

Abstract
The sodium leak channel, a Na+‐permeable, nonselective cation channel, is widely expressed in the nervous system, contributing a basal Na+‐leak conductance and regulating neuronal excitability. A 3‐year‐old girl, heterozygous for a de novo missense mutation in NALCN (c.956C>T; p.Ala319Val) predicted to be deleterious, presented from birth with: stimulus‐induced, episodic contractures of the limbs and face with associated respiratory distress; distal arthrogryposis; severe axial hypotonia; and severe global developmental delay (CLIFAHDD syndrome). In infancy, she manifested a reversed sleep‐wake rhythm, nocturnal life‐threatening respiratory rhythm disturbances with central apnea. Sevoflurane sensitivity caused respiratory depression and cardiac arrest.
Introduction
The sodium leak channel (NALCN) forms a channel complex, interacting directly with UNC80, and indirectly with UNC79.1, 2 The pore‐forming subunit, NALCN, comprises four homologous domains (I–IV) of six transmembrane segments (S1–S6). Four P‐loops, spanning from S5 to S6 in each domain, constitute an EEKE sodium‐ion selectivity filter. NALCN is highly conserved in mammals (98% identity between mouse and human) and is expressed highest in neurons of brain and spinal cord, less in heart and pancreas.3 NALCN forms a nonselective, voltage‐insensitive, noninactivating cation channel and contributes a background Na+ leak current to the neuronal resting membrane potentials, thereby modulating neuronal excitability.3 Background Na+ leak conductances are also implicated in the generation of the spontaneous rhythmic firing of pacemaker neurons, such as those involved in the neuronal network of respiratory activity.3, 4 Mice with targeted deletions of nalcn have severely disrupted respiratory rhythms and die within 24 h of birth.3 NALCN activity is regulated either by G protein‐coupled receptors, or by neuropeptides (substance P and neurotensin) through a G protein‐independent pathway involving the Src family of kinases and their interaction with UNC80.1, 2
In recent reports, three homozygous bi‐allelic NALCN loss‐of‐function mutations associated with a phenotype of severe infantile hypotonia, psychomotor retardation, and characteristic facies (IHPRF [MIM #615419]), plus variable difficulty feeding, failure to thrive, seizures, strabismus, and chronic constipation were described.5, 6, 7 Similar presentations were described with bi‐allelic deleterious UNC80 mutations.8, 9 Conversely, de novo dominant heterozygous NALCN missense mutations – all positioned in or near the pore‐forming S5 and S6 segments of domains I–IV – were associated with a phenotype of congenital contractures of limbs and face, hypotonia, and global developmental delay (CLIFAHDD syndrome).10, 11, 12, 13
We describe a 3‐year‐old child with a fully manifest CLIFAHDD syndrome caused by a de novo heterozygous NALCN missense mutation (c.956C>T; p.Ala319Val), predicted to be deleterious and positioned in the functionally important pore‐forming S6 segment of domain I of the protein. Reversed circadian rhythm, frequent episodes of disrupted respiratory rhythms, central and obstructive sleep apneas with recurrent hypoxia, and sensitivity to sevoflurane anesthetic threaten survival.
Ethics
The research was performed according to the guidelines of the Declaration of Helsinki. All participating subjects provided informed consent for genetic testing and publication.
Results
Methodologies and subject
Regular blood tests, thyroid and parathyroid hormones, urinary organic acids, plasma amino acids, lactate, very long‐chain fatty acids, acetylcarnitine profile, transferrin isoelectric focusing, and serum cholinesterase were unremarkable.
DNA analysis
Crisponi syndrome1993‐2016 was considered in differential diagnosis. Single gene sequencing of cytokine receptor‐like factor 1 (CRLF1), cardiotrophin‐like factor 1 (CLCF1), and leukemia inhibitory factor receptor (LIFR) revealed no variants. Trio‐exome sequencing, performed as described previously in Haugarvoll et al.15 revealed five genes with rare de novo or bi‐allelic mutations (Table 1). The most likely causal mutation was a de novo missense mutation in NALCN c.956C>T; p.Ala319Val (Fig. 1A), affecting a highly conserved amino acid residue (Fig. 1B), predicted to be damaging (Table 1), and to be positioned in the S6 transmembrane segment of domain I (Fig. S1). Previous reports have shown that mutations in this domain can cause the autosomal dominant CLIFAHDD syndrome.10, 11, 12, 13 The other four identified candidate genes were not supported by bio‐informatics evaluation and literature searches and thus, we do not consider them to be causative in this disease.
Table 1.
List of all variants in genes with rare de novo or bi‐allelic variants from whole‐exome sequencing of the proband and her parents
| Type | Gene | Chr | Positiona | cDNA | Protein | dbSNP | Exac Euro MAF2 (%) | SIFT/MT/PPb | Amino acid conservation | Relevance for phenotype |
|---|---|---|---|---|---|---|---|---|---|---|
| De novo | NALCN | 13 | 101944432 | c.956C>T | p.A319V | ND | ND | D/D/D | High | High: Gene implicated in CLIFAHDD syndrome:Chong et al10 |
| De novo | EPHA1 | 7 | 143097139 | c.440C>T | p.T147M | rs542068127 | 0.003 | D/D/D | High | Low: Gene possibly implicated in cancer and Alzheimer Disease |
| CompoundM | CASZ1 | 1 | 10718605 | c.1369G>A | p.V457I | rs148685497 | 0.02 | B/B/B | Low | Low |
| CompoundP | CASZ1 | 1 | 10720309 | c.790C>T | p.R264W | ND | ND | D/D/D | Low | Low |
| Homozygous | TES | 7 | 115889259 | c.299C>G | p.A100G | rs62621959 | 0.05 | B/D/P | High | Low |
| CompoundP | ADAMTS14 | 10 | 72468473 | c.809C>T | p.S270L | rs61749230 | 0.8 | D/D/D | High | Low |
| CompoundM | ADAMTS14 | 10 | 72493697 | c.1274G>A | p.G425D | rs760544573 | 0.008 | D/D/D | High | Low |
| CompoundM | ADAMTS14 | 10 | 72503400 | c.2030G>A | p.R677Q | rs141167300 | 0.03 | D/B/P | High | Low |
In total, 20895 exonic variants were found in the proband. Variant filtration against common variants (1000 Genomes and an in‐house database of variants, 0.5% allele frequency) yielded 324 rare coding variants that were compared against whole‐exome data from her parents. This revealed two true de novo missense variants, one homozygous variant and two genes with compound heterozygous variants. The only gene with strong a priori evidence of being implicated in this monogenic disease was NALCN. MAF, minor allele frequency; D, damaging; B, benign; P, possibly damaging; Superscript M = maternal or P = paternal indicates the derivation of the mutation.
Genome Reference Consortium Human Build 37 (GRCh37) http://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.13/
SIFT: http://sift.jcvi.org/, PP: PolyPhen‐2 http://genetics.bwh.havard.edu/pph2/bgi.shtml, MT: MutationTaster http://www.mutationtaster.org/
Figure 1.
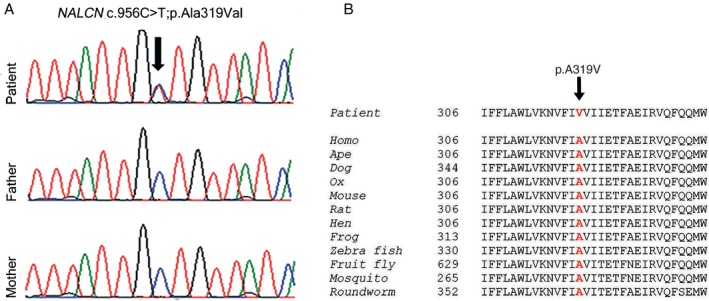
(A) The partial chromatograms from Sanger sequencing illustrate a heterozygous mutation (c.956C>T, p.Ala319Val) in exon 9 of NALCN, identified in the patient and normal sequences from healthy parents. The arrow indicates the mutation site. Computer programs, PolyPhen‐2, SIFT, and MutationTaster, show the mutation to be damaging. (B) Illustration of high evolutionary conservation of amino acid sequences in the region of the mutated residue (shown in red), supporting the concept that the mutation is functionally disruptive.
Clinical observations
Birth at 37 weeks gestation was uneventful (Apgar scores 9/9, birth weight 2890 g, length 47 cm, head circumference [OFC] 34 cm). The infant exhibited typical facial dysmorphisms and distal arthrogryposis (illustrated in Fig. 2). Suck and Moro reflexes were absent and tone markedly reduced. She appeared irritable with frequent episodes of facial and limb muscle contractions induced by noise, gentle touch, or handling but absent during sleep. Facial and oropharyngeal muscle contractions caused inspiratory stridor, perioral cyanosis, and transient hypoxemia (Fig. 2A); copious saliva frothed from pursed lips (Fig. 2B); she sweated in face, head and upper trunk (Fig. 2C). During such spells, arms flexed and abducted, while legs made cycling‐type movements. Repetitive movements, interpreted as seizures, were medicated with phenobarbital and/or clobazam, but these were discontinued because of drowsiness, absent EEG seizure activity, and a normal brain magnetic resonance imaging (MRI) scan. A CT scan of the pharynx confirmed micrognathia and excluded obstructive anomalies. Poor oral feeding was supplemented with nasogastric tube feedings.
Figure 2.
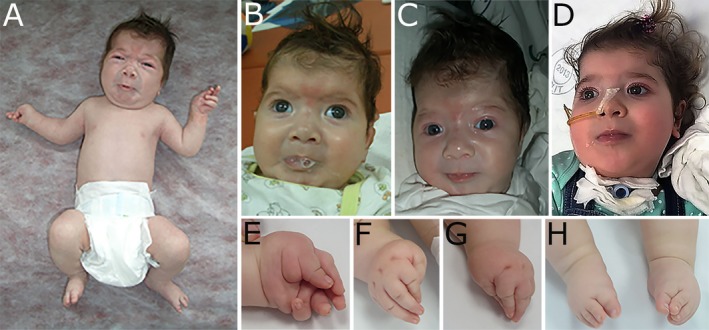
Typical facial characteristics of CLIFAHDD syndrome are shown in (A–C): broad nasal bridge, anteverted nasal tip and large nares, short columella, long philtrum, deep nasolabial folds, rounded cheeks, pursed lips, micrognathia, H‐shaped chin dimpling, low‐set ears, and short neck. (A) Illustrates the stimulus‐provoked facial and limb contractions, particularly frequent in the neonatal period; arms are flexed and abducted, legs flexed or cycling. Mild contractures were present at elbows, knees, and hips. (B) Copius foamy saliva extrudes from the pursed lips. (C) During the attack, the child sweats excessively in the face, head and upper trunk. (D) At age 33 months: the child is anxious, facial contractions are milder, drooling persists; she is dependent on nasogastric tube feeding and is treated with positive pressure ventilation via tracheostomy. (E–G) illustrate the distal arthrogryposis; with camptodactyly, adducted thumbs, and ulnar deviation. (H) Bilateral varus foot deformity.
The infant's precarious respiratory status required continuous monitoring and hospitalization for 3 months. Breathing and SpO2‐saturation were normal during daytime sleep. Arousal or handling provoked typical facial and oropharyngeal muscle contractions, demanding suction and supplementary oxygen. Care was particularly difficult at night, as she was often awake exhibiting stridorous breathing, disturbed respiratory rhythm with mixed central and obstructive sleep apnea (OSA) and SpO2‐desaturation, necessitating bag‐valve mask resuscitation and ventilation. At 2 months, one such episode was captured during attempted polysomnography (PSG): she had calmed down after the stress of EEG hook‐up and fallen asleep, breathing 72 breaths/min, SpO2‐saturation 85%, and heart rate 158/min. Five minutes into the recording, breathing suddenly became shallow, rapid and irregular; SpO2‐saturation dropped to 50%, and heart rate to 60/min; followed within seconds by central apnea, flat EEG and cardio‐respiratory arrest, that was reversed by immediate resuscitation. Nocturnal respiratory rhythm disturbances requiring bag‐valve mask ventilation continued to challenge caregivers.
At 7 months, she presented with acute laryngo‐bronchospasm, severe dyspnea, hypoxemia, tachyarrhythmia, and hyperthermia (39°C), without clinical or radiological signs of pulmonary infection or aspiration. She responded to inhaled β adrenergic‐2 agonists and corticosteroids; respiratory functions stabilized within 2 days.
From 15 months onward, respiratory functions deteriorated as she developed cervico‐thoracic kyphosis and “barrel chest” deformity. During induction of general anesthesia for chest MRI, sensitivity to sevoflurane caused acute respiratory depression and cardio‐respiratory arrest, prompting resuscitation and assisted ventilation in ICU. Likewise, sensitivity to barbiturate and benzodiazepine was suggested by excessive sedation with subtherapeutic doses.
By age 2 years, she required a tracheostomy and assisted ventilation due to chronic respiratory insufficiency. Observation of nocturnal insomnia and daytime somnolence and frequent napping suggested a reversed circadian rhythm, a sleep‐wake pattern recognized since early life.
At 33 months, a 2 h PSG recording documented normal sleep architecture and on average SpO2 saturation of 95%; in addition, there were 19 respiratory events, the longest lasting 109 sec: seven OSA with an oxygen desaturation index of 13.1 (Fig. 3A); three central apneas of maximally 35 sec duration, and SpO2 desaturation of 65% (Fig. 3B); periodic breathing and brief central apneas, with SpO2 desaturation of 74% (Fig. 3C); nine episodes of obstructive hypopneas and variants of altered respiratory rhythm (Fig. 3D). These observations signaled the continuation of severely disturbed respiratory rhythms, and were treated with positive pressure ventilation via tracheostomy.
Figure 3.
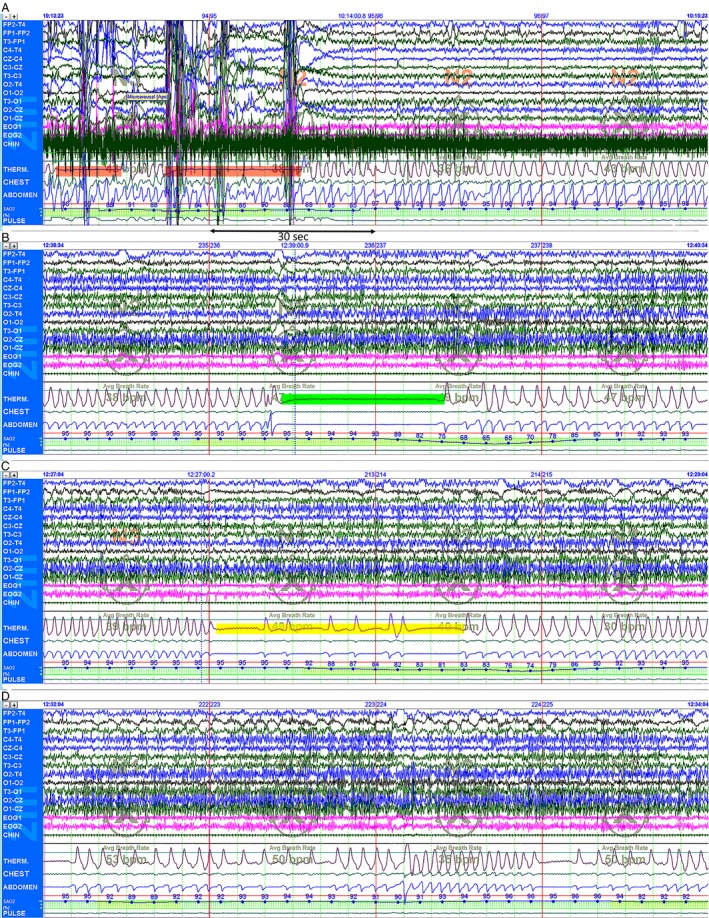
Representative polysomnography (PSG) recordings (each 120 sec) of changes in respiratory rhythm; baseline SpO2 saturation of 95%: (A) Two episodes of obstructive apnea (highlighted in red) lasting 12 and 25 sec, respectively, associated with paradoxical chest and abdominal wall motion, SpO2 desaturation of 84%, accompanied by increased chin EMG activity. Arousal response with change from stage 3 to stage 2 non‐rapid eye movement (NREM) NREM sleep. (B) The abrupt onset of central apnea lasting 35 sec (highlighted in green) accompanied by SpO2 desaturation of 65%. Note the lack of flow through the tracheostomy thermistor sensor, and complete absence of chest and abdominal wall movements; stage 2 NREM sleep. (C) Pathological periodic breathing and repeat brief central apneas (highlighted in yellow) and SpO2 desaturation of 74%. Arousal with change from stage 3 to stage 2 NREM sleep. (D) Variants of periodic breathing and altered respiratory rhythm. Recordings were performed with a tracheostomy in place, using standard digital PSG equipment: Polysmith Nihon Kohden America Inc., CA, USA. PSGs were scored as per the 2007 American Academy of Sleep Medicine (AASM) guidelines for the scoring of sleep and associated events.23
Periods of agitation, sweating, and excessive drooling persist. She has failed to thrive despite full caloric nasogastric tube feedings (weight 10 kg [−3.0 SD]) and suffers chronic constipation. One notes: growth retardation (length 82 cm [−2.9 SD], OFC 46 cm [−1.6 SD]), severe global developmental delay, cognitive impairment, absent speech, brief eye contact, severe axial hypotonia, no head control nor sitting. She can roll over, but lacks purposeful hand movements; muscle bulk is normal and tendon reflexes are preserved.
Discussion
NALCN is highly evolutionarily conserved. Studies of homolog gene mutations in invertebrates and mammals have elucidated some of NALCN's unique molecular and biophysical properties and its contributions to neuronal function and animal behavior (reviewed in16). Only recently, have the clinical consequences of disease‐causing mutations in NALCN, UNC80, and UNC79 been described in humans.
Chong and colleagues10 characterized the uniform phenotypic presentation, termed CLIFAHDD syndrome, of 14 de novo dominant pathogenic NALCN missense variants, all predicted to be positioned in or near the S5 and S6 transmembrane segments, the likely pore‐forming domains of NALCN.16 This observation was true for subsequently reported NALCN variants with a CLIFAHDD phenotype, including the one of this report (Fig. S1). Clustering of the mutations underscores the functional importance of these transmembrane segments. Molecular mechanisms by which NALCN variants cause the spectrum of clinical features are unknown. In preliminary functional studies, Chong et al.10 demonstrated that NALCN variants co‐transfected with wild‐type (WT) NALCN into HEK293T cells, nearly abolished detection of WT NALCN channels, while these were readily detectable with the co‐transfection of WT NALCN. The observations suggest that NALCN mutants may exhibit dominant‐negative effects, analogous to those documented with CACNA1A mutants (encoding the pore‐forming α 1A‐subunit of the Cav2.1 channel) associated with episodic ataxia type 2.17
Chong et al.10 list abnormal respiratory patterns in the newborn period and/or respiratory insufficiency for 2/3 individuals, but did not characterize the respiratory dysfunctions. Two infants had died of unknown causes. We report a 3‐year longitudinal observation in a child with severe CLIFAHDD syndrome caused by a novel de novo NALCN missense mutation (c.956C>T, p.Ala319Val), predicted to be deleterious and positioned in the pore‐forming S6 segment of domain I. From birth, she manifested severely disturbed respiratory rhythms with episodes of periodic breathing, mixed central and obstructive apnea, and occasional life‐threatening hypoxemia, requiring frequent bag‐valve mask resuscitation and ventilation. Micrognathia and episodic facial and oropharyngeal muscle contractions likely caused intermittent airway occlusion, manifesting as inspiratory stridor and significant SpO2 desaturation. Recent PSG recordings document (Fig. 3A–D) that respiratory disturbances, including brief episodes of OSA and chronic intermittent hypoxia persist into early childhood even after tracheostomy.
Respiratory rhythms were severely disrupted in nalcn −/−‐mutant mouse pups with a repetitive pattern of apnea for ~ 5 sec followed by bursts of deep breathing for ~5–10 sec; no pups survived beyond 24 h after birth. Rhythmic electrical discharges recorded from WT C4 phrenic nerves that innervate the diaphragm were absent in the nalcn mutants, consistent with a central origin of this respiratory defect.3 The pre‐Bötzinger complex (preBötC) network of the medulla is the substrate for inspiratory rhythm generation and the source of rhythmic excitatory output to cranial and spinal motor neurons. Respiratory rhythms are generated by inspiratory neurons in the preBötC through pacemaking and network mechanisms that are dynamically regulated by neuromodulators such as serotonin (5‐HT) and SP. The latter were shown to exert excitatory effects in preBötC neurons by modulating background cation conductances, including a nonselective cation channel with the properties of NALCN.18, 19 Future studies in mice, including NALCN knock out mutants, might confirm a role of NALCN in rhythmogenesis in preBötC inspiratory neuron.
Both fly and worm nalcn mutants exhibited increased sensitivity to volatile anesthetics, specifically halothane.20 During induction of sevoflurane anesthesia, our child unexpectedly exhibited high sensitivity to the drug at usual dosage, with severe respiratory depression culminating in cardio‐respiratory arrest. Resuscitation was successful; however, she required prolonged ventilator support.
Ever since the postnatal period, the child manifested a reversed circadian rhythm, being mostly awake at night, manifesting greater respiratory difficulty, and need for bag‐valve support and supplementary oxygen. Circadian clocks regulate membrane excitability in master pacemaker neurons, the mammalian suprachiasmatic nucleus (SCN), to control daily rhythms of sleep and wake. Patch‐clamp analyses of dissociated mice SCN neurons demonstrated the presence of an inward Na+ background current with the pharmacological profile of NALCN, responsible for the initial phase of the depolarizing drive during the interspike interval.21 By generating a forebrain specific knockout of nalcn in mice, a recent study confirmed that the majority of sodium leak current in SCN neurons is carried by NALCN and is under control of the circadian clock, whereby anti‐phase cycles of distinctly timed sodium and potassium currents drive clock neuron rhythms.22
Thus, many behavioral abnormalities observed with mutant NALCN in invertebrates and rodents occur in humans, indicating evolutionary preservation of NALCN functions. Chronic respiratory dysfunctions should be anticipated and treated aggressively in infants with confirmed NALCN mutations, as they may adversely affect neurodevelopmental outcome.
Author Contributions
All authors contributed equally to the conception and execution of the various aspects of the work and to the study as a whole. They collectively vouch for the accuracy and integrity of the data. Drs B. Lozic, J. Markic, S. L. Kojundzic, and A. F. Hahn were involved in the acquisition and interpretation of the clinical data. Drs H. Boman, S. Johansson, and P. M. Knappskog directed the acquisition, analysis, and interpretation of the genetic data. Drs Lozic, H. Boman, and A. F. Hahn drafted the manuscript, which was circulated, amended and approved by all authors.
Conflict of Interest
None declared.
Supporting information
Figure S1. Schematic diagram of the NALCN topology comprised of four domains (I–IV); each made up of six transmembrane segments (S1–S6). Segments S5 and S6 are predicted to form the central pore. Illustrated are the presently identified NALCN variants: 20 heterozygous de novo missense mutations associated with the CLIFAHDD phenotype are illustrated as black filled circles, the mutation of this report as red filled circle. Note that these mutations are positioned in or near the S5 and S6 segments of all domains, and are particularly clustered in the S6 segment of domain 1, the predicted position of the here reported variant. Reported homozygous NALCN variants associated with the autosomal recessive IHPRF phenotype (MIM: 615419) are illustrated as black squares. We have referred to http://www.uniprot.org/uniprot/Q8IZF0 for the amino acid topology of human NALCN.
Acknowledgments
We thank the parents of the child for their participation and cooperation in this study. The authors also thank Dr. Eugenija Marusic (Department of Pediatrics, University Hospital Centre Split) for providing the polysomnography data. This work was supported by a grant from the Regional Health Authority of Western Norway (grant no 911810). We also thank Kjell Petersen and Inge Jonassen at the Computational Biology Unit, Department of Informatics, University of Bergen, Norway for providing IT infrastructure for the Norwegian next generation sequencing data through the Elixir.no project.
References
- 1. Lu B, Su Y, Das S, et al. Peptide neurotransmitters activate a cation channel complex of NALCN and UNC‐80. Nature 2009;457:741–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu B, Zhang Q, Wang H, et al. Extracellular calcium controls background current and neuronal excitability via an UNC79‐UNC80‐NALCN cation channel complex. Neuron 2010;68:488–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lu B, Su Y, Das S, et al. The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell 2007;129:371–383. [DOI] [PubMed] [Google Scholar]
- 4. Lu TZ, Feng Z‐P. A sodium leak current regulates pacemaker activity of adult central pattern generator neurons in Lymnaea stagnalis . PLoS One 2011;6:e18745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Köroğlu Ç, Seven M, Tolun A. Recessive truncating NALCN mutation in infantile neuroaxonal dystrophy with facial dysmorphism. J Med Genet 2013;50:515–520. [DOI] [PubMed] [Google Scholar]
- 6. Al‐Sayed MD, Al‐Zaidan H, Albakheet A, et al. Mutations in NALCN cause an autosomal‐recessive syndrome with severe hypotonia, speech impairment, and cognitive delay. Am J Hum Genet 2013;93:721–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gal M, Magen D, Zahran Y, et al. A novel homozygous splice site mutation in NALCN identified in siblings with cachexia, strabismus, severe intellectual disability, epilepsy and abnormal respiratory rhythm. Eur J Med Genet 2016;59:204–209. [DOI] [PubMed] [Google Scholar]
- 8. Stray‐Pedersen A, Cobben J‐M, Prescott TE, et al. Biallelic mutations in UNC80 cause persistent hypotonia, encephalopathy, growth retardation, and severe intellectual disability. Am J Hum Genet 2016;98:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shamseldin HE, Faqeih E, Alasmari A, et al. Mutations in UNC80, encoding part of the UNC79‐UNC80‐NALCN channel complex, cause autosomal‐recessive severe infantile encephalopathy. Am J Hum Genet 2016;98:210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chong JX, McMillin MJ, Shively KM, et al. De novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay. Am J Hum Genet 2015;96:462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aoyagi K, Rossignol E, Hamdan FF, et al. A gain‐of‐function mutation in NALCN in a child with intellectual disability, ataxia, and arthrogryposis. Hum Mutat 2015;36:753–757. [DOI] [PubMed] [Google Scholar]
- 12. Fukai R, Saitsu H, Okamoto N, et al. De novo missense mutations in NALCN cause developmental and intellectual impairment with hypotonia. J Hum Genet 2016;61:451–455. [DOI] [PubMed] [Google Scholar]
- 13. Karakaya M, Heller R, Kunde V, et al. Novel mutations in the nonselective sodium leak channel (NALCN) lead to distal arthrogyposis with increased musle tone. Neuropediatrics 2016;47:273–277. [DOI] [PubMed] [Google Scholar]
- 14. Hahn AF, Boman H. Cold‐induced sweating syndrome including Crisponi syndrome. Updated March 2016 In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews [Internet]. Seattle, WA: University of Washington, Seattle, 1993. ‐2016. Available at: https://www.ncbi.nlm.nih.gov/books/NBK52917/ (accessed April 15, 2016). [Google Scholar]
- 15. Haugarvoll K, Johansson S, Tzoulis C, et al. MRI characterization of adult onset alpha‐methylacyl‐coA racemase deficiency diagnosed by exome sequencing. Orphanet J Rare Dis 2013;8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ren D. Sodium leak channels in neuronal excitability and rhythmic behaviors. Neuron 2011;72:899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jeng C‐J, Sun M‐C, Tang C‐Y. Dominant‐negative effects of episodic ataxia type 2 mutations involve disruption of membrane trafficking of human P/Q‐type Ca2+ channels. J Cell Physiol 2008;214:422–433. [DOI] [PubMed] [Google Scholar]
- 18. Peña F, Ramirez J‐M. Substance P‐mediated modulation of pacemaker properties in the mammalian respiratory network. J Neurosci 2004;24:7549–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ptak K, Yamanishi T, Aungst J, et al. Raphé neurons stimulate respiratory circuit activity by multiple mechanisms via endogenously released serotonin and substance P. J Neurosci 2009;29:3720–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Humphrey JA, Hamming KS, Thacker CM, et al. A putative cation channel and its novel regulator: cross‐species conservation of effects on general anesthesia. Curr Biol 2007;17:624–629. [DOI] [PubMed] [Google Scholar]
- 21. Jackson AC, Yao GL, Bean BP. Mechanism of spontaneous firing in dorsomedial suprachiasmatic nucleus neurons. J Neurosci 2004;24:7985–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Flourakis M, Kula‐Eversole E, Hutchinson AL, et al. A conserved bicycle model for circadian clock control of membrane excitability. Cell 2015;162:836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iber C, Ancoli‐Israel S, Chesson A, Quan SF. The AASM manual for the scoring of sleep and associated events: rules, terminology and technical specifications. Westchester, IL: American Academy of Sleep Medicine, 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Schematic diagram of the NALCN topology comprised of four domains (I–IV); each made up of six transmembrane segments (S1–S6). Segments S5 and S6 are predicted to form the central pore. Illustrated are the presently identified NALCN variants: 20 heterozygous de novo missense mutations associated with the CLIFAHDD phenotype are illustrated as black filled circles, the mutation of this report as red filled circle. Note that these mutations are positioned in or near the S5 and S6 segments of all domains, and are particularly clustered in the S6 segment of domain 1, the predicted position of the here reported variant. Reported homozygous NALCN variants associated with the autosomal recessive IHPRF phenotype (MIM: 615419) are illustrated as black squares. We have referred to http://www.uniprot.org/uniprot/Q8IZF0 for the amino acid topology of human NALCN.