

Abstract
Aims
Edoxaban, a novel factor Xa inhibitor, is a substrate of cytochrome P450 3 A4 (CYP3A4) and the efflux transporter P‐glycoprotein (P‐gp). Three edoxaban drug–drug interaction studies examined the effects of P‐gp inhibitors with varying degrees of CYP3A4 inhibition.
Methods
In each study, healthy subjects received a single oral dose of 60 mg edoxaban with or without an oral dual P‐gp/CYP3A4 inhibitor as follows: ketoconazole 400 mg once daily for 7 days, edoxaban on day 4; erythromycin 500 mg four times daily for 8 days, edoxaban on day 7; or single dose of cyclosporine 500 mg with edoxaban. Serial plasma samples were obtained for pharmacokinetics and pharmacodynamics. Safety was assessed throughout the study.
Results
Coadministration of ketoconazole, erythromycin, or cyclosporine increased edoxaban total exposure by 87%, 85%, and 73%, respectively, and the peak concentration by 89%, 68%, and 74%, respectively, compared with edoxaban alone. The half‐life did not change appreciably. Exposure of M4, the major active edoxaban metabolite, was consistent when edoxaban was administered alone or with ketoconazole and erythromycin. With cyclosporine, M4 total exposure increased by 6.9‐fold and peak exposure by 8.7‐fold, suggesting an additional interaction. Pharmacodynamic effects were reflective of increased edoxaban exposure. No clinically significant adverse events were observed.
Conclusions
Administration of dual inhibitors of P‐gp and CYP3A4 increased edoxaban exposure by less than two‐fold. This effect appears to be primarily due to inhibition of P‐gp. The impact of CYP3A4 inhibition appears to be less pronounced, and its contribution to total clearance appears limited in healthy subjects.
Keywords: CYP3A4, drug interactions, edoxaban, P‐glycoprotein, pharmacokinetics
What is Already Known about this Subject
Edoxaban, a direct factor Xa inhibitor, is a substrate of the efflux transporter P‐glycoprotein.
Edoxaban is a substrate of cytochrome P450 3 A4.
What this Study Adds
Mechanistic understanding into the contribution of cytochrome P450 3 A4 to total clearance of edoxaban vis‐à‐vis P‐glycoprotein.
Suggestion of the role of OATP1B1 in the distribution of the active metabolite.
Tables of Links
| TARGETS | |
|---|---|
| Enzymes 2 | OAT3 |
| Carboxylesterase 1 | P‐glycoprotein |
| CYP3A4 | OATP1B1 |
| Transporters 3 | OCT2 |
| OAT1 |
These Tables lists key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2, 3.
Introduction
Edoxaban is a factor Xa (FXa) inhibitor 4, approved in the USA for the prevention of stroke and systemic embolic events in patients with nonvalvular atrial fibrillation and a creatinine clearance ≤95 ml min–1, and for the treatment of venous thromboembolic events 5.
Edoxaban has an oral bioavailability of 62% 6. It is cleared through renal and nonrenal mechanisms, with each contributing equally to the total clearance in healthy subjects 7. Nonrenal clearance includes metabolism and biliary secretion. Metabolism occurs through carboxylesterase 1 (CES‐1), cytochrome P450 3 A4 (CYP3A4), and nonenzymatic hydrolysis. The most abundant and active metabolite, M4, is formed by CES‐1 and is present in human plasma at <10% of the total edoxaban exposure 7. Edoxaban inhibits FXa with the half‐maximal inhibitory concentration (IC50) of 3 nmol l–1. While the IC50 of M4 is 1.8 nmol l–1, it is not expected, in subjects with normal/moderate renal function, to contribute to the pharmacological effects of edoxaban, due to its low abundance and high protein binding (80%) 7. The other circulating metabolites include M1 (inactive, formed via nonenzymatic hydrolysis) and M6 and M8 (both pharmacologically active). Both M6 and M8 are formed through CYP3A4 metabolism of edoxaban and are present in plasma in lower abundance than M4 7.
P‐glycoprotein (P‐gp) is a membrane transporter that is expressed in many tissues and/or cell types including intestinal epithelia, hepatocytes, kidney proximal tubules, and the blood–brain barrier 8. In vitro experiments in human colon adenocarcinoma Caco‐2 cell monolayers expressing P‐gp revealed that edoxaban is transported via P‐gp and that this transport is strongly hindered by P‐gp inhibitors 9.
Edoxaban is not a substrate for uptake transporters such as organic anion transporting polypeptide (OATP1B1), organic anion transporters (OAT1 and OAT3), or organic cation transporter (OCT2) 3, 9. However, the metabolite M4 is a substrate for OATP1B1. Of note, cyclosporine additionally inhibits the uptake transporter, OATP1B1 10.
Based on in vitro data, it cannot be excluded that drugs that inhibit both P‐gp and CYP3A4 may affect the pharmacokinetics of edoxaban via both pathways. We have conducted clinical drug interaction studies with drugs that are dual inhibitors of P‐gp and CYP3A4 and have varying degrees of CYP3A4 inhibitory activity, with ketoconazole being a strong CYP3A4 inhibitor, erythromycin a moderate CYP3A4 inhibitor, and cyclosporine a weak CYP3A4 inhibitor 11, 12. P‐gp, being a pump, does not have a similar classification, although variability in response has been observed, probably due to heterogeneity in expression than potency of an inhibitor 13, 14. These studies were designed to delineate the relative contribution of P‐gp vs. CYP3A4 in the drug interactions with ketoconazole, erythromycin, and cyclosporine, as well as to characterise the overall magnitude of the interaction.
Methods
Subject eligibility
Subjects were eligible for the studies if they were aged between 18 and 45 years, had a body mass index (BMI) between 18 kg m–2 and 32 kg m–2, and were of nonchildbearing potential or using nonhormonal methods of contraception. Subjects were excluded if they used any prescribed or nonprescribed medication, used topical medication or herbal supplements, had taken St John's wort 30 days prior to day 1 of the study, used strong inhibitors or inducers of CYP enzymes 28 days prior to day 1 of the study, had a creatinine clearance <80 ml min–1, were known to be sensitive to any of the products administered during dosing, or were otherwise judged unsuitable to participate in the study. All subjects provided signed informed consent prior to enrolment.
Study design
Each of the three studies was an open‐label, randomised, two‐period, two‐treatment crossover study in healthy subjects. Each treatment period was separated by a washout period of at least 14 days for cyclosporine and ketoconazole and at least 7 days for erythromycin (Figure 1). All studies were approved by relevant Institutional Review Boards and the Edinburgh Independent Ethics Committee for Medical Research and were performed in accordance with the Declaration of Helsinki and International Conference on Harmonisation guidelines.
Figure 1.
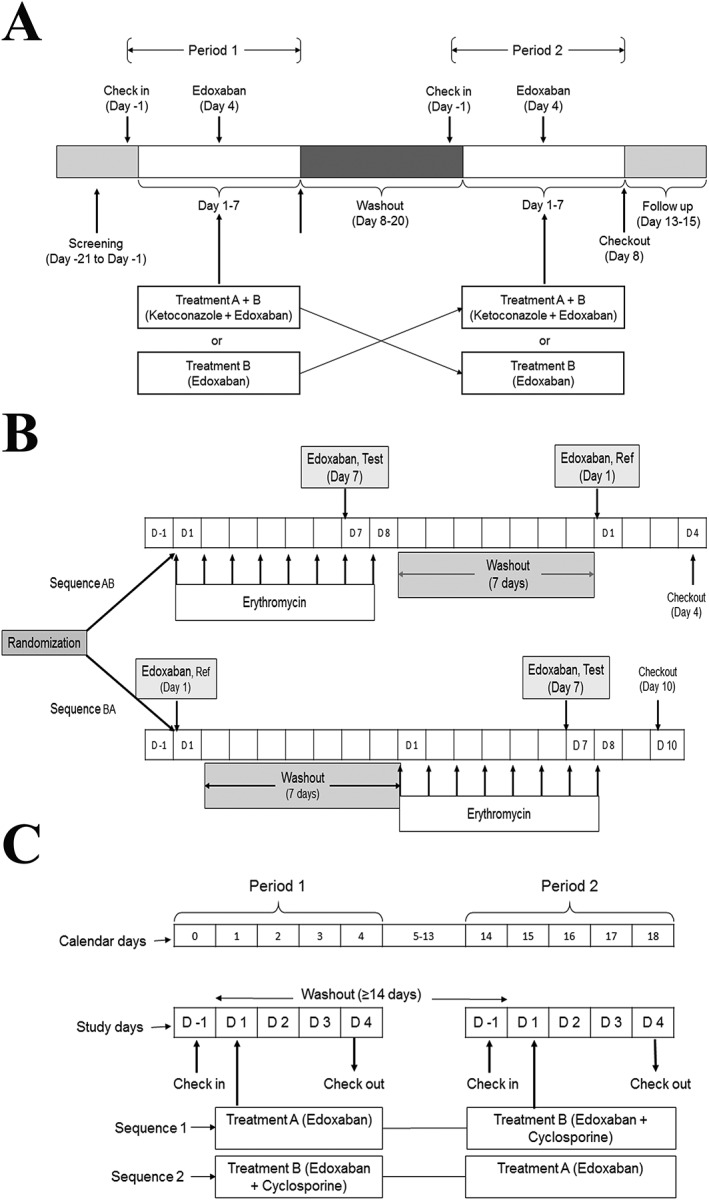
Study design. (A) Ketoconazole, (B) erythromycin, (C) cyclosporine
The primary objective of these studies was the comparison of the pharmacokinetic parameters of edoxaban alone and when coadministered with ketoconazole, erythromycin, or cyclosporine. Secondary endpoints included change from baseline in prothrombin time (PT) and activated partial thromboplastin time (aPTT) in the ketoconazole and erythromycin studies, inhibition of intrinsic factor X activity in the ketoconazole and cyclosporine studies, and assessments of safety and tolerability in all studies.
Serial blood samples for analysis of plasma concentration of edoxaban and its metabolites were collected for 96 h in the ketoconazole study and for 72 h in the erythromycin and cyclosporine studies. A few blood samples were also collected to measure inhibiting drug levels to ensure adequate and expected concentrations. Samples for biomarkers of coagulation were collected at baseline, for 24 h after edoxaban dosing in the ketoconazole and erythromycin studies, and over 72 h postdose in the cyclosporine study.
Ketoconazole
This study was conducted at MDS Pharma Services (Neptune, NJ, USA). Subjects were randomised to receive a single oral dose of edoxaban alone or an oral dose of ketoconazole 400 mg once daily for 7 days with concomitant dosing of edoxaban 60 mg and ketoconazole 400 mg on day 4 (Figure 1 A). Subjects were instructed to abstain from red meat and vitamin C in excess of 250 mg day–1 for the duration of the study to avoid a false positive reading for faecal occult blood. Morning study drug doses were administered following a 10‐h fast. Water was not permitted for a 1‐h period prior to dosing and a 1‐h period after dosing, with the exception of 240 ml administered with treatments; water was not restricted at any other time.
Sample size was determined based on the assumption of a geometric coefficient of variation for a maximum plasma concentration (Cmax) of 33.0 ng ml–1 and a minor effect (ratio <1.05) of ketoconazole on edoxaban pharmacokinetics. A sample size of 32 subjects was required to provide 85% power to conclude the absence of effect of ketoconazole for a 90% confidence interval (CI) no‐effect boundaries of 80–125%. A total of 40 subjects were enrolled to compensate for potential dropouts.
Erythromycin
The study was performed at MDS Pharma Services (Belfast, UK). Subjects were randomised to receive oral erythromycin 500 mg four times daily for 8 days along with a single oral dose of edoxaban 60 mg on day 7, and, in the next treatment sequence, subjects received a single oral dose of edoxaban 60 mg without erythromycin on day 1. Each subject participated in each treatment period once and received edoxaban on two occasions, with and without erythromycin (Figure 1B). Subjects were required to fast overnight for at least 10 h prior to edoxaban dosing and for at least 4 h afterwards. Erythromycin was administered four times daily with 240 ml of water. Doses were given with a meal or snack on days 1–8, with the exception of doses given during the 10‐h interval prior to edoxaban dosing.
A sample size of 28 subjects was required to provide at least 80% power to conclude an absence of effect of erythromycin on edoxaban pharmacokinetics using a 90% CI with no‐effect boundaries of 80–125%. A total of 36 subjects were enrolled to compensate for potential dropouts.
Cyclosporine
This study was conducted at MDS Pharma Services (Neptune, NJ, USA). Subjects were randomised to receive either a single oral dose of edoxaban 60 mg concomitant with a single oral dose of cyclosporine 500 mg or a single oral dose of edoxaban 60 mg. Each subject participated in each treatment period once and received edoxaban on two occasions, with and without cyclosporine (Figure 1C). Drugs were administered following a 10‐h fast, and no food was allowed for 4 h after drug administration. Water was not permitted during the hour before and the hour following dosing, with the exception of 240 ml administered with dosing; water was allowed at all other times.
A sample size of 28 subjects provided at least 80% power to conclude an absence of effect of cyclosporine on edoxaban pharmacokinetics using a 90% CI with no‐effect boundaries of 80–125%. A total of 34 subjects were enrolled to compensate for potential dropouts.
Bioanalytical methods
Plasma concentrations were analysed using validated assays employing liquid chromatographic separation followed by tandem mass spectrometric detection methods. Plasma concentrations of edoxaban and M4 in all three studies were analysed at Advion BioServices (Ithaca, NY, USA). The assay performance was linear within a concentration range of 0.764–382 ng ml–1 for edoxaban (intra‐assay coefficient of variation [CV] ≤11.0%, interassay CV ≤8%), and 0.0792–7.92 ng ml–1 for M4 (intra‐assay CV ≤12.3%, interassay CV ≤11.5%). Metabolites M1, M6, and M8 were assessed only in the ketoconazole study. The lower limit of quantification (LLOQ) for the detection of M1, M6, and M8 in plasma using liquid chromatography–tandem mass spectrometry was 0.100 ng ml–1, and the assay range was 0.100–5.00 ng ml–1. Ketoconazole concentration was analysed by Advion BioServices (Ithaca, NY, USA) within the concentration range of 0.10–20.0 μg ml–1. Erythromycin was analysed within the range of 10–10 000 ng ml–1 at Bioanalytical Systems, Inc. (West Lafayette, IN, USA). Whole blood cyclosporine concentrations were measured by Covance Bioanalytical Services (Indianapolis, IN, USA), with an LLOQ of 5 ng ml–1.
Clinical laboratory assessments
In the ketoconazole study, PT measurements were performed using Thromborel S and the Dade Behring technique. Prothrombin time and aPTT were assessed in the ketoconazole and erythromycin studies and analysed by Biomnis (formerly Laboratoire LCL; Ivry‐sur‐Seine, France), and MDS Pharma Services, respectively. Prothrombin time and aPTT were not assessed in the cyclosporine study. Blood samples for the determination of FXa activity were analysed using a chromogenic method by Biomnis in the ketoconazole study and by Medpace Reference Laboratories (Cincinnati, OH, USA) in the cyclosporine study. Intrinsic FXa activity was not assessed in the erythromycin study. Clinical laboratory analyses of haematology, serum chemistry, and urinalysis parameters were performed at MDS Pharma Services.
Pharmacokinetic and pharmacodynamic analyses
Plasma concentration–time data were analysed by noncompartmental methods using WinNonlin Professional Software (versions 4.0 or higher). The following parameters were estimated: maximum (peak) observed concentration in plasma (Cmax), time from dosing to the maximum (peak) observed concentration in plasma (tmax), area under the concentration‐time curve from time extrapolated to infinity (AUC), apparent terminal elimination half‐life (t1/2), and apparent clearance from plasma after oral administration (CL/F).
For statistical analysis of exposure parameters (Cmax, AUC), an analysis of variance was performed on log‐transformed values using sequence treatment and period as fixed effects, and subject nested within sequence as a random effect, using SAS® Proc Mixed. Absence of an effect of ketoconazole, erythromycin, or cyclosporine was concluded if the 90% CIs about the geometric least squares means ratios were contained within the interval of 80–125%. Nonparametric statistical analysis was performed using the Hodges–Lehmann estimator for differences in median values, and the Moses method was used to construct 90% CIs for tmax and t1/2 for edoxaban and its metabolites.
For assessment of pharmacodynamics, the following parameters were estimated using noncompartmental methods: maximum observed activity (Amax), change from baseline in Amax value (∆Amax), and percent change from baseline in Amax value (%∆Amax).
Safety analysis
Safety was assessed by adverse events monitored continuously throughout the study. Clinical laboratory tests, vital signs, 12‐lead electrocardiograms, physical examinations, and faecal occult blood tests were assessed at baseline, at scheduled times after dosing of the inhibitor, several times during the day when edoxaban was coadministered from time 0 to 24‐h postdose, and at the end of study. Data were summarised by descriptive statistics.
Results
Ketoconazole
A total of 40 subjects (35 men, 5 women) with a mean age of 30.9 years (range 20–45 years) and BMI ranging from 19.9 kg m–2 to 31.8 kg m–2 were enrolled in the study. There were 25 African Americans, 13 Caucasians, one Asian, and one American Indian/Alaskan. In total, 37 subjects completed both study periods.
Administration of edoxaban with ketoconazole increased both mean peak and total exposure of edoxaban by approximately 89% and 87%, respectively (Figure 2A; Table 1). The median time to peak concentration increased by about 30 min, while the terminal elimination half‐life was similar. The mean apparent oral clearance decreased from 38.9 l h–1 to 20.8 l h–1 (Table 1).
Figure 2.
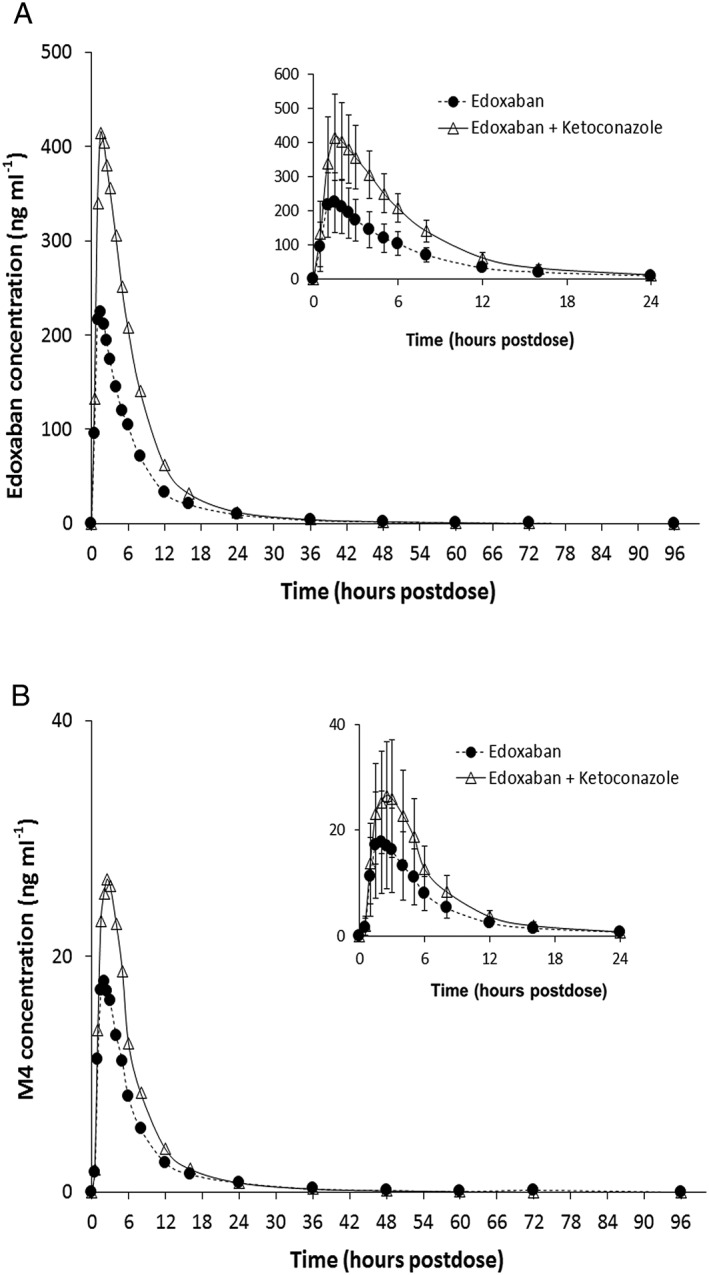
Mean plasma concentrations of (A) edoxaban and (B) metabolite M4 following administration with and without ketoconazole. Insets represent the first 24 h postdose; error bars represent the standard deviation
Table 1.
Effect of ketoconazole coadministration on edoxaban pharmacokinetics
| Edoxaban | M4 | |||
|---|---|---|---|---|
| Parameter | Edoxaban n = 39 | Edoxaban + ketoconazole n = 38 | Edoxaban n = 39 | Edoxaban + ketoconazole n = 38 |
| Cmax (ng ml–1) | 244.8 ± 86.89 | 440.6 ± 121.35 | 19.1 ± 9.82 | 28.4 ± 11.20 |
| tmax (h) a | 1.02 (0.48, 2.50) | 1.51 (0.98, 4.00) | 2.00 (0.98, 4.10) | 2.48 (1.48, 4.13) |
| AUC (ng h ml–1) | 1652 ± 412.1 | 3001 ± 617.5b | 129.1 ± 52.0 | 188.8 ± 65.7b |
| t1/2 (h) | 11.86 ± 4.11 | 12.27 ± 3.67b | 11.30 ± 3.78 | 11.10 ± 4.70b |
| CL/F (L h–1) | 38.90 ± 11.37 | 20.80 ± 4.18b | – | – |
| LSM ratio | 90% CI | LSM ratio | 90% CI | |
|---|---|---|---|---|
| Cmax | 189.11 | 168.32, 212.46 | 155.65 | 136.95, 176.91 |
| AUC | 186.77 | 176.04, 198.15 | 146.24 | 136.19, 157.03 |
| M1 | M6 | |||
|---|---|---|---|---|
| Cmax (ng ml–1) | 7.6 ± 3.39 | 13.0 ± 3.46 | 7.6 ± 3.61 | 3.5 ± 1.18 |
| tmax (h) a | 2.50 (1.48, 6.00) | 2.98 (1.50, 5.98) | 1.50 (0.48, 6.00) | 2.74 (1.48, 5.98) |
| AUC (ng h ml–1) | 65.5 ± 20.6 | 115.1 ± 26.6 | 75.0 ± 27.0 | 42.1 ± 13.6 |
| t1/2 (h) | 9.47 ± 4.61 | 9.14 ± 3.25 | 11.45 ± 4.03 | 10.42 ± 3.17 |
| CL/F (L h–1) | – | – | – | – |
| LSM ratio | 90% CI | LSM ratio | 90% CI | |
|---|---|---|---|---|
| Cmax | 184.20 | 163.45, 207.58 | 48.95 | 42.03, 57.00 |
| AUC | 179.79 | 168.53, 191.81 | 57.38 | 52.79, 62.38 |
Data presented as arithmetic mean ± standard deviation unless indicated otherwise.
AUC, area under the curve from the time of dosing extrapolated to infinity; CI, confidence interval; CL/F, apparent total body clearance; Cmax, maximum observed plasma drug concentration; LSM, least squares mean; t1/2, terminal half‐life; tmax, time of maximum observed concentration.
Median (min, max)
n = 37
Exposure to the metabolite M4 was higher when edoxaban was coadministered with ketoconazole, with approximately 56% and 46% higher peak and total exposures, respectively (Figure 2B). The mean terminal half‐life remained the same for both treatments. This increase in exposure for M4 could be the result of increased bioavailability of edoxaban as the metabolite‐to‐parent drug ratio (± standard deviation [SD]) remained similar (7.85 ± 2.21 for edoxaban dosed alone vs. 6.30 ± 1.63 for edoxaban dosed with ketoconazole). The average peak and total exposure for the metabolite M1 increased by 84% and 80%, respectively, with no change in mean terminal half‐life (Table 1). This increase is also consistent with the increased bioavailability of edoxaban in the presence of ketoconazole, as there was no change in the metabolite‐to‐parent drug ratio. On average, the peak and total exposure to the metabolite M6 decreased by 51% and 43%, respectively (Table 1). The metabolite‐to‐parent drug ratio decreased from 4.44 ± 1.18 to 1.45 ± 0.49, indicating the inhibitory effect of ketoconazole on CYP3A4, which mediates the formation of M6. Most concentrations of M8 were below the limit of quantification or close to the limit of quantification (0.100 ng ml–1); hence, pharmacokinetic parameters were not calculated for this metabolite. Nevertheless, as expected for a metabolite formed by CYP3A4, the detectable concentrations of M8 were lower when administered with ketoconazole.
The maximum percent change from baseline (SD) for PT and aPTT with ketoconazole (56.7% [15.5] and 50.3% [11.7], respectively) was greater than for edoxaban alone (31.8% [13.2] and 32.6% [8.0], respectively). The mean value of intrinsic FX inhibition was also increased for coadministration of edoxaban and ketoconazole (109.4%) compared with edoxaban alone (101.3%). These results were consistent with increased plasma concentrations following coadministration of edoxaban and ketoconazole 7.
Erythromycin
A total of 36 subjects were enrolled, and 33 completed the study. Three subjects discontinued due to adverse events related to erythromycin dosing. The enrolled subjects included 26 men and 10 women with a BMI range of 19.0–30.0 kg m–2. All subjects were Caucasian with a mean age of 25.3 years (range 18–45 years).
Coadministration with erythromycin resulted in an increase in both peak and total exposure of edoxaban, with no change in median tmax. (Figure 3A, Table 2). The peak and total exposure of edoxaban administered with erythromycin were approximately 68% and 85% higher, respectively, compared with edoxaban administered alone. Similarly, the peak and total exposure of M4 were approximately 75% and 78% higher, respectively, when administered with erythromycin (Figure 3B, Table 2). Erythromycin decreased the total apparent clearance by about 47% with no change in the formation of the M4 metabolite, as the metabolite‐to‐parent drug ratio remained similar (11.9 ± 2.72 for edoxaban dosed alone vs. 11.3 ± 3.40 for edoxaban dosed with erythromycin).
Figure 3.
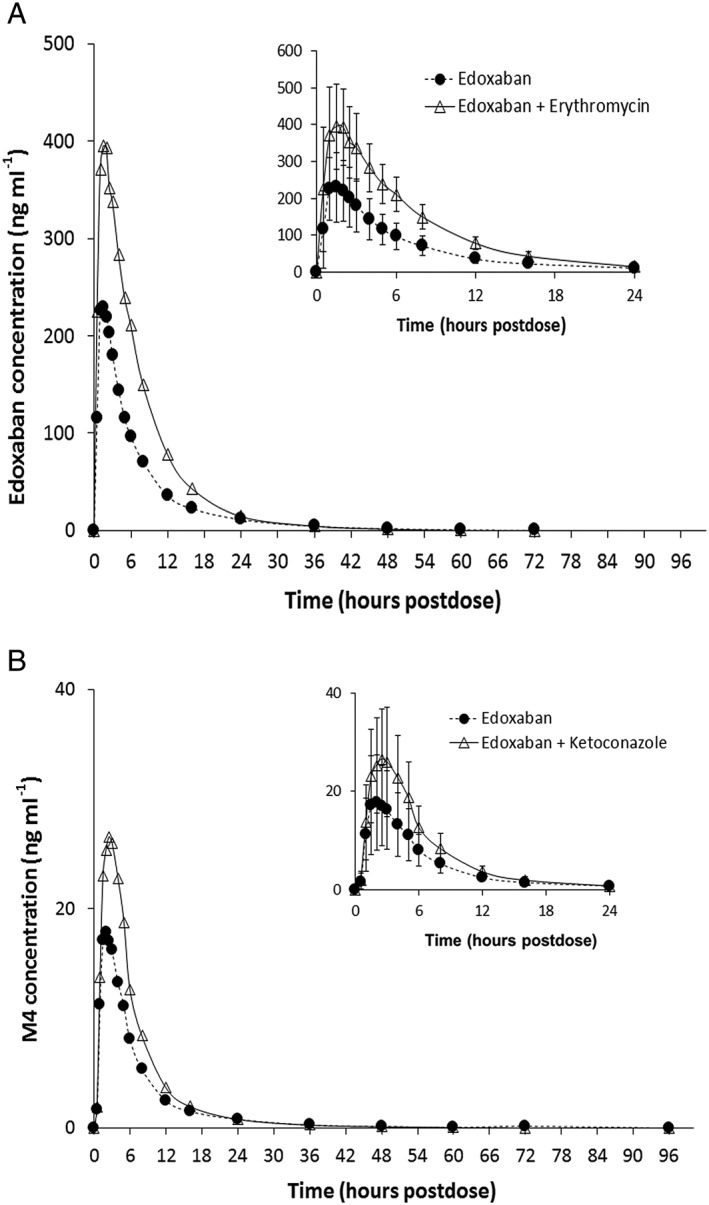
Mean plasma concentrations of (A) edoxaban (B) and metabolite M4 following administration with and without erythromycin. Insets represent the first 24 h postdose; error bars represent the standard deviation
Table 2.
Effect of erythromycin coadministration on edoxaban pharmacokinetics
| Edoxaban | M4 | |||
|---|---|---|---|---|
| Parameter | Edoxaban n = 33 | Edoxaban + erythromycin n = 33 | Edoxaban n = 33 | Edoxaban + erythromycin n = 33 |
| Cmax (ng ml–1) | 258.7 ± 100.46 | 421.9 ± 124.23 | 28.9 ± 12.63 | 49.0 ± 19.48 |
| tmax (h) a | 1.02 (0.50, 2.50) | 1.50 (0.48, 4.08) | 2.03 (1.48, 2.98) | 2.05 (1.48, 5.03) |
| AUC (ng h ml –1 ) | 1740 ± 443.7b | 3193 ± 617.1 | 207.8 ± 71.9b | 356.2 ± 112.6 |
| t 1/2 (h) | 10.24 ± 3.71b | 7.19 ± 1.56 | 12.56 ± 6.31b | 8.17 ± 2.22 |
| CL/F (L h –1 ) | 36.70 ± 9.40b | 19.45 ± 3.60 | – | – |
| LSM ratio | 90% CI | LSM ratio | 90% CI | |
|---|---|---|---|---|
| C max | 168.48 | 149.87, 189.40 | 175.13 | 155.84, 196.82 |
| AUC | 185.47 | 175.18, 196.37 | 177.69 | 165.10, 191.23 |
Data presented as arithmetic mean ± standard deviation unless indicated otherwise.
AUC, area under the curve from the time of dosing extrapolated to infinity; CI, confidence interval; CL/F, apparent total body clearance; Cmax, maximum observed plasma drug concentration; LSM, least squares mean; t1/2, terminal half‐life; tmax, time of maximum observed concentration.
Median (min, max)
n = 32
The maximum mean (± SD) observed activity for PT was greater when edoxaban was coadministered with erythromycin (16.1 ± 2.7 s) compared with edoxaban administration alone (13.8 ± 1.3 s). The maximum percent change from baseline (SD) in PT was 44.9% (21.6) for edoxaban coadministered with erythromycin compared with 24.8% (9.2) for edoxaban alone. Similarly, the maximum mean (± SD) observed activity for aPTT was greater when edoxaban was coadministered with erythromycin (41.5 ± 6.6 s vs. 35.9 ± 4.3 s), and the corresponding percent mean maximum change from baseline (SD) was 59.8% (22.8), compared with 37.5% (12.9) for edoxaban alone. The prolongation of PT and aPTT was consistent with the increased mean peak plasma concentrations of edoxaban, coadministered with erythromycin.
Cyclosporine
A total of 34 subjects were enrolled and 31 subjects completed the study. Of the subjects enrolled, 23 were African American, 10 Caucasian, and one of other race. The majority were men (23 men, 11 women). The mean age was 32.9 years, ranging from 18 years to 44 years, and BMI range was 21.2–32.0 kg m–2.
Single‐dose edoxaban peak and total exposure increased when edoxaban was coadministered with cyclosporine (Table 3; Figure 4A). Based on geometric least squares mean ratios, the increase was 73% and 74% (approximately 1.7‐fold) for total and peak exposure, respectively. The increase in the peak and total exposure of the human specific metabolite M4 was more pronounced, with 8.7‐ and 6.9‐fold increase in peak and total exposure, respectively, when edoxaban was coadministered with cyclosporine (Figure 4B, Table 3). The metabolite‐to‐parent drug ratio increased substantially from 9.5 ± 2.90 for edoxaban dosed alone vs. 35.9 ± 8.35 for edoxaban dosed with cyclosporine.
Table 3.
Effect of cyclosporine coadministration on edoxaban pharmacokinetics
| Edoxaban | M4 | |||
|---|---|---|---|---|
| Parameter | Edoxaban n = 33 | Edoxaban + cyclosporine n = 30 | Edoxaban n = 33 | Edoxaban + cyclosporine n = 30 |
| C max (ng ml –1 ) | 245.7 ± 77.64 | 410.9 ± 70.64 | 23.2 ± 9.34 | 191.1 ± 46.41 |
| t max (h) a | 1.50 (0.98, 3.02) | 2.99 (0.98, 4.02) | 2.00 (1.00, 3.02) | 3.03 (2.00, 4.02) |
| AUC (ng h ml –1 ) | 1833 ± 363.3b | 3131 ± 528.0 | 168.9 ± 51.1b | 1132 ± 268.0c |
| t 1/2 (h) | 14.50 ± 5.34b | 13.87 ± 5.75 | 14.64 ± 6.37b | 12.92 ± 6.23c |
| CL/F (L h –1 ) | 34.10 ± 7.37b | 19.68 ± 3.25 | – | – |
| LSM ratio | 90% CI | LSM ratio | 90% CI | |
|---|---|---|---|---|
| C max | 173.60 | 155.87, 193.36 | 870.93 | 781.94, 970.06 |
| AUC | 172.57 | 162.65, 183.11 | 687.00 | 636.71, 741.27 |
Data presented as arithmetic mean ± standard deviation unless indicated otherwise.
AUC, area under the curve from the time of dosing extrapolated to infinity; CI, confidence interval; CL/F, apparent total body clearance; Cmax, maximum observed plasma drug concentration; LSM, least squares mean; t1/2, terminal half‐life; tmax, time of maximum observed concentration.
Median (min, max)
n = 31
n = 28
Figure 4.
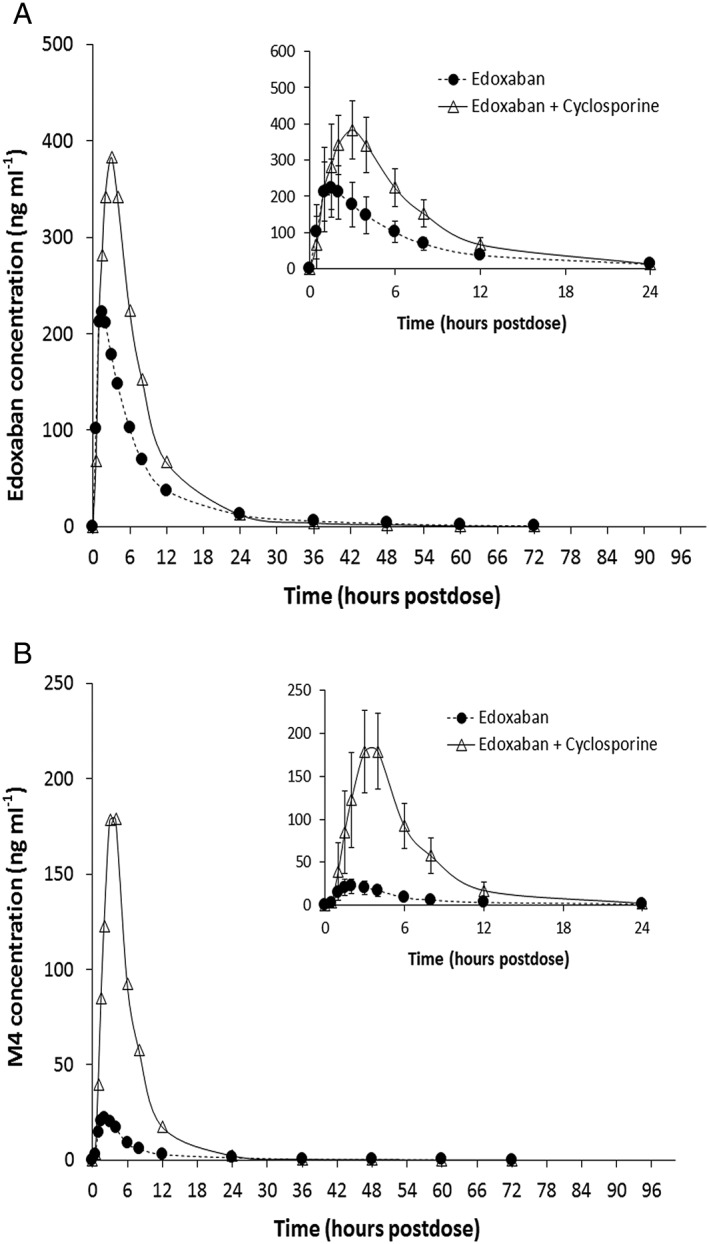
Mean plasma concentrations of (A) edoxaban and (B) metabolite M4 following administration with and without cyclosporine. Insets represent the first 24 h postdose; error bars represent the standard deviation
The percent mean maximum change in intrinsic FXa values from baseline was −81.8% after edoxaban was coadministered with cyclosporine vs. −64.3% when edoxaban was administered alone (approximately 1.3‐fold increase in inhibitory activity). These data are consistent with the increase in plasma concentrations of edoxaban when coadministered with cyclosporine.
Safety
Edoxaban was well tolerated in all studies, both when administered alone or in combination with ketoconazole, erythromycin, or cyclosporine. No serious adverse events were reported. The detailed description of the safety results is included in the Supporting Information (Appendix S1).
Discussion
The effect of dual P‐gp/CYP3A4 inhibitors ketoconazole, erythromycin, and cyclosporine on edoxaban pharmacokinetics was evaluated in three individual studies. Each of these inhibitors had a different extent of CYP3A4 inhibitory potential, but all of them are known P‐gp inhibitors 9. Each drug had a slightly different effect on the pharmacokinetic parameters of edoxaban. All three drugs increased the exposure (Cmax and AUC, respectively) of edoxaban: ketoconazole (89% and 87%), erythromycin (68% and 85%), and cyclosporine (74% and 73%).
The metabolites M6 and M8, formed by CYP3A4, were measured in the ketoconazole study to confirm the relative contribution of CYP3A4 inhibition on the increased edoxaban exposure. The CYP3A4 effects of the ketoconazole were observed in the four‐fold decrease in exposure of M6. Although levels of M8 were generally below the quantification limit, the concentration of M8, when detectable, was lower when administered with ketoconazole, indicating CYP3A4 inhibition. Since metabolism via CYP3A4 is a minor pathway for edoxaban (<4% of total clearance) 7, and M6 and M8 have a low relative abundance in plasma, these metabolites were not measured in studies with erythromycin and cyclosporine.
Results of prior drug interaction studies conducted with the P‐gp inhibitor quinidine and edoxaban dosed intravenously and orally indicate that the predominant mechanism for the increased edoxaban exposure by ketoconazole, erythromycin, and cyclosporine is mediated by the inhibition of gut P‐gp. 6, 15. With intravenous dosing of edoxaban, the increase in total exposure was only about 35%, whereas for oral edoxaban, the increase in exposure was about 68% 6. The results from our study also support this assertion. Thus, it can be concluded that P‐gp inhibition in the gut is the predominant mechanism for drug interaction with edoxaban, and drugs that are inhibitors of CYP3A4 alone are unlikely to affect the edoxaban pharmacokinetics appreciably.
In addition, results of the three studies described in this report provide further mechanistic insight into the distribution and elimination of edoxaban and its metabolites. For example, the ketoconazole interaction study shows the increase in total exposure cannot be fully accounted for by an increase in bioavailability alone, since the absolute bioavailability of edoxaban is 62% 6. Despite a minimal change in terminal half‐life, it appears that both clearance and volume of distribution changed by a similar magnitude, approximately 46%. Thus, ketoconazole appears to reduce both the clearance and volume of distribution of edoxaban in addition to increasing bioavailability by reducing intestinal P‐gp‐mediated efflux. However, the full effect of decreased clearance is probably underestimated from this study due to concomitant change in the volume parameter or inadequate sampling scheme. Since the half‐life of edoxaban is about 11.5 h, sampling should occur for about 60 h to characterize the terminal phase/clearance adequately. Collecting samples for shorter duration will, consequently, estimate the terminal phase and associated parameters inaccurately.
With erythromycin, the half‐life of edoxaban decreased with decreased clearance, in addition to an increase in bioavailability related to P‐gp inhibition in the gut. The approximate 47% decrease in apparent clearance was accompanied by a 63% decrease in apparent volume of distribution. This effect is consistent with published literature 16.
When edoxaban was administered with cyclosporine, the half‐life did not change appreciably, but the apparent clearance was almost half that of edoxaban dosed alone. Thus, the decrease in apparent clearance was probably balanced by a decrease in apparent volume of distribution, half‐life being a ratio of these two parameters. Given the decreases in both apparent clearance and volume of distribution, these data suggest that bioavailability increased, owing to inhibition of P‐gp in the gut by cyclosporine.
The relative exposure of the M4 metabolite with respect to the parent drug did not change when edoxaban was administered with ketoconazole or erythromycin, but increased substantially with cyclosporine. Since cyclosporine also inhibits OATP1B1 10, the disproportionate increase in exposure for M4 was postulated to be due to inhibition of OATP1B1. A drug‐interaction study with rifampin, an OATP1B1 inhibitor, also showed a similar increase in M4 exposure. Later in vitro studies confirmed that M4 is a substrate of OATP1B1 9.
The M1 metabolite is formed by nonenzymatic hydrolysis 7. Administration of ketoconazole, a P‐gp/CYP3A4 inhibitor, did not affect its relative abundance. M1 was not measured in other studies due to low circulating concentrations; it is inactive and not affected by coadministration of P‐gp/CYP3A4 inhibitors.
In vitro data show that M4, the most abundant metabolite, possesses comparable inhibitory activity of human FXa to edoxaban 17. However, the pharmacodynamic effects observed in this study were mostly consistent with exposure to edoxaban. Given the limited exposure to metabolites, with and without P‐gp inhibitors, the contribution of M4 or other less abundant metabolites to clinical efficacy or safety in most subjects is expected to be limited.
A single dose of edoxaban was well tolerated in healthy subjects when administered alone or in combination with ketoconazole, erythromycin, or cyclosporine. No unexpected or clinically concerning pattern of adverse experiences was noted.
Overall, results from the three studies demonstrate that CYP3A4 plays a minor role in the clearance of edoxaban in healthy subjects. Thus, inhibitors of CYP3A4 alone are unlikely to affect the exposure of edoxaban appreciably. The exposure of edoxaban is influenced by the activity of P‐gp as it is involved in limiting absorption and modulating distribution. Thus, inhibitors of P‐gp are expected to increase the exposure of edoxaban. As observed in these studies, P‐gp inhibitors with varying degree of CYP3A4 inhibition increased edoxaban exposure, but the increase was less than two‐fold.
Competing Interests
All authors of this manuscript have completed the Unified Competing Interest form at: http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any third party organization for the submitted work and no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years. All authors declare that they are, or were at the time this study was conducted, employees of Daiichi Sankyo, the sponsor of this study.
Contributors
J.M., N.M., M.S. and H.Z. participated in the study design and conduct. M.N. participated in data acquisition. Statistical analyses were performed by M.S., D.A.P., K.T., J.M., H.Z. and N.M. were involved in study design and conduct. D.A.P. and K.T. were involved in interpretation of study results and development of the manuscript. All authors revised the manuscript and approved the final version.
The authors would like to acknowledge the bioanalysis oversight by Ling He, and the writing assistance provided by Terri Schochet, PhD, and Ewa Wandzioch, PhD, of AlphaBioCom, LLC, and funded by Daiichi Sankyo. The authors also thank the clinical investigators Robert Noveck, MD, Brendan Colgan, MD, and Frank Lee, MD, who were responsible for the organization of the three clinical trials described in this manuscript.
Supporting information
Appendix S1 Supplemental information on the safety of edoxaban administered together with ketoconazole, erythromycin or cyclosporine
Supporting info item
Parasrampuria, D. A. , Mendell, J. , Shi, M. , Matsushima, N. , Zahir, H. , and Truitt, K. (2016) Edoxaban drug–drug interactions with ketoconazole, erythromycin, and cyclosporine. Br J Clin Pharmacol, 82: 1591–1600. doi: 10.1111/bcp.13092.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 2015; 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Furugohri T, Isobe K, Honda Y, Kamisato‐Matsumoto C, Sugiyama N, Nagahara T, et al. DU‐176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles. J Thromb Haemost 2008; 6: 1542–1549. [DOI] [PubMed] [Google Scholar]
- 5. SAVAYSA . (Edoxaban) Tablets for Oral Use. Highlights of Prescribing Information. Daiichi Sankyo Co: Parsippany, NJ, USA, 2015. [Google Scholar]
- 6. Matsushima N, Lee F, Sato T, Weiss D, Mendell J. Bioavailability and safety of the factor Xa inhibitor edoxaban and the effects of quinidine in healthy subjects. Clin Pharm Drug Dev 2013; 2: 358–366. [DOI] [PubMed] [Google Scholar]
- 7. Parasrampuria DA, Truitt KE. Pharmacokinetics and pharmacodynamics of edoxaban, a non‐vitamin K antagonist oral anticoagulant that inhibits clotting factor Xa. Clin Pharmacokinet 2015; 55: 641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. International Transporter Consortium , Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, et al. Membrane transporters in drug development. Nat Rev Drug Discov 2010; 9: 215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mikkaichi T, Yoshigae Y, Masumoto H, Imaoka T, Rozehnal V, Fischer T, et al. Edoxaban transport via P‐glycoprotein is a key factor for the drug's disposition. Drug Metab Dispos 2014; 42: 520–528. [DOI] [PubMed] [Google Scholar]
- 10. Amundsen R, Christensen H, Zabihyan B, Asberg A. Cyclosporine A, but not tacrolimus, shows relevant inhibition of organic anion‐transporting protein 1B1‐mediated transport of atorvastatin. Drug Metab Dispos 2010; 38: 1499–1504. [DOI] [PubMed] [Google Scholar]
- 11. FDA Center for Drug Evaluation Research . FDA Draft Guidance for Industry. Drug interaction studies – study design, data analysis, implications for dosing, and labeling recommendations. In: US Food and Drug Administration, 2012.
- 12. Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. In: US Food and Drug Administration, 2014.
- 13. Endicott JA, Ling V. The biochemistry of P‐glycoprotein‐mediated multidrug resistance. Annu Rev Biochem 1989; 58: 137–171. [DOI] [PubMed] [Google Scholar]
- 14. Shapiro AB, Ling V. Using purified P‐glycoprotein to understand multidrug resistance. J Bioenerg Biomembr 1995; 27: 7–13. [DOI] [PubMed] [Google Scholar]
- 15. Mendell J, Zahir H, Matsushima N, Noveck R, Lee F, Chen S, et al. Drug–drug interaction studies of cardiovascular drugs involving P‐glycoprotein, an efflux transporter, on the pharmacokinetics of edoxaban, an oral factor Xa inhibitor. Am J Cardiovasc Drugs 2013; 13: 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. University of Washington Department of Pharmaceutics . Metabolism & Transport Drug Interaction Database Version 3.0. 2012.
- 17. Mendell J, Johnson L, Chen S. An open‐label, phase 1 study to evaluate the effects of hepatic impairment on edoxaban pharmacokinetics and pharmacodynamics. J Clin Pharmacol 2015; 55: 1395–1405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supplemental information on the safety of edoxaban administered together with ketoconazole, erythromycin or cyclosporine
Supporting info item