

Abstract
Lung cancer is the leading cause of cancer-associated deaths worldwide. Surgery is the standard treatment for early-stage non-small cell lung cancer (NSCLC). However, 30% to 80% of these patients will die within 5 yearS of diagnosis. Circulating cell-free DNA (cfDNA) harbors pathologic characteristics of the original tumor, such as gene mutations or epigenetic alterations. Analysis of cfDNA has revolutionized the clinical care of advanced lung cancer patients undergoing targeted therapies. However, the low concentration of cfDNA in the blood of early-stage NSCLC patients has hampered its use for management of early disease. Continuing development of more specific and sensitive techniques for detection and analysis of cfDNA will soon enable its leverage in early stage and, perhaps, even screening settings. Therefore, cfDNA analysis may become a tool used for routine NSCLC diagnosis and for monitoring tumor burden, as well as for identifying hidden residual disease. In this review, we will focus on the current evidence of cfDNA in patients with early-stage NSCLC, new and upcoming approaches to identify circulating-tumor biomarkers, their clinical applications and future directions.
Keywords: Non-small cell lung cancer (NSCLC), circulating cell-free DNA (cfDNA), early detection, prognosis, cancer monitoring
Introduction
Lung cancer is the leading cause of cancer deaths in the world, accounting for ≈27% of all cancer-related deaths. It is also the second most diagnosed cancer type (after prostate and breast cancer), with an incidence rate of over 14% in both genders, and over 224,390 new cases and 158,080 deaths are expected to occur in the United States in 2016 (1). Among the two main histological types of lung cancer, non-small cell lung cancer (NSCLC) has the highest incidence, representing 80–85% of all lung cancer cases and being mostly locally advanced or metastatic at the time of diagnosis, according to the American Joint Committee on Cancer (AJCC) (2-4).
The standard treatment for early-stage NSCLC is surgery, which may be followed up by platinum-based chemotherapy in patients at high risk of recurrence. However, 30% to 80% of these patients will die within 5 years of diagnosis (5). Thus, the monitoring of tumor burden, minimal residual disease and tumor heterogeneity would lead to an improvement in the clinical management of early-stage NSCLC (6).
During the last years, there has been an increasing interest in the use of circulating cell-free DNA (cfDNA) as a diagnostic and prognostic factor in a diversity of clinical situations, such as cancer, autoimmune diseases and physical trauma (7-10). In cancer, it has been demonstrated that tumor cells may release genomic DNA into the blood, being apoptosis and necrosis of tumor cells the leading mechanisms of DNA release (11-13). Several studies have reported that NSCLC patients have higher levels of cfDNA in the blood compared to healthy controls or patients with benign disease (14,15). In fact, cfDNA plasma levels have been associated with severity of disease and poorer prognosis (16,17).
NSCLC diagnosis requires a diversity of pathologic analysis, among which tissue biopsy is the gold standard. Nevertheless, the diagnosis based on tumor biopsy presents several limitations, including the potential non-detection of early-stage tumor or residual lesions, and the inability to monitor response to treatment and prognosis (18,19). cfDNA harbors the pathologic characteristics of the original tumor, such as gene mutations or epigenetic alterations (20). Therefore, cfDNA isolated from the plasma or serum by non-invasive procedures has been suggested as a liquid biopsy, which may significantly improve the current system of cancer diagnosis, or even be used to detect early-stage tumors. Furthermore, several studies have found that genetic alterations detected in cfDNA are the same as those found in tumor tissue (21,22). Thus, cfDNA may be an effective method to analyze tumor DNA and obtain results useful in diagnosis, prognosis and follow-up of NSCLC patients.
In this review, we will focus on the evidence of cfDNA in patients with early-stage NSCLC, its clinical applications and future directions.
Methods of cfDNA detection
Nowadays, there are various methods to detect genetic alterations in cfDNA, including real-time polymerase chain reaction (PCR), digital PCR, amplification protocols with magnetic beads in oil emulsions (beads emulsion), amplification and magnetics (BEAMing), next-generation sequencing (NGS) and mass spectrometry (MS) genotyping (23-34). The limit of mutation detection of these techniques fluctuates from 15% to 0.01%, but one of the main problems is the absence of standardized methods for biospecimen collection, processing, cfDNA isolation and analysis. Thus, it is necessary to establish evidence-based guidelines in order to select the most reliable and cost-effective methodology for clinical applications if cfDNA analysis is to move forward.
Real-time PCR
Real-time PCR is the most widely used technology for detection of mutations in general, and that includes cfDNA. Three types of real-time PCR are routinely employed, based on the probes utilized: Taqman probes, Taqman Detection Mutation Assay and Scorpion probes. Taqman probes have a mutation detection limit of around 10%, whereas Taqman Detection Mutation Assay may detect mutations as low as 0.1% (23,24). These variations in sensitivity are due to the probe design: Taqman probes consist of a fluorophore covalently attached to the 5’-end of the oligonucleotide probe and a quencher at the 3’-end, whereas Taqman Detection Mutation Assay uses an allele-specific primer and an MGB blocker oligonucleotide that suppresses the wild-type background (35). Scorpion probes are covalently bound to a primer, a fluorophore and a quencher. In the absence of the mutation, during the Scorpion PCR reaction, the quencher close to the fluorophore absorbs its fluorescence. However, the presence of a mutation leads to separation of the quencher from the fluorophore, resulting in an increase of the emitted fluorescence (25).
In lung cancer, the majority of studies that analyzed cfDNA have utilized real-time PCR. Particularly, studies focused on the detection of epidermal growth factor receptor (EGFR) mutations in cfDNA have reported wide variability in their results (26,36). An Asian study that included 86 advanced NSCLC patients compared EGFR mutation status in pretreatment cfDNA and tissue samples and showed sensitivity of 43.1%, specificity of 100% (26). Another study in 76 Caucasian advanced NSCLC patients also evaluated the detection EGFR mutation in pretreatment blood samples with cfDNA extracted and reported greater sensitivity (78%) and equal specificity (100%) than the previous study (36). However, despite these encouraging results in some cases, real-time technology is not sensitive enough to detect all mutations in cfDNA, and mutations may be missed using this method.
Digital PCR
Digital PCR and real-time PCR share the same principle. However, the key difference between both techniques lies in the procedure to quantify nucleic acids. Real-time works with a unique solution and performs one reaction per single sample, whereas digital PCR produces thousands of replicates from a single sample and performs one reaction per each replicate. Therefore, the sensitivity of this technology is much higher, being capable to detect molecules of cfDNA within a germline DNA background in a ratio up to 1:100,000 (28). Nevertheless, the main disadvantage digital PCR is that standard thresholds for determining of presence and abundance of mutant cfDNA have not been established.
BEAMing
The principle of this technology lies in the association of digital PCR and flow cytometry, using beads, emulsification, amplification, and magnetics to achieve the necessary level of sensitivity. The BEAMing procedure begins with isolating and purifying the cfDNA, followed by pre-amplification by conventional PCR. These DNA templates are then amplified again by emulsion PCR using primers, which are covalently connected to magnetic microbeads via streptavidin-biotin interactions. At the end of the reaction, the amplicons generated in each emulsion droplet will remain physically attached to the microbeads, making it easier to separate and purify them using a magnet. Subsequently, the DNA attached to the microbeads is analyzed to evaluate the presence and the amount of mutations using flow cytometry. This technology is able to detect a low percentage of mutant DNA in a higher amount of fragments comprising wild-type DNA, approximately one single mutant allele in a background of 10,000 wild-type alleles, and it is also able to provide a digital readout of copy-number quantification (29). One study, which enrolled 44 advanced NSCLC with activating EGFR mutations in tumor tissue, detected EGFR status in plasma of 32 cases (72.7%; 95% CI, 58.0–83.6%) (37). Nevertheless, this is a complex technology, which limits its feasibility and reproducibility.
NGS
To date, NGS has demonstrated the highest detection sensitivity and specificity in clinical molecular oncology. Remarkably, published studies have demonstrated that NGS is a feasible, accurate, and sensitive technique for identifying tumor-derived mutations in cfDNA, with sensitivity and specificity of more than 85% for NSCLC (I–IV stages) (30,31). Nowadays there are many specialized and modified NGS techniques available, but they share the same principle (30-32). They are based on the production of short sequences from single molecules of DNA and their comparison to a reference sequence, which results in the sequencing of a significant portion of the genome. Deep sequencing also allows the targeted investigation of specific candidate loci, even if mutated alleles are highly diluted. Furthermore, the main advantage of this technology is that it may identify rearrangements and regions of copy number aberrations, genetic alterations that are not detectable with other techniques (30). However, this methodology is expensive to implement, requires expert personnel to comprehensively analyze and interpret the results, and a well-developed infrastructure for storage of large amounts of sequencing data. These limitations have hampered the application of NGS for routine clinical practice.
MS genotyping
To date, the leading method for detection of mutations using MS is matrix-assisted laser desorption-ionization-time-of-light (MALDI-TOF). This technology is able to detect different alleles based on the different masses of the primer extension products. The MALDI-TOF MS analysis process is based on the following steps: amplification, primer extension reaction and ionization, separation by size and detection of nucleic acids (33). At the end, a software provides a mass spectrum of the extension products (33). This methodology allows to analyze multiple mutations and was used to detect EGFR and KRAS (KRAS proto-oncogene, GTPase) mutations in 2,387 NSCLC patients, demonstrating that the MALDI-TOF platform reduced the rate of missed mutations compared with Sanger sequencing (38). However, it is more expensive and time consuming than other techniques and only returns genotypic data.
Clinical applications of cfDNA in early-stage NSCLC
Because cfDNA can be obtained from minimally invasive procedures and reflects the genetic alterations found in tumor tissue, cfDNA analysis is considered a potential tool for NSCLC diagnosis and monitoring. Hereafter we will review the main clinical applications of cfDNA in early-stage NSCLC (Figure 1).
Figure 1.
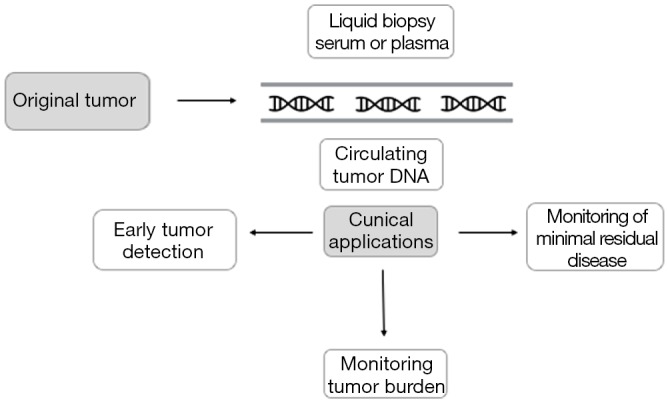
Clinical applications of cfDNA in early-stage NSCLC. cfDNA, circulating cell-free DNA; NSCLC, non-small cell lung cancer.
Detection of genetic alterations
Gene mutations
Several driver mutations have been found in NSCLC such as EGFR, KRAS, BRAF (B-Raf proto-oncogene, serine/threonine kinase) or HER2 (erb-b2 receptor tyrosine kinase 2).
EGFR is mutated in 16.7% of Caucasian patients with NSCLC. These mutations are more common in women, Asians and non-smokers with adenocarcinoma (39-42). The most frequent EGFR mutations are deletions in exon 19, a point mutation which replaces an arginine by a leucine at codon 858 (L858R) of exon 21, and at codon 719 (exon 18), a substitution of glycine by alanine, cysteine or serine (G719X) (43). According to the data of Murray et al. [2008], 3,303 mutations were found in 12,244 patients with advanced NSCLC, and the distribution of EGFR mutations was: 50% (1,662) of exon 19, 40% [1,291] of exon 21, 6% [213] exon 20, and 4% [137] of exon 18 (44). The discovery of mutations in the kinase domain of EGFR gene sparked great clinical interest because they are directly involved in tumor development. Recently, the detection of EGFR mutations in cfDNA has been associated with longer overall survival (OS) (P=0.005; HR =0.163; 95% CI, 0.046–0.571) and progression-free survival (PFS) (P=0.037; HR =0.345; 95% CI, 0.127–0.940) in a study that enrolled 388 surgically resected pathological stage I NSCLC patients (21). Interestingly, a recent phase IV, open label, single-arm clinical trial of patients with advanced NSCLC (NCT01203917), has led to the approval of the first cfDNA test for EGFR mutations. That study evaluated the efficacy and safety/tolerability of first-line gefitinib in Caucasian patients with stage III/IV and reported that cfDNA could be considered for analysis of sensitizing mutations if tumor tissue was unavailable (45).
The mitogen-activated protein kinases (MAPK) pathway is involved in cell proliferation (46). This pathway may be constitutively activated due to genetic alterations in their pathway effectors, which lead to an increased proliferation and cell division. KRAS is the precursor of this pathway and usually is mutated in lung cancer (47). Around 15% to 25% of NSCLC patients present mutations in this gene, more frequently in smokers and patients with adenocarcinoma histology (47). Most mutations are located in exons 2 and 3 (G12, G13 and Q61) (47). Although the detection of KRAS mutations in cfDNA has not been yet tested in early NSCLC, detectable cfDNA-KRAS mutations have been associated with lower OS and PFS (median OS 4.8 vs. 9.5 months; P=0.0002; HR =1.87; 95% CI, 1.23–2.84, and median PFS 3.0 vs. 5.6 months; P=0.0043; HR =1.60; 95% CI, 1.09–2.37) in 246 advanced NSCLC patients treated with platinum-based chemotherapy (48).
BRAF is a downstream effector of KRAS, which is frequently mutated in NSCLC. A thymine to adenine transversion mutation that results in the substitution of valine with Glutamate in codon 600 is the most common mutation in BRAF gene and leads to constitutively activated BRAF kinase (49). BRAF mutations occur between 1–3% of patients with NSCLC, more frequently in lung adenocarcinoma and former smokers (50). The detection of BRAF mutations in cfDNA has been associated with lower PFS and OS in various types of tumors, such as melanoma (P=0.021; 3.6 vs. 13.4 months for PFS and P=0.017; 7 vs. 21.8 months for OS) (51,52). However, to date the association between clinical outcomes of NSCLC patients and the detection of BRAF mutations in cfDNA has not been investigated.
Mutations in the kinase domain of HER2 gene identify a subset of lung adenocarcinomas with higher frequency in non-smokers, accounting for 4% of NSCLC patients (53). Most of HER2 mutations are insertions in exon 20 that varies from 3 to 12 base pairs. The detection of HER2 mutations in cfDNA has not been evaluated. However, in primary breast cancer (8/68; 12%) and metastatic breast cancer (5/30; 17%) patients, plasma HER2 amplification has been detected, although the follow-up time was insufficient to inform whether the presence of circulating amplified HER2 DNA was predictive of recurrent disease (54). Therefore, the detection of these genetic alterations in cfDNA may play a crucial role as prognostic biomarker for early-stage NSCLC.
Epigenetic alterations
Epigenetic alterations are not completely a specific tumor process; non-tumor tissue methylation also changes and is frequently age-associated. Therefore, it is essential to select suitable candidate genes if DNA methylation is to be used as a circulating biomarker. Despite this, the detection of epigenetic alterations in cfDNA is a potential biomarker of tumor burden, residual disease and even early detection. Methods and recent developments associated with the use of cfDNA for non-invasive detection of methylation changes in lung cancer patients are described elsewhere in this issue (55). Methylation changes are usually an early event in carcinogenesis and contribute to cancer initiation, progression and response to therapy (56). Remarkably, in early-stage NSCLC, the detection of aberrant methylation of nine genes [APC (APC, WNT signaling pathway regulator), cadherin 13 (CDH13), kallikrein related peptidase 10 (KLK10), deleted in lung and esophageal cancer 1 (DLEC1) Ras association domain family member 1A (RASSF1A), EGF containing fibulin like extracellular matrix protein 1 (EFEMP1), secreted frizzled related protein 1 (SFRP1), retinoic acid receptor beta (RARB) and cyclin dependent kinase inhibitor 2A (CDKN2A)] in cfDNA from 110 early-stage NSCLC patients have showed a significantly higher frequency of tumor-specific hypermethylation, as compared with the cancer-free plasma, and achieved a sensitivity of 83.64% and a specificity of 74.0% for cancer diagnosis (22). Tumor tissues and plasma samples were also compared, showing a satisfactory concordance between the nine gene methylation status in plasma samples and paired tumor tissues (22). Thus, the determination of the methylation patterns of these genes in cfDNA may be used as potential prognostic and diagnostic biomarkers for early-stage NSCLC.
Monitoring tumor burden
Carcinoembryonic antigen and cancer antigen 19-9 are the main circulating biomarkers used to evaluate tumor burden in NSCLC (57). Nevertheless, these proteins are also expressed in normal cells and may be elevated in benign conditions (58). Thus, the specificity of these tumor markers is low and other biomarkers to monitor tumor burden are required. Liquid biopsy based on cfDNA analysis has emerged as a potential method to monitor tumor burden. Remarkably, an ultrasensitive method for quantifying tumor cfDNA has been developed, known as Cancer Personalized Profiling by deep Sequencing (CAPP-Seq) (31). This methodology covers multiple types of genetic alterations and was applied in 17 patients with I–IV stage NSCLC (31). The results showed cfDNA detection in 100% of stage II–IV and 50% of stage I NSCLC patients, with 96% specificity for mutant allele fractions down to ≈0.02% (31). Levels of cfDNA were associated with tumor volume, distinguished between residual disease and treatment-related imaging changes, and provided earlier response evaluation than radiographic approaches (31). Moreover, a case-control study was able to differentiate radically resected NSCLC patients (stage I–III) from healthy controls (59). Higher mean values of cfDNA were detected even in stage IA patients (59). Quantification of cfDNA and analysis of microsatellite alterations was also associated with early recurrence (59). On the other hand, a separate study that evaluated the association between cfDNA and total tumor burden defined by positron emission tomography (PET) parameters in 53 advanced NSCLC patients, found no association between cfDNA and metabolic tumor volume (MTV) (r=0.1) or total lesion glycolysis (TLG) (r=0.1) (60). Thus, cfDNA may not be a simple measure of tumor burden (60). The reason why these studies showed different results may be because cfDNA is also elevated in other associated diseases, such as idiopathic pulmonary fibrosis and myocardial infarction (61,62). However, despite these discordant results, the development of effective technologies for the analysis of cfDNA should prompt the leverage of this non-invasive biomarker to assess tumor burden.
Monitoring of minimal residual disease
Methods that specifically classify patients based on the presence of minimal residual disease are not currently available. In practice, clinical and pathologic criteria are used to predict patients who probably harbor residual disease. The leading system parameter utilized for this purpose is the TNM classification of malignant tumors (TNM) (63). This method helps the clinician to assess the risk of a patient to develop recurrence and the possible benefit of adjuvant chemotherapy. Nevertheless, it does not inform if minimal residual disease is actually present. cfDNA can be a useful biomarker to detect residual disease after surgery and may provide evidence to select patients who are likely to suffer recurrence (63). In fact, cfDNA should be measured when the surgery is finished, but before the beginning of adjuvant therapy, which is usually administered 6–8 weeks after surgery (63). Thus, the cfDNA measurement may be a useful tool to improve clinical decisions.
The potential role of cfDNA in detecting minimal residual disease has been showed in a cohort of 18 patients with colorectal cancer undergoing resection with curative intent (64). cfDNA was detectable in all patients before resection and serial blood sampling revealed fluctuations in the cfDNA levels, that were associated with the extent of surgical resection (64). All the patients who had detectable cfDNA after surgery relapsed within 1 year, whereas all patients with undetectable cfDNA remained diseased free (64). These results revealed the clinical impact of cfDNA as a measure of tumor dynamics.
Future directions and conclusions
The sensitivity of cfDNA detection has been reported to be more than 85% in patients with early stage NSCLC patients, suggesting that cfDNA may be a diagnostic and prognostic biomarker fin this setting Although the level of cfDNA in the blood of early stage patients is generally low, the application of new, more sensitive and specific approaches, such as digital PCR and NGS may enable its use for the detection of tumor-associated genetic alterations. Furthermore, NGS may detect chromosomal abnormalities and copy number variations. Thus, based on these new technologies, cfDNA may be a potential tool for the early detection and monitoring of NSCLC.
In conclusion, cfDNA is a promising biomarker for the detection and follow-up of NSCLC patients. In clinical practice, cfDNA may serve as alternative for those patients who are unable to provide an accurate tissue-biopsy sample. It will also serve as invaluable source of information, complimentary to imaging, during the period of follow-up after surgery. Lastly, it will reduce the need for invasive sampling in the monitoring of cancer patients.
Acknowledgements
Funding: This work was partly supported by a research grant for Cristina Pérez-Ramírez (FPU12/04722), from Ministerio de Educación, Cultura y Deporte.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7-30. 10.3322/caac.21332 [DOI] [PubMed] [Google Scholar]
- 2.Byrd ES, Compton DR, Fritz CC, et al. editors. AJCC Cancer Staging Manual. 7th ed. Springer-Verlag New York, 2010. [Google Scholar]
- 3.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med 2008;359:1367-80. 10.1056/NEJMra0802714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molina JR, Yang P, Cassivi SD, et al. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc 2008;83:584-94. 10.1016/S0025-6196(11)60735-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chansky K, Sculier JP, Crowley JJ, et al. The International Association for the Study of Lung Cancer Staging Project: prognostic factors and pathologic TNM stage in surgically managed non-small cell lung cancer. J Thorac Oncol 2009;4:792-801. 10.1097/JTO.0b013e3181a7716e [DOI] [PubMed] [Google Scholar]
- 6.Francis G, Stein S. Circulating Cell-Free Tumour DNA in the Management of Cancer. Int J Mol Sci 2015;16:14122-42. 10.3390/ijms160614122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leon SA, Shapiro B, Sklaroff DM, et al. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 1977;37:646-50. [PubMed] [Google Scholar]
- 8.Shapiro B, Chakrabarty M, Cohn EM, et al. Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer 1983;51:2116-20. [DOI] [PubMed] [Google Scholar]
- 9.Steinman CR. Circulating DNA in systemic lupus erythematosus. Isolation and characterization. J Clin Invest 1984;73:832-41. 10.1172/JCI111278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lo YM, Rainer TH, Chan LY, et al. Plasma DNA as a prognostic marker in trauma patients. Clin Chem 2000;46:319-23. [PubMed] [Google Scholar]
- 11.Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer 2011;11:426-37. 10.1038/nrc3066 [DOI] [PubMed] [Google Scholar]
- 12.van der Vaart M, Pretorius PJ. The origin of circulating free DNA. Clin Chem 2007;53:2215. 10.1373/clinchem.2007.092734 [DOI] [PubMed] [Google Scholar]
- 13.Nie K, Jia Y, Zhang X. Cell-free circulating tumor DNA in plasma/serum of non-small cell lung cancer. Tumour Biol 2015;36:7-19. 10.1007/s13277-014-2758-3 [DOI] [PubMed] [Google Scholar]
- 14.Catarino R, Coelho A, Araujo A, et al. Circulating DNA: diagnostic tool and predictive marker for overall survival of NSCLC patients. PloS one 2012;7:e38559. 10.1371/journal.pone.0038559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ulivi P, Mercatali L, Casoni GL, et al. Multiple marker detection in peripheral blood for NSCLC diagnosis. PloS one 2013;8:e57401. 10.1371/journal.pone.0057401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paci M, Maramotti S, Bellesia E, et al. Circulating plasma DNA as diagnostic biomarker in non-small cell lung cancer. Lung Cancer 2009;64:92-7. 10.1016/j.lungcan.2008.07.012 [DOI] [PubMed] [Google Scholar]
- 17.Sozzi G, Conte D, Leon M, et al. Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol 2003;21:3902-8. 10.1200/JCO.2003.02.006 [DOI] [PubMed] [Google Scholar]
- 18.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883-92. 10.1056/NEJMoa1113205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yong E. Cancer biomarkers: Written in blood. Nature 2014;511:524-6. 10.1038/511524a [DOI] [PubMed] [Google Scholar]
- 20.Sorenson GD, Pribish DM, Valone FH, et al. Soluble normal and mutated DNA sequences from single-copy genes in human blood. Cancer Epidemiol Biomarkers Prev 1994;3:67-71. [PubMed] [Google Scholar]
- 21.Sorber L, Zwaenepoel K, Deschoolmeester V, et al. Circulating cell-free nucleic acids and platelets as a liquid biopsy in the provision of personalized therapy for lung cancer patients. Lung Cancer 2016. [Epub ahead of print]. 10.1016/j.lungcan.2016.04.026 [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Wang R, Song H, et al. Methylation of multiple genes as a candidate biomarker in non-small cell lung cancer. Cancer Lett 2011;303:21-8. 10.1016/j.canlet.2010.12.011 [DOI] [PubMed] [Google Scholar]
- 23.Wilkening S, Hemminki K, Thirumaran RK, et al. Determination of allele frequency in pooled DNA: comparison of three PCR-based methods. BioTechniques 2005;39:853-8. 10.2144/000112027 [DOI] [PubMed] [Google Scholar]
- 24.Bao Y, Ching B, Mouanoutoua M, et al. Cancer biomarker research using castPCR technology. Cancer Res 2012;72:Abstract nr 2100.
- 25.Scorpion Primers & Probes. Available online: http://www.premierbiosoft.com/tech_notes/Scorpion.html
- 26.Goto K, Ichinose Y, Ohe Y, et al. Epidermal growth factor receptor mutation status in circulating free DNA in serum: from IPASS, a phase III study of gefitinib or carboplatin/paclitaxel in non-small cell lung cancer. J Thorac Oncol 2012;7:115-21. 10.1097/JTO.0b013e3182307f98 [DOI] [PubMed] [Google Scholar]
- 27.Punnoose EA, Atwal S, Liu W, et al. Evaluation of circulating tumor cells and circulating tumor DNA in non-small cell lung cancer: association with clinical endpoints in a phase II clinical trial of pertuzumab and erlotinib. Clin Cancer Res 2012;18:2391-401. 10.1158/1078-0432.CCR-11-3148 [DOI] [PubMed] [Google Scholar]
- 28.Hindson BJ, Ness KD, Masquelier DA, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 2011;83:8604-10. 10.1021/ac202028g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diehl F, Li M, Dressman D, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A 2005;102:16368-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 2012;4:136ra68. 10.1126/scitranslmed.3003726 [DOI] [PubMed] [Google Scholar]
- 31.Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 2014;20:548-54. 10.1038/nm.3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kinde I, Wu J, Papadopoulos N, et al. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A 2011;108:9530-5. 10.1073/pnas.1105422108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Griffin TJ, Smith LM. Single-nucleotide polymorphism analysis by MALDI-TOF mass spectrometry. Trends Biotechnol 2000;18:77-84. 10.1016/S0167-7799(99)01401-8 [DOI] [PubMed] [Google Scholar]
- 34.Bordi P, Del Re M, Danesi R, et al. Circulating DNA in diagnosis and monitoring EGFR gene mutations in advanced non-small cell lung cancer. Transl Lung Cancer Res 2015;4:584-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.TaqMan® Probes. Available online: http://premierbiosoft.com/tech_notes/TaqMan.html
- 36.Karachaliou N, Mayo-de las Casas C, Queralt C, et al. Association of EGFR L858R Mutation in Circulating Free DNA With Survival in the EURTAC Trial. JAMA Oncol 2015;1:149-57. 10.1001/jamaoncol.2014.257 [DOI] [PubMed] [Google Scholar]
- 37.Taniguchi K, Uchida J, Nishino K, et al. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin Cancer Res 2011;17:7808-15. 10.1158/1078-0432.CCR-11-1712 [DOI] [PubMed] [Google Scholar]
- 38.Giannini R, Lupi C, Sensi E, et al. EGFR and KRAS mutational analysis in a large series of Italian non-small cell lung cancer patients: 2,387 cases from a single center. Oncol Rep 2016;36:1166-72. [DOI] [PubMed] [Google Scholar]
- 39.Cheng L, Alexander RE, Maclennan GT, et al. Molecular pathology of lung cancer: key to personalized medicine. Mod Pathol 2012;25:347-69. 10.1038/modpathol.2011.215 [DOI] [PubMed] [Google Scholar]
- 40.García-Foncillas J, Garrido P, Gómez J, et al. Guidelines for EGFR gene mutations testing in non-small cell lung carcinoma. Rev Esp Patol 2011;44:17-31. [Google Scholar]
- 41.Reungwetwattana T, Weroha SJ, Molina JR. Oncogenic pathways, molecularly targeted therapies, and highlighted clinical trials in non-small-cell lung cancer (NSCLC). Clin Lung Cancer 2012;13:252-66. 10.1016/j.cllc.2011.09.004 [DOI] [PubMed] [Google Scholar]
- 42.Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958-67. 10.1056/NEJMoa0904554 [DOI] [PubMed] [Google Scholar]
- 43.Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol 2012;13:e23-31. 10.1016/S1470-2045(11)70129-2 [DOI] [PubMed] [Google Scholar]
- 44.Murray S, Dahabreh IJ, Linardou H, et al. Somatic mutations of the tyrosine kinase domain of epidermal growth factor receptor and tyrosine kinase inhibitor response to TKIs in non-small cell lung cancer: an analytical database. J Thorac Oncol 2008;3:832-9. 10.1097/JTO.0b013e31818071f3 [DOI] [PubMed] [Google Scholar]
- 45.Douillard JY, Ostoros G, Cobo M, et al. First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer 2014;110:55-62. 10.1038/bjc.2013.721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 2002;12:9-18. 10.1038/sj.cr.7290105 [DOI] [PubMed] [Google Scholar]
- 47.Karachaliou N, Mayo C, Costa C, et al. KRAS mutations in lung cancer. Clin Lung Cancer 2013;14:205-14. 10.1016/j.cllc.2012.09.007 [DOI] [PubMed] [Google Scholar]
- 48.Nygaard AD, Garm Spindler KL, Pallisgaard N, et al. The prognostic value of KRAS mutated plasma DNA in advanced non-small cell lung cancer. Lung cancer 2013;79:312-7. 10.1016/j.lungcan.2012.11.016 [DOI] [PubMed] [Google Scholar]
- 49.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949-54. 10.1038/nature00766 [DOI] [PubMed] [Google Scholar]
- 50.Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046-51. 10.1200/JCO.2010.33.1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gonzalez-Cao M, Mayo-de-Las-Casas C, Molina-Vila MA, et al. BRAF mutation analysis in circulating free tumor DNA of melanoma patients treated with BRAF inhibitors. Melanoma Res 2015;25:486-95. 10.1097/CMR.0000000000000187 [DOI] [PubMed] [Google Scholar]
- 52.Molina-Vila MA, de-Las-Casas CM, Bertran-Alamillo J, et al. cfDNA analysis from blood in melanoma. Ann Transl Med 2015;3:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tomizawa K, Suda K, Onozato R, et al. Prognostic and predictive implications of HER2/ERBB2/neu gene mutations in lung cancers. Lung cancer 2011;74:139-44. 10.1016/j.lungcan.2011.01.014 [DOI] [PubMed] [Google Scholar]
- 54.Page K, Hava N, Ward B, et al. Detection of HER2 amplification in circulating free DNA in patients with breast cancer. Br J Cancer 2011;104:1342-8. 10.1038/bjc.2011.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lissa D, Robles AI. Methylation analyses in liquid biopsy. Transl Lung Cancer Res 2016;5:492-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Okayama H, Schetter AJ, Ishigame T, et al. The expression of four genes as a prognostic classifier for stage I lung adenocarcinoma in 12 independent cohorts. Cancer Epidemiol Biomarkers Prev 2014;23:2884-94. 10.1158/1055-9965.EPI-14-0182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Niklinski J, Furman M, Laudanski J, et al. Prognostic value of pretreatment CEA, SCC-Ag and CA 19-9 levels in sera of patients with non-small cell lung cancer. Eur J Cancer Prev 1992;1:401-6. 10.1097/00008469-199210000-00002 [DOI] [PubMed] [Google Scholar]
- 58.Perkins GL, Slater ED, Sanders GK, et al. Serum tumor markers. Am Fam Physician 2003;68:1075-82. [PubMed] [Google Scholar]
- 59.Sozzi G, Conte D, Mariani L, et al. Analysis of circulating tumor DNA in plasma at diagnosis and during follow-up of lung cancer patients. Cancer Res 2001;61:4675-8. [PubMed] [Google Scholar]
- 60.Nygaard AD, Holdgaard PC, Spindler KL, et al. The correlation between cell-free DNA and tumour burden was estimated by PET/CT in patients with advanced NSCLC. Br J Cancer 2014;110:363-8. 10.1038/bjc.2013.705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Casoni GL, Ulivi P, Mercatali L, et al. Increased levels of free circulating DNA in patients with idiopathic pulmonary fibrosis. Int J Biol Markers 2010;25:229-35. [PubMed] [Google Scholar]
- 62.Chang CP, Chia RH, Wu TL, et al. Elevated cell-free serum DNA detected in patients with myocardial infarction. Clin Chim Acta 2003;327:95-101. 10.1016/S0009-8981(02)00337-6 [DOI] [PubMed] [Google Scholar]
- 63.Diaz LA, Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 2014;32:579-86. 10.1200/JCO.2012.45.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Diehl F, Schmidt K, Choti MA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008;14:985-90. 10.1038/nm.1789 [DOI] [PMC free article] [PubMed] [Google Scholar]