

Abstract
Aims
We assessed the drug interaction profile of fermented red ginseng with respect to the activity of major cytochrome (CYP) P450 enzymes and of a drug transporter protein, P‐glycoprotein (P‐gp), in healthy volunteers.
Methods
This study was an open‐label crossover study. The CYP probe cocktail drugs caffeine, losartan, dextromethorphan, omeprazole, midazolam and fexofenadine were administered before and after 2 weeks of fermented red ginseng administration. Plasma samples were collected, and tolerability was assessed. Pharmacokinetic parameters were calculated, and the 90% confidence intervals (CIs) of the geometric mean ratios of the parameters were determined from logarithmically transformed data. Values were compared between before and after fermented red ginseng administration using analysis of variance (anova).
Results
Fifteen healthy male subjects were evaluated, none of whom were genetically defined as a poor CYP2C9, CYP2C19 or CYP2D6 metabolizer based on genotyping. Before and after fermented red ginseng administration, the geometric least‐square mean metabolic ratio (90% CI) was 0.901 (0.830–0.979) for caffeine (CYP1A2) to paraxanthine, 0.774 (0.720–0.831) for losartan (CYP2C9) to EXP3174, 1.052 (0.925–1.197) for omeprazole (CYP2C19) to 5‐hydroxyomeprazole, 1.150 (0.860–1.538) for dextromethorphan (CYP2D6) to dextrorphan, and 0.816 (0.673–0.990) for midazolam (CYP3A4) to 1‐hydroxymidazolam. The geometric mean ratio of the area under the curve of the last sampling time (AUClast) for fexofenadine (P‐gp) was 1.322 (1.112–1.571).
Conclusion
No significantly different drug interactions were observed between fermented red ginseng and the CYP probe substrates following the two‐week administration of concentrated fermented red ginseng. However, the inhibition of P‐gp was significantly different between fermented red ginseng and the CYP probe substrates. The use of fermented red ginseng requires close attention due to the potential for increased systemic exposure when it is used in combination with P‐gp substrate drugs.
Keywords: cytochrome P450, drug interaction, fermented red ginseng, P‐glycoprotein
What is Already Known about this Subject
The ginsenoside compositions of fermented and non‐fermented red ginseng are different. An investigation of the influence of fermented red ginseng on CYP enzymes and P‐glycoprotein has not been conducted.
What this Study Adds
Multiple doses of a fermented red ginseng product weakly inhibited CYP2C9, CYP3A4 and P‐glycoprotein.
No significantly different drug interactions were observed between fermented red ginseng and the CYP probe substrates.
The use of fermented red ginseng requires close attention due to the potential for increased systemic exposure when it is used in combination with P‐glycoprotein substrate drugs.
Tables of Links
| LIGANDS |
|---|
| caffeine http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3726 |
| dextromethorphan |
| fexofenadine |
| losartan |
| midazolam |
| omeprazole |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2, 3.
Introduction
Ginseng, the root of Panax ginseng C.A. Meyer (Araliaceae), is one of the most frequently used traditional medicines in Korea, China, Japan and other Asian countries 4. Ginseng is classified into three types depending on how it is processed: fresh ginseng (not processed), white ginseng (dried after peeling), and red ginseng (steamed and dried) 5. Red ginseng can be stored for extended periods of time because of the removal of humidity during the manufacturing process 6. Red ginseng is used clinically for various diseases in China, Korea and Japan, including cancer, erectile dysfunction, hypertension, liver dysfunction and post‐menopausal disorder 7, 8, 9, 10, 11, 12. Ginsenosides, the major ingredient responsible for the pharmacological activities of ginseng, are classified into the 20(S)‐protopanaxadiol (ginsenoside Rb1, Rb2, Rg3, Rc and Rd) and 20(S)‐protopanaxatriol (ginsenoside Re, Rg1, Rg2 and Rh1) groups based on their aglycone moieties 13, 14. In the majority of commercial ginseng‐derived products, the major and most abundant components are ginsenosides Rb1, Rb, Rc, Rd, Re, Rf and Rg1 15. Most of these components are poorly absorbed from the gastrointestinal tract due to their high molecular weights and bulky sugar moieties 16. However, the various methods used to manufacture ginseng‐derived products can alter the formation and concentration of the ginsenosides 17.
Recently, interest in compound K ([20‐O‐(β‐d‐glucopyranosyl)‐20(S)‐protopanaxadiol]) has increased because of its biological activities. Compound K has been reported to have anti‐cancer 18, 19, 20, anti‐inflammatory 21, 22, anti‐diabetes 23, 24 and anti‐allergenic effects 25, 26. Although the abundance of compound K is low in raw and red ginseng, in humans, orally administered protopanaxadiol (PPD)‐type ginsenosides, such as ginsenoside Rb1, Rb2, Rc and Rd, are primarily metabolized to compound K by intestinal microflora via the following hydrolytic transformation pathway: ginsenoside Rb1 → ginsenoside Rd → ginsenoside F2 → compound K 27. Ginseng is usually administered orally, and the amount of compound K absorbed into the body varies among individuals depending on the metabolic ability of the human intestinal microflora 28.
Fermented red ginseng was developed to increase the absorption rate of ginsenosides. The fermentation of red ginseng using intestinal microflora increases the content of ginsenoside metabolites, such as compound K, Rh1, Rg3, protopanaxatriol and protopanaxadiol. Because ginsenoside metabolites have relatively higher absorption rates than naturally occurring ginsenosides, the bioavailability of fermented red ginseng is dramatically improved compared to that of red ginseng 29. Fermented red ginseng has anti‐allergic and anti‐oxidant effects and improves nasal congestion symptoms 30. Due to the widespread use of ginseng in combination with medications, there is a strong possibility of interactions between ginseng and drugs 31. As such, there is continuing need to characterize the influence of ginseng on common metabolic and transport pathways. Some studies have assessed the influence of ginseng on a variety of cytochrome (CYP) enzymes in vitro and in vivo 31, 32. In addition, ginsenoside metabolites have also been reported to have the potential to inhibit P‐glycoprotein (P‐gp) in several studies 33, 34. In these studies, ginseng induced CYP3A activity, and P‐gp activity related to midazolam and fexofenadine was unaltered by ginseng administration.
Despite the differences in the ginsenoside composition of fermented and non‐fermented red ginseng, to the best of our knowledge, an investigation of the influence of fermented red ginseng on CYP enzymes and P‐gp has not yet been conducted. Therefore, this study was conducted to evaluate the influence of fermented red ginseng on CYP enzymes and P‐gp activity using probe substrates.
Herbal supplements may also alter the absorption or disposition of co‐administered medications secondary to the modulation of drug transporter proteins such as P‐gp. Recently, the possibility that ginsenoside metabolites inhibit P‐gp was presented 33. These results suggest that significant ginseng–drug interactions may be possible for ginseng‐derived products. However, the influence of fermented red ginseng on CYP enzyme and P‐gp activity has not been studied. Moreover, it is difficult to apply previous results to in vivo research because previous studies of ginsenoside–drug interactions have been conducted in vitro.
Although many case reports have reported on ginseng and drug interactions, the resulting evidence is weak because the studies involved do not provide sufficient information on the treatment combinations or the conditions of the participants. Because many people take drugs for health reasons and expect supplementary effects from ginseng, studies of ginseng and drug interactions, the activities of drug‐metabolizing enzymes, and drug transporters and pharmacokinetics are needed to provide information to ginseng consumers. The present study was conducted to characterize the influence of fermented red ginseng on the activity of CYP enzymes and P‐gp through drug interactions using CYP cocktail probe drugs, caffeine, losartan, dextromethorphan, omeprazole, midazolam and fexofenadine in healthy human subjects.
Methods
The study was approved by the Ministry of Food and Drug Safety and the Institutional Review Board of Chonbuk National University Hospital (Jeonju, Republic of Korea, IRB No. CUH 2013–04‐002) and was conducted in accordance with the Declaration of Helsinki regarding biomedical research involving human subjects and the Guidelines for Good Clinical Practice. A detailed explanation of the study was provided to all participants, and written informed consent was obtained prior to screening.
Subjects
Healthy volunteers aged 20–55 years were enrolled. Each subject's health was confirmed by physical examination, measurement of vital signs, 12‐lead electrocardiography (ECG), serology (hepatitis B virus surface antigen, hepatitis B virus surface antibody, hepatitis C virus antibody and anti‐HIV antibody), and routine laboratory assessments (haematology, chemistry and urinalysis). Subjects were excluded if they had consumed drugs known to significantly activate or inhibit drug‐metabolizing enzymes within the previous 30 days or if they had taken any prescription or over‐the‐counter drugs within the 10 days prior to the first administration of the product under investigation. The exclusion criteria were chosen to ensure that subjects with risks specific to the administration of the cocktail probe drugs or fexofenadine or with conditions that could impact the pharmacokinetic properties of the cocktail probe drugs or fexofenadine were not included.
Study design
The study was conducted with an open‐label, one‐sequence, two‐period crossover design at the Clinical Trial Center of Chonbuk National University Hospital (Jeonju, Republic of Korea). The following five CYP substrate drugs were used to assess interactions with the major drug‐metabolizing enzymes: caffeine (200 mg, Vivarin®, Meda Consumer Healthcare Inc., Marietta, USA) as a CYP1A2 substrate; losartan (50 mg, Cozaar®, MSD Korea Ltd., Seoul, Korea) as a CYP2C9 substrate; omeprazole (20 mg, Losec capsule®, AstraZeneca Korea, Seoul, Korea) as a CYP2C19 substrate; dextromethorphan (15 mg, Robitussin Long‐Acting CoughGels®, Pfizer Inc., New York, USA) as a CYP2D6 substrate; and midazolam (7.5 mg, Dormicum®, Roche Korea, Seoul, Korea) as a CYP3A4 substrate. To assess drug transporter protein‐mediated drug interactions, fexofenadine (30 mg, Allegra®, Handok Pharmaceutical Co., Ltd., Seoul, Korea) was administered as a P‐gp substrate.
During the pharmacokinetic phase, the fasting subjects received a CYP probe drug cocktail (200 mg caffeine +50 mg losartan +20 mg omeprazole +30 mg dextromethorphan +7.5 mg midazolam along with 240 ml of water) on Days 1 and 15 at 08:00. Blood samples for pharmacokinetic analysis of the five CYP probe drugs were collected before dosing (baseline) and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, and 24 h after dosing. On Days 2 and 16, each subject received a P‐gp probe drug (30 mg fexofenadine along with 240 ml of water) at 08:00. To determine the pharmacokinetics of fexofenadine, blood was sampled before dosing (baseline) and at 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, and 48 h after its administration. The blood samples were immediately centrifuged at 1800×g) for 10 minutes at 4°C, and the resulting plasma was stored at −70°C until analysis.
Over the subsequent 2 weeks (Days 4–17), one pouch (70 ml; the recommended daily dose) of concentrated fermented red ginseng liquid (70 ml, >3% of dried ginseng, Fermented Red Ginseng Liquid – Jin®, Woongjin Food Corp., Seoul, Korea) was administered once daily. The subjects were hospitalized from the evening of the day before administration of the study drugs (Day −1 and Day 14) to the mornings of Day 3 and Day 17. The subjects were continuously monitored by investigators throughout the study period. Adverse events (AEs) were identified by asking the subjects general health‐related questions and through self‐reporting by the subjects during the study. Physical examinations, routine laboratory assessments and vital sign measurements were performed at regular, predefined intervals throughout the study.
Bioanalytical methods
The plasma concentrations of the parents and metabolites of the five CYP probe drugs (caffeine and paraxanthine, losartan and EXP3174, omeprazole and 5‐hydroxyomeprazole, dextromethorphan and dextrorphan, and midazolam and 1‐hydroxymidazolam) and the P‐gp probe drug (fexofenadine) were determined using a validated liquid chromatography–tandem mass spectrometry method.
The plasma concentration of each probe drug and its metabolites were determined simultaneously for all samples from the subjects. The drugs in plasma were determined by liquid chromatography tandem mass spectrometry as described previously 35, with some modifications. All the analytes in this study generated a prominent, protonated molecular ion [M + H]+ in positive‐ion mode. The precursor‐to‐product ion reactions monitoring of the drugs, the extraction methods for the plasma samples, and the analytical conditions are described in Table 1. Aliquots of preparation samples were analysed in an API 4000 liquid chromatography–tandem mass spectrometry system (Applied Biosystems, Foster City, CA, USA) equipped with an Agilent 1200 series high‐performance liquid chromatography system.
Table 1.
Analytical conditions of the analytes and internal standard (IS)
| Analyte | Precursor (m/z) | Product (m/z) | Column | Mobile phase | Extraction method | LLOQ |
|---|---|---|---|---|---|---|
| Caffeine | 195.2 | 138.1 | Gemini‐NX C18 (100 × 2.0 mm, 3 μm) | 0.1% formic acid in water (A) 0.1% formic acid in ACN (B) gradient mode | LLE | 1.0 |
| Paraxanthine | 181.2 | 124.2 | 1.0 | |||
| Losartan | 423.1 | 207.2 | Zorbax C8 (50 × 2.1 mm, 3.5 μm) | 0.1% formic acid in water (A) 0.1% formic acid in methanol‐(B) gradient mode | Protein precipitation | 1.0 |
| EXP3174 | 437.1 | 235.2 | 1.0 | |||
| Omeprazole | 346.1 | 198.1 | Luna C18 (100 × 2.0 mm, 3 μm | 7.5 mM ammonium bicarbonate in water, pH 8.0 (A) 0.1% formic acid in acetonitrile (B) gradient mode | Protein precipitation | 5.0 |
| 5‐hydroxyomeprazole | 362.1 | 214.1 | 5.0 | |||
| Dextromethorphan | 272.2 | 171.2 | Luna C18 (100 × 2.0 mm, 3 μm) | 0.1% formic acid in water (A) 0.1% formic acid in ACN (B) gradient mode | Protein precipitation | 0.5 |
| Dextrorphan | 258.3 | 157.2 | 0.5 | |||
| Midazolam | 326.2 | 291.1 | Zorbax C8 (50 × 2.1 mm, 3.5 μm | 0.1% formic acid in water (A) 0.1% formic acid in methanol (B) gradient mode | Protein precipitation | 1.0 |
| 1‐hydroxymidazolam | 342.2 | 168.0 | 1.0 | |||
| Fexofenadine | 502.3 | 171.3 | ZORBAX SB‐C8 (50 × 2.1 mm, 3.5 μm) | 0.1% formic acid in water (A) 0.1% formic acid in ACN (B) gradient mode | Protein precipitation | 1.0 |
| Lansoprazole | 370.2 | 252.2 | IS for analysis of caffeine and metabolite, omeprazole and metabolite, and dextromethorphan and metabolite | |||
| Propranolol | 260.2 | 116.0 | IS for analysis of losartan and metabolite, midazolam and metabolite, and fexofenadine | |||
The intra‐ and inter‐day precision over the concentration ranges of the analytes were all lower than 6.39% and 5.85% (coefficient of variation, %CV), respectively, and the intra‐ and inter‐day accuracy was between 88.40% and 108.80% and between 91.56% and 110.20%, respectively.
Genotype analysis of CYP2C9, CYP2C19 and CYP2D6
CYP enzyme activity was measured with a PyroMark ID (Biotage AB, Uppsala, Sweden). The CYP2C9*2, CYP2C9*3, CYP2C19*2 and CYP2C19*3 enzymes were measured using a pyro‐sequencing method, and the CYP2D6*5 enzyme was measured using a polymerase chain reaction (PCR) method.
Pharmacokinetic and statistical analyses
Individual PK parameters were obtained by non‐compartmental methods using Phoenix WinNonlin 6.3 software (Pharsight Corporation, Sunnyvale, CA, USA). The maximum plasma concentration (C max) and time to C max (t max) were obtained directly from plasma concentration–time curves. The area under the curve of the last sampling time (AUClast) was calculated using the linear trapezoidal rule. The activity of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 was evaluated by calculating the metabolic ratio of caffeine, losartan, omeprazole, dextromethorphan and midazolam, respectively; the metabolic ratios were calculated by determining the corresponding metabolite/parent AUC ratio (AUClast, metabolite/AUClast, parent) in plasma.
Statistical analysis was performed using SAS® (Version 9.3, SAS Institute Inc., Cary, NC, USA). Descriptive statistics were used to summarize the pharmacokinetic data, and analysis of variance (anova) at a 5% significance level was used to compare the pharmacokinetic parameters. The point estimates and 90% confidence intervals (CIs) of the ratio of the geometric means (after fermented red ginseng intake/before fermented red ginseng intake) of the log‐transformed C max and AUClast were compared. Additionally, we used the Wilcoxon signed‐rank test due to the small number of subjects.
Sample size determination
Intra‐individual %CVs in AUC and C max of probe drugs were assumed not to exceed 30% 36. Lack of interaction was assumed if the 90% CIs for estimated mean ratio did not exceed a tolerance zone of 0.70–1.43. A total sample size of 15 achieves an 80% power at a 5% significance level when the true ratio of the means is 1.05.
Results
A total of 15 healthy subjects (with a mean ± SD age of 25.6 ± 2.6 years, a mean height of 177.2 ± 4.9 cm, and a mean weight of 71.9 ± 12.3 kg) were determined to be eligible for the study and enrolled. CYP enzyme genotyping was conducted in 15 subjects, and none of the participants were found to be genetically poor metabolizers of CYP2C9*2/*2, *2/*3 or *3/*3, of CYP2C19*2/*2, *2/*3 or *3/*3, or of CYP2D6*5/*5.
Effects of fermented red ginseng on CYP enzymes
The pharmacokinetic parameters of the 15 subjects who completed the study were evaluated according to the described protocols. The mean plasma concentration–time profiles of the CYP and P‐gp probe drugs are shown in Figure 1. The metabolic ratio between the parents and metabolites of each CYP probe drug are described in Table 2.
Figure 1.
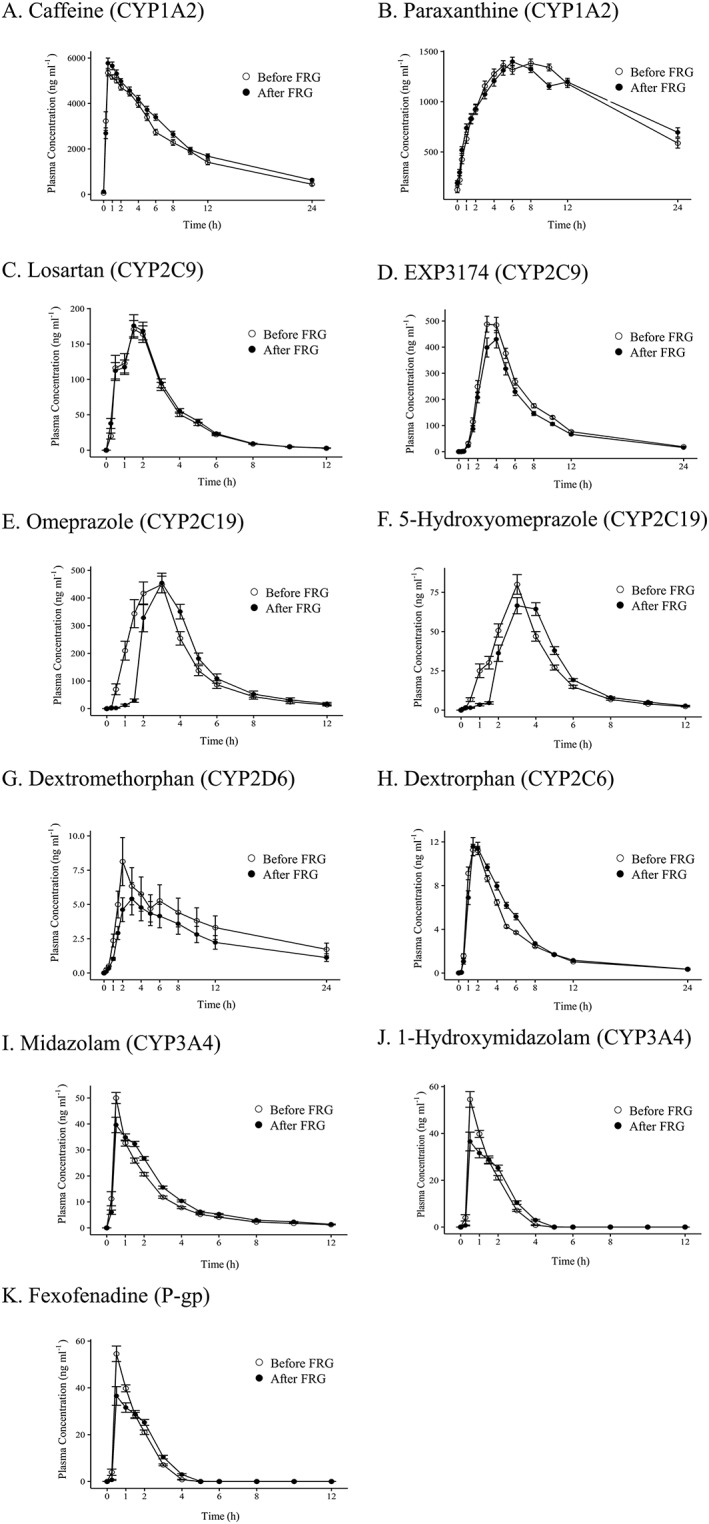
Mean (± standard error) plasma concentration–time profiles for the five probe drugs, their metabolites and fexofenadine before and after the administration of fermented red ginseng. P‐gp, P‐glycoprotein; FRG, fermented red ginseng‐derived product
Table 2.
Effects of fermented red ginseng on pharmacokinetics of probe drugs and their metabolites
| Analyte | Pharmacokinetic parameter | n | Geometric mean | Geometric mean ratio, After FRG: Before FRG (90% CI) | CV w (%) | P‐value b | |
|---|---|---|---|---|---|---|---|
| After FRG | Before FRG | ||||||
| Caffeine | C max (ng ml−1) | 15 | 6206.203 | 6193.919 | 1.002 (0.857–1.171) | 24.60 | 0.4212 |
| t max (h)a | 15 | 0.500 | 0.500 | ‐ | ‐ | 0.5024 | |
| AUClast (h ng ml−1) | 15 | 51314.971 | 45481.292 | 1.128 (0.997–1.277) | 19.39 | 0.2524 | |
| AUCinf (h ng ml−1) | 15 | 57490.588 | 49335.031 | 1.165 (1.002–1.356) | 23.87 | 0.2293 | |
| t 1/2 (h) | 15 | 6.939 | 5.497 | ‐ | ‐ | 0.0181 | |
| Paraxanthine | C max (ng ml−1) | 15 | 1464.896 | 1496.824 | 0.979 (0.906–1.057) | 11.97 | 0.9014 |
| t max (h)a | 15 | 6.000 | 8.000 | ‐ | ‐ | 0.4058 | |
| AUClast (h ng ml−1) | 15 | 24561.395 | 24151.161 | 1.017 (0.943–1.096) | 11.70 | 0.8469 | |
| AUCinf (h∙ng ml−1) | 15 | 53877.894 | 37622.878 | 1.432 (1.125–1.823) | 38.90 | 0.0479 | |
| t 1/2 (h) | 15 | 20.703 | 11.846 | ‐ | ‐ | 0.0067 | |
| Losartan | C max (ng ml−1) | 15 | 201.357 | 206.740 | 0.974 (0.797–1.190) | 31.97 | 0.9668 |
| t max (h)a | 15 | 1.500 | 1.500 | ‐ | ‐ | 0.4883 | |
| AUClast (h ng ml−1) | 15 | 541.687 | 516.699 | 1.048 (0.887–1.239) | 26.45 | 0.4212 | |
| AUCinf (h ng ml−1) | 15 | 548.613 | 523.857 | 1.047 (0.887–1.237) | 26.31 | 0.3894 | |
| t 1/2 (h) | 15 | 1.940 | 1.861 | ‐ | ‐ | 0.8469 | |
| EXP3174 | C max (ng ml−1) | 15 | 387.657 | 466.072 | 0.832 (0.694–0.997) | 28.76 | 0.2769 |
| t max (h)a | 15 | 4.000 | 3.000 | ‐ | ‐ | 0.2500 | |
| AUClast (h ng ml−1) | 15 | 2500.060 | 3082.080 | 0.811 (0.684–0.963) | 27.09 | 0.0413 | |
| AUCinf (h ng ml−1) | 15 | 2613.067 | 3212.424 | 0.813 (0.687–0.963) | 26.76 | 0.0479 | |
| t 1/2 (h) | 15 | 5.079 | 4.924 | ‐ | ‐ | 0.1514 | |
| Losartan | C max (ng ml−1) | 15 | 201.357 | 206.740 | 0.974 (0.797–1.190) | 31.97 | 0.9668 |
| t max (h)a | 15 | 1.500 | 1.500 | ‐ | ‐ | 0.4883 | |
| AUClast (h ng ml−1) | 15 | 541.687 | 516.699 | 1.048 (0.887–1.239) | 26.45 | 0.4212 | |
| AUCinf (h ng ml−1) | 15 | 548.613 | 523.857 | 1.047 (0.887–1.237) | 26.31 | 0.3894 | |
| t 1/2 (h) | 15 | 1.940 | 1.861 | ‐ | ‐ | 0.8469 | |
| EXP3174 | C max (ng ml−1) | 15 | 387.657 | 466.072 | 0.832 (0.694–0.997) | 28.76 | 0.2769 |
| t max (h)a | 15 | 4.000 | 3.000 | ‐ | ‐ | 0.2500 | |
| AUClast (h ng ml−1) | 15 | 2500.060 | 3082.080 | 0.811 (0.684–0.963) | 27.09 | 0.0413 | |
| AUCinf (h ng ml−1) | 15 | 2613.067 | 3212.424 | 0.813 (0.687–0.963) | 26.76 | 0.0479 | |
| t 1/2 (h) | 15 | 5.079 | 4.924 | ‐ | ‐ | 0.1514 | |
| Omeprazole | C max (ng ml−1) | 15 | 512.797 | 574.698 | 0.892 (0.747–1.066) | 28.18 | 0.3894 |
| t max (h)a | 15 | 3.000 | 2.000 | ‐ | ‐ | 0.0488 | |
| AUClast (h ng ml−1) | 15 | 1257.700 | 1351.108 | 0.931 (0.812–1.067) | 21.47 | 0.3894 | |
| AUCinf (h ng ml−1) | 15 | 1281.690 | 1369.503 | 0.936 (0.819–1.069) | 20.89 | 0.3894 | |
| t 1/2 (h) | 15 | 1.215 | 1.186 | ‐ | ‐ | 0.8040 | |
| 5‐hydroxyomeprazole | C max (ng ml−1) | 15 | 75.583 | 80.007 | 0.945 (0.859–1.039) | 14.90 | 0.3373 |
| t max (h)a | 15 | 3.000 | 3.000 | ‐ | ‐ | 0.0283 | |
| AUClast (h ng ml−1) | 15 | 239.033 | 244.051 | 0.979 (0.918–1.045) | 10.04 | 0.2769 | |
| AUCinf (h ng ml−1) | 15 | 283.859 | 270.368 | 1.050 (0.961–1.147) | 13.82 | 0.7615 | |
| t 1/2 (h) | 15 | 2.716 | 1.971 | ‐ | ‐ | 0.1070 | |
| Dextromethorphan | C max (ng ml−1) | 13 | 3.421 | 4.032 | 0.849 (0.688–1.047) | 30.77 | 0.0398 |
| t max (h)a | 13 | 2.000 | 2.000 | ‐ | ‐ | 0.3438 | |
| AUClast (h ng ml−1) | 13 | 17.140 | 18.890 | 0.907 (0.681–1.209) | 42.86 | 0.7354 | |
| AUCinf (h ng ml−1) | 11 | 43.514 | 50.077 | 0.869 (0.638–1.184) | 41.79 | 0.4131 | |
| t 1/2 (h) | 11 | 7.006 | 7.540 | ‐ | ‐ | 0.4648 | |
| Dextrorphan | C max (ng ml−1) | 13 | 12.005 | 11.721 | 1.024 (0.876–1.198) | 22.72 | 0.7998 |
| t max (h)a | 13 | 2.000 | 2.000 | ‐ | ‐ | 0.1309 | |
| AUClast (h ng ml−1) | 13 | 60.260 | 57.742 | 1.044 (0.926–1.176) | 17.20 | 0.4973 | |
| AUCinf (h∙ng ml−1) | 13 | 66.148 | 63.497 | 1.042 (0.932–1.164) | 15.98 | 0.7869 | |
| t 1/2 (h) | 13 | 3.432 | 3.920 | ‐ | ‐ | 0.7869 | |
| Midazolam | C max (ng ml−1) | 15 | 43.618 | 49.950 | 0.873 (0.732–1.041) | 27.84 | 0.2769 |
| t max (h)a | 15 | 0.500 | 0.500 | ‐ | ‐ | 0.0391 | |
| AUClast (h ng ml−1) | 15 | 118.749 | 102.676 | 1.157 (1.022–1.309) | 19.38 | 0.0637 | |
| AUCinf (h ng ml−1) | 15 | 125.665 | 110.472 | 1.138 (1.007–1.285) | 19.08 | 0.0833 | |
| t 1/2 (h) | 15 | 2.955 | 3.304 | ‐ | ‐ | 0.3591 | |
| 1‐hydroxymidazolam | C max (ng ml−1) | 15 | 42.235 | 56.292 | 0.750 (0.583–0.966) | 40.86 | 0.0754 |
| t max (h)a | 15 | 0.517 | 0.500 | ‐ | ‐ | 0.1758 | |
| AUClast (h ng ml−1) | 15 | 69.474 | 73.587 | 0.944 (0.805–1.107) | 25.09 | 0.6387 | |
| AUCinf (h ng ml−1) | 15 | 74.221 | 80.311 | 0.924 (0.791–1.080) | 24.63 | 0.4543 | |
| t 1/2 (h) | 15 | 0.678 | 0.728 | ‐ | ‐ | 0.5614 | |
| Fexofenadine | C max (ng ml−1) | 15 | 84.767 | 71.851 | 1.180 (0.979–1.422) | 29.62 | 0.2583 |
| t max (h)a | 15 | 2.000 | 2.000 | ‐ | ‐ | 0.7771 | |
| AUClast (h ng ml−1) | 15 | 594.590 | 449.875 | 1.322 (1.112–1.571) | 27.33 | 0.0181 | |
| AUCinf (h ng ml−1) | 15 | 611.566 | 467.806 | 1.307 (1.110–1.540) | 25.86 | 0.0181 | |
| t 1/2 (h) | 15 | 4.725 | 4.899 | ‐ | ‐ | 0.4543 | |
Values are presented by geometric mean and geometric mean ratio (90% CI)
AUCinf, area under the plasma concentration–time curve from 0 extrapolated to infinity; AUClast, area under the plasma concentration–time curve from time 0 to the last measurable time; CI, confidence interval; C max, maximum plasma concentration; CVw, intra‐individual coefficient of variation; FRG, fermented red ginseng; P‐gp, P‐glycoprotein; t 1/2, elimination half‐life; t max, time to maximum plasma concentration
t max was presented by median
Wilcoxon Signed‐Rank test
The point estimate (90% CI) of the ratio of the geometric means (after fermented red ginseng intake/before fermented red ginseng intake) of the C max and AUClast of caffeine was 1.002 (0.857–1.171) and 1.128 (0.997–1.277), respectively, and the corresponding point estimate (90% CI) of the caffeine metabolic ratio (i.e., paraxanthine AUC/caffeine AUC), which was used to evaluate CYP1A2 enzyme activity, was 0.901 (0.830–0.979).
The point estimate (90% CI) of the ratio of the geometric means (after fermented red ginseng intake/before fermented red ginseng intake) of the C max and AUClast of losartan was 0.974 (0.797–1.190) and 1.048 (0.887–1.239), respectively, and that of the losartan metabolic ratio, i.e., EXP3174 AUC/losartan AUC, which was used to evaluate CYP2C9 enzyme activity, was 0.774 (0.720–0.831).
For omeprazole, the point estimate (90% CI) of the ratio of the geometric means (after fermented red ginseng intake/before fermented red ginseng intake) of the C max and AUClast was 0.892 (0.747–1.066) and 1351.108, respectively. The corresponding point estimate (90% CI) of the omeprazole metabolic ratio, i.e., 5‐hydroxyomeprazole AUC/omeprazole AUC, which was used to evaluate CYP2C19 enzyme activity, was 1.052 (0.925–1.197).
The point estimate (90% CI) of the ratio of the geometric means (after fermented red ginseng intake/before fermented red ginseng intake) of the C max and AUClast of dextromethorphan was 0.849 (0.688–1.047) and 0.907 (0.681–1.209), respectively, and that of the dextromethorphan metabolic ratio, i.e., dextrorphan AUC/dextromethorphan AUC, which was used to evaluate CYP2D6 enzyme activity, was 1.150 (0.861–1.538).
The point estimate (90% CI) of the ratio of the geometric means (after fermented red ginseng intake/before fermented red ginseng intake) of the C max and AUClast of midazolam was 0.873 (0.732–1.041) and 1.157 (1.022–1.309), respectively, and that of the midazolam metabolic ratio, i.e., 1‐hydroxymidazolam AUC/midazolam AUC, which was used to evaluate CYP3A4 enzyme activity, was 0.816 (0.673–0.990).
Effects of fermented red ginseng on P‐glycoprotein
The mean C max and AUClast of fexofenadine were 71.9 ng ml−1 and 449.9 h∙ng ml−1, respectively, between Days 2 and 3 (before fermented red ginseng intake); between Days 16 and 17 (after fermented red ginseng intake), these values were 84.8 ng ml−1 and 594.6 h∙ng ml−1. The point estimate (90% CI) of the ratio of the geometric means (after fermented red ginseng intake/before fermented red ginseng intake) of the C max and AUClast of fexofenadine was 1.180 (0.979–1.422) and 1.322 (1.112–1.571), respectively.
Adverse event profile
The adverse events of fermented red ginseng and the probe drugs were evaluated in all 15 subjects. A total of 23 AEs was reported by 11 of the participants; these AEs included dizziness (eight events), euphoric mood (13 events), somnolence (one event), and xerophthalmia (one event). Symptoms such as dizziness, euphoric mood and somnolence are known to result from the use of midazolam. All AEs were mild, and all the test subjects who showed adverse sequelae recovered; serious AEs were not observed.
The severity of all AEs was either mild or moderate, and all were resolved without medical intervention. There were no significant differences in the occurrence or severity of AEs, including changes in vital signs or clinical laboratory evaluations during the clinical trials.
Discussion
We evaluated the effects of fermented red ginseng on the activity of CYP enzymes and P‐gp using the cocktail approach, which is commonly used to assess the effect of drugs on the activities of the main CYP450 enzymes in vivo 37.
In most cocktail studies, only the metabolite to parent drug concentration ratio at a specific time point is used to determine interactions. However, the metabolite to parent drug concentration ratio does not provide an accurate quantification of interaction effects on enzyme activity. The European Medicines Agency (EMA) guideline on the investigation of drug interactions recommends that drug interaction studies that use the cocktail approach conduct a full characterization of the plasma concentration–time curves of the probe drug to estimate the effect on (oral) clearance or AUC; use of the ratio of metabolite to parent drug concentration at a specific time point is generally not recommended. Therefore, in this study, the pharmacokinetic parameters were calculated based on the plasma concentration–time profiles. Furthermore, to ensure the treatment was sufficient to induce or inhibit the activity of the enzyme or transporter, the subjects were administered the recommended daily requirement of the fermented red ginseng product for two weeks. According to the EMA guidelines for cocktail studies, the use of a cocktail with an appropriate composition is extremely important, and studies should be conducted to determine the effects of the cocktail on CYP450 enzymes (or transporters). The probe drugs used here were selected according to previous cocktail studies adopting the EMA and FDA guidelines.
Since the ‘Pittsburgh cocktail’ was developed 38, many other cocktail methods have followed, including the ‘Cooperstown cocktail’ 39, the ‘Karolinska cocktail’ 40 and the ‘Inje cocktail’ 37. In this study, we used the Inje cocktail combination and added drugs for P‐gp to evaluate transport proteins along with the metabolic enzymes. All these drugs are established probe substrates that meet the criteria for cocktail drugs, including selectivity towards the respective CYP enzyme(s), the absence of interference with the metabolism and clearance of other drugs in the cocktail, safety and good tolerability. Caffeine has been studied as a probe drug to evaluate CYP1A2 enzyme activity because over 95% of it is metabolized to paraxanthine by CYP1A2 41. Losartan has been suggested as a highly specific and sensitive probe for CYP2C9 activity because EXP3174, a carboxylic acid metabolite of losartan, is specifically produced by CYP2C9‐mediated metabolism of losartan 42, 43. 5‐Hydroxyomeprazole and 5‐hydroxyomeprazole sulfone are produced by the metabolism of omeprazole by CYP2C19 and CYP3A4 44, 45. However, omeprazole has been widely used as a probe drug for CYP2C19. Dextromethorphan has been used as a probe drug for CYP2D6, and midazolam is the well‐established drug that is commonly used for the phenotyping of CYP3A activity 46, 47.
In a previous study, Yu et al. compared the effects of American ginseng (Panax quinquefolius) and Asian ginseng (Panax ginseng) extracts on gene expression of the hepatic P450 enzyme in adult rats and primary cultures or rat hepatocytes 48. They found no evidence of the induction of CYP2B1, CYP3A23 or CYP1A2 in rat cultures. In another study, ginseng had no effect on several CYP isoforms, including CYP3A4, CYP1A2, CYP2E1 and CYP2D6 49, 50. However, in elderly humans, CYP2D was found to be slightly inhibited by ginseng 49. Gurley et al. found that ginseng administration (500 mg three times daily for 28 days) had no apparent effect on CYP1A2, CYP2D6, CYP2E1 and CYP3A4 activity in 12 healthy participants 49, 50. In a study of ginseng administration (4% ginsenosides, 100 mg twice daily for 24 days) in healthy volunteers, Anderson et al. also reported that ginseng did not modulate CYP3A activity 51. However, the investigation by Malati et al. of 12 healthy volunteers showed that ginseng administration (5% ginsenosides, 500 mg twice daily for 28 days) induced CYP3A activity 31.
In the current study, we found that fermented red ginseng administration inhibited CYP1A2, CYP2C9 and CYP3A4 activity but not CYP2C19 or CYP2D6 activity. Discrepancies between the results of this study and others may be because the content of absorbable ginsenosides such as ginsenoside Rh2, compound K, PPD and PPT is higher in fermented red ginseng than in ginseng. Liu et al. found that PPT and PPD, the deglycosylation products of naturally occurring ginsenosides, exhibited potent inhibition against CYP3A4 activity, and compound K and ginsenoside Rh2, which are more concentrated in fermented red ginseng than in red ginseng or fresh ginseng, also exhibited moderate inhibition of CYP2C9 and CYP3A4 activity 52. Moreover, many in vitro studies have supported the idea that the ginsenosides, including ginsenoside Rb1, Rb2, Rc and Rg1, in fresh ginseng do not affect CYP enzyme activity but that metabolites of these inhibit CYP1A2, CYP2C9 and CYP3A4 48, 52. However, in this study, oral administration of fermented red ginseng inhibited CYP1A2 enzyme activity, but this effect is unlikely to cause significant interactions. Moreover, fermented red ginseng did not significantly affect CYP2C19. CYP2D6 enzyme activity may have been induced, but the corresponding coefficient of variation was large, and the results of the nonparametric tests did not reach significance. Because, unlike most other CYP450 enzymes, CYP2D6 is not very susceptible to enzyme induction, the probability of CYP2D6 enzyme induction is likely very low 53, 54. CYP3A4 enzyme activity was inhibited by fermented red ginseng, but the effect is unlikely to be significantly different because of its small magnitude. We observed a statistically significant decrease in losartan mean plasma ratios after fermented red ginseng administration, indicating that fermented red ginseng intake inhibited CYP2C9 enzyme activity; however, this minor inhibitory effect on CYP2C9 is not likely to be significantly different because of its small magnitude. Therefore, fermented red ginseng is not likely to influence concomitantly administered CYP probe drugs in a significantly meaningful way because its inhibitory effects were small when it was administered according to dietary recommendations.
Fexofenadine has been used as a probe to assess P‐gp transport and provides a broad measure of membrane transporter activity. In this study, the AUClast value of fexofenadine after the administration of fermented red ginseng was greater than that before. We have shown that the administration of fermented red ginseng can increase exposure to the concomitantly administered drug. Such increased exposure might alter the drug's effectiveness. The administration of fermented red ginseng is therefore likely to alter the effects of fexofenadine. However, in this study, the possible influence of other substrates, including organic anion‐transporting polypeptides (OATPs), over that of fexofenadine could not be ruled out. Differences between the results of previous studies and the current findings were expected due to the differences between fresh ginseng and fermented red ginseng products that result from the processing of ginseng and because of differences in the race of the study subjects.
A limitation of this study is the small sample size of healthy subjects. Studies with a large sample size of patient subjects are needed to confirm the pharmacokinetic and pharmacodynamic effects of ginseng–drug interactions.
This study was able to accurately assess potential drug interactions and minimize confounding factors, such as comorbidities and concomitant medications, because it was conducted with healthy volunteers. However, due to variation in the composition of fermented red ginseng products that result from the manufacturing process, it may not be possible to expect the same results for all ginseng products. Moreover, unlike medicines, fermented red ginseng is generally consumed at levels higher than the recommended dose. Therefore, it is important to assess ginseng–drug interactions in humans where high doses of ginseng are involved.
Conclusion
In summary, the results showed that multiple doses of a fermented red ginseng product weakly inhibited CYP2C9, CYP3A4 and P‐gp. However, no significant drug interactions between fermented red ginseng and the CYP and P‐gp probe substrates were observed. Therefore, the potential of this product to cause metabolic and transport drug interactions is low.
This study was supported by funding from the Biomedical Research Institute of Chonbuk National University Hospital. Clinical trial registration number (ClinicalTrials.gov): NCT02056743.
Competing Interests
The authors have declared that there are no conflicts of interest.
Kim, M. ‐G. , Kim, Y. , Jeon, J. ‐Y. , and Kim, D. ‐S. (2016) Effect of fermented red ginseng on cytochrome P450 and P‐glycoprotein activity in healthy subjects, as evaluated using the cocktail approach. Br J Clin Pharmacol, 82: 1580–1590. doi: 10.1111/bcp.13080.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al. The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 2015; 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jia L, Zhao Y. Current evaluation of the millennium phytomedicine – ginseng (I): etymology, pharmacognosy, phytochemistry, market and regulations. Curr Med Chem 2009; 16: 2475–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yun TK. Brief introduction of Panax ginseng C.A. Meyer. J Korean Med Sci 2001; 16 (Suppl): S3–S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Park JD. Recent studies on the chemical constituents of Korean ginseng (Panax ginseng C. A. Meyer). J Ginseng Res 1996; 20: 389–415. [Google Scholar]
- 7. Wang CZ, Anderson S, Du W, He TC, Yuan CS. Red ginseng and cancer treatment. Chin J Nat Med 2016; 14: 7–16. [DOI] [PubMed] [Google Scholar]
- 8. Sun S, Qi LW, Du GJ, Mehendale SR, Wang CZ, Yuan CS. Red notoginseng: higher ginsenoside content and stronger anticancer potential than Asian and American ginseng. Food Chem 2011; 125: 1299–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Andrade E, de Mesquita AA, Claro Jde A, de Andrade PM, Ortiz V, Paranhos M, et al. Study of the efficacy of Korean Red Ginseng in the treatment of erectile dysfunction. Asian J Androl 2007; 9: 241–244. [DOI] [PubMed] [Google Scholar]
- 10. Sung J, Han KH, Zo JH, Park HJ, Kim CH, Oh BH. Effects of red ginseng upon vascular endothelial function in patients with essential hypertension. Am J Chin Med 2000; 28: 205–216. [DOI] [PubMed] [Google Scholar]
- 11. Lee MS, Kim CT, Kim IH, Kim Y. Effects of Korean Red Ginseng extract on hepatic lipid accumulation in HepG2 cells. Biosci Biotechnol Biochem 2015; 79: 816–819. [DOI] [PubMed] [Google Scholar]
- 12. Cheema D, Coomarasamy A, El‐Toukhy T. Non‐hormonal therapy of post‐menopausal vasomotor symptoms: a structured evidence‐based review. Arch Gynecol Obstet 2007; 276: 463–469. [DOI] [PubMed] [Google Scholar]
- 13. Kim DS, Chang YJ, Zedk U, Zhao P, Liu YQ, Yang CR. Dammarane saponins from Panax ginseng . Phytochemistry 1995; 40: 1493–1497. [DOI] [PubMed] [Google Scholar]
- 14. Attele AS, Wu JA, Yuan CS. Ginseng pharmacology: multiple constituents and multiple actions. Biochem Pharmacol 1999; 58: 1685–1693. [DOI] [PubMed] [Google Scholar]
- 15. Harkey MR, Henderson GL, Gershwin ME, Stern JS, Hackman RM. Variability in commercial ginseng products: an analysis of 25 preparations. Am J Clin Nutr 2001; 73: 1101–1106. [DOI] [PubMed] [Google Scholar]
- 16. Liu H, Yang J, Du F, Gao X, Ma X, Huang Y, et al. Absorption and disposition of ginsenosides after oral administration of Panax notoginseng extract to rats. Drug Metab Dispos 2009; 37: 2290–2298. [DOI] [PubMed] [Google Scholar]
- 17. Nocerino E, Amato M, Izzo AA. The aphrodisiac and adaptogenic properties of ginseng. Fitoterapia 2000; 71 (Suppl 1): S1–S5. [DOI] [PubMed] [Google Scholar]
- 18. Wakabayashi C, Murakami K, Hasegawa H, Murata J, Saiki I. An intestinal bacterial metabolite of ginseng protopanaxadiol saponins has the ability to induce apoptosis in tumor cells. Biochem Biophys Res Commun 1998; 246: 725–730. [DOI] [PubMed] [Google Scholar]
- 19. Lee JY, Shin JW, Chun KS, Park KK, Chung WY, Bang YJ, et al. Antitumor promotional effects of a novel intestinal bacterial metabolite (IH‐901) derived from the protopanaxadiol‐type ginsenosides in mouse skin. Carcinogenesis 2005; 26: 359–367. [DOI] [PubMed] [Google Scholar]
- 20. Matsunaga H, Katano M, Yamamoto H, Mori M, Takata K. Studies on the panaxytriol of Panax ginseng C. A. Meyer: isolation, determination and antitumor activity. Chem Pharm Bull 1989; 37: 1279–1281. [DOI] [PubMed] [Google Scholar]
- 21. Park EK, Shin YW, Lee HU, Kim SS, Lee YC, Lee BY, et al. Inhibitory effect of ginsenoside Rb1 and compound K on NO and prostaglandin E2 biosyntheses of RAW264.7 cells induced by lipopolysaccharide. Biol Pharm Bull 2005; 28: 652–656. [DOI] [PubMed] [Google Scholar]
- 22. Cuong TT, Yang CS, Yuk JM, Lee HM, Ko SR, Cho BG, et al. Glucocorticoid receptor agonist compound K regulates Dectin‐1‐dependent inflammatory signaling through inhibition of reactive oxygen species. Life Sci 2009; 85: 625–633. [DOI] [PubMed] [Google Scholar]
- 23. Han GC, Ko SK, Sung JH, Chung SH. Compound K enhances insulin secretion with beneficial metabolic effects in db/db mice. J Agric Food Chem 2007; 55: 10641–10648. [DOI] [PubMed] [Google Scholar]
- 24. Yoon SH, Han EJ, Sung JH, Chung SH. Anti‐diabetic effects of compound K versus metformin versus compound K‐metformin combination therapy in diabetic db/db mice. Biol Pharm Bull 2007; 30: 2196–2200. [DOI] [PubMed] [Google Scholar]
- 25. Bae EA, Choo MK, Park EK, Park SY, Shin HY, Kim DH. Metabolism of ginsenoside R(c) by human intestinal bacteria and its related antiallergic activity. Biol Pharm Bull 2002; 25: 743–747. [DOI] [PubMed] [Google Scholar]
- 26. Choo MK, Park EK, Han MJ, Kim DH. Antiallergic activity of ginseng and its ginsenosides. Planta Med 2003; 69: 518–522. [DOI] [PubMed] [Google Scholar]
- 27. Quan LH, Piao JY, Min JW, Kim HB, Kim SR, Yang DU, et al. Biotransformation of ginsenoside Rb1 to prosapogenins, gypenoside XVII, ginsenoside Rd, ginsenoside F2, and compound K by Leuconostoc mesenteroides DC102. J Ginseng Res 2011; 35: 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee J, Lee E, Kim D, Lee J, Yoo J, Koh B. Studies on absorption, distribution and metabolism of ginseng in humans after oral administration. J Ethnopharmacol 2009; 122: 143–148. [DOI] [PubMed] [Google Scholar]
- 29. Lee HS, Kim MR, Park Y, Park HJ, Chang UJ, Kim SY, et al. Fermenting red ginseng enhances its safety and efficacy as a novel skin care anti‐aging ingredient: in vitro and animal study. J Med Food 2012; 15: 1015–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee EJ, Song MJ, Kwon HS, Ji GE, Sung MK. Oral administration of fermented red ginseng suppressed ovalbumin‐induced allergic responses in female BALB/c mice. Phytomedicine 2012; 19: 896–903. [DOI] [PubMed] [Google Scholar]
- 31. Malati CY, Robertson SM, Hunt JD, Chairez C, Alfaro RM, Kovacs JA, et al. Influence of Panax ginseng on cytochrome P450 (CYP)3 A and P‐glycoprotein (P‐gp) activity in healthy participants. J Clin Pharmacol 2012; 52: 932–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Budzinski JW, Foster BC, Vandenhoek S, Arnason JT. An in vitro evaluation of human cytochrome P450 3 A4 inhibition by selected commercial herbal extracts and tinctures. Phytomedicine 2000; 7: 273–282. [DOI] [PubMed] [Google Scholar]
- 33. Li N, Wang D, Ge G, Wang X, Liu Y, Yang L. Ginsenoside metabolites inhibit P‐glycoprotein in vitro and in situ using three absorption models. Planta Med 2014; 80: 290–296. [DOI] [PubMed] [Google Scholar]
- 34. Zhang J, Zhou F, Wu X, Gu Y, Ai H, Zheng Y, et al. 20(S)‐ginsenoside Rh2 noncompetitively inhibits P‐glycoprotein in vitro and in vivo: a case for herb–drug interactions. Drug Metab Dispos 2010; 38: 2179–2187. [DOI] [PubMed] [Google Scholar]
- 35. Oh KS, Park SJ, Shinde DD, Shin JG, Kim DH. High‐sensitivity liquid chromatography–tandem mass spectrometry for the simultaneous determination of five drugs and their cytochrome P450‐specific probe metabolites in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 2012; 895‐896: 56–64. [DOI] [PubMed] [Google Scholar]
- 36. Fuhr U, Jetter A, Kirchheiner J. Appropriate phenotyping procedures for drug metabolizing enzymes and transporters in humans and their simultaneous use in the ‘cocktail’ approach. Clin Pharmacol Ther 2007; 81: 270–283. [DOI] [PubMed] [Google Scholar]
- 37. Ryu JY, Song IS, Sunwoo YE, Shon JH, Liu KH, Cha IJ, et al. Development of the ‘Inje cocktail’ for high‐throughput evaluation of five human cytochrome P450 isoforms in vivo . Clin Pharmacol Ther 2007; 82: 531–540. [DOI] [PubMed] [Google Scholar]
- 38. Frye RF, Matzke GR, Adedoyin A, Porter JA, Branch RA. Validation of the five‐drug ‘Pittsburgh cocktail’ approach for assessment of selective regulation of drug‐metabolizing enzymes. Clin Pharmacol Ther 1997; 62: 365–376. [DOI] [PubMed] [Google Scholar]
- 39. Streetman DS, Bleakley JF, Kim JS, Nafziger AN, Leeder JS, Gaedigk A, et al. Combined phenotypic assessment of CYP1A2, CYP2C19, CYP2D6, CYP3A, N‐acetyltransferase‐2, and xanthine oxidase with the ‘Cooperstown cocktail’. Clin Pharmacol Ther 2000; 68: 375–383. [DOI] [PubMed] [Google Scholar]
- 40. Zhu B, Ou‐Yang DS, Chen XP, Huang SL, Tan ZR, He N, et al. Assessment of cytochrome P450 activity by a five‐drug cocktail approach. Clin Pharmacol Ther 2001; 70: 455–461. [DOI] [PubMed] [Google Scholar]
- 41. Hakooz NM. Caffeine metabolic ratios for the in vivo evaluation of CYP1A2, N‐acetyltransferase 2, xanthine oxidase and CYP2A6 enzymatic activities. Curr Drug Metab 2009; 10: 329–338. [DOI] [PubMed] [Google Scholar]
- 42. Sica DA, Gehr TW, Ghosh S. Clinical pharmacokinetics of losartan. Clin Pharmacokinet 2005; 44: 797–814. [DOI] [PubMed] [Google Scholar]
- 43. Yasar U, Tybring G, Hidestrand M, Oscarson M, Ingelman‐Sundberg M, Dahl ML, et al. Role of CYP2C9 polymorphism in losartan oxidation. Drug Metab Dispos 2001; 29: 1051–1056. [PubMed] [Google Scholar]
- 44. Karam WG, Goldstein JA, Lasker JM, Ghanayem BI. Human CYP2C19 is a major omeprazole 5‐hydroxylase, as demonstrated with recombinant cytochrome P450 enzymes. Drug Metab Dispos 1996; 24: 1081–1087. [PubMed] [Google Scholar]
- 45. Andersson T, Miners JO, Veronese ME, Tassaneeyakul W, Tassaneeyakul W, Meyer UA, et al. Identification of human liver cytochrome P450 isoforms mediating omeprazole metabolism. Br J Clin Pharmacol 1993; 36: 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mäenpää J, Hall SD, Ring BJ, Strom SC, Wrighton SA. Human cytochrome P450 3A (CYP3A) mediated midazolam metabolism: the effect of assay conditions and regioselective stimulation by alpha‐naphthoflavone, terfenadine and testosterone. Pharmacogenetics 1998; 8: 137–155. [PubMed] [Google Scholar]
- 47. Jacqz‐Aigrain E, Funck‐Brentano C, Cresteil T. CYP2D6‐ and CYP3A‐dependent metabolism of dextromethorphan in humans. Pharmacogenetics 1993; 3: 197–204. [DOI] [PubMed] [Google Scholar]
- 48. Yu CT, Chen J, Teng XW, Tong V, Chang TK. Lack of evidence for induction of CYP2B1, CYP3A23, and CYP1A2 gene expression by Panax ginseng and Panax quinquefolius extracts in adult rats and primary cultures of rat hepatocytes. Drug Metab Dispos 2005; 33: 19–22. [DOI] [PubMed] [Google Scholar]
- 49. Gurley BJ, Gardner SF, Hubbard MA, Williams DK, Gentry WB, Cui Y, et al. Clinical assessment of effects of botanical supplementation on cytochrome P450 phenotypes in the elderly: St John's wort, garlic oil, Panax ginseng and Ginkgo biloba . Drugs Aging 2005; 22: 525–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gurley BJ, Gardner SF, Hubbard MA, Williams DK, Gentry WB, Cui Y, et al. Cytochrome P450 phenotypic ratios for predicting herb–drug interactions in humans. Clin Pharmacol Ther 2002; 72: 276–287. [DOI] [PubMed] [Google Scholar]
- 51. Anderson GD, Rosito G, Mohustsy MA, Elmer GW. Drug interaction potential of soy extract and Panax ginseng . J Clin Pharmacol 2003; 43: 643–648. [PubMed] [Google Scholar]
- 52. Liu Y, Zhang JW, Li W, Ma H, Sun J, Deng MC, et al. Ginsenoside metabolites, rather than naturally occurring ginsenosides, lead to inhibition of human cytochrome P450 enzymes. Toxicol Sci 2006; 91: 356–364. [DOI] [PubMed] [Google Scholar]
- 53. Eichelbaum M, Mineshita S, Ohnhaus EE, Zekorn C. The influence of enzyme induction on polymorphic sparteine oxidation. Br J Clin Pharmacol 1986; 22: 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schellens JH, van der Wart JH, Brugman M, Breimer DD. Influence of enzyme induction and inhibition on the oxidation of nifedipine, sparteine, mephenytoin and antipyrine in humans as assessed by a ‘cocktail’ study design. J Pharmacol Exp Ther 1989; 249: 638–645. [PubMed] [Google Scholar]