

Abstract
Aim
In 2006, Omnitrope (by Sandoz) was the first approved biosimilar in Europe. To date, 21 biosimilars for seven different biologics are on the market. The present study compared the clinical trials undertaken to obtain market authorization.
Methods
We summarized the findings of a comprehensive review of all clinical trials up to market authorization of approved biosimilars, using the European public assessment reports (EPARs) published by the European Medicines Agency (EMA). The features compared were, among others, the number of patients enrolled, the number of trials, the types of trial design, choice of endpoints and equivalence margins for pharmacokinetic (PK)/pharmacodynamic (PD) and phase III trials.
Results
The variability between the clinical development strategies is high. Some differences are explainable by the characteristics of the product; if, for example, the PD marker can be assumed to predict the clinical outcome, no efficacy trials might be necessary. However, even for products with the same reference product, the sample size, endpoints and statistical models are not always the same.
Conclusions
There seems to be flexibility for sponsors regarding the decision as to how best to prove biosimilarity.
Keywords: biosimilar drug development programmes, biosimilarity, biosimilars, EMA, EPAR, trial design
Tables of Links
| TARGETS | |
|---|---|
| Catalytic receptors 2 | Enzymes 3 |
| Erythropoietin receptor | Insulin receptor |
| Growth hormone receptor | G protein‐coupled receptors 4 |
| Tumour necrosis factor receptor | Follicle stimulating hormone receptor |
| Granulocyte colony‐stimulating factor |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2, 3, 4.
Introduction
Biologics have revolutionized treatment in important disease areas, such as cancer, diabetes and inflammatory diseases. The downside of the use of biologics is the high cost; in 2002, $46 billion were spent on biologics worldwide and it is expected that this will increase to $221 billion in 2017 5.
Due to the high prices and the first expiry of patents of biologics over the last few years, the development of copies of biologics, so‐called biosimilars, has recently gained much attention. The European Medicines Agency (EMA) is the leading regulator in this regard, having approved the first biosimilar in 2006 (Omnitrope, by Sandoz), and since then, the landscape of authorized biosimilars in Europe has widened considerably. Currently, there are 21 products for seven different biologics on the market. It is predicted that the use of biosimilars may lead to a $250 billion reduction in spending on biologics in the US between 2014 and 2024 6.
So far, there is no unified definition of biosimilars that is accepted by all regulatory agencies. The EMA 7 defines a biosimilar as: ‘a biological medicinal product that contains a version of the active substance of an already authorized original biological medicinal product (reference medicinal product). A biosimilar demonstrates similarity to the reference medicinal product in terms of quality characteristics, biological activity, safety and efficacy based on a comprehensive comparability exercise’. There is also a need to distinguish between biologics and nonbiological complex medicinal products, as described recently 8.
Unlike chemically defined ‘simple’ molecules, as usually employed in e.g. oral generics, biosimilar active substances do not meet this general definition of the ‘same active substance’ because some characteristics are very sensitive to the manufacturing process, which cannot be completely duplicated. This was also stated by the Directive 2001/83/EC of the European Parliament and of the Council (Article 10) 9. The more complex structure and the larger molecule size of biologics makes them more complicated to develop. This complexity results in an extended approval process, which involves large clinical trials in addition to a comprehensive analytical and nonclinical analysis. In contrast to the approval process for generics, for which showing bioequivalence of pharmacokinetic (PK) parameters is considered most sensitive to substantiate therapeutic equivalence, in most cases phase III trials are additionally demanded for showing biosimilarity 10. From a regulatory perspective, biosimilars are therefore not accepted as generic applications.
This particularity has also led the EMA to consider that the standardized approach that is used for generics is not feasible for biosimilars 7. Therefore, it can be expected that the development programmes of the drug sponsors will differ. Wang and Chow 11 compared the properties of the PK and pharmacodynamic (PD) studies involved in the approval of the six biosimilars that were available at that time. They found considerable variability in the equivalence assessments – e.g. the amount of PD analysis needed ranged from no analysis at all to complex analyses involving formal testing procedures. However, the focus of their paper was on PK and PD studies only. Several review papers have also focused on biologics that are biosimilar to a specific active substance – e.g. on epoetin 12, 13, 14, 15, filgrastim 16 and infliximab 17. To our knowledge, no overall comparison of the clinical development programmes for all 21 marketed biosimilars has yet been published. In the current review, we present the results of a comparison of the PK, PD and phase III trials for all marketed biosimilars in Europe.
Methods
The main resources for this project were the European public assessment reports (EPARs) that were published by the EMA 18 and are available online 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39. If not stated otherwise, the information presented here is taken from these reports. Only studies up to the date of market authorization were considered; postmarketing commitments were not analyzed.
If information was missing in the EPARs or details of the studies were not clear, an online search was conducted using PubMed, ISI's web of science, clinicaltrials.gov, EudraCT and Google Scholar, using keywords such as the drug name and the sponsor, the international nonproprietary name and the sponsor or the trial identifier of the sponsor.
The properties of the submitted studies were compared with those described in the available EMA guidelines for the assessment of biosimilarity 7, 40 and with the product‐specific guidelines. The product‐specific guidelines take into account the characteristics of the substance and give detailed recommendations for the trial design 41.
The present study analyzed the clinical trials of authorized biosimilars only. Neither products that were withdrawn before the decision of the Committee for Medicinal Products for Human Use (CHMP) (e.g. Isomarv, Marvel LifeSciences Ltd 42) nor products that were withdrawn after market authorization (e.g. Filgrastim ratiopharm, Ratiopharm GmbH 43) were considered. Products for which the CHMP refused approval were also not analyzed. At the time of the study, this was the case for two products: Solumarv (Marvel Lifesciences Ltd 44) and Alpheon (BioPartners GmbH 45). This left a total of 21 applications of biosimilars to be considered.
Not all of these 21 biosimilars are, in fact, different products; some companies conducted a joint development programme but marketed a product under different brand names. For example, for the active substance infliximab, Stada Arzneimittel AG and Hospira UK Ltd submitted a joint application. This product is sold by Stada Arzneimittel AG as Silapo, whereas the Hospira UK Ltd product is called Retacrit. As the submitted clinical trials were identical, the application was considered as a single project in the current study, and the different products are separated with a slash – e.g. Silapo/Retacrit.
For assessing the number of trials, it should be noted that if a trial had an extension using exactly the same set of patients, this was not counted in the current study as a separate trial. Studies that were listed in the EPAR but did not involve the test product (e.g. studies comparing a US and an EU product – i.e. for the biosimilars Benepali and Abasaglar) were not considered in the current study.
Results
At the time of the study, there were 21 biosimilars (resulting from 13 different applications) on seven different biologics available on the European Market. Table 1 shows an overview of the reference products for the approved biosimilars and their mechanisms of action. These drugs act endocrinologically, to stimulate body growth or oozyte maturation, and include insulin‐like growth factor to boost erythropoiesis or granulopoiesis, and the more recently developed immunosuppressive antibodies that inhibit the cytokine tumour necrosis factor‐alpha.
Table 1.
Overview of biologics for which a biosimilar is authorized in Europe
| Active substance | Originator drug name | Originator company | Mechanism of action |
|---|---|---|---|
| Haematopoietic growth factors | |||
| Epoetin alfa/zeta | Eprex (EU), Erypo (Germany) | Janssen/Ortho Biotech | The active substance, epoetin alfa, is a copy of a hormone called erythropoietin, and works in exactly the same way as the natural hormone to stimulate the production of red blood cells from the bone marrow |
| Filgrastim | Neupogen | Amgen/Roche | Filgrastim acts in the same way as naturally produced granulocyte colony‐stimulating factor by encouraging the bone marrow to produce more white blood cells |
| Endocrinologically acting drugs | |||
| Follitropin alfa | Gonal‐f | Merck Serono Europe | The active substance in Gonal‐f, follitropin alfa, is a copy of the natural hormone, follicle‐stimulating hormone (FSH). In the body, FSH controls reproductive function: in women, it stimulates the production of eggs; and in men, it stimulates the production of sperm in the testicles |
| Insulin glargine | Lantus | Sanofi‐Aventis | This is a replacement insulin that is very similar to the insulin made by the body. The replacement insulin acts in the same way as naturally produced insulin and helps glucose to enter cells from the blood |
| Somatropin | Genotropin | Pfizer | This promotes growth during childhood and adolescence, and also affects the way that the body handles proteins, fat and carbohydrates. The active substance, somatropin, is identical to the human growth hormone |
| Anti‐inflammatory blockers of tumour necrosis factor alpha | |||
| Etanercept | Enbrel | Pfizer | The active substance, etanercept, is a protein that has been designed to block the activity of a chemical messenger in the body called tumour necrosis factor (TNF) |
| Infliximab | Remicade | Janssen | The active substance in Remicade, infliximab, is a monoclonal antibody. A monoclonal antibody is an antibody (a type of protein) that has been designed to recognize and attach to a specific structure (called an antigen) in the body. Infliximab has been designed to attach to a chemical messenger in the body called TNF‐alpha. This messenger is involved in causing inflammation and is found at high levels in patients with the diseases that Remicade is used to treat. By blocking TNF‐alpha, infliximab improves the inflammation and other symptoms of these diseases |
The mechanism of action is quoted with only minor modifications from the ‘EPAR – Summaries for the public’ available in the EPAR database of EMA 18
Indications and extrapolation
In most cases, the indications applied for are essentially the same as those of the reference product. Some restrictions were stated when considering the active substances epoetin alfa and epoetin zeta, which are both types of erythropoietin; for Silapo/Retacrit (biosimilar to epoetin zeta), the indication of ‘reduction in allogeneic blood transfusions in adult non‐iron‐deficient patients prior to major elective orthopaedic surgery’ was not granted owing to the lack of shown equivalence for the subcutaneous (SC) administration route. Epoetin Alfa Hexal/Abseamed/Binocrit (biosimilar to epoetin alfa) is not indicated for ‘increasing the yield of autologous blood from patients in a predonation programme’. More detailed information on the indications is available in Table S1.
In most cases, only one phase III trial in one indication was submitted for authorization and it was assumed that the results could be generalized to all other indications with the same route of administration. For example, for Remsima/Inflectra (active substance infliximab), the phase III trial was undertaken in patients with active rheumatoid arthritis 46, but the product is licensed for all indications of the reference product, including rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis, adult and paediatric Crohn's disease, and adult and paediatric ulcerative colitis. In the EPAR, it is stated that the sponsor provided a literature review of the therapeutic indication and also of the mechanism of action of the drug. According to the report, the sponsor also presented preliminary data on 23 patients with either Crohn's disease or ulcerative colitis (no details were given in the EPAR) and agreed to conduct additional postmarketing studies. Using all submitted information, the regulators allowed the company an extension to all indications. More background information about the extrapolation of Remsima/Inflectra can be found in the study by Reinisch et al. 47.
Although two active substances (filgrastim, somatropin) received orphan drug designations for specific indications 48, none of the biosimilars had orphan drug status according to the EPAR database of the EMA 18.
PK/PD‐ vs. phase III trials
Table 2 shows an overview of the study populations enrolled both in the PK or PD trials and in the phase III trials. A trial was counted as a PK/PD trial in the current study only if the primary endpoint was a measure of PK or PD. A trial with efficacy as the primary endpoint and additional PK assessments was therefore not listed as a PK/PD trial.
Table 2.
Population size and number of trials for assessing biosimilarity
| Active substance | Product | Company | Number of subjects in PK/PD studies (P: patients, V: volunteers) | Number of PK/PD studies | Number of patients in phase III studies | Number of phase III studies | Compared with reference product in phase III studies? |
|---|---|---|---|---|---|---|---|
| Epoetin alfa/zeta | Silapo/Retacrit | Stada Arzneimittel AG/ Hospira UK Limited | 72 (V) | 2 | 1272 | 3 | Yes |
| Epoetin Alfa Hexal/Abseamed/Binocrit | Hexal/Medice Arzneimittel Puetter/Sandoz | 234 (V) | 5 | 592 | 2 | Yes | |
| Filgrastim | Zarzio/Filgrastim Hexal | Sandoz/Hexal | 146 (V) | 4 | 170 | 1 | No |
| Tevagrastim/Ratiograstim/Biograstim | Teva Generics/Ratiopharm/ABZ‐Pharma | 200 (V) | 2 | 677 | 3 | Yes | |
| Nivestim | Hospira UK Ltd | 92 (V) | 2 | 279 | 1 | Yes | |
| Grastofil/Accofil | Apotex Europe BV/Accord Healthcare | 215 (V) | 4 | 120 | 1 | No | |
| Follitropin alfa | Ovaleap | Teva Pharma | 76 (V) | 2 | 299 | 1 | Yes |
| Bemfola | Finox Biotech | 24 (V) | 1 | 273 | 1 | Yes | |
| Insulin glargine | Abasaglar | Eli Lily | 211, 20 (V, P) | 5 | 1295 | 2 | Yes |
| Somatropin | Omnitrope | Sandoz | 61 (V) | 3 | 140 | 2 | Yes |
| Etanercept | Benepali | Samsung Bioepis UK Limited | 138 (V) | 1 | 596 | 1 | Yes |
| Infliximab | Remsima/Inflectra | Celltrion Healthcare/Hospira UK Limited | 269 (P) | 2 | 606 | 1 | Yes |
| Flixabi | Samsung Bioepis UK Limited | 159 (V) | 1 | 584 | 1 | Yes |
The sponsors conducted between one and five PK/PD trials with, in total, 24–269 patients involved. This is shown in Figure 1. The PK/PD trials were mostly undertaken in healthy volunteers; for the application of Remsima/Inflectra, only patients were used, and for insulin glargine, there were 20 patients enrolled, in addition to 211 volunteers. For insulin glargine, the additional use of patients was requested by the CHMP according to the EPAR. While no specific reasons are mentioned in the publically available documents for Remsima/Inflectra, this product has specific and potentially severe adverse effects, which make extended testing of infliximab in healthy volunteers appear undesirable. For example, infliximab may induce anaphylaxis 49 and reactivate tuberculosis 50, 51.
Figure 1.
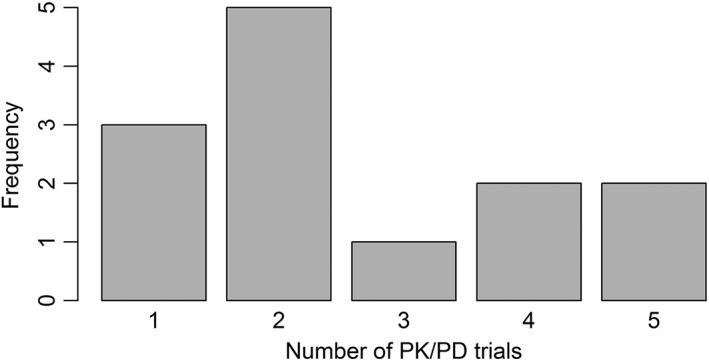
Overview of the number of pharmacokinetic/pharmacodynamic (PK/PD) trials undertaken for obtaining approval as a biosimilar. A trial is counted as a PK/PD trial if the primary endpoint is PK or PD
Between one and three phase III trials were submitted, with between 120 and 1295 patients enrolled. Particularly small sample sizes were observed for Zarzio/Filgrastim Hexal (170 subjects), Grastofil/Accofil (120 subjects) and Omnitrope (140 subjects). While the applications for the first two were focused on PK/PD‐trials (see discussion below), the studies for the latter were done in children, which might explain the small sample size.
The relative size of the PK/PD trials in comparison with phase III trials also varied considerably – i.e. the ratio of the number of patients and volunteers in PK/PD trials to the number of patients in phase III trials ranged from 1.79 (for Grastofil/Accofil) to 0.06 (for Silapo/Retacrit). For the approval of Grastofil/Accofil (active substance filgrastim), 215 subjects took part in four PK/PD studies 52, whereas only 72 volunteers participated in two trials with Silapo/Retacrit (active substance epoetin zeta) 53, 54. By contrast, in phase III studies, the sponsors conducted three efficacy trials for Silapo/Retacrit, with more than 1000 patients, but only a single‐arm trial (no comparison with reference), with 120 subjects, for Grastofil/Accofil.
It should be taken into account that Silapo/Retacrit and Grastofil/Accofil have different reference products, with different indications. One explanation for the variation in sample size is that if the PD marker is predictive for the clinical efficacy outcome, the product‐specific guidelines allow the possibility of replacing larger phase III trials with PK/PD trials. The product‐specific guideline for Grastofil/Accofil is about products containing granulocyte colony‐stimulating factor 55. There, it is said in the context of the assessment of efficacy that: ‘alternative models, including PD studies in healthy volunteers, may be pursued for the demonstration of comparability if justified’. This will also be reiterated more specifically in the planned revision of that guideline 56.
However, there are also product‐specific guidelines describing the surrogate markers that should not be used for efficacy comparison. For example, the guidelines for products containing recombinant erythropoietins 57 mention that the recommended PD marker reticulocyte count ‘is not an established surrogate marker for the efficacy of epoetin and therefore not a suitable endpoint in clinical trials’.
Even for one active substance, the splitting of resources into phase III and PK/PD studies may not be the same, which is illustrated by the four filgrastim applications Grastofil/Accofil, Zarzio/Filgrastim Hexal, Tevagrastim/Ratiograstim/Biograstim and Nivestim. For Grastofil/Accofil and Zarzio/Filgrastim Hexal, only single‐arm trials were undertaken in phase III. Therefore, no direct head‐to‐head comparison of clinical efficacy with a reference product was performed. The applications for Tevagrastim/Ratiograstim/Biograstim and Nivestim contained comparative trials in phase III – e.g. three phase III trials, involving a total of 677 subjects, were carried out for Tevagrastim/Ratiograstim/Biograstim. Therefore, the EMA allows some flexibility for the sponsors to decide which approach they can choose for proving biosimilarity.
Trial design
Table 3 shows the details of the submitted studies for all products. It indicates the assessments for which they were used (efficacy, safety, PK, PD). A trial is considered as part of the assessments in these categories if the results of the trial are discussed in the relevant sections in the EPAR. As safety is evaluated in nearly all studies, we focused on the efficacy, PK and PD studies.
Table 3.
Overview of study designs that were used in the clinical development programme for obtaining approval as a biosimilar. Only studies undertaken prior to market authorization are listed
| Active substance | Product | Study design | N | Single/multiple dose | Route of administration, dose | PK | PD | E | S |
|---|---|---|---|---|---|---|---|---|---|
| Epoetin alfa/zeta | Silapo/Retacrit | 2 × 2 crossover | 24 | Single | IV, dose n.s. | X | – | – | – |
| 3‐period crossover | 48 | Single | IV, SC, dose n.s. | X | – | – | – | ||
| Parallel groupa | 609 | Multiple | IV, 1000 IU or 2000 IU 3 t.i.w.13 | – | – | X | X | ||
| 2 × 2 crossovera | 402 | Multiple | IV, 1000 IU or 2000 IU 1–3 t.i.w. | – | – | X | X | ||
| Single‐arma | 261 | Multiple | n.s., individual doses | – | – | X | X | ||
| Epoetin Alfa Hexal/ Abseamed/Binocrit | 2 × 2 crossover | 6 | Single | IV, SC, 100 μg kg−1 | X | X | – | X | |
| Parallel group | 76 | Multiple | IV, 100 μg kg−1 3 t.i.w. | X | X | – | X | ||
| Parallel group | 74 | Multiple | SC, 100 μg kg−1 3 t.i.w. | X | X | – | X | ||
| Test vs. NeoRecormon (epoetin beta), trial design not given | 72 | Single, multiple | SC, 100 μg kg−1 3 t.i.w. | – | – | – | – | ||
| Single‐arm | 6 | Multiple | SC,100 μg kg−1 3 t.i.w. | – | – | – | – | ||
| Parallel groupa | 478 | Multiple | IV, 3 t.i.w., dose before randomization14 | – | – | X | X | ||
| Parallel groupa | 114 | Multiple | SC, dose n.s. | – | – | X | X | ||
| Filgrastim | Zarzio/Filgrastim Hexal | 2 × 2 crossover | 40 | Multiple | SC, 10 μg kg−1 day−1 | X | X | – | X |
| 2 × 2 crossover | 26 | Single | IV, 5 μg kg−1 day−1 | X | X | – | X | ||
| 2 × 2 crossover | 56 | Multiple | SC, 2.5 μg kg−1 day−1 or 5 μg kg−1 day−1 | X | X | – | X | ||
| 2 × 2 cross‐over | 24 | Single | SC, 1 μg kg−1 day−1 | X | X | – | X | ||
| Single‐arma | 170 | Multiple | SC, 30 MIU for <60 kg, 48 MIU for ≥60 kg | – | – | X | X | ||
| Tevagrastim/Ratiograstim/Biograstim | 2 × 2 crossover | 56 | Single | SC, 5 μg kg−1 or 10 μg kg−1 | X | X | – | X | |
| 2 × 2 crossover | 144 | Single | IV with 5 μg kg−1 or 10 μg kg−1 or SC with 5 μg kg−1 or 10 μg kg−1 | X | X | – | X | ||
| Placebo and active control parallel group1,17, a | 348 | Multiple | SC, 5 μg kg−1 day−1 | X | – | X | X | ||
| Parallel group in first cycle, switch to test afterwards2, a | 237 | Multiple | SC, 5 μg kg−1 day−1 | X | – | X | X | ||
| Parallel group in first cycle, switch to test afterwards3, a | 92 | Multiple | SC, 5 μg kg−1 day−1 | X | – | X | X | ||
| Nivestim | 2 × 2 crossover | 44 | Single | IV or SC, 10 μg kg−1 day−1 | X | X | – | X | |
| 2 × 2 crossover | 48 | Multiple | SC, 5 μg kg−1 day−1 or 10 μg kg−1 day−1 | X | X | ‐ | X | ||
| Parallel group4, a | 279 | Multiple | SC, 5 μg kg−1/day | – | – | X | X | ||
| Grastofil/Accofil | 2 × 2 crossover | 35 | Single | IV, 5 μg kg−1 | X | X | – | X | |
| 2 × 2 crossover | 68 | Single | SC, 75 μg or 150 μg | X | X | – | X | ||
| Placebo and active control parallel group | 69 | Multiple | SC, 5 μg kg−1 | X | X | – | X | ||
| 3‐period crossover (EU reference vs. US reference vs. test) | 43 | Single | SC, 300 μg | X | X | – | X | ||
| Single‐arm5, a | 120 | Multiple | SC, 5 μg kg−1 | – | X | X | X | ||
| Follitropin alfa | Ovaleap | Single‐arm | 40 | Single | SC, 37.5 IU, 75 IU, 150 IU or 300 IU | X | – | – | X |
| 2 × 2 crossover | 36 | Single | SC, 300 IU | X | – | – | X | ||
| Parallel group6, a | 299 | Multiple | SC, start dose: 150 IU15 | – | X | X | X | ||
| Bemfola | 2 × 2 crossover | 24 | Single | SC, 225 IU | X | – | – | X | |
| Parallel group7, a | 273 | Multiple | SC, start dose: 150 IU15 | – | X | X | X | ||
| Insulin glargine | Abasaglar | 4‐period crossover (no details on sequences) | 80 | Single | SC, 0.5 U kg−1 | X | X | – | X |
| 4‐period crossover (no details on sequences) | 91 | Single | SC, 0.5 U kg−1 | X | X | – | X | ||
| 2 × 2 crossover | 16 | Single | SC, 0.5 U kg−1 | X | X | – | X | ||
| 4‐period crossover (test and reference in two different doses, no information on sequences) | 24 | Single | SC, 0.3 U kg−1 and 0.6 U kg−1 | X | X | – | X | ||
| 2 × 2 crossover | 20 | Single | SC, 0.3 U kg−1 | – | X | – | X | ||
| Parallel group8, a | 536 | Multiple | SC, previous dose | – | – | X | X | ||
| Parallel group9, a | 759 | Multiple | SC, previous dose | – | – | X | X | ||
| Somatropin | Omnitrope | 2 × 2 crossover (Somatropin Sandoz powder vs. placebo) | 12 | Single | SC, 5 mg | X | X | – | – |
| 2 × 2 cross‐over (Somatropin Sandoz powder vs. US reference) | 25 | Single | SC, 5 mg | X | X | – | – | ||
| 2 × 2 crossover (Somatropin Sandoz vs. Somatropin Sandoz Liquid) | 24 | Single | SC, 5 mg | X | X | – | – | ||
| Parallel group18, a | 89 | Multiple | SC, 0.1 IU kg−1 day−1 | X | – | X | X | ||
| Single‐arma | 51 | Multiple | SC, 0.03 mg kg−1 day−1 | – | – | X | X | ||
| Etanercept | Benepali | 2 2 × 2 crossover (test vs. US reference, test vs. EU reference) | 138 | Single | SC, 50 mg | X | – | – | X |
| Parallel group10, a | 596 | Multiple | SC, 50 mg | X | – | X | X | ||
| Infliximab | Remsima/Inflectra | Parallel group | 19 | Multiple | IV, 3 mg kg−1 | X | X | X | X |
| Parallel group | 250 | Multiple | IV, 5 mg kg−1 | X | – | X | X | ||
| Parallel group11, a | 606 | Multiple | IV, 3 mg kg−1 | X | X | X | X | ||
| Flixabi | 3‐arm parallel group (test, EU reference, US reference) | 159 | Single | IV, 5 mg kg−1 | X | – | – | X | |
| Parallel group12, a | 584 | Multiple | IV, 3 mg kg(−16) | X | – | X | X |
E, efficacy; IV, intravenous; N, number of subjects; n.s., not specified; PD, pharmacodynamic; PK, pharmacokinetic; S, safety; SC, subcutaneous; t.i.w., times a week; X, data from the study were discussed in this part of the EPAR
EudraCT‐ID: 1: 2004–001 452‐36, 2: 2004–001 450‐84, 3: 2004–001 449‐13, 4: 2007–000 394‐36, 5: 2007–005 034‐36, 6: 2009–017 674‐20, 7: 2010–019 287‐37, 8: 2011–000 829‐73, 9: 2011–000 828‐15, 10: 2012–005 026‐30, 11: 2010–018 646‐31, 12: 2012–005 733‐37
Further dosing details: 13: If a lower dose was needed it was given less frequently (e.g. twice or once every week). 14: Dose adjustments were allowed every three weeks. 15: Individual adjustments were possible. 16: Dose increments were allowed after week 30 by 1.5 mg kg−1 up to 7.5 mg kg−1 per visit
Further study design details: 17: After the first cycle, the placebo group switches to test; the primary endpoint was after cycle 1. 18: First part: Somatropin Sandoz powder vs. EU reference; second part: Somatropin Sandoz powder and Somatropin Sandoz Liquid; third part: Somatropin Sandoz Liquid
In the 2 × 2 crossover design, each subject receives both test (T) and reference (R) products and is randomly assigned to receive the products either in the order RT or in the order TR, with the constraint that an equal number of subjects are assigned to each sequence. In a parallel group trial, each subject is randomly assigned to T or R whereas the number of subjects assigned to these categories do not necessarily need to be the same.
In the guideline on similar biological medicinal products containing biotechnology‐derived proteins as an active substance 40, it is recommended that a single‐dose 2 × 2 crossover trial be used for the assessment of PK and PD. This is in line with the bioequivalence guideline 58, which recommends that a randomized, single‐dose, crossover design (with sequence groups RT and TR, as above) is used to compare two formulations. Crossover trials have the advantage that the effect of confounding variables is reduced because every subject acts as his or her own control. This also leads to a more precise comparison of R and T, and, as a result, a smaller sample size as compared with a parallel group trial 59.
Most sponsors followed the advice in the above‐mentioned guideline and, in all but one case, at least one trial with a 2 × 2 crossover design was submitted. The only exceptions were the two biosimilars with the active substance infliximab, for which only parallel group trials were used. Nonetheless, this is in line with the product‐specific guideline on similar biological medicinal products containing monoclonal antibodies (mAbs), in which it is stated that: ‘a parallel group design may be necessary due to the long half‐life of mAbs and the potential influence of immunogenicity’ 60.
For Epoetin Alfa Hexal/Abseamed/Binocrit, the crossover trial consisted of only six subjects and was therefore not the main trial. The pivotal PK/PD trials were two parallel group trials, with a total of 150 volunteers. This contradicts the product‐specific guidelines on similar biological medicinal products containing recombinant erythropoietins, which recommend using single‐dose crossover trials 57. There is no comment in the EPAR or in the papers related to the trial 61, 62 on this deviation. However, it should be noted that the earliest product‐specific guideline on erythropoietins was published in 2006 63 and, as the drug was approved in 2007, the negotiation for the trial with the EMA most likely started before the guidance was released. In the application of Silapo/Retacrit (also a biosimilar to epoetin and approved in 2007), the PK/PD trials were conducted using a crossover design. Interestingly, one of the trials was not a 2 × 2 crossover but a three‐period, three‐treatment crossover design comparing the reference product after a single SC dose with the test product after intravenous (IV) and SC administration, which is a deviation from the standard approach. Therefore it seems that, at the time that the studies for both applications were planned, the sponsors had some flexibility in the choice of design.
In phase III, in most cases at least one parallel group design was used for comparing efficacy and safety. The only exception were the already discussed applications related to the active substance filgrastim. In this case, the guideline allowed the omission of an efficacy comparison in phase III. Therefore, some sponsors primarily used single‐arm trials in order to evaluate safety.
Route of administration and single vs. multiple dose
For the route of administration in PK/PD trials, the overarching guideline 40 states that it is sufficient to use the SC route alone, although the reference product can be administered by both IV and SC routes. In the conducted trials, for all but the biosimilars with infliximab as active substance, at least one trial using the SC route of administration was performed. It should be noted that the reference product Remicade should only be administered by the IV route 64, so the assessment of the IV route of administration alone is fully justified.
The guidance 40 recommends a single‐dose administration. However, many sponsors also used multiple doses for their PK/PD assessment. In general, a multiple‐dose trial in patients is acceptable if a single‐dose trial cannot be conducted in healthy volunteers for tolerability reasons, and a single‐dose trial is not feasible in patients. This may have determined the trial design for Remsima/Inflectra. Alternatively, a signal in some endpoints can only be measured after repeated administration, such as the increase in CD34+ cell counts, which requires multiple doses of filgrastim 65.
Endpoints, equivalence margins and statistical methods
Table 4 provides an overview of the endpoints and equivalence margins used for PK, PD and efficacy assessment. For PK and PD, the margins used were compared with the recommended margins in the product‐specific guidelines. The use of 90% confidence intervals for PK trials is recommended in the guidelines for the assessment of bioequivalence 58. For PD assessment, 95% confidence intervals were reported in the EPARs, which might be explained by the fact that PD parameters are already considered as efficacy parameters. Thus, the same standard as outlined in ICH E9 66 was applied as would be expected for any efficacy parameter in a clinical trial.
Table 4.
Chosen endpoint and margins used in clinical trials
| Active substance | Product | PK endpoints | PK limits: 80–125%, CI: 90% | PD endpoints | PD limits: 80–125%, CI: 95% | Primary efficacy endpoint (type, measurement, margins) |
|---|---|---|---|---|---|---|
| Epoetin alfa/zeta | Silapo/Retacrit | Yes | No (wider margins for Cmax) | – | – | Mean dosage [continuous, (−45, 45)]; mean haemogloblin level [continuous, (−1, 1) or (−0.6, 0.6)] |
| Epoetin Alfa Hexal/Abseamed/Binocrit | Yes | Yes | Yes | No (tighter margins used) | Absolute change in haemogloblin levels between the screening/ baseline [continuous, (−0.5,0.5)] | |
| Filgrastim | Zarzio/Filgrastim Hexal | Yes | Yes | Yes | No (tighter margins used) | Incidence (binary) and duration of severe neutropenia in cycle 1 in days (count) (−, −) |
| Tevagrastim/Ratiograstim/Biograstim | Yes | Yes | Yes | Margins, yes; confidence level not given | The duration of severe neutropenia in cycle 1 in days (count) (−1 day, 1 day) | |
| Nivestim | Yes | Yes | Yes | Yes | The duration of severe neutropenia in cycle 1 in days (count) (−1 day, 1 day) | |
| Grastofil/Accofil | Yes | Yes | Yes | Yes | The duration of severe neutropenia in cycle 1 in days (count) (−, −) | |
| Follitropin alfa | Ovaleap | No details | No details | X | No (only descriptive) | Number of oocytes retrieved [count, (−3, 3)] |
| Bemfola | Yes | Yes | X | No (only descriptive) | Number of oocytes retrieved [count, (−2.9, 2.9)] | |
| Insulin glargine | Abasaglar | Yes | Yes | Yes | Yes | Change in HbA1c from baseline to 24 weeks (continuous, non‐inferiority margin: 0.4%) |
| Somatropin | Omnitrope | Yes | Yes | Yes | No (no formal comparison, no results shown) | Height standardized by age and gender, Height velocity standard deviation score [continuous, (− 2.8, 2.8)] |
| Etanercept | Benepali | Yes | No details | – | – | ACR20 responders (binary, (−15%, 15%) |
| Infliximab | Remsima/Inflectra | Yes | Yes | X | No (no margins defined) | ACR20 responders (binary, (−15%, 15%)) |
| Flixabi | Yes | Yes | – | – | ACR20 responders (binary, (−15%, 15%)) |
Pharmacokinetic/pharmacodynamic (PK/PD) endpoints: are the endpoints used as described in the guidelines in at least one PK/PD study?; ACR, American College of Rheumatology; CI, confidence interval level; Cmax, maximum concentration; HbA1c, glycosylated haemoglobin; X = no recommendation given in guidelines, − = no assessment in application; (−, −) = no margins given. All information is taken from the EPARs 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39
The endpoints and equivalence margins used for PK were mostly in line with the recommendations. Wider margins were used only for Silapo/Retacrit (code name SB309 in the EPAR) for the maximum concentration (Cmax) (70–143% instead of 80–125%). In the EPAR, it is explained that the sponsor ‘referred to the scientific advice given by CHMP in April 2004 that stated that the concept of “comparability” cannot use bioequivalence but that similar PK profiles of SB309 and the reference product would strengthen the choice of reference in the clinical trials. The advice concluded that for this purpose descriptive statistics will suffice’. Nonetheless, in the EPAR, 90% confidence intervals for the primary endpoints – area under the curve (AUC) and Cmax – were compared with the equivalence margins, which were, as stated in the EPAR, only defined post hoc. In the application for the other biosimilar to epoetin, Epoetin Alfa Hexal/Abseamed/Binocrit, the standard bioequivalence margins were used. Therefore, the deviation from the guidelines can, in this case, not be explained exclusively by the characteristics of the active substance.
For Ovaleap and Benepali, no margins and confidence levels were reported. Neither the EPAR nor the related publication 67 gives more than descriptive summary statistics for Ovaleap, so it is not clear if any formal testing was performed. In the EPAR for Benepali, it is stated that ‘the primary endpoints were well within the predefined acceptance range’, but neither confidence intervals nor acceptance ranges are stated.
Interestingly, even if the bioequivalence criteria are not fulfilled, the product might still be approved. For example, for Zarzio/Filgrastim Hexal, the sponsor submitted four PK/PD studies with a total of four different doses, one dose using both the IV and SC routes of administration (1 μg kg−1, 2.5 μg kg−1, 5 μg kg−1 and 10 μg kg−1 SC route; 5 μg kg−1 IV route). For the lower doses and after multiple SC doses, neither Cmax nor AUC met the acceptances ranges. In the EPAR, it is stated that: ‘the applicant claimed that the observed differences were due to differences in the levels of purity of the two products, leading to a systematic bias toward an apparently increased bioavailability for the reference product’. That is why the PK parameters were adjusted to the enzyme‐linked immunosorbent assay‐detectable dose and, consequently, the AUC then fulfilled the criterion. For Cmax, the values were still outside of the equivalence interval for a single dose of 2.5 μg kg−1 and multiple doses of both 2.5 μg kg−1 and 5 μg kg−1. The sponsor provided modelling results and an explanation of the mechanism of action. Overall, it was concluded that: ‘the small differences observed in the PK profile of filgrastim are not expected to translate into significant differences in the PD response’.
On the other hand, even if the equivalence criteria based on the confidence intervals are met, the CHMP might not be fully satisfied – e.g. for Grastofil/Accofil, the 90% confidence intervals for the ratios of all primary PK endpoints were in the predefined equivalence range of 80–125%. In the EPAR, it is acknowledged that the sponsor used the methods and equivalence limits requested by the CHMP. Nonetheless, as the AUC0–32 and the AUC0–∞ showed that the point estimates for test and reference were different, with a very low P‐value (P ≤ 0.0001) in two studies, the sponsor had to provide further analysis and justifications.
The recommendations concerning PD endpoints and equivalence margins are less concrete in the product‐specific guidelines than for PK. For two active substances (infliximab, follitropin alfa), no endpoints are named at all. In some product‐specific guidelines, it is mentioned that the known PD marker cannot predict the clinical outcome (described above for epoetin alfa/zeta). However, it is also stated that: ‘it is recommended that PD markers are added to the PK studies whenever feasible’ 40. Nonetheless, there are three applications without any PD assessment at all (Silapo/Retacrit, Benepali and Flixabi). However, these were applications which included large phase III trials with 1272, 596 and 584 patients, respectively. For Silapo/Retacrit, it was noted in the EPAR that the PD mechanism is known in the literature and it is reported that: ‘although PD studies should be part of the development programme for a biosimilar epoetin, the lack of such studies is not critical since demonstration of similar efficacy and safety between the new and the reference product is required anyway’. Therefore, it seems to be possible to get approval if the guidelines are not followed in detail, if the overall application is convincing.
In all cases in which advice in product‐specific guidelines was given and PD was evaluated in the application, the recommended endpoints were used. If margins were used, in all cases 80–125% or narrower margins were used. The narrower margins were likely to have been chosen to fit the efficacy equivalence margins in the phase III trials. For example, in the EPAR for Epoetin Alfa Hexal/Abseamed/Binocrit, it is stated that: ‘the acceptance range was then changed to 97–103% by protocol amendment based on haemoglobin (Hb) concentration changes defined as equivalence margins in phase III studies’.
In a way similar to the PK assessment, it is possible to achieve a positive opinion from the CHMP, even though not all PD parameters are found completely within the equivalence margins. For example, only one out of five relevant PD studies for Abasaglar met all criteria. The sponsor explained this by reference to the small sample sizes used and the presence of outliers that caused the criteria not to be met.
In the efficacy assessment in the phase III trials, in most cases there is one chosen primary endpoint, which is well defined in the EPAR. The only exceptions were Silapo/Retacrit, where two primary endpoints were used and Omnitrope, with four different primary endpoints. In both cases, no adjustment for multiplicity was mentioned. For Zarzio/Filgrastim Hexal, again, two efficacy endpoints were used but efficacy was not the primary endpoint in the phase III studies because it was a single‐arm trial with only comparison with historical data.
The chosen endpoints vary according to the indication. The observed types of endpoint were binary, continuous and count data, with continuous and count data being the most common (used nine and eight times, respectively) and binary data being less frequently used (four times). The different types of data also lead to different statistical models. For example, the predominantly used endpoint for biosimilars to filgrastim is the count of the number of days during the first cycle of chemotherapy, when neutropenia is severe. Methods of analysis for such an endpoint include standard approaches, such as Poisson regression 68. For the biosimilar to infliximab, it was assessed if the patients were responders or nonresponders, which is a binary variable. A responder is here defined according to the American College of Rheumatology 20% improvement criteria (ACR20) endpoint 69, which is one of the recommended scores by the EMA for the assessment of rheumatoid arthritis 70.
Obviously, the statistical methodology used has to be adapted to the chosen endpoint. However, even for biosimilars referring to the same reference product, the statistical model does not have to be the same. This is the case for Ovaleap and Bemfola, which are both biosimilars to follitropin alfa. For Ovaleap, a zero‐inflated Poisson (ZIP) regression model 71, with adjustment for age and country, was used as the primary analysis. The primary analysis for Bemfola was, depending on the data, either a Mann‐Whitney two one‐sided test (TOST) and a Bootstrap‐T for non‐normal data or a Schuirmann's TOST test 72. In the related product‐specific guidelines, it is stated that: ‘it should be taken into account that over‐stimulation as well as understimulation can result in cycle cancellation and a number of zero oocytes retrieved (primary endpoint)’ 73. Both approaches take this into account, so they seem to be in line with the guidelines, although the approach for Bemfola only considers the number of zero oocytes indirectly. Therefore, the ZIP model might be closer to the guidelines.
For biosimilars with the same reference product, in most cases identical endpoints were used. One exception is the substance epoetin alfa/zeta, for which, for the product Silapo/Retacrit, the mean dosage and the mean Hb level were used. In the efficacy assessment for the approval of Epoetin Alfa Hexal/Abseamed/Binocrit, the absolute change in Hb level was the primary endpoint. The reason for that might be that the studies were potentially planned before the first product‐specific guideline 63 was published. Therefore, the process was probably less standardized.
The equivalence margins that were used also depended on the indication and the chosen endpoint. Often, neither the publication nor the EPAR offer any explanation for the choice of margins. For biosimilars referring to the same reference product and with the same endpoint, the chosen margins were identical, except for Ovaleap and Bemfola, for which a minor deviation was observed: (−3, 3) and (−2.9, 2.9), respectively, were used. The difference is negligible but it shows that the decision regarding which equivalence margins to use is done individually for each sponsor.
Discussion
The approval of biosimilars is more complex than for generics. Extensive clinical trials must be undertaken in order to prove the therapeutic equivalence of a biosimilar before the product can be authorized. This particularly includes analysis of PK and PD parameters and efficacy comparisons in phase III trials.
The present literature review of the EPARs published by the EMA and additional publications related to clinical trials shows that there is a large variability between the submitted applications. For example, the overall sample size in PK/PD trials varies considerably, from 24 to 269 subjects. In phase III trials, between 120 and 1295 patients were used. This variability can be partially explained by the different characteristics of the reference products.
In addition, the different types of endpoint in the efficacy assessment make it difficult to standardize the assessment method. For some products, binary data were used, whereas for other drugs the endpoint was continuous. This leads to different statistical models and methods, which need to be negotiated with the authorities. These different types of endpoint make it impossible to define general equivalence margins as these margins have to fit the therapeutic range of the drug. Interestingly, even for biosimilars with the same reference product, the endpoints, statistical model and equivalence margins, and also the sample size, were not in all cases comparable. This shows that there seems to be some flexibility for the sponsor on the decision as to how best to conduct the development plan.
The recommendations in the product‐specific guidelines were mostly followed. In some cases, there were also deviations. However, the product was approved in the end. It therefore seems to be possible to get approval for a product without following all regulatory guidelines in the assessment, as long as the overall application is convincing.
We believe that the EPARs should offer detailed information on the approval process, in order to make the decisions more transparent. Nonetheless, there are some aspects that could be further improved from our point of view. Firstly, the reports do not seem to be standardized. The length varies enormously; the shortest EPAR was published for Omnitrope (25 pages), whereas the longest was for Inflectra/Remsima (105 pages). The depth of information also differs; in some reports, the studies are described in detail, whereas in others, not even the trial doses are given. Furthermore, there is no unified structure, which makes it difficult to quickly identify the relevant information in the trial. For example, it would be easier to find information on the PK/PD trials if the details of the trial were summarized in an overview table, as is done in some of the newer EPARs for efficacy assessment (e.g. Bemfola). Secondly, in many cases no explanation is offered for the choice of equivalence margins in phase III trials. As the margins are crucial for the success of the trial, it would be desirable to understand the reasons for them. In addition, the EudraCT number that was introduced in 2004 can simplify the search for information about a specific trial. However, this number is not a part of the EPAR and, moreover, the database search for this number is complicated and often not successful. This makes it difficult to link the results in the EPARs to the more detailed information about a specific clinical trial in the database.
Conclusions
There is high variability between the submitted applications. This is partially explainable by the different reference products, but sponsors can also negotiate the biosimilar development package with the regulatory agency. This was confirmed by the observation that, even for biosimilars with the same reference product, the relative weight put on PK/PD and phase III trials, the endpoints and the statistical models vary. Furthermore, it seems to be possible to get approval even if not all aspects in the overarching guidelines and the product‐specific guidelines are followed. It is also possible to achieve a opinion of the regulators if the equivalence criteria are not fulfilled. By contrast, it can be necessary to provide further justification, even though all equivalence criteria have been met. In conclusion, it seems that there is a fair degree of flexibility in the packages that sponsors can conduct to gain regulatory approval for a biosimilar. We note that some concerns on the part of the sponsor can be addressed by seeking early scientific advice from the EMA when planning the trials 74.
Competing Interests
There are no competing interests to declare.
Johanna Mielke was supported by the Swiss State Secretariat for Education, Research and Innovation (SERI) under contract number 999754557. The opinions expressed and arguments employed herein do not necessarily reflect the official views of the Swiss Government. The project is part of the IDEAS European training network ( http://www.ideas‐itn.eu /) from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska‐Curie grant agreement No 633567. Bernd Jilma was supported by the Austrian Science Fund SFB54‐P04.
Supporting information
Table S1 Summary of indications of reference product and biosimilars.
Supporting info item
Mielke, J. , Jilma, B. , Koenig, F. , and Jones, B. (2016) Clinical trials for authorized biosimilars in the European Union: a systematic review. Br J Clin Pharmacol, 82: 1444–1457. doi: 10.1111/bcp.13076.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 2016; 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 2015; 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 2015; 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rickwood S, Kleinrock M, Nunez‐Gaviria M, Sakhrani S, Aitken M. The global use of medicines: outlook through 2017. IMS Health: IMS Institute for Healthcare Informatics, 2013. [Google Scholar]
- 6. Miller S. The $250 billion potential of biosimilars; Express Scripts, 2013. [online]. Available at http://lab.express‐scripts.com/lab/insights/industry‐updates/the‐$250‐billion‐potential‐of‐biosimilars (last accessed 01 February 2016).
- 7. CHMP . Guideline on similar biological medicinal products [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf (last accessed 02 November 2015).
- 8. Ehmann F, Pita R. The EU is ready for non‐biological complex medicinal products. GaBi J 2016; 5: 30–35. [Google Scholar]
- 9. European Commission . Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the community code relating to medicinal products for human use [online]. Available at http://ec.europa.eu/health/files/eudralex/vol‐1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf (last accessed 15 June 2016).
- 10. Chow S‐C. Biosimilars. Design and analysis of follow‐on biologics. Boca Raton: CRC Press/Chapman & Hall, 2014. [Google Scholar]
- 11. Wang Y‐MC, Chow AT. Development of biosimilars – pharmacokinetic and pharmacodynamics considerations. J Biopharm Stat 2009; 20: 46–61. [DOI] [PubMed] [Google Scholar]
- 12. Combe C, Tedree RL, Schellekens H. Biosimilar epoetins: an analysis based on recently implemented European Medicines Evaluation agency guidelines on comparability of biopharmaceutical proteins. Pharmacotherapy 2005; 25: 954–962. [DOI] [PubMed] [Google Scholar]
- 13. Schellekens H. The first biosimilar epoetin: but how similar is it? Clin J Am Soc Nephrol 2008; 3: 174–178. [DOI] [PubMed] [Google Scholar]
- 14. Jelkmann W. Biosimilar recombinant human erythropoietins (‘epoetins’) and future erythropoiesis‐stimulating treatments. Expert Opin Biol Ther 2012; 12: 581–592. [DOI] [PubMed] [Google Scholar]
- 15. Mikhall A, Farouk M. Epoetin biosimilars: five years on. Adv Ther 2013; 30: 28–40. [DOI] [PubMed] [Google Scholar]
- 16. Gascon P. Presently available biosimilars in hematology‐oncology: G‐CSF. Target Oncol 2012; 7 (Suppl. 1): 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rinaudo‐Gaujos M, Paul S, Tedesco ED, Genin C, Roblin X, Peyrin‐Biroulet L. Review article: biosimilars are the next generation of drugs for liver and gastrointestinal diseases. Aliment Pharmacol Ther 2013; 38: 914–924. [DOI] [PubMed] [Google Scholar]
- 18. EPAR database of EMA [online]. Available at http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp&mid=WC0b01ac058001d124 (last accessed 15 June 2016).
- 19. CHMP . Omnitrope: EPAR – scientific discussion; 2006. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Scientific_Discussion/human/000607/WC500043692.pdf (last accessed 27 October 2015).
- 20. CHMP . Epoetin Alfa Hexal: EPAR – scientific discussion; 2007. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Scientific_Discussion/human/000726/WC500028287.pdf (last accessed 28 October 2015).
- 21. CHMP . Binocrit: EPAR – scientific discussion; 2007. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_ _Scientific_Discussion/human/000725/WC500053615.pdf (last accessed 28 October 2015).
- 22. CHMP . Abseamed: EPAR – scientific discussion; 2007. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Scientific_Discussion/human/000725/WC500053615.pdf (last accessed 28 October 2015).
- 23. CHMP . Silapo: EPAR – scientific discussion; 2007. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Scientific_Discussion/human/000760/WC500050914.pdf (last accessed 27 October 2015).
- 24. CHMP . Retacrit: EPAR – scientific discussion; 2007. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Scientific_Discussion/human/000872/WC500054374.pdf (last accessed 27 October 2015).
- 25. CHMP . Tevagrastim: EPAR – public assessment report; 2008. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/000827/WC500036667.pdf (last accessed 26 October 2015).
- 26. CHMP . Ratiograstim: EPAR – public assessment report; 2008. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/000825/WC500047793.pdf (last accessed 26 October 2015).
- 27. CHMP . Biograstim: EPAR – public assessment report; 2008. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/000826/WC500053904.pdf (last accessed 26 October 2015).
- 28. CHMP . Zarzio: EPAR – public assessment report; 2008. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/000917/WC500046528.pdf (last accessed 26 October 2015).
- 29. CHMP . Filgrastim Hexal: EPAR – public assessment report; 2008. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/000918/WC500022471.pdf (last accessed 26 October 2015).
- 30. CHMP . Nivestim: EPAR – public assessment report; 2010. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/001142/WC500093664.pdf (last accessed 28 October 2015).
- 31. CHMP . Inflectra: EPAR – public assessment report; 2013. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002778/WC500151490.pdf (last accessed 27 October 2015).
- 32. CHMP . Remsima: EPAR – public assessment report; 2013. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002778/WC500151490.pdf (last accessed 27 October 2015).
- 33. CHMP . Ovaleap: EPAR – public assessment report; 2013. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002608/WC500152908.pdf (last accessed 27 October 2015).
- 34. CHMP . Grastofil: EPAR – public assessment report; 2013. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002150/WC500154066.pdf (last accessed 29 October 2015).
- 35. CHMP . Abasria: EPAR ‐ public assessment report; 2014. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002835/WC500175383.pdf (last accessed 29 October 2015).
- 36. CHMP . Absaglar: EPAR – public assessment report; 2014. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002835/WC500175383.pdf (last accessed 29 October 2015).
- 37. CHMP . Bemfola: EPAR – public assessment report; 2014. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002615/WC500166820.pdf (last accessed 29 October 2015).
- 38. CHMP . Benepali: EPAR – public assessment report; 2015. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/004007/WC500200380.pdf (last accessed 16 February 2016).
- 39. CHMP . Flixabi: EPAR – public assessment report; 2016. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/004020/WC500208358.pdf (last accessed 14 June 2016).
- 40. CHMP . Guideline on similar biological medicinal products containing biotechnology‐derived proteins as active substance: non‐clinical and clinical issues; 2014. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf (last accessed 02 November 2015).
- 41. Warren JB. Generics, chemisimilars and biosimilars: is clinical testing fit for purpose? Br J Clin Pharmacol 2013; 75: 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. EMA Press office . Press release: Marvel LifeSciences Ltd withdraws its marketing authorisation applications for Insulin Human Rapid Marvel, Insulin Human Long Marvel and Insulin Human 30/70 Mix Marvel; 2008. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2009/11/WC500015335.pdf (last accessed 11 December 2015).
- 43. Human Medicines Development and Evaluation . Public statement on Filgrastim ratiopharm (filgrastim) – withdrawal of the marketing authorisation in the European Union; 2011. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Public_statement/2011/07/WC500109157.pdf (last accessed 22 February 2016).
- 44. CHMP . Solumarv: EPAR – public assessment report; 2015. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/003858/WC500201969.pdf (last accessed 22 February 2016).
- 45. CHMP . Refusal assessment report for Alpheon; 2006. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/000585/WC500070792.pdf (last accessed 22 February 2016).
- 46. Yoo DH, Hrycaj P, Miranda P, Ramiterre E, Piotrowski M, Shevchuk S, et al. A randomised, double‐blind, parallel group study to demonstrate equivalence in efficacy and safety of CT‐P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis 2013; 72: 1613–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reinisch W, Louis E, Danese S. The scientific and regulatory rationale for indication extrapolation: a case study based on the infliximab biosimilar CT‐P13. Expert Rev Gastroenterol Hepatol 2015; 9 (Suppl. 1): 17–26. [DOI] [PubMed] [Google Scholar]
- 48. Rare disease (orphan) designations database of EMA [online]. Available at http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/orphan_search.jsp (last accessed 15 June 2016).
- 49. Hwang SH, Yoo HS, Yoon MK, Park HS. Detection of IgE binding component to infliximab in a patient with infliximab‐induced anaphylaxis. Ann Allergy Asthma Immunol 2014; 112: 393–394. [DOI] [PubMed] [Google Scholar]
- 50. Harris J, Keane J. How tumour necrosis factor blockers interfere with tuberculosis immunity. Clin Exp Immunol 2010; 161: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cantini F, Niccoli L, Goletti D. Adalimumab, etanercept, infliximab, and the risk of tuberculosis: data from clinical trials, national registries, and postmarketing surveillance. J Rheumatol Suppl 2014; 91: 47–55. [DOI] [PubMed] [Google Scholar]
- 52. Jilma B, Jagiello‐Gruszfeld A, Tomczak P, Gadgil H, Orlik G, Desai K, et al. Demonstration of clinical comparability of the biosimilar filgrastim to Neupogen, in terms of safety and efficacy, in healthy volunteers and patients receiving myelosuppressive chemotherapy. Eur Oncol Haematol 2014; 10: 107–115. [Google Scholar]
- 53. Kirkov V, Dimitrova V, Siebert‐Weigel M, Wolf‐Pflugmann M, Koytchev R, Richter W, et al. Evaluation of the pharmacokinetics of two recombinant human erythropoietin preparations: epoetin zeta and epoetin alfa. 1st Communication: a monocentric, open, randomized, single dose, two‐period crossover trial in healthy volunteers. Arzneimittelforschung 2008; 58: 215–219. [DOI] [PubMed] [Google Scholar]
- 54. Kirkov V, Dimitrova V, Siebert‐Weigel M, Wolf‐Pflugmann M, Koytchev R, Richter W, et al. Evaluation of the pharmacokinetics of two recombinant human erythropoietin preparations: epoetin zeta and epoetin alfa. 2nd Communication: a monocentric, double‐blind, randomized, single dose, three‐period crossover trial in healthy volunteers. Arzneimittelforschung 2008; 58: 220–224. [DOI] [PubMed] [Google Scholar]
- 55. CHMP . Guidance on similar medicinal products containing recombinant granulocyte‐colony stimulating factor; 2006. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003955.pdf (last accessed 04 November 2015).
- 56. CHMP . Concept paper on the revision of the guideline on non‐clinical and clinical development of similar biological medicinal products containing recombinant granulocyte‐colony stimulating factor; 2015. [online]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/07/WC500190635.pdf (last accessed 28 January 2016).
- 57. CHMP . Guideline on non‐clinical and clinical development of similar biological medicinal products containing recombinant erythropoietins (Revision); 2010. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/04/WC500089474.pdf (last accessed 01 December 2015).
- 58. CHMP . Guidelines on the investigation of bioequivalence; 2010. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf (last accessed 28 January 2016).
- 59. Jones B, Kenward MG. Design and analysis of cross‐over trials, 3th edn. Boca Raton: CRC Press/Chapman & Hall, 2015. [Google Scholar]
- 60. CHMP . Guideline on similar biological medicinal products containing monoclonal antibodies – non‐clinical and clinical issues; 2012. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf (last accessed 04 November 2015).
- 61. Soergel F, Thyroff‐Friesinger U, Vetter A, Vens‐Cappell B, Kinzig M. Bioequivalence of HX575 (recombinant human epoetin alfa) and a comparator epoetin alfa after multiple intravenous administrations: an open‐label randomised controlled trial. BMC Clin Pharmacol 2009; 9: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Soergel F, Thyroff‐Friesinger U, Vetter A, Vens‐Cappell B, Kinzig M. Bioequivalence of HX575 (recombinant human epoetin alfa) and a comparator epoetin alfa after multiple subcutaneous administrations. Pharmacology 2009; 83: 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. CHMP . Guideline on non‐clinical and clinical development of similar biological medicinal products containing recombinant erythropoietins; 2006. [online]. Available at https://www3.bio.org/healthcare/followonbkg/AnnexGuidanceonSimilarMedicinalProductsContainingRecombinantErythropoietins.pdf (last accessed 01 December 2015).
- 64. CHMP . Remicade: EPAR summary for the public; 2009. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Summary_for_the_public/human/000240/WC500050883.pdf (last accessed 10 January 2016).
- 65. Gascon P, Fuhr U, Soergel F, Kinzig‐Schippers M, Makhson A, Balser S, et al. Development of a new G‐CSF product based on biosimilarity assessment. Ann Oncol 2010; 21: 1419–1429. [DOI] [PubMed] [Google Scholar]
- 66. CHMP . ICH Topic E9: Statistical principles for clinical trials; 1998. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002928.pdf (last accessed 17 February 2016).
- 67. Lammerich A, Bias P, Gertz B. Phase 1 safety, tolerability, and pharmacokinetic study of single ascending doses of XM17 (recombinant human follicle‐stimulating hormone) in downregulated healthy women. Int J Womens Health 2015; 7: 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cameron AC, Trivedi PK. Regression analysis of count data, 2nd edn. Cambridge: Cambridge University Press, 2013. [Google Scholar]
- 69. Felson DT, Anderson JJ, Boers M, Bombardier C, Chernoff M, Fried B, et al. The American College of Rheumatology preliminary core set of disease activity measures for rheumatoid arthritis clinical trials. Arthritis Rheum 1993; 36: 729–740. [DOI] [PubMed] [Google Scholar]
- 70. CHMP . Points to consider on clinical investigations of medicinal products other than NSAIDS for treatment of rheumatoid arthritis; 2003. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003439.pdf (last accessed 01 February 2016).
- 71. Lambert D. Zero‐inflated Poisson regression, with an application to defects in manufacturing. Technometrics 1992; 34: 1–14. [Google Scholar]
- 72. Schuirmann D. A comparison of the two one‐sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm 1987; 15: 657–680. [DOI] [PubMed] [Google Scholar]
- 73. CHMP . Guideline on non‐clinical and clinical development of similar biological medicinal products containing recombinant human follicle stimulating hormone (r‐hFSH); 2013. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/03/WC500139624.pdf (last accessed 04 November 2015).
- 74. Regnstrom J, Koenig F, Aronsson B, Reimer T, Svendsen K, Tsigkos S, et al. Factors associated with success of market authorisation applications for pharmaceutical drugs submitted to the European Medicines Agency. Eur J Clin Pharmacol 2010; 66: 39–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Summary of indications of reference product and biosimilars.
Supporting info item