

Abstract
Objective
Charcot–Marie–Tooth disease type X1 (CMTX1), which is caused by mutations in the gap junction (GJ) protein beta‐1 gene (GJB1), is the second most common form of Charcot–Marie–Tooth disease (CMT). GJB1 encodes the GJ beta‐1 protein (GJB1), which forms GJs within the myelin sheaths of peripheral nerves. The process by which GJB1 mutants cause neuropathy has not been fully elucidated. This study evaluated the biophysical characteristics of GJB1 mutants and their correlations with the clinical features of CMTX1 patients.
Methods
All patients with a validated GJB1 mutation were assessed using the Charcot–Marie–Tooth disease neuropathy score version 2 (CMTNS). The impacts of the mutations on the biophysical functions of GJB1 were characterized by assessing intracellular localization, expression patterns, and GJ Ca2+ permeability.
Result
Nineteen GJB1 mutations were identified in 24 patients with a clinical diagnosis of CMT. Six are novel mutations: p.L6S, p.I20F, p.I101Rfs*8, p.F153L, p.R215P, and p.D278V. Diverse pathological effects of the mutations were demonstrated, including reduced GJB1 expression, intracellular mislocalization, and altered GJ functions. GJB1 mutations that caused a complete loss of GJ Ca2+ permeability appeared to be associated with an earlier disease onset, whereas those resulting in preservation of GJ permeability and with predominant cell membrane expression tended to have a later onset and a milder phenotype.
Interpretation
This study demonstrated that the degree of loss of GJ function caused by the GJB1 mutations might contribute to the onset and severity of neuropathic symptoms in CMTX1.
Introduction
Charcot–Marie–Tooth disease (CMT) is a clinically and genetically heterogeneous group of inherited neuropathies characterized by distal muscle weakness and atrophy, foot deformities, distal sensory loss and depressed tendon reflexes. CMT type X1 (CMTX1), caused by a mutation in the gap junction protein beta‐1 gene (GJB1), is the second most common subtype of CMT, accounting for 6.2~15% of all CMT cases.1, 2, 3, 4 Heterozygous females are usually latently affected and have a milder phenotype. Median or ulnar motor nerve conduction velocities (MNCVs) are typically 30–40 m/sec in affected males and 30–50 m/sec in affected females.5, 6, 7, 8 To date, more than 400 GJB1 mutations have been reported (www.molgen.ua.ac.be/CMTMutations/), indicating that almost all of the residues are required for the normal functioning of gap junction (GJ) beta‐1 protein (GJB1).8
GJB1, also known as connexin32 (Cx32), is expressed in Schwann cells and forms GJs via the interaction of two apposed hemichannels (connexons).9 These GJs allow for the diffusion or exchange of ions across apposed cell membranes of noncompact regions of peripheral myelin and thus connect these regions to the perinuclear cytoplasm of Schwann cells.8 GJs play an important role in the homeostasis of myelinated axons because they are involved in many vital biological functions, including metabolic cooperation, spatial buffering, electrical coupling, growth control, and cellular differentiation.10 Many GJB1 mutants fail to form functional GJs, and most GJB1 mutations are presumed to cause loss of function.8, 11 Although some studies have investigated the clinical and electrophysiological characteristics of different GJB1 mutants,7 correlations between the biological functions and patients' clinical phenotypes have not yet been fully elucidated.
This study identified 19 GJB1 mutations, including six novel mutations, in a cohort of 24 unrelated Taiwanese patients with CMTX1 and assessed the localization, protein expression, and GJ permeability of each GJB1‐mutant protein. In addition, the clinical and molecular features of these GJB1 mutants were evaluated, and the correlations of their biophysical characteristics with the corresponding clinical features of patients with GJB1 mutations were examined.
Materials and Methods
Patients
Patients were diagnosed with CMTX1 based on identification of a GJB1 mutation, the presence of characteristic clinical features, and the results of electrophysiological analyses. The clinical manifestations of the CMTX1 patients were evaluated using the Charcot–Marie–Tooth disease neuropathy score version 2 (CMTNS).12 The nerve conduction analyses were performed using standard techniques. Written informed consent was obtained from the participants. This study was approved by the Institutional Review Board of Taipei Veterans General Hospital.
GJB1 mutation analyses
Genomic DNA was extracted from peripheral blood using standard protocols. Mutation analyses were performed using Sanger sequencing and the Big Dye 3.1 dideoxy terminator method (Applied Biosystems, Foster City, CA) on an ABI Prism 3730XI Genetic Analyzer (Applied Biosystems). Amplicon sequences were compared with the reference GJB1 coding sequence (GRCh38, NM_00109764). The standard sequence of the human GJB1 protein (NP_001091111) was used to detect amino acid changes. Putative pathogenic variants were further distinguished according to their absences in both Exome Aggregation Consortium database (ExAC; http://exac.broadinstitute.org) and 1000 control chromosomes from individuals of Han Chinese origin.
Expression plasmids
The full‐length coding region of human GJB1 was cloned into pcDNA3.1/myc‐His (Invitrogen, Grand Island, NY) to generate wild‐type (WT) GJB1 expression plasmids. The GJB1 mutations identified in this study were separately introduced into the WT expression plasmids using a QuikChange Site‐Directed Mutagenesis Kit according to the manufacturer's recommendations (Agilent, Santa Clara, CA). The transfection marker pDsRed1‐N1, the endoplasmic reticulum marker pDsRed‐ER, and the Golgi marker pDsRed‐Monomer‐Golgi were all purchased from Clontech (Mountain View, CA).
Cell culture and transfection
HeLa and HEK293T cells were maintained in DMEM medium supplemented with 10% fetal bovine serum in a humidified incubator at 37°C under 5% CO2. Transient transfection was performed using Lipofectamine 2000 (Invitrogen).
Immunofluorescence studies
HeLa cells were transfected to express WT or mutant GJB1 proteins alone or together with pDsRed‐ER or pDsRed‐Monomer‐Golgi. At 48 h after transfection, the cells were fixed in 4% paraformaldehyde, permeabilized with 0.2% Tween‐20, blocked with 1% BSA, and incubated with anti‐GJB1 antibody overnight at 4°C. The primary and secondary antibodies used are listed in Table S1. Images were captured using an Olympus FluoView FV10i confocal laser scanning fluorescence microscopy system with a 60 × oil immersion objective (Olympus, Tokyo, Japan).
Analysis of GJB1 protein expression by western blotting
HeLa cells transfected with WT or mutant GJB1 constructs were lysed with RIPA buffer supplemented with a protease inhibitor cocktail (Millipore, Darmstadt, Germany) at 48 h after transfection. The total protein concentrations in the lysates were quantified using a Bradford‐based assay kit (Bio‐Rad, Hercules, CA), and 50 μg of proteins from each cell lysate were used for western blotting (Table S1). Detection was performed using a standard enhanced chemiluminescence method. Densitometric analyses of the protein bands were performed using NIH ImageJ software.
Ionic permeability of GJB1 GJ channels
Ca2+ imaging analyses were performed to assess the abilities of these mutated GJB1 GJ channels to mediate the intercellular propagation of Ca2+ signaling as previously described.13, 14 HEK293T cells were cotransfected with GJB1 expression plasmids and the transfection marker pDsRed1‐N1. Then, the cells were washed with Hanks' buffered salt solution (HBSS) at 48 h after transfection and loaded with the Ca2+ indicator fura‐2/AM by incubation in HBSS supplemented with 5 μmol/L fura‐2/AM and Pluronic F‐227 detergent (0.02%) at 37°C for 40 min. Next, the cells were washed and saturated with 20 μmol/L ATP in HBSS for 20 min to block the intercellular Ca2+ signaling mediated by purinergic receptors; thus, GJs were the only available pathway for Ca2+ wave propagation. One transfected cell was stimulated mechanically by temporarily distorting the plasma membrane with a glass micropipette (tip diameter, <1 μm) mounted on a vertical microinjection system (Narishige MHW‐3, Tokyo, Japan). An Olympus IX71 microscope equipped with an Olympus UApo/340 40 × /1.35 NA objective, a Polychrome V monochromator (Till Photonics, FEI, Hillsboro, Oregon), and a charge‐coupled device camera (Hamamatsu ORCA‐AG, Shizuoka Pref, Japan) controlled by SimplePCI software (Hamamatsu) was used to monitor changes in the intracellular Ca2+ concentrations ([Ca2+]i) in the stimulated and surrounding cells with transfection markers. Fura‐2 ratio images were acquired at excitation wavelengths of 340 and 380 nm, and the 340/380 nm ratio was calculated to reflect the [Ca2+]i changes.
Results
Clinical features of CMTX1 patients with GJB1 mutations
Nineteen GJB1 mutations were identified in 24 unrelated patients with CMTX1. These mutations were predicted to be located throughout all of the functional domains of the GJB1 protein (Fig. 1). Six were novel mutations, including p.L6S, p.I20F, p.I101Rfs*8, p.F153L, p.R215P, and p.D278V. With the exception of p.I101Rfs*8, p.L144del, and p.R220X, they were all missense mutations. Table S2 lists the in silico predictions of the functional effects of the five novel missense mutations.
Figure 1.
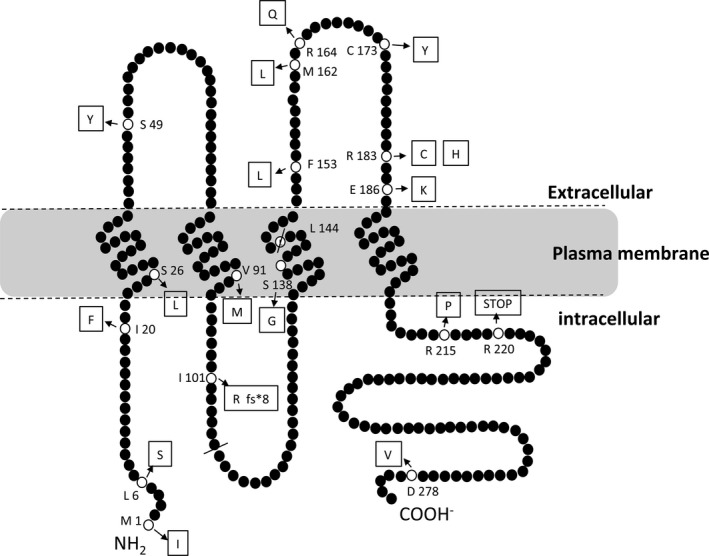
Schematic representations of the membrane topologies of GJB1 and the mutations identified in this study. GJB1 spans the plasma membrane four times with cytoplasmic C‐ and N‐termini, a cytoplasmic loop and two extracellular loops. The p.M1I, p.L6S, and p.I20F mutations are in the N‐terminal domain. The p.S26L, p.V91M, p.S138G, and p.L144del mutations are in the transmembrane domains. The p.I101Rfs*8 mutation leads to a premature truncation in the cytoplasmic loop. The p.S49Y mutation is located in the first extracellular loop. Furthermore, the p.F153L, p.M162L, p.R164Q, p.C173Y, p.R183C, p.R183H, and p.E186K mutations are located in the second extracellular loop, and the p.R215P, p.R220X, and p.D278V mutations are located in the C‐terminal domain.
Table 1 summarizes the clinical features of these patients. The age of onset (AO) ranged from 7 to 45 years. Twenty‐one (87.5%) of the patients were males. The male patients had an earlier AO (mean AO, male: female = 21.3 ± 10.75: 30.7 ± 3.05 years). At the age of examination (AE), the male patients also had a slower median MNCV (mean median MNCV, male: female = 35.3 ± 5.68: 39.5 ± 3.19 m/sec) and higher CMTNS (mean CMTNS, male: female = 12.8 ± 4.43: 9.0 ± 2.00). The above findings were compatible with the more severe phenotypes of the male patients with an X‐linked disease. The majority of our patients (19/24, 79.2%) had demyelinating CMT and the rest of them (5/24, 20.8%) had axonal neuropathy, according to the cut‐off median MNCV of 38 m/sec at the AE.15 Twenty of the patients (20/24, 83.3%) had intermediate MNCVs with their median MNCV falling into the range of 30–50 m/sec. Clinically, most of the patients (22/24, 91.7%) exhibited weakness of the distal limb muscles at initial presentation, whereas only two patients complained of distal paresthesia at presentation. Notably, the female patient with a p.I101Rfs*8 mutation had asymmetric weakness and atrophy of the hand muscles and was initially diagnosed with hereditary neuropathy with liability to pressure palsy (HNPP). Patchy demyelination is occasionally observed in CMTX1.16 These findings indicate that a GJB1 mutation should be considered in patients with asymmetric neuropathy without obvious acquired etiologies.
Table 1.
Biophysical characteristics of mutant GJB1 proteins and clinical features of the patients with these GJB1 mutations
| GJB1 mutations | Clinical features of patients | Biophysical characteristics of mutated proteins | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A.A. change | Nucleotide change | Pat. no. | Sex | Initial symptoms | AO (yr) | AE (yr) | CMTNS | CMAP (mV) | MNCV (m/sec) | GJB1 localization | GJ Ca2+ permeability | dT | τ | GJB1 expression |
| Significantly compromised plasma membrane expression of GJB1 and GJ plaque formation with impaired GJ permeability (Group A) | ||||||||||||||
| M1Ia | c.3G>T | 1 | M | LW | 16 | 18 | 11 | 0.3 | 29.3 | U | L | – | – | U |
| I101Rfs*8a | c.302_303delTA | 2 | F | HNPP | 28 | 30 | 11 | 2.1 | 42.9 | U | L | – | – | U |
| L144del | c.432_434delGTT | 3 | M | LW | 21 | 23 | 9 | 3.2 | 37.0 | Cyt | L | – | – | ↓ |
| C173Y | c.518G>A | 4 | M | FW | 18 | 27 | 13 | 1.3 | 32.2 | Cyt | L | – | – | ↓ |
| R183C | c.547C>T | 5 | M | LW | 20 | 33 | 18 | 0.5 | 37.5 | Cyt | L | – | – | ↓ |
| E186K | c.556G>A | 6 | M | LW | 7 | 24 | 10 | 8.9 | 33.3 | Cyt | L | – | – | ↓ |
| L6Sa | c.17T>C | 7 | M | LW | 18 | 22 | 10 | 3.1 | 34.4 | Cyt, Mem | I | NL | ↑ | ↓ |
| I20Fa | c.58A>T | 8 | M | FW | 30 | 69 | 18 | 1.1 | 30.8 | Cyt, Mem | I | NL | ↑ | ↓ |
| 9 | M | FW | 14 | 19 | 6 | 0.6 | 34.3 | |||||||
| S26L | c.77C>T | 10 | M | FW | 28 | 34 | 19 | 0.4 | 37.9 | Cyt, Mem | I | NL | ↑ | NL |
| 11 | M | UW | 14 | 20 | 13 | 0.8 | 38.9 | |||||||
| 12 | F | FW | 30 | 45 | 9 | 4 | 38.9 | |||||||
| S49Y | c.146C>A | 13 | M | FW | 35 | 58 | 16 | 2.4 | 36.4 | Cyt, Mem | I | NL | ↑ | ↓ |
| V91M | c.271G>A | 14 | M | FW | 10 | 40 | 14 | 0.4 | 33.7 | Cyt, Mem | I | NL | ↑ | ↓ |
| 15 | M | LN | 15 | 42 | 13 | 1.3 | 32.3 | |||||||
| R164Q | c.491G>A | 16 | M | LW | 13 | 22 | 15 | 9.1 | 33 | Cyt, Mem | I | ↑ | ↑ | NL |
| R215Pa | c.644G>C | 17 | M | LW | 15 | 43 | 15 | A | A | Cyt, Mem | I | NL | ↑ | ↓ |
| 18 | M | LW | 23 | 26 | 15 | 0.7 | 29.2 | |||||||
| R220X | c.658C>T | 19 | M | LW | 7 | 60 | 18 | 0.2 | 40.5 | Cyt, Mem | I | ↑ | NL | NL |
| Mean values for the 17 male patients | 19 | 34.6 | 13.4 | 2.2 | 35.1 | |||||||||
| Predominant plasma membrane expression of GJB1 with formation of abundant GJ plaques, or normal GJ Ca2+ permeability (Group B) | ||||||||||||||
| S138G | c.412A>G | 20 | F | FW | 34 | 43 | 7 | 5 | 36.6 | Cyt, Mem | NL | NL | NL | ↓ |
| F153La | c.457T>C | 21 | M | LW | 21 | 27 | 9 | 1.2 | 31.6 | Cyt, Mem | NL | NL | NL | NL |
| M162L | c.484A>T | 22 | M | LW | 40 | 61 | 10 | 3.9 | 33.8 | Mem | I | ↑ | NL | NL |
| R183H | c.548G>A | 23 | M | FW | 37 | 52 | 15 | 0.8 | 35.1 | Mem | NL | NL | NL | ↓ |
| D278Va | c.833A>T | 24 | M | LN | 45 | 50 | 1 | 8.8 | 55.7 | Mem | I | ↑ | ↑ | ↓ |
| Mean values for the four male patients | 35.8 | 47.5 | 8.8 | 3.7 | 39.1 | |||||||||
GJB1 mutations: A.A = amino acid;
First reported in patients with hereditary peripheral neuropathy.
Clinical features of patients: A = absent evoked response; AO = age of onset; AE = age at examination; CMAP = median nerve compound motor action potential at AE; CMTNS = Charcot–Marie–Tooth neuropathy score version 2 at AE; F = female; M = male; MNCV = median nerve motor nerve conduction velocity at AE; Pat. no. = identify number for each patient.
Initial symptoms: FW = distal four limbs weakness; LW = distal lower limbs weakness; LN = distal lower limbs numbness; UW = distal upper limbs weakness; UN = distal upper limbs numbness; HNPP = hereditary neuropathy with liability to pressure palsy.
Biophysical characteristics of mutated proteins: Cyt = cytoplasmic protein retention; GJ = gap junction; L = loss completely; Mem = protein expressed in plasma membrane; NL = normal; I = impaired; U = undetectable; dT = delayed onset of Ca2+ increase in follower cells; τ = time from onset of Ca2+ increase to peak in the follower cells; ↓ = significantly decreased protein expression level; ↑ = significantly prolonged; – not measured.
Intracellular localization of the GJB1 mutants
HeLa cells‐expressing WT‐GJB1 exhibited punctate cytoplasmic staining and large GJ plaques in the intercellular contact regions (Fig. 2A). Aberrant intracellular localization was a common biophysical dysfunction of the GJB1 mutants, occurring in 16 out of the 19 mutants (84.2%) in our study. The M1I mutant was expressed in a very weak, diffuse cytoplasmic pattern (Fig. 2B). Fewer GJ plaques at intercellular boundaries with predominantly cytoplasmic GJB1 staining were observed in cells expressing the following mutants (10/19, 52.6%): L6S, I20F, S26L, S49Y, V91M, S138G, F153L, R164Q, R215P, and R220X (Fig. 2C, D, E, F, G, I, K, M, R, and S). The GJB1 immunoreactivities of the L144del, C173Y, R183C, and E186K mutants (Fig. 2J, N, O, Q) revealed that they were entirely localized to the cytoplasm and failed to reach the cell membrane. In vitro double immunofluorescence staining using a Golgi apparatus marker revealed that these four mutants were retained primarily in the Golgi apparatus (Fig. 3A, B, C, and D). Notably, the p.I101Rfs*8 mutation led to premature truncation in the cytoplasmic loop, and this mutant was not labeled with a loop antibody. Therefore, we constructed a distinct plasmid expressing the I101Rfs*8 mutant GJB1 fused with a Myc tag at the C‐terminus and reprobed the transfectants using a Myc antibody. This approach revealed very weak, diffuse, and exclusively intracellular staining similar to that of the M1I mutant (Fig. 2H). Only cells expressing the M162L, R183H, and D278V GJB1 mutants (Fig. 2L, P, T) exhibited subcellular localization patterns similar to WT‐GJB1.
Figure 2.
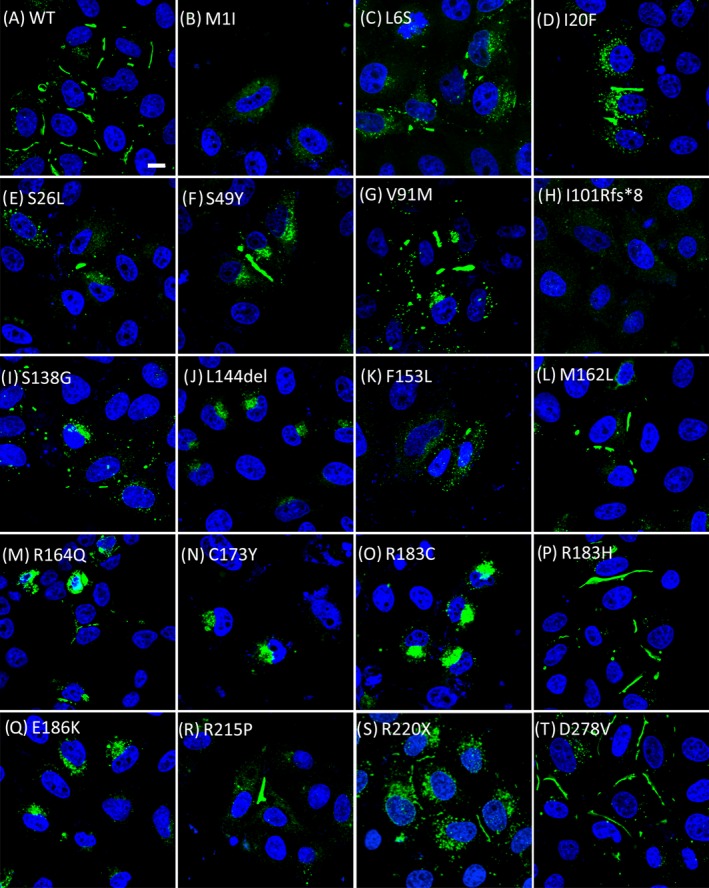
Immunolocalization of wild‐type and mutant GJB1 in transfected HeLa cells. Confocal fluorescence images of transfected cells labeled with an Alexa 488‐conjugated (Green) anti‐GJB1 antibody (in all figures, except H) or an anti‐Myc antibody (Figure 2H). Numerous large gap junction plaques (GJPs) with a small amount of punctate GJB1‐immunoreactive cytoplasmic staining were observed in cells expressing wild‐type GJB1 (A) and in those expressing the mutant M162L (L), R183H (P), or D278V (T). In cells expressing the I20F (D), S49Y (F), V91M (G), or R215P (R) GJB1 mutant, large‐sized but smaller amounts of GJPs were observed at intercellular boundaries with increased GJB1‐immunoreactive cytoplasmic staining. Smaller GJPs with larger amounts of GJB1‐immunoreactive cytoplasmic staining were observed in cells expressing the L6S (C), S26L (E), S138G (I), F153L (K), R164Q (M), or R220X (S) mutant. In contrast, the mutants C173Y (N), R183C (O), E186K (Q), and L144del (J) were retained intracellularly and were incapable of forming detectable GJPs. M1I (B) and I101Rfs*8 (H) appeared to be localized to the cytoplasm, with very weak and diffuse staining patterns. Cell nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI; blue). Scale bar, 10 μm.
Figure 3.
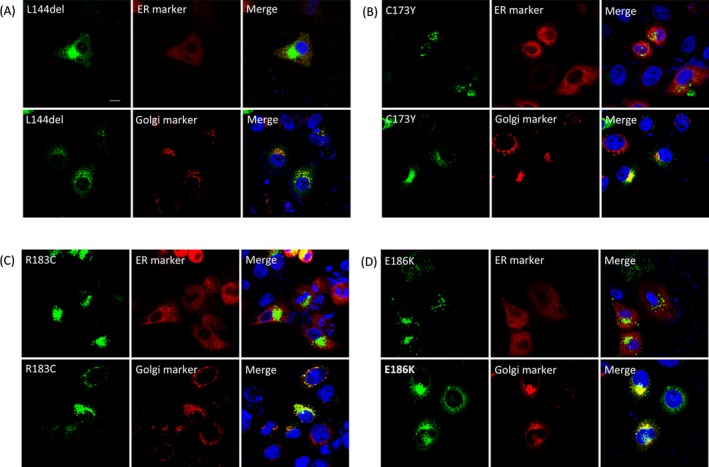
Subcellular localization of intracellularly trapped GJB1 mutants. Confocal fluorescence images of HeLa cells cotransfected with plasmids encoding GJB1 (labeled with a GJB1‐specific antibody; green) and markers of the ER (DsRed‐ER) or the Golgi (DsRed‐Monomer‐Golgi). Similar staining patterns were observed in cells expressing L144del (A), C173Y (B), R183C (C), and E186K (D); GJB1 staining was mainly observed in the perinuclear region and exhibited colocalization with Golgi markers. Cell nuclei were stained with DAPI (blue). Scale bar, 10 μm.
Expression levels of the GJB1 mutants
Fourteen mutants (73.7%) were expressed at significantly lower levels than WT‐GJB1 (Fig. 4A and B), including those with a similar intracellular localization as WT‐GJB1 (R183H and D278V), those partially expressed on cell membrane (L6S, I20F, S49Y, V91M, S138G, and R215P), and those completely intracellularly trapped (L144del, C173Y, R183C, and E186K). In particular, the expression levels of the M1I and I101Rfs*8 mutants were too low to be precisely detected (Fig. 4A, upper part). Only five mutants (S26L, F153L, M162L, R164Q, and R220X) had comparable expression levels to that of WT‐GJB1. The R220X mutant expressed a truncated protein at the expected size of 25 kDa.
Figure 4.
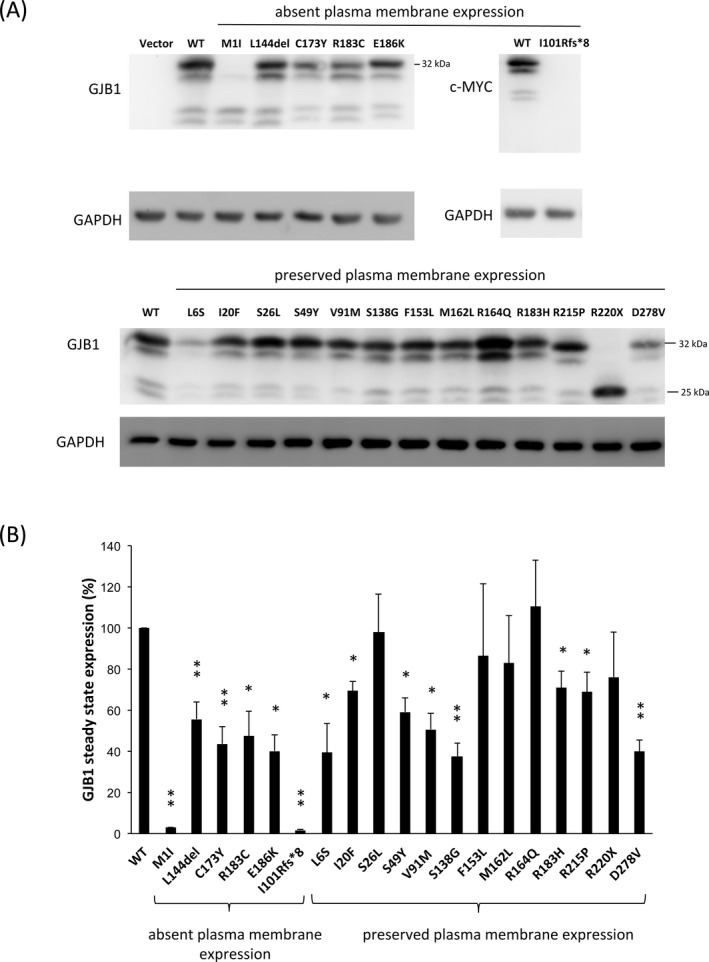
In vitro expression of GJB1 in HeLa cells. (A) Representative images from immunoblotting analyses of GJB1 expression in HeLa cells transfected with plasmids expressing wild‐type (WT) or mutant GJB1, as indicated. The GJB1 mutants were categorized into “Absent plasma membrane expression” and “Preserved plasma membrane expression” according to the findings of the immunofluorescence study. Cells transfected with WT‐GJB1 or the indicated nontruncated mutant showed a major band at 32 kDa, and the truncated mutant R220X produced a band at the expected size of 25 kDa. Two different antibodies were used: a monoclonal GJB1 antibody, which recognized the cytoplasmic loop, and a c‐Myc antibody, which reacted to the Myc‐fused GJB1‐WT or I101Rfs*8 mutant. The GAPDH loading controls used are shown below. (B) Densitometric quantification of the GJB1 protein expression levels in cells transfected with plasmids expressing WT or mutant GJB1, as indicated. The GJB1 expression levels were normalized to GAPDH expression and were expressed as a fraction of the WT expression, which was set to 100%. The error bars indicate the standard error of the mean (SEM) from four independent experiments. The asterisk indicates a significant difference (*, P < 0.05; **, P < 0.01).
Ca2+ signaling propagation of the mutant GJ channels
Mechanical stimulation of a single WT‐GJB1‐expressing cell induced an increase in [Ca2+]i in the stimulated cell (cell 1); this increase was rapidly transferred to adjacent GJ‐coupled cells (cell 2) through GJ channels (Fig. 5A, video S1). In contrast, cells expressing the intracellularly trapped mutants (L144del, C173Y, R183C, and E186K) exhibited an inward Ca2+ oscillation only but failed to transfer Ca2+ to neighboring cells, indicating that the mutants had failed to form functional GJ channels and that the cells had lost Ca2+ permeability (as exemplified by C173Y in Fig. 5B, video S2). For cells with detectable GJ permeability, the Ca2+ propagation between two GJ‐coupled cells was characterized by two parameters using this approach: (1) the time delay in the onset of the increase in the [Ca2+]i in follower cells (dT);13 and (2) the onset‐to‐peak duration of Ca2+ signaling in follower cells (τ), as exemplified by WT‐GJB1 in Fig. 5C. Summaries of the GJ permeabilities for a total of 13 mutants with detectable GJ permeability are presented in Figures 5D (dT) and 5E (τ). The onset latencies in follower cells (dT) were significantly increased in those expressing the M162L, R164Q, R220X, or D278V mutant, and the onset‐to‐peak durations (τ) were significantly longer in the cells expressing one of the eight GJB1 mutants: L6S, I20F, S26L, S49Y, V91M, R164Q, R215P, and D278V. Only the GJs composed of S138G, F153L, or R183H mutants (3/19, 15.8%) had similar permeabilities to those formed by WT‐GJB1.
Figure 5.
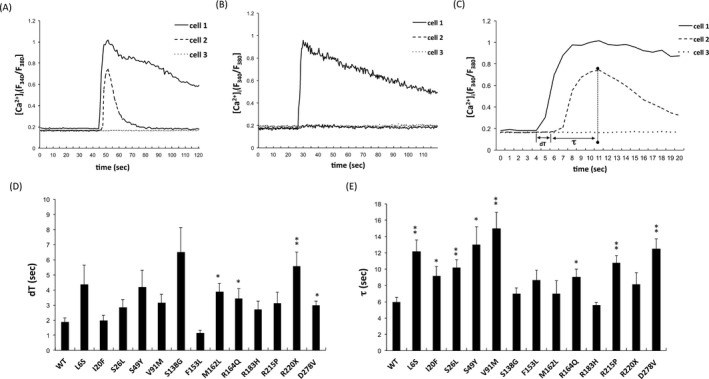
Quantitative measurement of intercellular Ca2+ signaling in HEK293T cells expressing wild‐type and mutant GJB1 gap junctions. (A) Representative Ca2+ traces (fura‐2 ratio, excitation at 340 nm and 380 nm) obtained from fura‐2‐loaded cells expressing wild‐type GJB1 in response to mechanical stimulation. The intracellular Ca2+ concentrations ([Ca2+]i) in a stimulated cell (labeled cell 1), gap junction‐coupled cell (labeled cell 2), and uncoupled cell (labeled cell 3) were plotted. (B) Representative Ca2+ traces obtained in response to mechanical stimulation of cells expressing the intracellularly trapped C173Y mutant. The stimulus induced an increase in the [Ca2+]i in the stimulated cell (labeled cell 1), but this increase failed to transfer to neighboring cells (labeled cells 2 and 3). (C) Parameters describing intercellular [Ca2+]i fluxes across GJB1 gap junctions. Mechanical stimulation increased the [Ca2+]i in the stimulated cell (cell 1, touched) and subsequently induced the amplitude of Ca2+ transients that spread to the adjacent gap junction‐coupled cell (cell 2, follower cell). dT, the time delay in the onset of the [Ca2+]i increase in the follower cells; and τ, the onset‐to‐peak duration in the follower cells. (D), (E) Comparisons of the average dT and τ between the wild‐type GJB1 and mutants. The error bars indicate standard error of the mean (SEM) from six independent experiments. The asterisk indicates a significant difference (*, P < 0.05; **, P < 0.01).
Correlating the biophysical dysfunctions of GJB1 mutants with the clinical features of CMTX1 patients
The above biophysical characteristics of the mutant GJB1 proteins and the clinical features of the affected patients are summarized in Table 1. To correlate the biophysical changes caused by the genotypes with the patients' phenotypes, we classified the GJB1 mutants into two groups based on their biophysical dysfunctions (Table 1). The first group (A) encompassed mutants with significantly compromised plasma membrane expression of GJB1 and GJ plaque formation and also impaired GJ permeability, including the mutants with complete loss of GJ permeability due to undetectable GJB1 expression (M1I, I101Rfs*8), those with cytoplasmic retention (L144del, C173Y, R183C, and E186K), and those with a reduced plasma membrane expression combined with predominant cytoplasmic retention of GJB1 (L6S, I20F, S26L, S49Y, V91M, R164Q, R215P, and R220X). The mutants in the second group (B), which included S138G, F153L, M162L, R183H, and D278V, were featured by normal GJ Ca2+ permeability in our study or predominant plasma membrane expression of GJB1 with abundant GJ plaque formation.
Several clinical parameters of the patients with CMTX1 are listed in the Table 1, including the AO, AE, CMTNS, and median nerve compound motor action potential (CMAP) and MNCV at the AE. Only male patients were included for the comparison because gender substantially influences the clinical and neurophysiological presentations. Among the clinical parameters, only AO and CMTNS were significantly different among the two groups. For individuals carrying mutations with preserved plasma membrane locations of GJB1 and GJ functions (group B), the AO was near 17 years later in average than those carrying mutations with compromised plasma membrane expression of GJB1 and GJ permeability (group A) (Student's t‐test, P = 0.001). The average CMTNS was significantly lower in group B than group A (Student's t‐test, P = 0.04), suggesting a milder phenotype in this group. These observations suggested that the GJB1 mutants with intense biophysical dysfunctions might be associated with more severe phenotypes. In contrast, the corresponding phenotypes in the group B mutants were relatively milder with a later disease onset.
Discussion
This study delineated the pathological effects of 19 GJB1 mutations by characterizing multiple biophysical dysfunctions of the GJB1 protein, including abnormalities in intracellular trafficking, protein expression, and GJ function. These mutations were evenly distributed over all functional domains of the protein in a cohort of patients with the classic CMTX1 phenotype. Therefore, the comprehensive determination of the biophysical characteristics and clinical presentations of these patients in this study deepens our understanding of the pathogenic mechanisms of CMTX1.
Several studies have reported that alterations in intracellular trafficking, protein expression, and GJ function are the main abnormalities of mutant GJB1 proteins associated with CMTX1.8, 10, 17, 18 Our results support these notions because all of the GJB1 mutations examined in this study resulted in at least one of these biophysical changes. These altered functions may interact with each other and result in complex pathological effects, contributing to the diverse phenotypes and wide variations in the AO and clinical severity. However, how these cellular dysfunctions are implicated in the onset and progression of the disease is not known. In this study, we correlated the biophysical changes in the protein levels with the clinical parameters of the patients to explore the key factors affecting the disease progression.
We categorized these GJB1 mutants according to the severity of the associated biophysical dysfunctions. Analyses of the patients' clinical features and the mutants' biophysical dysfunctions revealed that the mutants with severe molecular defects and impaired GJ permeability (group A) appeared to be associated with an earlier AO and a severe phenotype. These mutants obviously cause development of the disease through a severe loss of GJ function. The observed phenotype–genotype association emphasizes the important role of abnormal GJ function in the pathogenesis of CMTX1, supporting the notion that GJB1, as a GJ protein, is essential for peripheral nerve functioning. Among the 19 GJB1 mutants in this study, the majority (16/19, 84.2%) had abnormal GJ function. Therefore, our results demonstrated that compromised GJ permeability may be an important mechanism for many GJB1 mutants in the development of peripheral neuropathy.
We found that the four mutated GJB1 proteins (p.L144del, p.C173Y, p.R183C, and p.E186K) with completely cytoplasmic retention were predominantly retained in the Golgi apparatus (Fig. 3). GJB1 proteins are normally assembled into connexons (hemichannels) in the Golgi apparatus,19 but the mutant proteins are retained in the Golgi and are not assembled properly.20 Therefore, these proteins do not reach the plasma membrane, and cell pairs expressing these mutants are not functionally coupled. The mislocalized proteins are likely to be degraded through lysosomal proteolysis or the proteasome pathway, leading to a significant reduction of the steady‐state protein expression levels of all of these mutants. The four mutations causing completely cytoplasmic retention of the protein are localized to the third transmembrane domain (L144del) or the second extracellular domain (C173Y, R183C, and E186K) of GJB1, suggesting that these two domains play important roles in GJB1 trafficking.
There are some limitations of this study. First, the in vitro non‐Schwann cell system cannot fully mimic in vivo myelinating Schwann cells and the in vivo pathologies derived from GJB1 mutations, such as demyelinating or axonal degeneration,21 cannot be evaluated in this study. Since the gap junction permeability is unlikely the only determinant for clinical presentations of CMTX1, it is not surprising that we cannot perfectly correlate the biophysical changes with clinical parameters of the 19 GJB1 mutations. Next, the number of patients was not sufficiently large. Then, GJ permeability was evaluated by performing a mechanical stimulation to induce Ca2+ propagation. Although mechanical stimuli have been widely used to trigger calcium wave propagation as a method to study gap junctions,22 they still differ from the physiological stimuli in peripheral nerves. Finally, differences between the heterotypic and homotypic effects were not investigated. However, this study clearly demonstrated the biophysical and clinical features of the 19 GJB1 mutants and suggested the presence of a trend between different biophysical characteristics of the specific GJB1 mutants and the patients' clinical phenotypes. Our results revealed that the GJB1 mutations resulting in preserved GJ permeability and those with predominant cell membrane expression were likely to have a later AO and a milder phenotype. Promoting GJB1 GJ function may be a potential therapeutic target in the treatment of CMTX1. In fact, this rationale for developing CMTX1 therapy has been supported by recent animal studies, demonstrating that restoration of Schwann cell‐specific GJB1 expression in Gjb1‐null mice by intrathecal lentiviral gene delivery can lead to improvements in motor performance, nerve conduction velocities, and neuropathology of peripheral nerves.23, 24 Since different GJB1 mutations may have different molecular consequences, elucidating the molecular and clinical phenotypes of each GJB1 mutation may help future gene therapy of CMTX1 with an optimal and mutation‐tailored regimen.
Conflict of Interest
None declared.
Supporting information
Table S1. Antibodies used for analysis
Table S2. In silico prediction of the functional effects of the five novel missense GJB1 mutations
Video S1. Ca2+ signaling propagation of the wild‐type GJB1 gap junction channels. Before mechanical stimulation, most cells displayed relatively uniform resting intracellular Ca2+ concentrations ([Ca2+]i) (green). For cells expressing WT‐GJB1, mechanical stimulation of a single cell induced an increase in the [Ca2+]i at the point of contact, and a wave of increased [Ca2+]i spread throughout the cell (dark red trace). After a short delay (~1.5–2 sec), waves of Ca2+ were rapidly transferred to the adjacent GJ‐coupled cells through GJ channels and spread outward to the more distal neighboring cells.
Video S2. Failure of Ca2+ signaling propagation in the C173Y mutant GJB1 gap junction channels. In contrast, for cells expressing the intracellularly trapped C173Y GJB1 mutant, mechanical stimulation of a single cell provoked a [Ca2+]i elevation in the stimulated cell, but the inward Ca2+ current did not propagate outward to neighboring cells
Acknowledgments
We thank the patients who participated in this study and the High‐throughput Genome Analysis Core Facility of the National Core Facility Program for Biotechnology of Taiwan (NSC‐101‐2319‐B‐010‐001) for providing nucleotide sequencing services. We also thank the Ministry of Science and Technology, Taiwan (102‐2628‐B‐075‐006‐MY3, 105‐2628‐B‐075‐002‐MY3), and Taipei Veterans General Hospital (V104C‐041) for providing funding.
Contributor Information
Yo‐Tsen Liu, Email: ytliu2@vghtpe.gov.tw.
Yi‐Chung Lee, Email: ycli@vghtpe.gov.tw.
References
- 1. Braathen GJ, Sand JC, Lobato A, et al. Genetic epidemiology of Charcot‐Marie‐Tooth in the general population. Eur J Neurol 2011;18:39–48. [DOI] [PubMed] [Google Scholar]
- 2. Gutmann L, Shy M. Update on Charcot‐Marie‐Tooth disease. Curr Opin Neurol 2015;28:462–467. [DOI] [PubMed] [Google Scholar]
- 3. Saporta MA, Shy ME. Inherited peripheral neuropathies. Neurol Clin 2013;31:597–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Timmerman V, Strickland AV, Züchner S. Genetics of Charcot‐Marie‐Tooth (CMT) disease within the frame of the human genome project success. Genes (Basel) 2014;5:13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Birouk N, LeGuern E, Maisonobe T, et al. X‐linked Charcot‐Marie‐Tooth disease with connexin 32 mutations: clinical and electrophysiologic study. Neurololgy 1998;50:1074–1082. [DOI] [PubMed] [Google Scholar]
- 6. Braathen GJ, Sand JC, Bukholm G, Russell MB. Two novel connexin32 mutations cause early onset X‐linked Charcot‐Marie‐Tooth disease. BMC Neurol 2007;7:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dubourg O, Tardieu S, Birouk N, et al. Clinical, electrophysiological and molecular genetic characteristics of 93 patients with X‐linked Charcot‐Marie‐Tooth disease. Brain 2001;124:1958–1967. [DOI] [PubMed] [Google Scholar]
- 8. Kleopa KA, Abrams CK, Scherer SS. How do mutations in GJB1 cause X‐linked Charcot‐Marie‐Tooth disease? Brain Res 2012;1487:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakagawa S, Maeda S, Tsukihara T. Structural and functional studies of gap junction channels. Curr Opin Struct Biol 2010;20:423–430. [DOI] [PubMed] [Google Scholar]
- 10. Kleopa KA, Sargiannidou I. Connexins, gap junctions and peripheral neuropathy. Neurosci Lett 2015;596:27–32. [DOI] [PubMed] [Google Scholar]
- 11. Jerath NU, Shy ME. Hereditary motor and sensory neuropathies: understanding molecular pathogenesis could lead to future treatment strategies. Biochim Biophys Acta 2015;1852:667–678. [DOI] [PubMed] [Google Scholar]
- 12. Murphy SM, Herrmann DN, McDermott MP, et al. Reliability of the CMT neuropathy score (second version) in Charcot‐Marie‐Tooth disease. J Peripher Nerv Syst 2011;16:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sun J, Ahmad S, Chen S, et al. Cochlear gap junctions coassembled from Cx26 and 30 show faster intercellular Ca2 + signaling than homomeric counterparts. Am J Physiol Cell Physiol 2005;288:C613–C623. [DOI] [PubMed] [Google Scholar]
- 14. Zhang Y, Hao H. Conserved glycine at position 45 of major cochlear connexins constitutes a vital component of the Ca2+ sensor for gating of gap junction hemichannels. Biochem Biophys Res Commun 2013;436:424–429. [DOI] [PubMed] [Google Scholar]
- 15. Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980;103:259–280. [DOI] [PubMed] [Google Scholar]
- 16. Michell AW, Laura M, Blake J, et al. GJB1 gene mutations in suspected inflammatory demyelinating neuropathies not responding to treatment. J Neurol Neurosurg Psychiatry 2009;80:699–700. [DOI] [PubMed] [Google Scholar]
- 17. Yum SW, Kleopa KA, Shumas S, Scherer SS. Diverse trafficking abnormalities of connexin32 mutants causing CMTX. Neurobiol Dis 2002;11:43–52. [DOI] [PubMed] [Google Scholar]
- 18. Kleopa KA, Scherer SS. Molecular genetics of X‐linked Charcot‐Marie‐Tooth disease. Neuromolecular Med 2006;8:107–122. [DOI] [PubMed] [Google Scholar]
- 19. Musil LS, Goodenough DA. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell 1993;74:1065–1077. [DOI] [PubMed] [Google Scholar]
- 20. Martin PE, Mambetisaeva ET, Archer DA, et al. Analysis of gap junction assembly using mutated connexins detected in Charcot‐Marie‐Tooth X‐linked disease. J Neurochem 2000;74:711–720. [DOI] [PubMed] [Google Scholar]
- 21. Vavlitou N, Sargiannidou L, Markoullis K, et al. Axonal pathology precedes demyelination in a mouse model of X‐linked demyelinating/type I Charcot‐Marie‐Tooth neuropathy. J Neuropathol Exp Neurol 2010;69:945–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iyyathurai J, Himpens B, Bultynck G, D'hondt C. Calcium Wave Propagation Triggered by Local Mechanical Stimulation as a Method for Studying Gap Junctions and Hemichannels. Methods Mol Biol 2016;1437:203–211. [DOI] [PubMed] [Google Scholar]
- 23. Sargiannidou L, Kagiava A, Bashiardes S, et al. Intraneural GJB1 gene delivery improves nerve pathology in a model of X‐linked Charcot‐Marie‐Tooth disease. Ann Neurol 2015;78:303–316. [DOI] [PubMed] [Google Scholar]
- 24. Kagiava A, Sargiannidou I, Theophilidis G, et al. Intrathecal gene therapy rescues a model of demyelinating peripheral neuropathy. Proc Natl Acad Sci USA 2016;113:E2421–E2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Antibodies used for analysis
Table S2. In silico prediction of the functional effects of the five novel missense GJB1 mutations
Video S1. Ca2+ signaling propagation of the wild‐type GJB1 gap junction channels. Before mechanical stimulation, most cells displayed relatively uniform resting intracellular Ca2+ concentrations ([Ca2+]i) (green). For cells expressing WT‐GJB1, mechanical stimulation of a single cell induced an increase in the [Ca2+]i at the point of contact, and a wave of increased [Ca2+]i spread throughout the cell (dark red trace). After a short delay (~1.5–2 sec), waves of Ca2+ were rapidly transferred to the adjacent GJ‐coupled cells through GJ channels and spread outward to the more distal neighboring cells.
Video S2. Failure of Ca2+ signaling propagation in the C173Y mutant GJB1 gap junction channels. In contrast, for cells expressing the intracellularly trapped C173Y GJB1 mutant, mechanical stimulation of a single cell provoked a [Ca2+]i elevation in the stimulated cell, but the inward Ca2+ current did not propagate outward to neighboring cells