodic memory retrieval. Curr Opin Neurobiol 2013;23:255–60. 10.1016/j.conb.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim H. Dissociating the roles of the default-mode, dorsal, and ventral networks in episodic memory retrieval. Neuroimage 2010;50:1648–57. 10.1016/j.neuroimage.2010.01.051 [DOI] [PubMed] [Google Scholar]
- 51.Simons JS, Spiers HJ. Prefrontal and medial temporal lobe interactions in long-term memory. Nat Rev Neurosci 2003;4:637–48. 10.1038/nrn1178 [DOI] [PubMed] [Google Scholar]
- 52.Rohrer JD, Paviour D, Bronstein AM, et al. Progressive supranuclear palsy syndrome presenting as progressive nonfluent aphasia: a neuropsychological and neuroimaging analysis. Mov Disord 2010;25:179–88. 10.1002/mds.22946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmidt R, Verstraete E, de Reus MA, et al. Correlation between structural and functional connectivity impairment in amyotrophic lateral sclerosis. Hum Brain Mapp 2014;35:4386–95. 10.1002/hbm.22481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Filippini N, Douaud G, Mackay CE, et al. Corpus callosum involvement is a consistent feature of amyotrophic lateral sclerosis. Neurology 2010;75:1645–52. 10.1212/WNL.0b013e3181fb84d1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hornberger M, Savage S, Hsieh S, et al. Orbitofrontal dysfunction discriminates behavioral variant frontotemporal dementia from Alzheimer's disease. Dement Geriatr Cogn Disord 2010;30:547–52. 10.1159/000321670 [DOI] [PubMed] [Google Scholar]
- 56.Chang JL, Lomen-Hoerth C, Murphy J, et al. A voxel-based morphometry study of patterns of brain atrophy in ALS and ALS/FTLD. Neurology 2005;65:75–80. 10.1212/01.wnl.0000167602.38643.29 [DOI] [PubMed] [Google Scholar]
- 57.Boxer AL, Geschwind MD, Belfor N, et al. Patterns of brain atrophy that differentiate corticobasal degeneration syndrome from progressive supranuclear palsy. Arch Neurol 2006;63:81–6. 10.1001/archneur.63.1.81 [DOI] [PubMed] [Google Scholar]
- 58.Josephs KA, Whitwell JL, Dickson DW, et al. Voxel-based morphometry in autopsy proven PSP and CBD. Neurobiol Aging 2008;29:280–9. 10.1016/j.neurobiolaging.2006.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Josephs KA. Key emerging issues in progressive supranuclear palsy and corticobasal degeneration. J Neurol 2015;262:783–8. 10.1007/s00415-015-7682-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whitwell JL, Avula R, Master A, et al. Disrupted thalamocortical connectivity in PSP: a resting-state fMRI, DTI, and VBM study. Parkinsonism Relat Disord. 2011;17:599–605. 10.1016/j.parkreldis.2011.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gardner RC, Boxer AL, Trujillo A, et al. Intrinsic connectivity network disruption in progressive supranuclear palsy. Ann Neurol 2013;73:603–16. 10.1002/ana.23844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Whitwell JL, Schwarz CG, Reid RI, et al. Diffusion tensor imaging comparison of progressive supranuclear palsy and corticobasal syndromes. Parkinsonism Relat Disord. 2014;20:493–8. 10.1016/j.parkreldis.2014.01.023 [DOI] [PubMed] [Google Scholar]
- 63.Worker A, Blain C, Jarosz J, et al. Diffusion tensor imaging of Parkinson's disease, multiple system atrophy and progressive supranuclear palsy: a tract-based spatial statistics study. PLoS ONE 2014;9:e112638 10.1371/journal.pone.0112638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arvanitakis Z, Wilson RS, Bienias JL, et al. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol 2004;61:661–6. 10.1001/archneur.61.5.661 [DOI] [PubMed] [Google Scholar]
- 65.Ott A, Stolk RP, van Harskamp F, et al. Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology 1999;53:1937–42. 10.1212/WNL.53.9.1937 [DOI] [PubMed] [Google Scholar]
- 66.Sandyk R. The relationship between diabetes mellitus and Parkinson's disease. Int J Neurosci 1993;69:125–30. 10.3109/00207459309003322 [DOI] [PubMed] [Google Scholar]
- 67.Lalić NM, Marić J, Svetel M, et al. Glucose homeostasis in Huntington disease: abnormalities in insulin sensitivity and early-phase insulin secretion. Arch Neurol 2008;65:476–80. 10.1001/archneur.65.4.476 [DOI] [PubMed] [Google Scholar]
- 68.Petersén A, Björkqvist M. Hypothalamic-endocrine aspects in Huntington's disease. Eur J Neurosci 2006;24:961–7. 10.1111/j.1460-9568.2006.04985.x [DOI] [PubMed] [Google Scholar]
- 69.Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3:169–78. 10.1016/S1474-4422(04)00681-7 [DOI] [PubMed] [Google Scholar]
- 70.Aziz NA, van der Marck MA, Pijl H, et al. Weight loss in neurodegenerative disorders. J Neurol 2008;255:1872–80. 10.1007/s00415-009-0062-8 [DOI] [PubMed] [Google Scholar]
- 71.Ahmed RM, Irish M, Kam J, et al. Quantifying the eating abnormalities in frontotemporal dementia. JAMA Neurol 2014;71:1540–6. 10.1001/jamaneurol.2014.1931 [DOI] [PubMed] [Google Scholar]
- 72.Ahmed RM, Latheef S, Bartley L, et al. Eating behavior in frontotemporal dementia: Peripheral hormones vs hypothalamic pathology. Neurology 2015;85: 1310–17. 10.1212/WNL.0000000000002018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cykowski MD, Takei H, Schulz PE, et al. TDP-43 pathology in the basal forebrain and hypothalamus of patients with amyotrophic lateral sclerosis. Acta Neuropathol Commun 2014;2:171 10.1186/s40478-014-0171-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goedert M. NEURODEGENERATION. Alzheimer's and Parkinson's diseases: the prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015;349:1255555 10.1126/science.1255555 [DOI] [PubMed] [Google Scholar]
- 75.Clavaguera F, Hench J, Goedert M, et al. Invited review: Prion-like transmission and spreading of tau pathology. Neuropathol Appl Neurobiol 2015;41:47–58. 10.1111/nan.12197 [DOI] [PubMed] [Google Scholar]
- 76.Thal DR, Rüb U, Orantes M, et al. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791–800. 10.1212/WNL.58.12.1791 [DOI] [PubMed] [Google Scholar]
- 77.Grothe MJ, Teipel SJ, Alzheimer's Disease Neuroimaging Initiative. Spatial patterns of atrophy, hypometabolism, and amyloid deposition in Alzheimer's disease correspond to dissociable functional brain networks. Hum Brain Mapp 2016;37:35–53. 10.1002/hbm.23018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lehmann M, Ghosh PM, Madison C, et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer's disease. Brain 2013;136:844–58. 10.1093/brain/aws327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brettschneider J, Del Tredici K, Toledo JB, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 2013;74:20–38. 10.1002/ana.23937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brettschneider J, Del Tredici K, Irwin DJ, et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol 2014;127:423–39. 10.1007/s00401-013-1238-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Clavaguera F, Akatsu H, Fraser G, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA 2013;110:9535–40. 10.1073/pnas.1301175110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gajdusek DC, Gibbs CJ. Attempts to Demonstrate a Transmissible Agent in Kuru, Amyotrophic Lateral Sclerosis, and Other Sub-Acute and Chronic Nervous System Degenerations of Man. Nature 1964;204:257–9. 10.1038/204257a0 [DOI] [PubMed] [Google Scholar]
- 83.Swash M. How does ALS spread between neurones in the CNS? J Neurol Neurosurg Psychiatr 2013;84:116–17. 10.1136/jnnp-2012-303992 [DOI] [PubMed] [Google Scholar]
- 84.Cavanagh JB, Nolan CC, Brown AW. Glial cell intrusions actively remove detritus due to toxic chemicals from within nerve cells. Neurotoxicology. 1990; 11:1–12. [PubMed] [Google Scholar]
- 85.Fatima M, Tan R, Halliday GM, et al. Spread of pathology in amyotrophic lateral sclerosis: assessment of phosphorylated TDP-43 along axonal pathways. Acta Neuropathol Commun 2015;3:47 10.1186/s40478-015-0226-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gerson JE, Sengupta U, Lasagna-Reeves CA, et al. Characterization of tau oligomeric seeds in progressive supranuclear palsy. Acta Neuropathol Commun 2014;2:73 10.1186/2051-5960-2-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mackenzie IR, Bigio EH, Ince PG, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 2007;61:427–34. 10.1002/ana.21147 [DOI] [PubMed] [Google Scholar]
- 88.Soto-Ortolaza AI, Ross OA. Genetic susceptibility variants in parkinsonism. Parkinsonism Relat Disord 2016;22(Suppl 1):S7–11. 10.1016/j.parkreldis.2015.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee SE, Khazenzon AM, Trujillo AJ, et al. Altered network connectivity in frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. Brain 2014;137:3047–60. 10.1093/brain/awu248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Premi E, Cauda F, Gasparotti R, et al. Multimodal FMRI resting-state functional connectivity in granulin mutations: the case of fronto-parietal dementia. PLoS ONE 2014;9:e106500 10.1371/journal.pone.0106500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hall AM, Throesch BT, Buckingham SC, et al. Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer's disease. J Neurosci 2015;35:6221–30. 10.1523/JNEUROSCI.2552-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Busche MA, Eichhoff G, Adelsberger H, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science 2008;321:1686–9. 10.1126/science.1162844 [DOI] [PubMed] [Google Scholar]
- 93.Born HA, Kim JY, Savjani RR, et al. Genetic suppression of transgenic APP rescues Hypersynchronous network activity in a mouse model of Alzeimer's disease. J Neurosci 2014;34:3826–40. 10.1523/JNEUROSCI.5171-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Buzsáki G, Moser EI. Memory, navigation and theta rhythm in the hippocampal-entorhinal system. Nat Neurosci 2013;16:130–8. 10.1038/nn.3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.García-Cabrero AM, Guerrero-López R, Giráldez BG, et al. Hyperexcitability and epileptic seizures in a model of frontotemporal dementia. Neurobiol Dis 2013;58:200–8. 10.1016/j.nbd.2013.06.005 [DOI] [PubMed] [Google Scholar]
- 96.Marton TF, Sohal VS. Of Mice, Men, and Microbial Opsins: How Optogenetics Can Help Hone Mouse Models of Mental Illness. Biol Psychiatry 2016;79:47–52. 10.1016/j.biopsych.2015.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]