

Abstract
Aims
Low blood concentrations of the naturally occurring amino acid L‐homoarginine (L‐hArg) are related to impaired cardiovascular outcome and mortality in humans and animals. L‐hArg is a weak substrate of nitric oxide synthase and an inhibitor of arginases in vitro. The aim of our study was to obtain kinetic and dynamic data after oral L‐hArg supplementation.
Methods
In a double‐blind, randomized, placebo‐controlled crossover study, 20 young volunteers received 125 mg L‐hArg once daily for 4 weeks. Kinetic parameters (C max, T max and AUC0‐24h) were calculated after ingestion of single and multiple doses of oral supplementation as primary endpoint. Secondary endpoints that were evaluated were routine laboratory, L‐arginine, asymmetric dimethylarginine (ADMA), pulse wave velocity (PWV), augmentation index (AIx), flow‐mediated vasodilatation (FMD), corticospinal excitability, i.e. motor threshold (MT), and cortical excitability, i.e. intracortical inhibition (ICI) and facilitation (ICF).
Results
One hour after ingestion (T max), L‐hArg increased the baseline L‐hArg plasma concentration (2.87 ± 0.91 μmol l−1, mean ± SD) by 8.74 ± 4.46 [95% confidence intervals 6.65; 10.9] and 17.3 ± 4.97 [14.9; 19.6] μmol l−1 (C max), after single and multiple doses, respectively. Once‐only and 4 weeks of supplementation resulted in AUCs0‐24h of 63.5 ± 28.8 [50.0; 76.9] and 225 ± 78.5 [188; 2624] μmol l−1*h, for single and multiple doses, respectively. Routine laboratory parameters, L‐arginine, ADMA, PWV, AIx, FMD, MT, ICI and ICF did not change by L‐hArg supplementation compared to baseline.
Conclusion
Once daily orally applied 125 mg L‐hArg raises plasma L‐hArg four‐ and sevenfold after single dose and 4 weeks of supplementation, respectively, and is safe and well tolerated in young volunteers.
Keywords: asymmetric dimethylarginine, L‐arginine, L‐homoarginine, nitric oxide, vascular function
What is Already Known about this Subject
L‐Homoarginine (L‐hArg) is an amino acid found in pea pulses (Lathyrus sativus and cicera). It is a weak substrate for nitric oxide (NO) synthases (NOS) and an inhibitor of arginases.
In clinical and epidemiological studies, low circulating L‐hArg is associated with impaired cerebrovascular and cardiovascular outcome.
Orally administered L‐hArg is readily absorbed in rats and pigs with a recovery of >95% of unmetabolized L‐hArg in urine.
What this Study Adds
Our data show that oral supplementation with 125 mg L‐hArg raises plasma L‐hArg concentrations four‐ and sevenfold after single and multiple dosing in humans, respectively.
Four weeks of supplementation did not change vascular and neuronal function in young volunteers, nor did any toxic side‐effects occur.
Tables of Links
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 3, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 1, 2.
Introduction
The L‐arginine/nitric oxide (NO) pathway plays an important role in vascular haemostasis and L‐arginine has been supplemented to optimize health and welfare 4. However, after oral administration, L‐arginine is subject to extensive pre‐systemic and systemic elimination by arginases and oral doses required to increase plasma concentrations range from 3 to 8 g/day 5, 6. The non‐proteinogenic amino acid L‐homoarginine (L‐hArg) interferes with the L‐arginine/NO pathway and has been identified as a risk marker for cardiovascular, cerebrovascular and kidney diseases as well as for cardiovascular and all‐cause mortality (reviewed in 7, 8). L‐hArg is a weak substrate of NO synthase (NOS) and arginase. The maximal activity (V max) for murine NOS‐dependent NO formation is similar for L‐hArg and L‐arginine, but the Michaelis–Menten constant (K m) is 10–20 times higher for L‐hArg 9. The K m value for L‐hArg of rat liver arginase is 7.2 mmol l−1 which is about 70‐fold above its actual concentration in rat liver 10, 11. The V max of rat liver arginase is 130 times faster for L‐arginine compared with L‐hArg 10. These properties make L‐hArg a competitive inhibitor of rat liver arginase and render unlikely a strong catabolism in mammals. L‐hArg is a substrate for the y+‐transporter system which is responsible for stereoselective uptake and secretion of cationic amino acids in many cells 12. Furthermore, in vitro data identified high L‐hArg as a non‐competitive inhibitor of alkaline phosphatases (ALP, 13, 14, 15). Of note, L‐hArg originating from guanidated lysine residues in casein and soya‐bean proteins is readily absorbed in the jejunum and ileum of Göttingen minipigs and broiler chickens 16, 17. In line with these in vitro and in vivo data, oral supplementation of 1 and 10 mg kg−1 body weight L‐hArg were almost quantitatively (>95%) excreted unmetabolized in urine in pigs and rats, respectively 18. The aim of this study was to investigate the kinetic and dynamic properties of single and multiple oral doses of 125 mg L‐hArg in young humans and the effect of multiple doses on the endothelial and vascular function and cortical excitability.
Methods
Subjects
Twenty‐four healthy volunteers (15 female, 9 male) without evidence of disease were found eligible for this study (Figure 1). They were recruited from the participating departments and from students at the University Medical Centre Hamburg‐Eppendorf. Exclusion criteria were sitting blood pressure ≥ 160/100 or ≤90/60 mmHg, sitting heart rate ≥ 99 bpm or ≤50 beats per minute (bpm), a history of clinically significant hypotensive episodes or symptoms of fainting, dizziness, or light‐headedness, a body mass index (BMI) ≥32 or ≤16 at screening, a history or symptoms of cardiovascular disease, particularly coronary artery disease, arrhythmias, or congestive heart failure, a history of significant central nervous system disease, including transient ischemic attack, stroke, seizure disorder, or behavioural disturbances, the use of any drugs, a history of hepatitis B or C, and/or human immunodeficiency virus (HIV 1 + 2), participation in an investigational drug or medical device study within 30 days of first dosing, donation of blood or blood products within the last 2 months (male) or 3 months (female) prior to study, and pregnancy (female). Written informed consent was obtained from all participants. The study was planned as a non‐drug study and the study protocol was approved by the Ethics Committee of the Hamburg Board of Physicians (PV4038) accordingly. The investigation was conducted in accordance with the Declaration of Helsinki and registered at clinicaltrials.gov (NCT02675660).
Figure 1.
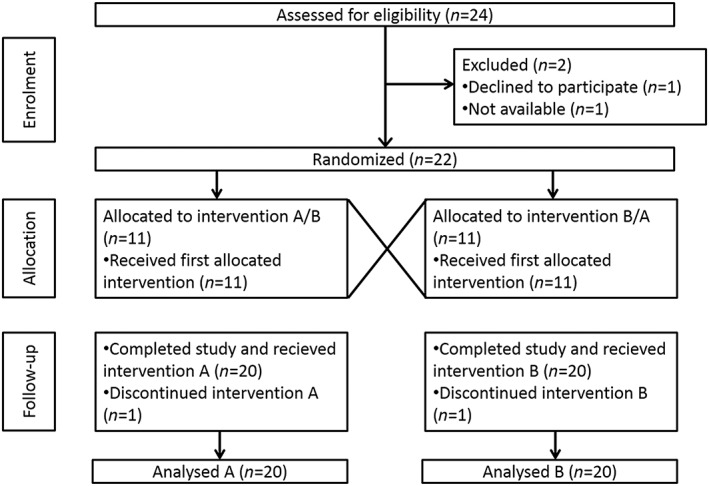
CONSORT diagram
Study design
In a double‐blind, placebo‐controlled crossover design, 22 participants were randomized to receive either 125 mg L‐hArg or placebo once daily in the morning for 4 weeks each (Figure S1). Placebo and L‐hArg capsules were provided by Wellnest International Ltd. (West Sussex, UK), the latter being marketed as a dietary supplement. Cellulose capsules contained lactose or 119 ± 13 mg L‐hArg (mean ± SD, n = 7) and cornstarch as excipient. The study periods were separated by a washout phase of 4 weeks, and the sequence of the medications was randomly chosen in each participant. The study was preceded by a run‐in phase, where all participants received a single dose of 125 mg L‐hArg. Blood samples (2.7 ml EDTA vacutainer) for plasma L‐hArg determinations were drawn at time points 0, 15, 30 min, 1, 2, 4, 8, 24, 48, 72 and 120 h after single and multiple doses of L‐hArg and placebo, respectively. At baseline, after each supplementation period (L‐hArg and placebo) and after 4 weeks of follow‐up, biochemical analyses including L‐arginine and asymmetric dimethylarginine (ADMA) determinations were performed and adverse events were evaluated. At baseline and after each supplementation period (L‐hArg and placebo), dynamic analyses applying plethysmography [i.e. pulse wave velocity (PWV) and augmentation index (AIx)], ultrasound [i.e. flow‐mediated vasodilatation (FMD)], and transcranial magnetic stimulation [TMS, i.e. motor threshold (MT), intracortical inhibition (ICI), intracortical facilitation (ICF)] were recorded. Two individuals abandoned study participation without statement of reasons.
Biochemical analyses
Plasma L‐hArg, L‐arginine and ADMA concentrations were determined in 20 participants by liquid chromatography (LC)‐tandem mass spectrometry (MS) analysis as described previously 19, 20. Briefly, 25 μL aliquots of plasma were spiked with stable isotope‐labelled L‐hArg, L‐arginine and ADMA, which served as internal standards. Proteins were precipitated with 100 μL of methanol, filtrated through a 0.22 μm hydrophilic membrane (Multiscreen HTS™, Millipore, Molsheim, France), derivatized with butanolic 1 N HCl, and analysed by LC‐tandem MS (Varian 1200 MS, Agilent Technologies, Santa Clara, USA). Quantification was performed by calculation of peak area ratios and calibration with known concentrations of analytes in dialysed EDTA plasma. Limits of quantification were 0.01 μmol l−1 for L‐hArg, 0.25 μmol l−1 for L‐arginine and 0.005 μmol l−1 for ADMA. For all arginine metabolites, coefficients of variation were ≤7.5% 19, 20. Blood counts, blood glucose, serum creatinine, glutamic oxaloacetic transaminase (GOT), glutamic pyruvic transaminase (GPT), ALP, and high sensitive C‐reactive protein (hsCRP) were determined with routine laboratory assays. Estimated glomerular filtration rate (eGFR) was calculated with the CKD‐EPI formula 21.
Kinetic analyses
Kinetic parameters, i.e. maximum plasma concentration (C max), time to maximum plasma concentration (T max) and area under the plasma concentration–time curve (AUC0‐24h), were calculated for L‐hArg after single dose and multiple doses. AUCs were calculated for up to 24 h. To account for possible circadian rhythms of endogenous L‐hArg, plasma concentrations following L‐hArg administration at each time point were corrected for individual baseline (time point zero of single and multiple measurement) and placebo data prior to calculation of C max, T max, and AUC0‐24h values. Even for corrected data, the calculation of half‐life was still not possible. All kinetic calculations were performed using Excel (version 2010, Microsoft Corporation, Redmont, USA).
Dynamic analyses
Systolic (SBP) and diastolic blood pressure (DBP) was measured at baseline in three independent examinations at supine position after 5 min of resting, and results were averaged. PWV and AIx were obtained in supine position by plethysmography, applying the vascular explorer system (Enverdis, Jena, Germany). Augmented pressure was calculated as the difference between the second systolic peak and the first systolic peak, and AIx was calculated as the ratio between augmented pressure and pulse pressure. Values were normalized to a heart rate of 75 bpm. Central PWV was assessed recording waveforms at the femoral and carotid site, sequentially. FMD was assessed in the volunteers' right arm by high resolution ultrasound (12 MHz linear array transducer, Sienna, Siemens, Munich, Germany) as described previously 22. In brief, longitudinal echo scans of the brachial artery were obtained before and after reactive hyperaemia. FMD was calculated as the percent in artery diameter 1 min after cuff release relative to the diameter before cuff release. Corticospinal excitability, i.e. MT, and cortical excitability, i.e. ICI and ICF, were evaluated during rest with well‐established single and paired‐pulse TMS protocols using a 7 cm diameter figure of 8 shaped coil and two Magstim 200 stimulators (Magstim Co., Whitland, Carmarthenshire, UK) and Signal software 4.05 and a CED1902‐amplifier (both Cambridge Electronic Design, Cambridge, UK) for TMS data recording and processing 23, 24. Two subjects were excluded from ICI and ICF measurement; due to high MT and low recruitment, no stable test stimulus motor evoked potential >0.2 mA could be achieved (and with low amplitude test stimuli no reliable ICI nor ICF can be elicited 25).
Statistical analyses
All data are given as mean ± standard deviation (SD) [95% confidence intervals, if appropriate] or median [25th; 75th percentile]. Statistical comparisons were made by Student's t‐test (two‐tailed) for unpaired or paired data of two groups and repeated measures anova with Newman–Keuls post hoc test for paired data of four groups. Statistical analysis was performed with GraphPad Prism (version 5 for Windows, La Jolla, USA).
Results
Baseline characteristics of investigated subjects are listed in Table 1. All participants were healthy Asian‐Caucasian with no history or symptoms of cardiovascular disease, particularly coronary artery disease, arrhythmias, congestive heart failure, transient ischemic attack, stroke, seizure disorder or behavioural disturbances. Baseline L‐hArg concentration was 2.87 ± 0.91 μmol l−1, mean ± SD, with no difference between women and men, i.e. 2.66 and 3.13 μmol l−1, respectively (P = 0.26, Student's t‐test for unpaired data). Oral supplementation with 125 mg L‐hArg increased the plasma concentrations of L‐hArg (C max) after single and multiple doses by 8.74 ± 4.46 [95% confidence intervals 6.65; 10.9] and 17.3 ± 4.97 [14.9; 19.6] μmol l−1, respectively (Table 2). The AUC0‐24h was 3.5‐fold higher after multiple dosing compared with a single dose of L‐hArg. C max was reached after 1 h irrespectively of the dosing regimen.
Table 1.
Baseline characteristics of participantsa
| Mean | Standard deviation | |
|---|---|---|
| Age (years) | 35 | 14 |
| Gender (n, %) | 11 females (55%) | |
| Smoker (n, %) | 5 (25%) | |
| BMI (kg m −2 ) | 24 | 2.9 |
| Blood pressure (mmHg) b | ||
| Systolic | 119 | 9.3 |
| Diastolic | 75 | 6.8 |
| Blood counts/clinical chemistry | ||
| Leukocytes (c/nL) | 6.0 | 1.3 |
| Thrombocytes (c/nL) | 255 | 62 |
| GOT (U/L) | 25.8 | 9.8 |
| GPT (U/L) | 21.3 | 8.9 |
| ALP (U/L) | 46 | 18 |
| Blood glucose (mg/dL) | 76 | 18 |
| hsCRP (mg/dL) c | 0.9 | [0.9; 2.2] |
| eGFR (ml min −1 ) | 100 | 15 |
| L‐hArg (μmol l −1 ) | 2.87 | 0.91 |
| L‐Arginine (μmol l −1 ) | 80 | 20 |
| ADMA (μmol l −1 ) | 0.60 | 0.08 |
Data are given as mean ± standard deviation unless otherwise indicated.
Average of three independent measurements.
Median [25th; 75th percentile].
ADMA, asymmetric dimethylarginine; ALP, alkaline phosphatase; BMI, body mass index; eGFR, estimated glomerular filtration rate computed using the CKD‐EPI formula, GOT, glutamic oxaloacetic transaminase; GPT, glutamic pyruvic transaminase; L‐hArg, L‐homoarginine, hsCRP, high‐sensitivity C‐reactive protein.
Table 2.
Kinetic characteristics of L‐homoarginine in human plasma after a single dose and four weeks of 125 mg oral supplementationa
| Single dose | Multiple dose | P value b | |
|---|---|---|---|
| C max [μmol l −1 ] | 8.74 ± 4.46 [6.65; 10.9] | 17.3 ± 4.97 [14.9; 19.6] | <0.001 |
| T max [h] | 1.15 ± 0.54 [0.90; 1.40] | 1.28 ± 0.50 [1.05; 1.51] | 0.22 |
| AUC 0‐24h [μmol l −1 *h] | 63.5 ± 28.8 [50.0; 76.9] | 225 ± 78.5 [188; 2624] | <0.001 |
Kinetic parameters are calculated for baseline‐placebo corrected data. Data are given as mean ± standard deviation [95% confidence intervals].
P‐value: single vs. multiple dose, Student's t‐test (paired, two‐tailed). C max indicates maximum plasma concentration; T max, time to maximum plasma concentration; AUC0‐24h, area under the plasma concentration–time curve (24 h).
Baseline characteristics given in Table 1 were not altered by supplementation, except for blood glucose, which was increased after L‐hArg supplementation and ALP activity, which was increased after placebo and at follow‐up (P < 0.05 for all; Table S1). L‐hArg supplementation for 4 weeks did not change L‐arginine or ADMA plasma concentrations. Adverse events were equally distributed in both study arms (Table S2). Supplementation with L‐hArg had no impact on PWV and AIx as compared with baseline or placebo (Figure 2). FMD, ICI and ICF were also unchanged after placebo and L‐hArg supplementation (Figure 3). MT was slightly increased after placebo supplementation compared with baseline (P < 0.05), but no significant difference was observed between L‐hArg and placebo.
Figure 2.
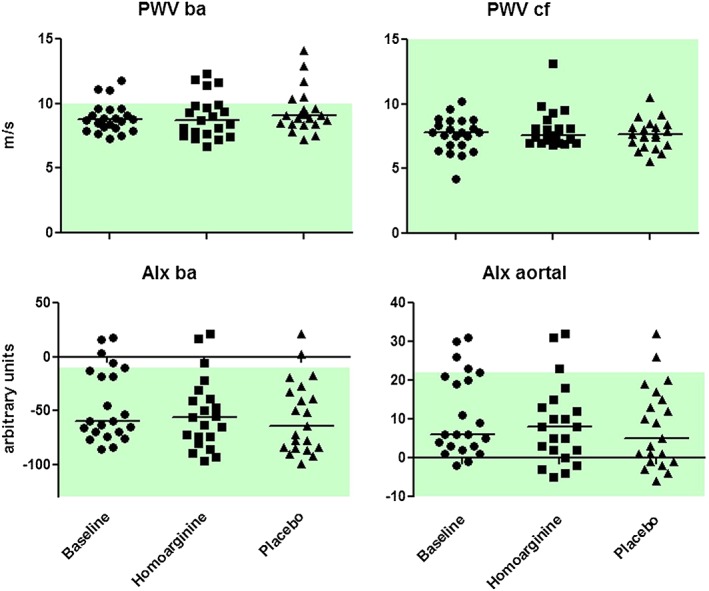
Pulse wave velocity (PWV) and augmentation index (AIx) data at baseline and after 4 week of supplementation with L‐homoarginine and placebo (n = 20). PWV was calculated for the A. brachialis (ba) and the carotid‐femoralis (cf). AIx was calculated for the aorta and the A. brachialis (ba). Normal range values according to the vascular explorer system (Enverdis, Jena, Germany) are depicted in green
Figure 3.
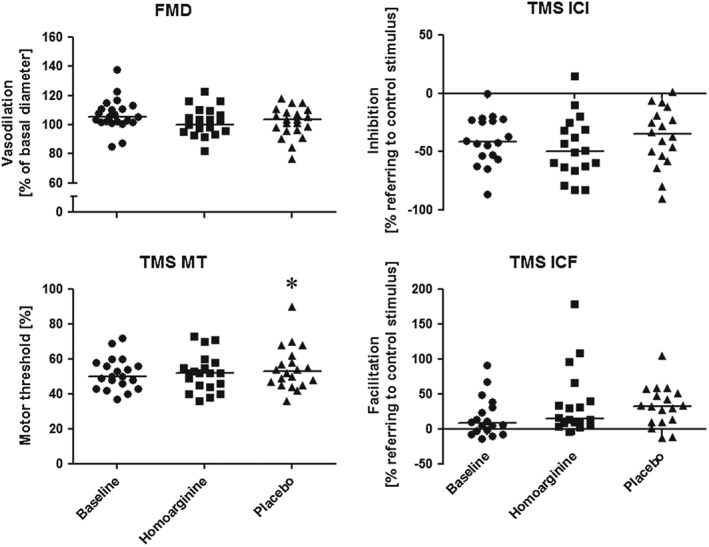
Transcranial magnetic stimulation (TMS) and flow‐mediated vasodilatation (FMD) were performed at baseline and after 4 weeks of supplementation with L‐homoarginine and placebo (n = 20 for FMD, n = 18–20 for TMS). Corticospinal excitability, i.e. motor threshold (MT), cortical excitability, i.e. intracortical inhibition (ICI) and facilitation (ICF), were evaluated with single and paired‐pulse TMS protocols. *P < 0.05 vs. baseline, repeated measures ANOVA with Newman–Keuls post hoc test
Discussion
The major finding of our study is that oral administration of 125 mg L‐hArg once daily increases L‐hArg plasma concentrations in healthy humans four‐ and sevenfold over baseline levels, after single and 28 daily doses, respectively. A median plasma concentration of 1.88 μmol l−1 L‐hArg has previously been determined in 786 healthy individuals (aged from 35 to 54 years) with a tendency of higher L‐hArg concentrations in younger participants 26. In our study group of young individuals (mean age 35 years), we observed a mean L‐hArg concentration of 2.87 μmol l−1 at baseline. The higher L‐hArg concentration at baseline might be attributed to the younger age of our participants. Although we did not observe a difference in L‐hArg concentrations between women and men, 125 mg L‐hArg tended to increase C max more in women compared with men, i.e. by 18.6 and 15.7 μmol l−1, respectively (P = 0.20, Student's t‐test for unpaired data). The sex difference was most likely due to the lower body weight in women, i.e. 63 vs. 82 kg.
Multiple doses of 125 mg L‐hArg increased the C max sevenfold over baseline as compared with a fourfold increase after a single dose. We did not perform serial blood collections to evaluate C max, T max, and AUC0‐24h at different time‐points after multiple dosing regimens and thus do not know if the tripling of AUC0‐24h represents the steady state kinetics. As a substrate for the y+‐transporter system, L‐hArg is likely to be taken up by several organs 12. Even though we corrected our data for baseline and placebo concentrations to account for diurnal variation in L‐hArg plasma concentrations, we were not able to calculate the terminal half‐life in our study.
In line with previous observations made for the oral supplementation with L‐arginine, L‐hArg did not follow first‐order elimination kinetics in a single compartment model 22. L‐hArg is a competitive inhibitor of rat liver arginase 10. We therefore investigated whether 4 weeks of 125 mg L‐hArg supplementation increases L‐arginine plasma concentrations or its endogenous methylation product ADMA 27. Even though we did not find increased L‐arginine nor ADMA concentrations in our investigation, we cannot rule out that alternative dosing regimens, i.e. higher doses or shorter dosing intervals, might influence L‐arginine metabolism. L‐Arginine is not only subject to systemic but also to extensive pre‐systemic elimination by arginases, and the latter might not sufficiently be inhibited by a single dose once daily of a weak arginase inhibitor 5. Experimental data showed that L‐hArg is an inhibitor of ALP 13, 14, 15 and clinical observations revealed a negative correlation between circulating L‐hArg and ALP levels in patients undergoing coronary angiography 28. However, physiological concentrations of L‐hArg, i.e. 0.2–3 μmol l−1 do not inhibit ALP, but induce osteogenic transformation of vascular smooth muscle cells, augmenting vascular calcification in experimental atherosclerosis 29. In the present investigation, the applied dosing regime of L‐hArg did not alter ALP activity or vascular phenotypes.
Lower plasma concentrations of L‐hArg have been reported in a variety of clinical conditions, among them coronary artery disease, congestive heart failure, ischemic stroke and myocardial infarction 28, 30, 31, 32, 33, 34, 35. Low L‐hArg has been linked to a worsened prognosis in patients with renal and cardiovascular disease as well as to cardiovascular and all‐cause mortality in the general population 28, 36, 37, 38. These retrospective and prognostic cohort studies provide no experimental evidence in favour of a supplementation with L‐hArg.
To date, L‐hArg has only been supplemented in several animal models 16, 17, 18, 32, 39, 40. In all studied species (i.e. chickens, pigs, rats and mice) orally supplemented L‐hArg was readily absorbed in the intestine, it increased L‐hArg plasma concentrations and was excreted almost quantitatively and unmetabolized into the urine. Oral supplementation of C57BL/6 mice for 4 weeks with 14 mg l−1 L‐hArg in drinking water (approx. 2 mg kg−1 body weight) resulted in a threefold increase in L‐hArg plasma concentration from 0.14 to 0.46 μmol l−1 32. Supplementation of 125 mg L‐hArg once daily (approx. 2 mg kg−1 body weight) for 4 weeks in humans resulted in a sevenfold increase (C max) in L‐hArg plasma concentration. These data clearly indicate that metabolism of L‐hArg is different between mice and men and needs further investigation. Although the increase in L‐hArg observed in mice was rather moderate, the applied dose significantly improved neurological outcome in an experimental model of stroke 32. So far, murine and human data indicate that genetic alterations of L‐arginine:glycine amidinotransferase (AGAT) are responsible for changes in L‐hArg levels 32, 41. Therefore, AGAT itself might represent a possible target for future interventions to regulate L‐hArg levels. Furthermore, it is still unknown whether the therapeutic potential of L‐hArg supplementation is translatable to humans. However, our data show that L‐hArg supplementation in humans is feasible.
In experimental and clinical studies L‐hArg was associated with endothelial function, e.g. FMD 42, kidney function, e.g. eGFR 38, neurotoxicity, e.g. altered neuronal excitability 43, blood pressure 44 and glucose metabolism 39. Supplementing 20 young individuals with 125 mg L‐hArg once daily did not change FMD, eGFR, ICI, ICF, SBP or DBP. We observed a moderate increase in blood glucose after L‐hArg supplementation (Table S1). This could be an adverse reaction to the supplement, but experimental findings in obese mice have shown an opposite effect of L‐hArg supplementation on blood glucose. Oral supplementation of C57BL/6 mice for 16 weeks with 14 and 28 mg l−1 L‐hArg in drinking water blunted a metabolic phenotype induced by a high‐fat diet; i.e. L‐hArg fostered insulin secretion and ameliorated blood glucose levels 39. MT seemed significantly increased after placebo compared with baseline, but one outlier contributed to this effect. Furthermore, MT of placebo and L‐hArg supplementation groups did not differ significantly from each other. Despite the clinical studies showing associations between L‐hArg and clinical phenotypes, no evidence for a direct effect is given. At least for the dosing period applied to healthy individuals in the current study, no impact, neither harm nor benefit, was observed.
Na+/K+‐ATPase is a crucial enzyme responsible for the active transport of sodium and potassium ions in the nervous system necessary to maintain the ionic gradient for neuronal excitability. In vitro studies showed an inhibitory effect of L‐hArg on Na+/K+‐ATPase in the synaptic plasma membrane from cerebral cortex of young rats at concentrations of 5–20 μmol l−1 43. However, in our study we did not observe any alterations of the cortical excitability by hArg supplementation, i.e. neither ICI nor ICF were changed. Given its polarity, transport of L‐hArg across the blood–brain barrier is likely to require an active transport system. L‐hArg is a substrate for the y+‐transporter system 12 and has been reported to act as a competitive inhibitor of L‐arginine uptake by porcine endothelial cells 45. However, in mice, L‐hArg was reported to be taken up into the brain 32. Thus, it can only be concluded from the present data that supplementation of 125 mg L‐hArg for 4 weeks does not seem to interfere with cortical excitability in healthy individuals.
AGAT is expressed predominantly in the kidney and liver, L‐hArg and GFR are positively associated in cohort studies, and L‐hArg plasma concentrations decline with progression of chronic kidney disease 38, 46. At baseline we observed relatively high L‐hArg plasma concentrations possibly attributed to the young age and normal kidney function in our study population (Table 1). We did not investigate urinary excretion of L‐hArg or AGAT expression, which might be altered upon L‐hArg supplementation. Previously it was shown that L‐arginine is extensively metabolized by arginase in the gut wall and liver 5, 47. This limits its oral bioavailability as a substrate for NOS and subsequent effect on vascular function. L‐hArg is an alternative substrate for NOS not subjected to elimination by arginase 9, 10. However, to date it is still speculative whether the beneficial effects of L‐hArg are solely due to interactions with L‐arginine/NOS metabolism. The ratio of L‐arginine over the endogenous NOS inhibitor ADMA is one predictor for the substrate availability for NOS 48. In our study, supplementation with L‐hArg did not change L‐arginine, ADMA or the L‐arginine/ADMA ratio. In line with this, we did not observe any improvement of endothelial function; neither FMD, nor PWI or AIx were changed. This does not render changes in endothelial function impossible after longer supplementation period or in subjects with pre‐existing cardiovascular diseases. Nevertheless, the primary endpoint of our study was the determination of kinetic parameters and our study was sufficiently powered for this purpose.
In conclusion, the results of the present study provide a rationale for larger, prospective clinical studies with longer treatment periods to investigate the effects of oral supplementation with 125 mg L‐hArg in patients with cardiovascular or metabolic disease.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: DA had a grant from the European Community in the previous 3 years; CG had grants and personal fees with Bayer Healthcare, Boehringer Ingelheim, GlaxoSmithKline, Lundbeck, Pfizer, Sanofi Aventis, UCB, Merck Serono, EBS technologies, Silk Road Medical, German Research Council, German Ministry of Science and Education and the European Community in the previous 3 years; CUC had a grant with the Else Kröner‐Fresenius Stiftung in the previous 3 years; there are no other relationships or activities that could appear to have influenced the submitted work.
The excellent medical and technical assistance of A. Dehn, S. Griesbach, M. Kastner, J. Lockowandt A. Steenpass and J. Wiener is appreciated. Dr Atzler acknowledges the support of the European Community under a Marie Curie Intra‐European Fellowship for Career Development and Dr Choe was funded by an Else Kröner Memorial Stipendium from the Else Kröner‐Fresenius Stiftung. This publication was funded by LMU Munich's Institutional Strategy LMUexcellent within the framework of the German Excellence Initiative (DA). The contributions to sample and data collection made by volunteers are gratefully acknowledged.
Contributors
Conception and design of the work: DA, CUC, ES. Analysis and interpretation of data: DA, MS, KC, IO, JH, CUC, ES. Drafting or revising the manuscript: DA, MS, KC, IO, UJ, CUC, AJ, ES. Final approval of the manuscript: DA, MS, KC, IO, JH, FCH, CG, UJ, AJ, RHB, CUC, ES. DA, MS, ES, and CUC contributed equally.
Supporting information
Figure S1 Study design. Time points indicated are days. Kinetic and dynamic parameters were evaluated after single dose and at the end of multiple doses (L‐homoarginine and placebo).
Table S1 Laboratory and anthropometric phenotypes at baseline, after 4 weeks of supplementation (L‐homoarginine and placebo), and after four weeks of follow‐up.
Table S2 Treatment‐emergent adverse events experienced by one or more participants during the treatment period.
Supporting info item
{kind=link}
Atzler, D. , Schönhoff, M. , Cordts, K. , Ortland, I. , Hoppe, J. , Hummel, F. C. , Gerloff, C. , Jaehde, U. , Jagodzinski, A. , Böger, R. H. , Choe, C. , and Schwedhelm, E. (2016) Oral supplementation with L‐homoarginine in young volunteers. Br J Clin Pharmacol, 82: 1477–1485. doi: 10.1111/bcp.13068.
References
- 1. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al. The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 2015; 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu G, Meininger CJ. Arginine nutrition and cardiovascular function. J Nutr 2000; 130: 2626–2629. [DOI] [PubMed] [Google Scholar]
- 5. Morris SM Jr. Enzymes of arginine metabolism. J Nutr 2004; 134 (10 Suppl): 2743S–2747S ;discussion 65S–7S. [DOI] [PubMed] [Google Scholar]
- 6. Böger RH. The pharmacodynamics of L‐arginine. Altern Ther Health Med 2014; 20: 48–54. [PubMed] [Google Scholar]
- 7. Atzler D, Schwedhelm E, Choe CU. L‐homoarginine and cardiovascular disease. Curr Opin Clin Nutr Metab Care 2015; 18: 83–88. [DOI] [PubMed] [Google Scholar]
- 8. Pilz S, Meinitzer A, Gaksch M, Grübler M, Verheyen N, Drechsler C, et al. Homoarginine in the renal and cardiovascular systems. Amino Acids 2015; 47: 1703–1713. [DOI] [PubMed] [Google Scholar]
- 9. Moali C, Boucher JL, Sari MA, Stuehr DJ, Mansuy D. Substrate specificity of NO synthases: detailed comparison of L‐arginine, homo‐L‐arginine, their N omega‐hydroxy derivatives, and N omega‐hydroxynor‐L‐arginine. Biochemistry 1998; 37: 10453–10460. [DOI] [PubMed] [Google Scholar]
- 10. Reczkowski RS, Ash DE. Rat liver arginase: kinetic mechanism, alternate substrates, and inhibitors. Arch Biochem Biophys 1994; 312: 31–37. [DOI] [PubMed] [Google Scholar]
- 11. Yang Y, Wu Z, Jia S, Dahanayaka S, Feng S, Meininger CJ, et al. Safety of long‐term dietary supplementation with L‐arginine in rats. Amino Acids 2015; 47: 1909–1920. [DOI] [PubMed] [Google Scholar]
- 12. White MF, Gazzola GC, Christensen HN. Cationic amino acid transport into cultured animal cells. I. Influx into cultured human fibroblasts. J Biol Chem 1982; 257: 4443–4449. [PubMed] [Google Scholar]
- 13. Lin CW, Fishman WH. L‐Homoarginine. An organ‐specific, uncompetitive inhibitor of human liver and bone alkaline phosphohydrolases. J Biol Chem 1972; 247: 3082–3087. [PubMed] [Google Scholar]
- 14. Rufo MB, Fishman WH. L‐homoarginine, a specific inhibitor of liver‐type alkaline phosphatase, applied to the recognition of liver‐type enzyme activity in rat intestine. J Histochem Cytochem 1972; 20: 336–343. [DOI] [PubMed] [Google Scholar]
- 15. Suzuki K, Yoshimura Y, Hisada Y, Matsumoto A. Sensitivity of intestinal alkaline phosphatase to L‐homoarginine and its regulation by subunit–subunit interaction. Jpn J Pharmacol 1994; 64: 97–102. [DOI] [PubMed] [Google Scholar]
- 16. Schmitz M, Hagemeister H, Erbersdobler HF. Homoarginine labeling is suitable for determination of protein absorption in miniature pigs. J Nutr 1991; 121: 1575–1580. [DOI] [PubMed] [Google Scholar]
- 17. Siriwan P, Bryden WL, Annison EF. Use of guanidinated dietary protein to measure losses of endogenous amino acids in poultry. Br J Nutr 1994; 71: 515–529. [DOI] [PubMed] [Google Scholar]
- 18. Hou Y, Hu S, Jia S, Nawaratna G, Che D, Wang F, et al. Whole‐body synthesis of L‐homoarginine in pigs and rats supplemented with L‐arginine. Amino Acids 2016; 48: 993–1001. [DOI] [PubMed] [Google Scholar]
- 19. Atzler D, Mieth M, Maas R, Böger RH, Schwedhelm E. Stable isotope dilution assay for liquid chromatography‐tandem mass spectrometric determination of L‐homoarginine in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 2011; 879: 2294–2298. [DOI] [PubMed] [Google Scholar]
- 20. Schwedhelm E, Maas R, Tan‐Andresen J, Schulze F, Riederer U, Böger RH. High‐throughput liquid chromatographic‐tandem mass spectrometric determination of arginine and dimethylated arginine derivatives in human and mouse plasma. J Chromatogr B Analyt Technol Biomed Life Sci 2007; 851: 211–219. [DOI] [PubMed] [Google Scholar]
- 21. Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009; 150: 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schwedhelm E, Maas R, Freese R, Jung D, Lukacs Z, Jambrecina A, et al. Pharmacokinetic and pharmacodynamic properties of oral L‐citrulline and L‐arginine: impact on nitric oxide metabolism. Br J Clin Pharmacol 2008; 65: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zimerman M, Heise KF, Hoppe J, Cohen LG, Gerloff C, Hummel FC. Modulation of training by single‐session transcranial direct current stimulation to the intact motor cortex enhances motor skill acquisition of the paretic hand. Stroke 2012; 43: 2185–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ziemann U, Reis J, Schwenkreis P, Rosanova M, Strafella A, Badawy R, et al. TMS and drugs revisited 2014. Clin Neurophysiol 2015; 126: 1847–1868. [DOI] [PubMed] [Google Scholar]
- 25. Kujirai T, Caramia MD, Rothwell JC, Day BL, Thompson PD, Ferbert A, et al. Corticocortical inhibition in human motor cortex. J Physiol 1993; 471: 501–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Atzler D, Appelbaum S, Cordts K, Ojeda FM, Wild PS, Münzel T, et al. Reference intervals of plasma homoarginine from the German Gutenberg Health Study. Clin Chem Lab Med 2016; 54: 1231–1237. [DOI] [PubMed] [Google Scholar]
- 27. Tsikas D, Wu G. Homoarginine, arginine, and relatives: analysis, metabolism, transport, physiology, and pathology. Amino Acids 2015; 47: 1697–1702. [DOI] [PubMed] [Google Scholar]
- 28. März W, Meinitzer A, Drechsler C, Pilz S, Krane V, Kleber ME, et al. Homoarginine, cardiovascular risk, and mortality. Circulation 2010; 122: 967–975. [DOI] [PubMed] [Google Scholar]
- 29. Alesutan I, Feger M, Tuffaha R, Castor T, Musculus K, Buehling SS, et al. Augmentation of phosphate‐induced osteo‐/chondrogenic transformation of vascular smooth muscle cells by homoarginine. Cardiovasc Res 2016; 110: 408–418. [DOI] [PubMed] [Google Scholar]
- 30. Atzler D, Baum C, Ojeda F, Keller T, Cordts K, Schnabel RB, et al. Low homoarginine levels in the prognosis of patients with acute chest pain. J Am Heart Assoc 2016; 5: e002565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Atzler D, Rosenberg M, Anderssohn M, Choe CU, Lutz M, Zugck C, et al. Homoarginine – an independent marker of mortality in heart failure. Int J Cardiol 2013; 168: 4907–4909. [DOI] [PubMed] [Google Scholar]
- 32. Choe CU, Atzler D, Wild PS, Carter AM, Böger RH, Ojeda F, et al. Homoarginine levels are regulated by L‐arginine:glycine amidinotransferase and affect stroke outcome: results from human and murine studies. Circulation 2013; 128: 1451–1461. [DOI] [PubMed] [Google Scholar]
- 33. Drechsler C, Meinitzer A, Pilz S, Krane V, Tomaschitz A, Ritz E, et al. Homoarginine, heart failure, and sudden cardiac death in haemodialysis patients. Eur J Heart Fail 2011; 13: 852–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pilz S, Meinitzer A, Tomaschitz A, Drechsler C, Ritz E, Krane V, et al. Low homoarginine concentration is a novel risk factor for heart disease. Heart 2011; 97: 1222–1227. [DOI] [PubMed] [Google Scholar]
- 35. Pilz S, Tomaschitz A, Meinitzer A, Drechsler C, Ritz E, Krane V, et al. Low serum homoarginine is a novel risk factor for fatal strokes in patients undergoing coronary angiography. Stroke 2011; 42: 1132–1134. [DOI] [PubMed] [Google Scholar]
- 36. Atzler D, Gore MO, Ayers CR, Choe CU, Böger RH, de Lemos JA, et al. Homoarginine and cardiovascular outcome in the population‐based Dallas Heart Study. Arterioscler Thromb Vasc Biol 2014; 34: 2501–2507. [DOI] [PubMed] [Google Scholar]
- 37. Pilz S, Teerlink T, Scheffer PG, Meinitzer A, Rutters F, Tomaschitz A, et al. Homoarginine and mortality in an older population: the Hoorn study. Eur J Clin Invest 2014; 44: 200–208. [DOI] [PubMed] [Google Scholar]
- 38. Tomaschitz A, Meinitzer A, Pilz S, Rus‐Machan J, Genser B, Drechsler C, et al. Homoarginine, kidney function and cardiovascular mortality risk. Nephrol Dial Transplant 2014; 29: 663–671. [DOI] [PubMed] [Google Scholar]
- 39. Stockebrand M, Hornig S, Neu A, Atzler D, Cordts K, Böger RH, et al. Homoarginine supplementation improves blood glucose in diet‐induced obese mice. Amino Acids 2015; 47: 1921–1927. [DOI] [PubMed] [Google Scholar]
- 40. Pentyala J, Rao SLN. Sustained nitric oxide generation with L‐homoarginine. Res Commun Biochem Cell Mol Biol 1999; 3: 223–232. [Google Scholar]
- 41. Davids M, Ndika JD, Salomons GS, Blom HJ, Teerlink T. Promiscuous activity of arginine:glycine amidinotransferase is responsible for the synthesis of the novel cardiovascular risk factor homoarginine. FEBS Lett 2012; 586: 3653–3657. [DOI] [PubMed] [Google Scholar]
- 42. Valtonen P, Laitinen T, Lyyra‐Laitinen T, Raitakari OT, Juonala M, Viikari JS, et al. Serum L‐homoarginine concentration is elevated during normal pregnancy and is related to flow‐mediated vasodilatation. Circ J 2008; 72: 1879–1884. [DOI] [PubMed] [Google Scholar]
- 43. da Silva CG, Parolo E, Streck EL, Wajner M, Wannmacher CM, Wyse AT. In vitro inhibition of Na+,K(+)‐ATPase activity from rat cerebral cortex by guanidino compounds accumulating in hyperargininemia. Brain Res 1999; 838: 78–84. [DOI] [PubMed] [Google Scholar]
- 44. van der Zwan LP, Davids M, Scheffer PG, Dekker JM, Stehouwer CD, Teerlink T. L‐Homoarginine and L‐arginine are antagonistically related to blood pressure in an elderly population: the Hoorn study. J Hypertens 2013; 31: 1114–1123. [DOI] [PubMed] [Google Scholar]
- 45. Bogle RG, Moncada S, Pearson JD, Mann GE. Identification of inhibitors of nitric oxide synthase that do not interact with the endothelial cell L‐arginine transporter. Br J Pharmacol 1992; 105: 768–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Drechsler C, Kollerits B, Meinitzer A, März W, Ritz E, König P, et al. Homoarginine and progression of chronic kidney disease: results from the Mild to Moderate Kidney Disease Study. PLoS One 2013; 8: e63560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Castillo L, deRojas TC, Chapman TE, Vogt J, Burke JF, Tannenbaum SR, et al. Splanchnic metabolism of dietary arginine in relation to nitric oxide synthesis in normal adult man. Proc Natl Acad Sci U S A 1993; 90: 193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Böger RH, Vallance P, Cooke JP. Asymmetric dimethylarginine (ADMA): a key regulator of nitric oxide synthase. Atheroscler Suppl 2003; 4: 1–3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Study design. Time points indicated are days. Kinetic and dynamic parameters were evaluated after single dose and at the end of multiple doses (L‐homoarginine and placebo).
Table S1 Laboratory and anthropometric phenotypes at baseline, after 4 weeks of supplementation (L‐homoarginine and placebo), and after four weeks of follow‐up.
Table S2 Treatment‐emergent adverse events experienced by one or more participants during the treatment period.
Supporting info item