

Abstract
Aims
The aims of the study were: (i) to characterize the pharmacokinetics (PK) of doxorubicin (DOX) and doxorubicinol (DOXol) in patients diagnosed with non‐Hodgkin's lymphoma (NHL) using a population approach; (ii) to evaluate the influence of various covariates on the PK of DOX; and (iii) to evaluate the role of DOX and DOXol exposure in haematological toxicity.
Methods
Population PK modelling (using NONMEM) was performed using DOX and DOXol plasma concentration–time data from 45 NHL patients treated with R‐CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone). The influence of drug exposure on haematological toxicity was analysed using the Mann–Whitney‐Wilcoxon test.
Results
A five‐compartment model, three for DOX and two for DOXol, with first‐order distribution and elimination for both entities best described the data. Population estimates for parent drug (CL) and metabolite (CLm) clearance were 62 l h−1 and 27 l h−1, respectively. The fraction metabolized to DOXol (Fm) was estimated at 0.22. While bilirubin and aspartate aminotransferase showed an influence on the CL and CLm, the objective function value decrease was not statistically significant. A trend towards an association between the total area under the concentration–time curve (AUCtotal), the area under the concentration–time curve for DOX (AUC) plus the area under the concentration–time curve for DOXol (AUCm), and the neutropenia grade (P = 0.068) and the neutrophil counts (P = 0.089) was observed, according to an exponential relationship.
Conclusions
The PK of DOX and DOXol were well characterized by the model developed, which could be used as a helpful tool to optimize the dosage of this drug. The results suggest that the main active metabolite of DOX, DOXol, is involved in the haematological toxicity of the parent drug.
Keywords: doxorubicin, doxorubicinol, non‐Hodgkin's lymphoma, population pharmacokinetics
What Is Already Known about this Subject
Doxorubicin is used in antineoplastic therapy at a fixed dosage standardized to body surface area.
A wide variability in doxorubicin pharmacokinetics has been reported in population pharmacokinetic models developed in heterogeneous populations.
The use of doxorubicin is limited by its safety; doxorubicinol could be implied in the toxicity mechanisms.
What this Study Adds
The population pharmacokinetics of doxorubicin and doxorubicinol in adult patients diagnosed with non‐Hodgkin's lymphoma were characterized.
Doxorubicinol exposure seems to be involved in the haematological toxicity of doxorubicin.
This population pharmacokinetic model can be used as a helpful tool for doxorubicin dosage adjustment in patients diagnosed with non‐Hodgkin's lymphoma.
Table of Links
| LIGANDS |
|---|
| Doxorubicin |
This Table lists key ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1.
Introduction
Doxorubicin (DOX) is widely used in clinical practice for the treatment of solid tumours and haematological malignancies. However, its clinical activity is limited by its toxicity. Acute myelosuppression and chronic cardiomyopathy are dose‐limiting adverse effects 2. DOX efficacy and toxicity show wide interindividual variability. Thus, knowing the pharmacokinetic–pharmacodynamic (PK/PD) relationship could be an interesting approach to optimizing treatment with DOX.
This antineoplastic drug is mainly metabolized in the liver by a NADPH‐dependent aldoketo reductase present in all cell types, and particularly in erythrocytes and cells of the liver and kidney. Carbonyl reductase 1 and 3 also play an important role in the transformation to its main active metabolite, doxorubicinol (DOXol), the activity of which is around 10% of the parent drug. It has previously been suggested that DOXol exposure contributes to the efficacy and toxicity of the treatment 3, 4, 5, 6, 7, 8, 9.
In the common range of doses administered, this antineoplastic agent shows linear kinetic profiles. According to its complex distribution, the evolution of DOX plasma concentrations has been described by two‐ 10, 11, 12, 13, 14 or three‐ 8, 13, 15, 16, 17, 18, 19, 20, 21, 22, 23 compartment models, which is compatible with a wide distribution into the peripheral tissues. The clearance (CL) of DOX has previously been estimated at around 60 l h−1 8, 10, 11, 13, 15, 24, being altered in older patients, children 19, 20, pregnant women 25 and obese patients 26, 27. Moreover, the concomitant administration of P‐glycoprotein inhibitors 8, 28, the use of high doses of DOX (higher than 50 mg m−2) 26 or the tumour entity 11, 27 have been reported to influence CL.
The clinical response to DOX shows wide interindividual variability (IIV). Therefore, characterization of an appropriate PK/PD relationship in specific populations can be an interesting tool for preventing toxicity and optimizing efficacy. The relevance of the main active metabolite in terms of its probable relationship with the efficacy and toxicity of this drug has been reported in a large number of publications 4, 5, 6, 7, 13, 24, 28, 29, 30, 31.
Although several population PK (popPK) studies of DOX have included DOXol 8, 10, 11, 12, 15, 18, there have been no studies in a uniform and specific non‐Hodgkin's lymphoma (NHL) population. The objectives of the current analysis were to develop a popPK model for DOX and DOXol in patients diagnosed with NHL, to evaluate the influence of covariates (subject demographics, baseline clinical laboratory values, etcetera) on the PK estimates and to assess the role of exposure to the parent drug and main active metabolite on the haematological toxicity.
Methods
Patients
The study was carried out between June 2009 and June 2015 in patients diagnosed with NHL and treated with R‐CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone) every 21 days for six cycles. Prophylaxis of neutropenia with granulocyte colony‐stimulating factor (GCSF) was performed according to standard guidelines 32. DOX was administered by a continuous infusion of 0.5 h duration at 50 mg m−2. A total of 45 adult patients were included in the study, with 30 patients recruited by the Department of Haematology at the University Hospital of Salamanca and 15 patients enrolled in the clinical trial GEL‐R‐COMP‐2013. Blood samples were taken after written informed consent had been obtained from the patients. Patients' characteristics are shown in Table 1. The following covariates were registered on the day of administration and 21 days later (next cycle): age; gender; body weight; height; body surface area (BSA); body mass index (BMI); lean body weight (LBW); Eastern Cooperative Oncology Group performance status (ECOG); International Prognostic Index (IPI); creatinine clearance (CLcr); aspartate aminotransferase (AST), alanine aminotransferase (ALT) and bilirubin levels; and leucocyte, neutrophil and platelet counts.
Table 1.
Summary of patient demographics, doxorubicin treatment and baseline clinical characteristics of patients included in the pharmacokinetic modelling
| n | Mean (SD) | Range | |
|---|---|---|---|
| Patients | 45 | ||
| Male | 23 | ||
| Female | 22 | ||
| Type of lymphoma | |||
| Burkitt‐like lymphoma | 2 | ||
| Diffuse large B cell lymphoma | 36 | ||
| Follicular lymphoma | 2 | ||
| Other | 5 | 66 (15) | 26–84 |
| Weight (kg) | 71 (12) | 43–110 | |
| Height (m) | 1.64 (0.01) | 1.43–1.92 | |
| Body surface area ( m −2 ) | 1.8 (0.2) | 1.3–2.3 | |
| Body mass index ( kg m −2 ) | 26.5 (3.9) | 19.9–37.6 | |
| Lean body weight (kg) a | 47.9 (10.0) | 28.7–69.5 | |
| Dose of DOX ( mg m −2 ) | 51 (7) | 25–71 | |
| Dose of DOX (mg) | 89 (14) | 53–130 | |
| Infusion duration (h) | 0.5 (0.2) | 0.2–1.3 | |
| Creatinine clearance ( ml min −1 ) b | 91 (40) | 40–201 | |
| Bilirubin ( mg dl −1 ) | 0.44 (0.19) | 0.10–0.70 | |
| ALT (IU l −1 ) | 23 (18) | 7–88 | |
| AST (IU l −1 ) | 25 (13) | 12–64 | |
| Leucocyte count (× 10 9 l −1 ) | 6.3 (2.7) | 2.5–15.5 | |
| Neutrophil count (× 10 9 l −1 ) | 4.1 (2.4) | 1.0–14.1 | |
| Platelet count (× 10 9 l −1 ) | 280 (118) | 52–648 |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; ECOG, Eastern Cooperative Oncology Group performance status; DOX, doxorubicin; F, female; M, male; IPI, International Prognostic Index; n, number of patients; SD, standard deviation
Lean body weight estimated using the Janmahasatian 33
Creatinine clearance estimated using the Cockcroft–Gault formula
Study site and ethical approval
A total of 15 of the patients included in the current study were enrolled in the GEL‐R‐COMP‐2013 clinical trial. This is an open‐label, multicentre, randomized phase II study, performed in accordance with the Declaration of Helsinki and good clinical practice guidelines. It was approved by the national authorities and the institutional ethics committee of each participating centre. The trial was registered in http://clinicaltrials.gov (identifier: NCT02012088). All patients signed a specific informed consent to participate in the present study.
Drug sampling and collection
Sparse PK data were collected from 45 NHL patients. For each patient, blood samples (5 ml) were drawn from a peripheral vein and collected at sampling times of 0, 30, 90 and 180 min after the end of a DOX infusion. Plasma was separated from the blood by centrifugation at 627 g for 10 min and frozen at −80°C. The best sampling times adapted to the clinical routine were selected according to the PK properties of DOX and DOXol using D‐optimal methodology in WinNonlin® software version 5.3 (Pharsight Corporation, Mountain View, CA, USA).
Quantification of drug concentrations
Plasma concentrations of DOX and DOXol were quantified using a validated ultra‐high‐performance liquid chromatography (UHPLC) method coupled to a fluorescence detector previously developed in our laboratory 34. The lower limits of quantification (LLOQ) were 8 ng ml−1 and 3 ng ml−1 for DOX and DOXol, respectively. The mean coefficient of variation of the intra‐ and interday variability of the method were 7.6% and 9.2% for DOX and 6.3% and 7.8% for DOXol, respectively. In terms of accuracy, the means ± standard deviation of the percentage of recoveries were 100.3 ± 8.1 for DOX and 99.5 ± 6.4 for DOXol. The method was validated following the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines for bioanalytical methods for the requirements of linearity, sensitivity, selectivity, accuracy, and intra‐ and interday precision and stability 35, 36.
popPK analysis
All analyses were performed in accordance with the FDA and EMA guidelines for popPK analyses 37, 38. NONMEM® version 7.3 (ICON Development Solutions, Hanover, MD, USA) was used to develop the popPK model. Parameters were estimated using the first‐order conditional estimation method. Additional software used for the development and evaluation of the model were Perl‐speaks‐NONMEM (PsN, version 4.4.0, http://psn.sourceforge.net) 39 and R software (version 3.1.0, http://www.r‐project.org), with the specific PK packages Xpose (version 4.5.0, http://xpose.sourceforge.net) and NPDE (version 2.0, www.npde.biostat.fr). Pirana (version 2.9.2, http://www.pirana‐software.com) 40, 41 and RStudio® version 0.98.976. (RStudio Inc., Boston, MA, USA) were used as a workbench for the previous described software.
The model building procedure was as follows. Firstly, a model to describe DOX PK was developed. When this model had been fully established, the DOXol data were added, and a joint model was developed, estimating all the parameters together 15. Two‐ and three‐compartment models with first‐order distribution and elimination were fitted to the parent drug plasma concentrations. A systematic strategy to solve the sparse data information was designed by fixing the volumes of distribution or the parameters related to the most extensive peripheral compartment to those previously reported in the literature. The popPK models of Wilde et al. 10 and Kontny et al. 15 were selected as reference models to describe the evolution of DOX plasma concentrations, using two‐ and three‐compartments, respectively. Exponential, proportional, additive and combined proportional and additive error models were explored to describe random effects, both IIV and residual variability.
The minimum objective function value (OFV) was used to select the best model for hierarchical models. The Akaike information criterion (AIC) and the Bayesian information criterion were applied for non hierarquical models 42. Residual standard error (RSE), η‐ and ε‐ shrinkage were taken into account as precision and bias metrics, respectively. The ability of the base model to describe the data was assessed by inspection of goodness‐of‐fit plots (e.g. population and individual predictions vs. observations; residuals vs. time after dose and observations), prediction‐corrected visual predictive check (pcVPC) and normalized prediction distribution error (NPDE). Individuals with samples associated with an absolute conditional weighted residual |CWRES| > 4 were not taken into account in the dataset 43.
Once an appropriate structural model had been developed for DOX, the following covariates were explored on PK parameters, based on the stepwise covariate model (SCM) building procedure (forward selection, α = 0.05; backward elimination, α = 0.01) implemented in PsN: age, gender, body weight, height, BSA, BMI, LBW, CLcr, and AST, ALT and bilirubin levels. The relationships between continuous covariates (e.g. age, AST) and PK parameters were evaluated using linear, exponential and power functions, the covariates being centred or scaled to their median values. The influence of gender as a categorical covariate was evaluated by a linear relationship. This DOX PK model was used as a starting point for adding the metabolite PK model. The metabolite data were fitted to a one‐or two‐compartment model with first‐order distribution and elimination. The same model building process and evaluation as previously described was applied for the development of the DOX and DOXol joint model.
The internal model evaluation was carried out using bootstrapping and Monte Carlo simulation diagnostics. A 1000‐sample bootstrap was conducted. The mean and 95% confidence interval (CI) was obtained for each PK parameter estimated. The relative errors between the mean parameters obtained by bootstrapping and by the popPK model were calculated and expressed as percentage of error. Simulation‐based diagnostics were conducted using pcVPC 44 and NPDE 45, simulating the dataset 1000 times for each. The pcVPCs were generated using PsN. The 95% CI for the 10th, 50th and 90th percentiles of the simulations at different times were calculated and compared with the prediction‐ and variability‐corrected observations. The data were binned automatically and stratified by compartment to differentiate between parent drug and metabolite 46. NPDE results were summarized graphically and statistically using the NPDE add‐on package. According to the null hypothesis, the NPDE results follow a standard normal distribution N (0,1).
Drug exposure and haematological toxicity
The leucocyte, neutrophil and platelet counts were recorded prior to all cycles of DOX treatment. These values were classified according to the Common Terminology Criteria for Adverse Events (CTCAE) version 3.0. Haematological toxicity was classified as severe (grade 3–4) or nonsevere (0–2). The area under the concentration–time curve for DOX (AUC) and for DOXol (AUCm) were estimated as the ratio between the dose administered and the CL of each entity (taking into account the DOXol fraction, Fm). DOX and DOXol exposures (AUC, AUCm and the sum of both, AUCtotal), and also the maximum experimental concentration of the parent drug (Cmax) and metabolite (Cmax,m), were compared between the two groups of patients (severe and nonsevere toxicity). The Mann–Whitney–Wilcoxon test (with the level of significance set at P < 0.05) was performed for each haematological parameter (leucocyte, neutrophil and platelet count). In addition, a correlation study between these haematological parameters and selected exposure PK parameters was carried out using a Pearson correlation analysis for the linear and exponential relationship (with the level of significance set at P < 0.05). Patients treated with GCSF between the two study cycles of treatment or without haematological information were removed from the analysis.
Results
Study population and samples
The study enrolled a total of 45 NHL patients treated with DOX (R‐CHOP) from six Spanish Hospitals. The majority of patients had a diffuse large B‐cell lymphoma (n = 36). All of the patients were adults with normal hepatic, renal and cardiac functions (Table 1). A total of 125 observations of DOX plasma concentrations and 120 of DOXol were analysed in a popPK model.
Final popPK model
The final popPK model comprised five compartments, three for DOX and two for DOXol, with first‐order distribution and elimination. For the systematic strategy to solve the sparse information, fixing the volumes of distribution to values previously reported was found to be better than fixing the parameters related to the largest peripheral compartment. Thus, the volumes of distribution of DOX were fixed to those proposed by Kontny et al. 26. The volumes of distribution of DOXol were fixed to the values obtained from a sensitivity analysis carried out on these parameters for this purpose. An exponential and proportional error model, for both DOX and DOXol, was used to estimate the IIV and the residual variability, respectively. The IIV of the intercompartmental clearances of DOX (Q2 and Q3) and CLm were fixed to the values obtained in the model‐building process. No correlation between the random‐effect parameters was found. Residual variability was modelled using a proportional error term for both entities. While bilirubin and AST showed influence on both CL and CLm, none of them achieved statistically significant decrease OFV. Thus, no covariate was included in our model. One patient was considered as an outlier (|CWRES| > 4) and removed from the analyses. The PK parameters, RSE and shrinkages estimated by the final model are provided in Table 2. The PK parameters were estimated with good precision and unbiased. The ability of our model to describe the observed data was evaluated using the goodness‐of‐fit plots for DOX and DOXol (Figure 1).
Table 2.
Population pharmacokinetic and bootstrap parameters of the final model
| Parameter | Final model (n = 44) | Bootstrap (n = 1000) | ||||
|---|---|---|---|---|---|---|
| Mean | RSE (%) | Shrinkage (%) | Mean | REP (%) | CI 95% | |
| CL (l h−1) | 62.4 | 11.5 | – | 63.4 | 1.7 | 50.3 – 79.1 |
| V 1 (l) | 17.7 | – | – | 17.7 | – | 17.7 – 17.7 |
| Q2 (l h−1) | 50.7 | 18.4 | – | 52.4 | 3.4 | 31.4 – 72.4 |
| V 2 (l) | 1830 | – | – | 1830 | – | 1830 – 1830 |
| Q3 (l h−1) | 28.4 | 13.5 | – | 29.9 | 5.4 | 21.9 – 44.8 |
| V 3 (l) | 71 | – | – | 71.0 | – | 71.0 – 71.0 |
| V 4 (l) | 79.8 | – | – | 79.8 | – | 79.8 – 79.8 |
| CLm (l h−1) | 26.8 | 42.9 | – | 37.0 | 38.1 | 14.0 – 88.2 |
| Fm | 0.22 | 14.7 | – | 0.232 | 5.3 | 0.165 – 0.333 |
| V 5 (l) | 653 | – | – | 653 | – | 653 – 653 |
| Q5 (l h−1) | 424 | 18.0 | – | 468.6 | 10.5 | 309.0 – 694.3 |
| IIVCL (%) | 22.9 | 32.7 | 40 | 22.3 | 2.6 | 7.3 – 36.2 |
| IIV Q2 (%) | 64.1 | – | – | 64.1 | – | 64.1 – 64.1 |
| IIV Q3 (%) | 28.2 | – | – | 28.2 | – | 28.2 – 28.2 |
| IIV CL,m (%) | 47.2 | – | – | 47.2 | – | 47.2 – 47.2 |
| IIVF,m (%) | 41.7 | 19.6 | 22 | 39.4 | 5.5 | 16.7 – 58.2 |
| IIVQ5 (%) | 58.9 | 39.4 | 35 | 82.6 | 40.2 | 15.7 – 162.8 |
| IIVResidual (%) | 37.1 | 8.3 | 15 | 37.1 | 0.0 | 30.5 – 43.5 |
| IIVResidual,m (%) | 32.1 | 10.4 | 21 | 28.8 | 14.9 | 14.0 – 39.6 |
CI, confidence interval; CL, clearance of doxorubicin (DOX); CLm, clearance of doxorubicinol (DOXol); Fm, conversion rate to DOXol; IIV, interindividual variability, exponential error in all the cases; OFV, objective function value; REP, relative error prediction (coefficient of variation between the typical value of a parameter estimated with the model and with the bootstrap); Qn, intercompartmental clearance of the n‐th compartment; Residual and Residualm, residual variability of DOX and DOXol, respectively, proportional error model; RSE, relative standard error; Vn, volume of distribution of the n‐th compartment. The values of random‐effect parameters have been expressed as %. Bootstrap of 1000 replicates. Bold font represents fixed parameters
Figure 1.
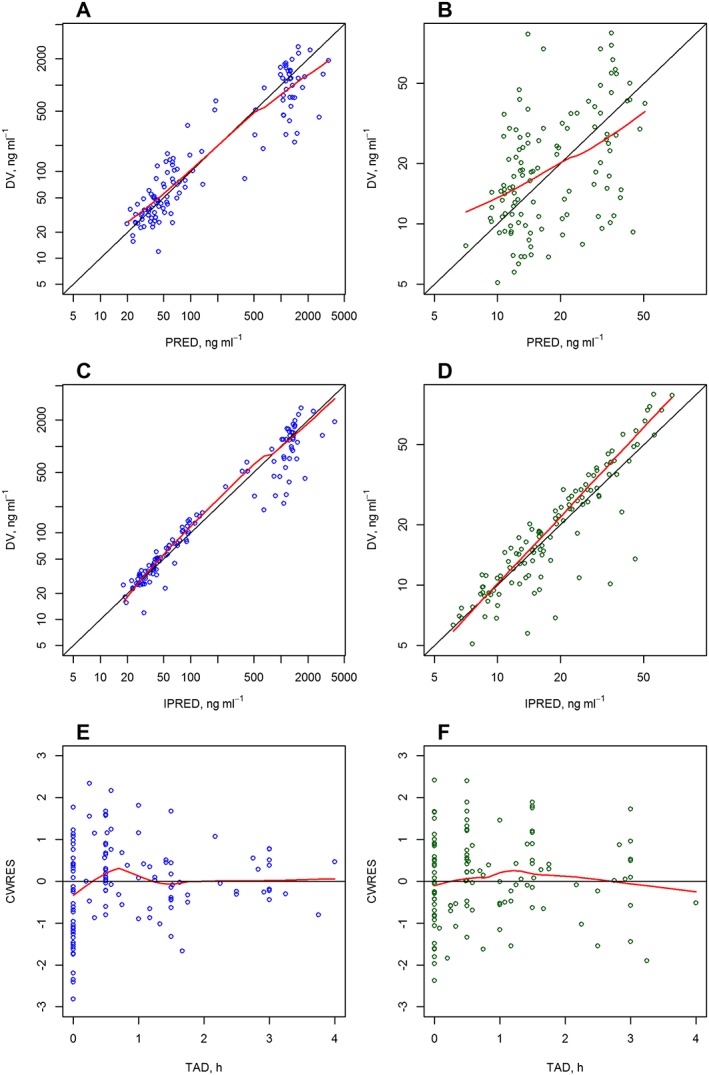
Goodness‐of‐fit plots for the final population pharmacokinetic model for doxorubicin (A, C, D) and doxorubicinol (B, D, F). CWRES, conditional weighted residual; DV, dependent variable (drug plasma concentration); IPRED, individual prediction; PRED, population prediction; TAD, time after dose. Black solid line, reference line in the diagnostic; red solid line, observed trend line in the plots (locally‐weighted polynomial regression)
The PK parameters estimated by the bootstrap analysis are provided in Table 2. Both fixed and random‐effect parameters estimated using the final popPK model developed were consistent with the bootstrap estimates and were within the 95% CI of the mean bootstrap estimates. The pcVPC of the final model (Figure 2) also supported the predictive ability of the model to describe the observed data for DOX and DOXol. The NPDE (Figure 3) statistical analysis showed, both for DOX and DOXol, that the mean and the variance were not significantly different from 0 and 1, respectively (P > 0.05). Furthermore, their distribution was no different from a normal one, and the global test supports the nonrejection of the null hypothesis (P > 0.05). The above three evaluations indicated that the model developed was appropriate for prediction‐ and simulation‐based applications.
Figure 2.
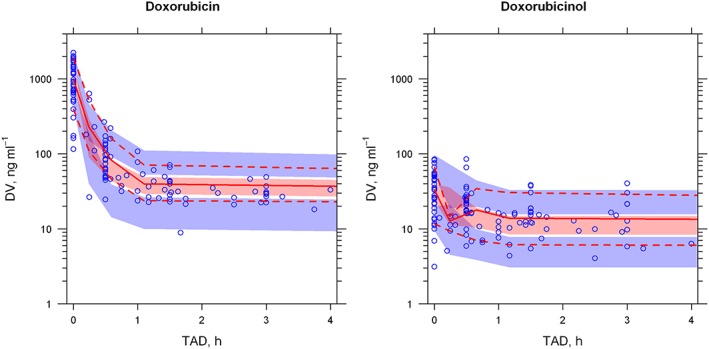
Prediction‐ and variability‐corrected visual predictive check (pcVPC) plot of the final population pharmacokinetic model. DV, dependent variable (drug plasma concentration); TAD: time after dose. The blue circles represent prediction‐ and variability‐corrected observations for doxorubicin (left) and doxorubicinol (right); the red solid line represents the 50th percentile of the prediction‐ and variability‐corrected observations; the red‐shaded area represents the 95% confidence interval (CI) for the 50th percentile of the simulated data; the red dashed line represents the 10th and 90th percentiles for the DOX observations; the blue‐shaded area represent the 95% CI for the 10th and 90th percentiles of the simulated data
Figure 3.
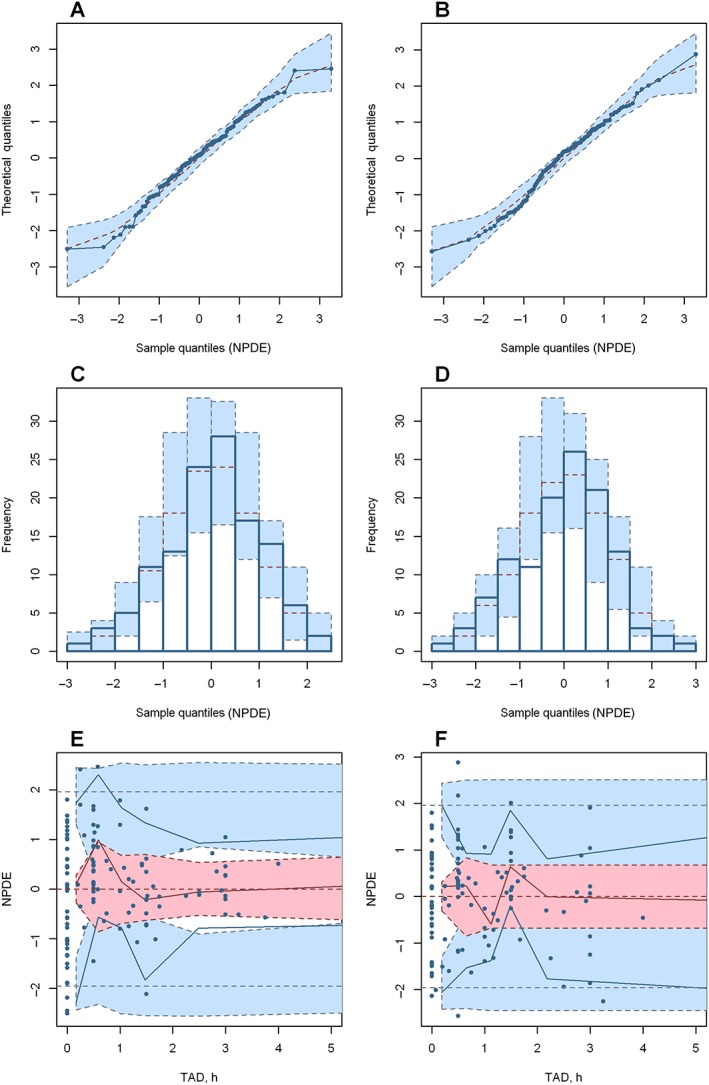
Normalized prediction distribution error (NPDE) plots of the final model for doxorubicin (A, C, E) and doxorubicinol (B, D, F). (A) and (B) are quantile–quantile plots of the distribution of NPDE against theoretical distribution (blue‐shaded area). (C) and (D) are histograms of the distribution of NPDE against theoretical distribution (blue‐shaded area). (E) and (F) are plots of NPDE vs. time after dose (TAD). In plots (E) and (F), the red solid line represents the median NPDE of the observations; the red‐shaded area represents the simulation‐based 95% confidence interval (CI) for the median; the blue solid line represents the NPDE of the observed 5th and 95th percentiles; the blue‐shaded area represent simulation‐based 95% CI for the corresponding model‐predicted percentiles; the blue circles represent NPDE of the observations
Drug exposure and haematological toxicity
For six of the 44 patients included in the final dataset, haematological information was not recorded. Furthermore, two patients were treated with GCSF within the period evaluated. Therefore, these eight patients were removed from the analysis (n = 36). Accordingly to the criteria for leucopenia and neutropenia, two and four patients, respectively, were classified in the severe toxicity group. The results of the influence of DOX and DOXol exposure (AUC, AUCm and AUCtotal) on the haematological parameters evaluated are provided in Table 3. The mean values of the different PK parameters categorized by haematological toxicity group are presented. In addition, the results of the correlation analysis between the previously mentioned PK parameters and leucocyte and neutrophil counts, reported 21 days after the studied DOX administration, are shown.
Table 3.
Drug exposure and haematological toxicity analysis
| Categorical analysis (Mann–Whitney–Wilcoxon test) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AUC (mg h l−1) | AUCm (mg h l−1) | AUCtotal (ng ml−1) | ||||||||
| G0‐G2 | G3‐G4 | P‐value | G0‐G2 | G3‐G4 | P‐value | G0‐G2 | G3‐G4 | P‐value | ||
| Leukopenia | 1.44 | 1.78 | 0.120 | 0.75 | 0.63 | 0.604 | 2.18 | 2.41 | 0.419 | |
| Neutropenia | 1.45 | 1.55 | 0.563 | 0.70 | 1.08 | 0.378 | 2.14 | 2.62 | 0.065 | |
| Continuous analysis (Pearson correlation) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AUC (mg h l−1) | AUCm (mg h l−1) | AUCtotal (ng ml−1) | ||||||||
| Relationship | r | P‐value | r | P‐value | r | P‐value | ||||
| Leucocyte count ( 10 9 l −1 ) | Exponential | –0.202 | 0.237 | –0.100 | 0.562 | –0.246 | 0.148 | |||
| Neutrophil count ( 10 9 l −1 ) | Exponential | –0.170 | 0.322 | –0.182 | 0.288 | –0.287 | 0.089 | |||
| Leucocyte count ( 10 9 l −1 ) | Linear | –0.112 | 0.514 | –0.051 | 0.769 | –0.133 | 0.440 | |||
| Neutrophil count ( 10 9 l −1 ) | Lineal | –0.094 | 0.585 | –0.089 | 0.606 | –0.150 | 0.384 | |||
AUC, area under the concentration–time curve for doxorubicin (DOX); AUCm, area under the concentration–time curve for doxorubicinol (DOXol); AUCtotal, sum of AUC plus AUCm; G0‐G4, grade of haematological toxicity according to Common Terminology Criteria for Adverse Events version 3.0; r, Pearson product‐moment correlation coefficient (with its associated P‐value); P‐value for the Mann–Whitney test and Pearson correlation (categorical and continuous analysis, respectively)
Discussion
In the present study, DOX and DOXol PK were well described by a five‐compartment model with first‐order distribution and elimination. Our results are consistent with the previous popPK studies conducted in adults 8, 11, 15, 16. Although the DOXol plasma concentration profile has been described by a biexponential distribution both in humans and in mice 18, 47, a one‐compartment model was reported in all the previous popPK studies 8, 10, 12, 15, 18, 19, 20. This discrepancy could be explained by the difficulty in estimating all the parameters for a two‐compartment model. However, the use of a one‐compartment model instead of a two‐compartment model to describe DOXol PK could lead to inaccurate AUCm estimations, hiding its possible pharmacodynamic effect. Thus, the current popPK model is the first to describe the PK of DOXol using a two‐compartment model.
Different strategies have been studied for the dosage optimization of DOX and other antineoplastic agents 28, 48, 49 with Gao 49 concluding that in specific cases ‘it is totally erroneous to continue to use BSA alone for dose calculation’. According to this fact, the different measures of body size (e.g. BMI, BSA), in addition to clinical variables (e.g. ECOG, IPI) and biochemistry covariates (e.g. AST, ALT levels), were evaluated to explain the variability in CL. Previously published DOX popPK models have included few and different covariates: gender 26, 27, age 19, pregnancy 25, obesity 26, 27 and doses higher than 50 mg m−2 26, as well as concomitant administration of P‐glycoprotein inhibitors 8, 28 or tumour entity 11, 27. The covariates tested did not show a significant influence on DOX CL. This could be explained by the high homogeneity of the population studied.
Thus, three‐ and two‐compartment models for DOX and DOXol, respectively, were developed successfully. The stochastic models were exponential error for the IIV and proportional for residual variability in both active entities. Moreover, it was necessary to fix the IIV of Q2, Q3 and CLm to avoid the over‐parameterization of the model. The PK parameters of our model, shown in Table 2, were well estimated, with a good precision for all the fixed‐effect parameter estimates, with the RSE lower than 25%, except for CLm (42.9%). The difficulty of estimating the latter parameter was due to the paucity of the data available and the sampling schedule, adapted to the clinical routine (up to 4 h), which does not allow precise modelling of the distribution and elimination in the terminal phase. The RSEs obtained for random‐effects parameters and shrinkage values were lower than 40%, which were appropriate according to the accepted criteria of precision and bias.
The use of previous kinetic knowledge for the drug, coupled with the observed data, also contributed to the development of a popPK model with sparse data to avoid over‐parameterization. NONMEM software conducts this analysis using the $PRIOR subroutine. This methodology is useful for stabilizing the parameter estimates when the available data are very sparse 50. However, it is very sensitive to the a priori information, and requires a detailed knowledge of fixed‐ and random‐effect parameter distributions in a similar population. In addition, the use of this methodology assumes that the prior parametrization and structural model were reliable. These assumptions can affect the identifiability of the parameters 51 and did not allow us to evaluate different structural models to that described previously. For these reasons, and mainly because no data for a specific population were available, we discarded the prior approach. We were therefore able to remove or fix parameters and make sure that our model was not over‐parameterized, as well as ensure the identifiability of the parameters. Moreover, we were able to evaluate different structural models from previous studies in the literature.
The values estimated for DOX and DOXol PK parameters were similar to those previously reported 8, 11, 15, 16. In addition, the model was successfully evaluated by bootstrap, pcVPC and NPDE as being the best simulation‐based diagnostic for the models with shrinkages higher than 20–30% estimated from sparse data 52. These results showed the model developed for DOX and DOXol to have adequate descriptive and predictive ability, and support it as a useful and valid tool to optimize the DOX dosage.
More than 50% of the patients treated with the recommended doses of DOX suffer a dose‐dependent neutropenia at the nadir, two weeks after drug administration. This toxicity can be delayed by up to four weeks. This pathological situation is frequently resolved before the next dose of DOX is administered 2, 13. In cases where this does not resolve, administration of GCSF or a delay in administering the next dose of DOX is needed. The ability to predict this situation could be helpful for the early identification of patients requiring an additional treatment with GCSF or a dose adjustment in the next cycle administered. Several relationships between DOX and/or DOXol plasma concentrations and bone marrow depletion at the nadir (e.g. survival factor, leucocyte count) have been established in various anticancer protocols, including CHOP, according to the classical exponential and sigmoidal maximum‐effect models (Emax) 13, 24, 28, 53, 54.
The analysis carried out did not show a statistically significant relationship (P > 0.05) between the PK parameters reflecting the parent drug or metabolite exposure and the bone marrow depletion, according to linear and exponential models. Nonlinearity relationships (e.g. Emax) were not tested because of the lack of haematological information within the study period. In addition, there were no statistically differences in the derived PK exposure parameters between the severe and nonsevere haematological toxicity groups. However, a slight influence of AUCtotal on neutropenia (P = 0.065) and neutrophils count (P = 0.085) was observed (Table 3). The small number of patients in the severe haematological toxicity group and the lack of power could explain why significance was not achieved. In addition, the elimination of patients treated with GCSF, who were likely to have had higher haematological toxicity, could have led to a selection bias in the analysis, and was a limitation of the results obtained. However, the study showed the possible contribution of DOXol in the severe haematological toxicity following DOX administration and the importance of taking account of the main metabolite of DOX in toxicological analysis.
In conclusion, a popPK model of DOX and DOXol was developed, describing adequately the evolution of plasma concentration for both entities. This is the first two‐compartment model for predicting DOXol PK. The accurate prediction and simulation abilities of the model were demonstrated by its successful internal evaluation (goodness‐of‐fit, pcVPC, NPDE, bootstrap). The data available in the studied population did not permit us to establish significant relationships between drug exposure and haematological toxicity. Thus, although a trend towards an association between AUCtotal and neutropenia was observed, suggesting the importance of taking DOXol into account to assess haematological toxicity, it did not show statistical significance, probably because of the sparsity of the data available. Nevertheless, the model proposed might be useful for DOX dosage adjustments in patients diagnosed with NHL.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare this work was supported in part by GELTAMO group (Grupo Español de Linfomas/Trasplante Autólogo de Médula Ósea). There are no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
We acknowledge the Biobank of Castilla y Leon for the plasma provided. We greatly thank all the patients and staff of the haematology services who participated in the study, especially Antonio Salar, Juan Manuel Sancho, Carlos Grande, Jorge Gayoso and José María Guinea de Castro.
Pérez‐Blanco, J. S. , Santos‐Buelga, D. , Fernández de Gatta, M. M. , Hernández‐Rivas, J. M. , Martín, A. , and García, M. J. (2016) Population pharmacokinetics of doxorubicin and doxorubicinol in patients diagnosed with non‐Hodgkin's lymphoma. Br J Clin Pharmacol, 82: 1517–1527. doi: 10.1111/bcp.13070.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl. Acids Res 2016; 44 (Database Issue): D1054–D68.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hortobágyi GN. Anthracyclines in the treatment of cancer. An overview. Drugs 1997; 54 (Suppl. 4): 1–7. [DOI] [PubMed] [Google Scholar]
- 3. Olson RD, Mushlin PS, Brenner DE, Fleischer S, Cusack BJ, Chang BK, et al. Doxorubicin cardiotoxicity may be caused by its metabolite, doxorubicinol. Proc Natl Acad Sci U S A 1988; 85: 3585–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stewart DJ, Grewaal D, Green RM, Mikhael N, Goel R, Montpetit VA, et al. Concentrations of doxorubicin and its metabolites in human autopsy heart and other tissues. Anticancer Res 1993; 13: 1945–1952. [PubMed] [Google Scholar]
- 5. Mushlin PS, Cusack BJ, Boucek RJ, Andrejuk T, Li X, Olson RD. Time‐related increases in cardiac concentrations of doxorubicinol could interact with doxorubicin to depress myocardial contractile function. Br J Pharmacol 1993; 110: 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Palle J, Frost B‐M, Peterson C, Gustafsson G, Hellebostad M, Kanerva J, et al. Doxorubicin pharmacokinetics is correlated to the effect of induction therapy in children with acute myeloid leukemia. Anticancer Drugs 2006; 17: 385–392. [DOI] [PubMed] [Google Scholar]
- 7. Thorn CF, Oshiro C, Marsh S, Hernandez‐Boussard T, McLeod H, Klein TE, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics 2011; 21: 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Callies S, de Alwis DP, Wright JG, Sandler A, Burgess M, Aarons L. A population pharmacokinetic model for doxorubicin and doxorubicinol in the presence of a novel MDR modulator, zosuquidar trihydrochloride (LY335979). Cancer Chemother Pharmacol 2003; 51: 107–118. [DOI] [PubMed] [Google Scholar]
- 9. Boucek RJ, Olson RD, Brenner DE, Ogunbunmi EM, Inui M, Fleischer S. The major metabolite of doxorubicin is a potent inhibitor of membrane‐associated ion pumps. A correlative study of cardiac muscle with isolated membrane fractions. J Biol Chem 1987; 262: 15851–15856. [PubMed] [Google Scholar]
- 10. Wilde S, Jetter A, Rietbrock S, Kasel D, Engert A, Josting A, et al. Population pharmacokinetics of the BEACOPP polychemotherapy regimen in Hodgkin's lymphoma and its effect on myelotoxicity. Clin Pharmacokinet 2007; 46: 319–333. [DOI] [PubMed] [Google Scholar]
- 11. Joerger M, Huitema ADR, Meenhorst PL, Schellens JHM, Beijnen JH. Pharmacokinetics of low‐dose doxorubicin and metabolites in patients with AIDS‐related Kaposi sarcoma. Cancer Chemother Pharmacol 2005; 55: 488–496. [DOI] [PubMed] [Google Scholar]
- 12. Wong AL, Seng KY, Ong EM, Wang LZ, Oscar H, Cordero MT, et al. Body fat composition impacts the hematologic toxicities and pharmacokinetics of doxorubicin in Asian breast cancer patients. Breast Cancer Res Treat 2014; 144: 143–152. [DOI] [PubMed] [Google Scholar]
- 13. Piscitelli SC, Rodvold KA, Rushing DA, Tewksbury DA. Pharmacokinetics and pharmacodynamics of doxorubicin in patients with small cell lung cancer. Clin Pharmacol Ther 1993; 53: 555–561. [DOI] [PubMed] [Google Scholar]
- 14. Wilde S, Jetter A, Zaigier M, Rietbrock S, Menzel H, Sieber M, et al. Population pharmacokinetics of cyclophosphamide, doxorubicin and etoposide in 30 patients with BEACOPP chemotherapy. Int J Clin Pharmacol Ther 2002; 40: 586–588. [DOI] [PubMed] [Google Scholar]
- 15. Kontny NE, Würthwein G, Joachim B, Boddy AV, Krischke M, Fuhr U, et al. Population pharmacokinetics of doxorubicin: establishment of a NONMEM model for adults and children older than 3 years. Cancer Chemother Pharmacol 2013; 71: 749–763. [DOI] [PubMed] [Google Scholar]
- 16. Escudero‐Ortiz V, Ramón‐López A, Duart MAJ, Pérez‐Ruixo JJ, Valenzuela B. Farmacocinética poblacional de doxorubicina aplicada a la personalización de su dosificación en pacientes oncológicos. Farm Hosp 2012; 36: 282–291. [DOI] [PubMed] [Google Scholar]
- 17. Danesi R, Fogli S, Gennari A, Conte P, Del Tacca M. Pharmacokinetic‐pharmacodynamic relationships of the anthracycline anticancer drugs. Clin Pharmacokinet 2002; 41: 431–444. [DOI] [PubMed] [Google Scholar]
- 18. Joerger M, Huitema AD, Richel DJ, Dittrich C, Pavlidis N, Briasoulis E, et al. Population pharmacokinetics and pharmacodynamics of doxorubicin and cyclophosphamide in breast cancer patients: a study by the EORTC‐PAMM‐NDDG. Clin Pharmacokinet 2007; 46: 1051–1068. [DOI] [PubMed] [Google Scholar]
- 19. Völler S, Boos J, Krischke M, Würthwein G, Kontny NE, Boddy AV, et al. Age‐dependent pharmacokinetics of doxorubicin in children with cancer. Clin Pharmacokinet 2015; 54: 1139–1149. [DOI] [PubMed] [Google Scholar]
- 20. Thompson PA, Rosner GL, Matthay KK, Moore TB, Bomgaars LR, Ellis KJ, et al. Impact of body composition on pharmacokinetics of doxorubicin in children: a Glaser Pediatric Research Network study. Cancer Chemother Pharmacol 2009; 64: 243–251. [DOI] [PubMed] [Google Scholar]
- 21. Robert J, Bui NB, Vrignaud P. Pharmacokinetics of doxorubicin in sarcoma patients. Eur J Clin Pharmacol 1987; 31: 695–699. [DOI] [PubMed] [Google Scholar]
- 22. Freyer G, Tranchand B, Ligneau B, Ardiet C, Souquet PJ, Court‐Fortune I, et al. Population pharmacokinetics of doxorubicin, etoposide and ifosfamide in small cell lung cancer patients: results of a multicentre study. Br J Clin Pharmacol 2000; 50: 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robert J, Hoerni B. Age dependence of the early‐phase pharmacokinetics of doxorubicin. Cancer Res 1983; 43: 4467–4469. [PubMed] [Google Scholar]
- 24. Toffoli G, Corona G, Cattarossi G, Boiocchi M, Di Gennaro G, Tirelli U, et al. Effect of highly active antiretroviral therapy (HAART) on pharmacokinetics and pharmacodynamics of doxorubicin in patients with HIV‐associated non‐Hodgkin's lymphoma. Ann Oncol 2004; 15: 1805–1809. [DOI] [PubMed] [Google Scholar]
- 25. Ryu RJ, Eyal S, Kaplan HG, Akbarzadeh A, Hays K, Puhl K, et al. Pharmacokinetics of doxorubicin in pregnant women. Cancer Chemother Pharmacol 2014; 73: 789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rudek MA, Sparreboom A, Garrett‐Mayer ES, Armstrong DK, Wolff AC, Verweij J, et al. Factors affecting pharmacokinetic variability following doxorubicin and docetaxel‐based therapy. Eur J Cancer 2004; 40: 1170–1178. [DOI] [PubMed] [Google Scholar]
- 27. Dobbs NA, Twelves CJ, Gillies H, James CA, Harper PG, Rubens RD. Gender affects doxorubicin pharmacokinetics in patients with normal liver biochemistry. Cancer Chemother Pharmacol 1995; 36: 473–476. [DOI] [PubMed] [Google Scholar]
- 28. Sparreboom A, Planting AS, Jewell RC, van der Burg ME, van der Gaast A, de Bruijn P, et al. Clinical pharmacokinetics of doxorubicin in combination with GF120918, a potent inhibitor of MDR1 P‐glycoprotein. Anticancer Drugs 1999; 10: 719–728. [DOI] [PubMed] [Google Scholar]
- 29. Elis A, Lishner M, Walker S, Atias D, Korenberg A, Koren G. Doxorubicin in lymphoma: association between pharmacokinetic variability and clinical response. Ther Drug Monit 2010; 32: 50–52. [DOI] [PubMed] [Google Scholar]
- 30. Leca F, Marchiset‐Leca D, Noble A, Antonetti M. New data on the pharmacokinetics of adriamycin and its major metabolite, adriamycinol. Eur J Drug Metab Pharmacokinet 1991; 16: 107–111. [DOI] [PubMed] [Google Scholar]
- 31. Legha SS, Benjamin RS, Mackay B, Ewer M, Wallace S, Valdivieso M, et al. Reduction of doxorubicin cardiotoxicity by prolonged continuous intravenous infusion. Ann Intern Med 1982; 96: 133–139. [DOI] [PubMed] [Google Scholar]
- 32. Smith TJ, Bohlke K, Lyman GH, Carson KR, Crawford J, Cross SJ, et al. Recommendations for the use of WBC growth factors: American Society of Clinical Oncology Clinical Practice guideline update. J Clin Oncol 2015; 33: 3199–3212. [DOI] [PubMed] [Google Scholar]
- 33. Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. Quantification of lean body weight. Clin Pharmacokinet 2005; 44: 1051–1065. [DOI] [PubMed] [Google Scholar]
- 34. Pérez‐Blanco JS, del Mar Fernández de Gatta M, Hernández‐Rivas JM, Sánchez MJG, Marinero MLS, López FG. Validation and clinical evaluation of a UHPLC method with fluorescence detector for plasma quantification of doxorubicin and doxorubicinol in haematological patients. J Chromatogr B 2014; 955–956: 93–97. [DOI] [PubMed] [Google Scholar]
- 35. European Medicines Agency (EMA) . Guideline on bioanalytical method validation; 2011. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf (last accessed 1 May 2016).
- 36. Food and Drug Administration (FDA) . Guidance for industry: bioanalytical method validation; 2001. U.S. Department of Health and Human Services [online]. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070107.pdf (last accessed 1 May 2016).
- 37. European Medicines Agency (EMA) , Committee for Medicinal Products for Human Use (CHMP) . Guideline on reporting the results of population pharmacokinetic analyses; 2008. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003067.pdf (last accessed 2 May 2016).
- 38. US Department of Health and Human Services , US Food and Drug Administration , Center for Drug Evaluation and Research (CDER) , Center for Biologics Evaluation and Research (CBER) . Guidance for industry: population pharmacokinetics; 1999. [online]. Available at http://www.GuidanceComplianceRegulatoryInformation/Guidances/ucm072137.pdf (last accessed 2 May 2016).
- 39. Lindbom L, Pihlgren P, Jonsson N. PsN‐Toolkit – a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed 2005; 79: 241–257. [DOI] [PubMed] [Google Scholar]
- 40. Keizer RJ, van Benten M, Beijnen JH, Schellens JHM, Huitema ADR. PiraÑA and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed 2011; 101: 72–79. [DOI] [PubMed] [Google Scholar]
- 41. Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol 2013; 2: e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hutmacher MM, Kowalski KG. Covariate selection in pharmacometric analyses: a review of methods. Br J Clin Pharmacol 2014; 79: 132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Byon W, Smith MK, Chan P, Tortorici MA, Riley S, Dai H, et al Establishing best practices and guidance in population modeling: an experience with an internal population pharmacokinetic analysis guidance. CPT Pharmacometrics Syst Pharmacol. 2013; 2: e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J 2011; 13: 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Comets E, Brendel K, Mentré F. Computing normalised prediction distribution errors to evaluate nonlinear mixed‐effect models: the NPDE add‐on package for R. Comput Methods Programs Biomed 2008; 90: 154–166. [DOI] [PubMed] [Google Scholar]
- 46. Karlsson MO, Holford N. A tutorial on visual predictive checks [online]. PAGE, 2008. Available at http://www.page‐meeting.org/pdf_assets/8694‐Karlsson_Holford_VPC_Tutorial_hires.pdf (last accessed 2 May 2016).
- 47. Urva SR, Shin BS, Yang VC, Balthasar JP. Sensitive high performance liquid chromatographic assay for assessment of doxorubicin pharmacokinetics in mouse plasma and tissues. J Chromatogr B Analyt Technol Biomed Life Sci 2009; 877: 837–841. [DOI] [PubMed] [Google Scholar]
- 48. Rosner GL, Hargis JB, Hollis DR, Budman DR, Weiss RB, Henderson IC, et al. Relationship between toxicity and obesity in women receiving adjuvant chemotherapy for breast cancer: results from cancer and leukemia group B study 8541. J Clin Oncol 1996; 14: 3000–3008. [DOI] [PubMed] [Google Scholar]
- 49. Gao B, Klumpen H‐J, Gurney H. Dose calculation of anticancer drugs. Expert Opin Drug Metab Toxicol 2008; 4: 1307–1319. [DOI] [PubMed] [Google Scholar]
- 50. Gisleskog PO, Karlsson MO, Beal SL. Use of prior information to stabilize a population data analysis. J Pharmacokinet Pharmacodyn 2002; 29: 473–505. [DOI] [PubMed] [Google Scholar]
- 51. Shivva V, Korell J, Tucker IG, Duffull SB. An approach for identifiability of population pharmacokinetic–pharmacodynamic models. CPT Pharmacometrics Syst Pharmacol 2013; 2: e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Savic RM, Karlsson MO. Importance of shrinkage in empirical bayes estimates for diagnostics: problems and solutions. AAPS J 2009; 11: 558–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rushing DA, Raber SR, Rodvold KA, Piscitelli SC, Plank GS, Tewksbury DA. The effects of cyclosporine on the pharmacokinetics of doxorubicin in patients with small cell lung cancer. Cancer 1994; 74: 834–841. [DOI] [PubMed] [Google Scholar]
- 54. Ackland SP, Ratain MJ, Vogelzang NJ, Choi KE, Ruane M, Sinkule JA. Pharmacokinetics and pharmacodynamics of long‐term continuous‐infusion doxorubicin. Clin Pharmacol Ther 1989; 45: 340–347. [DOI] [PubMed] [Google Scholar]