

Abstract
Tungsten disulfide (WS2) nanowires have been synthesized through a microwave-assisted chemical route that uses tungstic acid, elemental sulfur and monoethanolamine as starting reagents for obtaining a precursor solution of tetrathiotungstate ions. Acidification of the precursor solution yields amorphous precipitates, which lead to the formation of nanowires of WS2 with thickness of about 5–10 nm when heated at 750 °C under argon atmosphere for 1.5 h. Phase and the microstructure of the prepared powders have been investigated through x-ray powder diffraction and high-resolution transmission electron microscopy, respectively. Optical absorption of the WS2 powders reveals a red shift of the exciton bands compared to bulk WS2.
Keywords: tungsten disulfide, monoethanolamine, microwave heating, nanowires
Introduction
One-dimensional (1D) nanostructures of layered WS2, such as nanorods, nanotubes, nanowires and nanofibers, have drawn substantial research interest from different fields of technology ever since Iijima reported the formation of carbon nanotubes from graphite in 1991 [1]. Nanostructured WS2 finds application as hydrogen and lithium storage material [2, 3]; as material for solid-state secondary lithium battery cathodes [4]; in inorganic–organic nanocomposites for application in batteries and other electrochemical devices [5]; as catalyst in hydrodesulfurization of crude oil [6, 7] and as solid lubricants [8–10].
Tenne et al carried out extensive studies on various layered materials and have been pioneers in the preparation of concentric polyhedra and/or nanotubes of MS2(M=Mo and W) layered materials through gas-phase reaction of H2S with the respective metal-oxides at elevated temperatures and under reducing atmosphere [11–15]. Production of macroscopic quantities of WS2 nanotubes had later been reported through modification of the gas-phase reaction, where W/WOx was heated under a mixture of Ar and H2S gases [16]. Beside the gas-phase reaction processes, there are various solution-based and a few alternative methods that have gained recognition for the preparation of 1D nanostructures of WS2. Hydrothermal method [17–19] is one of the most widely reported solution-based routes. Nanotubes of WS2 have been synthesized by autoclaving a solution mixture of ammonium tungstate, citric acid and hexadecylamine at 180 °C for 7 days followed by heat treatment of the precursors under H2S flow [18]. Shang et al adopted a two-step hydrothermal process using tungstic acid, sodium sulfate and thiourea as the starting reagents to obtain quasi-1D WS2 nanocrystals with width and thickness of around 140 nm and 30 nm, respectively [19]. Another well-established solution-based method of the preparation of WS2 nanotubes is thermal decomposition of ammonium tetrathiotungstate at ∼\!1300 °C in a flow of hydrogen gas [20] or direct decomposition of various tetraalkylammonium tetrathiotungstate precursors under nitrogen atmosphere [21]. Later, Li et al modified the thermal decomposition route of tetrathiotungstate ion to prepare WS2 nanotubes (with diameters of 5–37 nm and lengths between 0.2 and 5 μm) through pyrolysis of co-condensed self-assemblies of anionic tungstates (WS42−) and cationic surfactant molecules at 850 °C under argon atmosphere [22]. WS2 microtubes, microribbons and ropes have also been grown through alternative methods, such as the iodine-transport reaction close to equilibrium [23] at 1060 K, when heated in an evacuated silica ampoule for 22 days at a pressure of 10−3 Pa. Most of these synthesis strategies are constrained either by their prolonged and tedious processing at high temperature or by pressure conditions or by the technical skill required for the preparation of the precursor material. Recently, Gedanken and co-workers [24] have reported an alternative and technically proficient method of growing WS2 nanorods through sonochemical reaction between W(CO)6 and sulfur in diphenylmethane at 90 °C under argon atmosphere, followed by heat treatment of the precursors at 800 °C under argon flow. Microwave-assisted method is another significant alternative that has gained attention in recent years as a simple, rapid and efficient process for growth of nanostructured oxides [25, 26] and sulfides [27–29]. Despite the recognition of the microwave-assisted method for the preparation of various sulfides, we are unaware of reports on the preparation of nanostructured WS2 through this method. Motivated by this observation, we have attempted to synthesize 1D nanostructures of WS2 through microwave irradiation induced reaction between tungstic acid and elemental sulfur in dilute monoethanolamine (MEA). The process leads to the formation of tetrathiotungstate ions in-situ, which on acidification results in precipitates that are separated and heat-treated at 750 °C for 1.5 h under argon atmosphere. Microwave irradiation facilitates instantaneous generation of the tetrathiotungstate precursors in solution, which in consequence makes the process of preparation of nanostructured WS2 much faster and less cumbersome compared to the other reported chemical routes [30]. The developed process is technically simple and makes use of a domestic microwave oven.
Experimental
Preparation method
All the chemicals were used as received without further purification. In the preparation, tungstic acid was chosen as the source of the tungsten, instead of sodium tungstate, in order to avoid sodium ion contamination in the final product. Tungstic acid was freshly prepared by acidification of an aqueous solution of 1 g of sodium tungstate ( , Merck India, 98%) with concentrated HCl. The prepared precipitate of tungstic acid was mixed with 10 ml of MEA (HOCH2CH2NH2, Merck India, 98%) in presence of 1 ml of distilled water to obtain a whitish colloid, which transformed to a clear solution on stirring through formation of MEA chelated complex of tungstate [(MEA−H+)2WO4]. In a separate container, 0.8 g of elemental sulfur (Merck India, 99.6%) was solubilized in 10 ml of MEA under microwave irradiation through formation of monoethanolaminium sulfides, bisulfides and polysulfides (i.e. Sdissolved) [31, 32]. The clear solutions of tungstic acid and sulfur (in MEA) were then mixed together, and the resultant homogenous red color precursor solution was subjected to microwave irradiation for 5–6 min in cycles of 30 s at 800 W. The precursor solution was then acidified with concentrated HCl to obtain a brown colored precipitate. The brown precipitate, on filtration and washing with water for several times, transformed to a yellowish-brown colored precursor mass when dried in a vacuum desiccator. Annealing the dried mass at 750 °C for 1.5 h in argon atmosphere resulted in the black colored powders of WS2. A domestic microwave oven (LG MG-396WA/397WB, 800 W, 2.45 GHz) was used for the microwave irradiation.
, Merck India, 98%) with concentrated HCl. The prepared precipitate of tungstic acid was mixed with 10 ml of MEA (HOCH2CH2NH2, Merck India, 98%) in presence of 1 ml of distilled water to obtain a whitish colloid, which transformed to a clear solution on stirring through formation of MEA chelated complex of tungstate [(MEA−H+)2WO4]. In a separate container, 0.8 g of elemental sulfur (Merck India, 99.6%) was solubilized in 10 ml of MEA under microwave irradiation through formation of monoethanolaminium sulfides, bisulfides and polysulfides (i.e. Sdissolved) [31, 32]. The clear solutions of tungstic acid and sulfur (in MEA) were then mixed together, and the resultant homogenous red color precursor solution was subjected to microwave irradiation for 5–6 min in cycles of 30 s at 800 W. The precursor solution was then acidified with concentrated HCl to obtain a brown colored precipitate. The brown precipitate, on filtration and washing with water for several times, transformed to a yellowish-brown colored precursor mass when dried in a vacuum desiccator. Annealing the dried mass at 750 °C for 1.5 h in argon atmosphere resulted in the black colored powders of WS2. A domestic microwave oven (LG MG-396WA/397WB, 800 W, 2.45 GHz) was used for the microwave irradiation.
In the developed process, the amount of sulfur (and MEA) in the starting solution was always taken in excess to the stoichiometric requirement of W : S mole ratio of 1 : 2. This ensured adequate concentration of S2− ions in the microwave irradiated precursor solution.
Characterization
The optical absorption spectra of the microwave irradiated precursor solution as well as the annealed powders (dispersed in ethanol) were recorded at room temperature using Shimadzu UV-1601 UV-vis spectrophotometer. The phase analysis of the prepared powders was carried out using X'pert Pro 3040/60 high-resolution x-ray diffractometer with CuKα radiation (λ=1.5418 Å) at a rate of 5° per min. The simultaneously recorded thermogravimetry (TG) and differential thermal analysis (DTA) of the precipitate obtained through acidification of the microwave irradiated precursor solution, was carried out on NETZSCH STA 409 PC instrument at a heating rate of  under argon atmosphere using alumina crucible. Composition of the as-prepared precipitate as well as the heat-treated (at 750 °C for 1.5 h) powders obtained through acidification of the microwave irradiated precursor solution, was analyzed through Fourier transform infra-red (FTIR) spectroscopy using KBr pellets and Perkin–Elmer Spectrum RXI instrument. The chemical composition of the final powders was confirmed through energy dispersive x-ray (EDX) analysis using OXFORD ISIS-300 electron microprobe. JEOL JEM-2100 high-resolution transmission electron microscope (HRTEM) was used to obtain the detailed microstructure of the annealed powders. The sample for the HRTEM studies was prepared by dispersing the powders in ethanol through ultra-sonication and drop casting the dispersion onto a carbon coated copper grid.
under argon atmosphere using alumina crucible. Composition of the as-prepared precipitate as well as the heat-treated (at 750 °C for 1.5 h) powders obtained through acidification of the microwave irradiated precursor solution, was analyzed through Fourier transform infra-red (FTIR) spectroscopy using KBr pellets and Perkin–Elmer Spectrum RXI instrument. The chemical composition of the final powders was confirmed through energy dispersive x-ray (EDX) analysis using OXFORD ISIS-300 electron microprobe. JEOL JEM-2100 high-resolution transmission electron microscope (HRTEM) was used to obtain the detailed microstructure of the annealed powders. The sample for the HRTEM studies was prepared by dispersing the powders in ethanol through ultra-sonication and drop casting the dispersion onto a carbon coated copper grid.
Results and discussion
UV-vis spectra of the microwave irradiated, red colored precursor solution of MEA chelated complex of tungstate and sulfur in MEA revealed absorption bands centered at 217, 278, 394 and 335 nm, as shown in figure 1(a). The first three absorption bands matched with those of the tetrathiotungstate ions (WS42−) [30, 33], which might have formed in the precursor solution through microwave-induced exchange reaction. This reaction may involve the successive replacement of the O2− ions in the MEA chelated complex of tungstate (i.e. WO4) by the S2− ions in the solution, and it is validated by the presence of a small peak of WOS32− ion at 335 nm. This ion possibly forms as the intermediate in the transformation of WO42− to WS42−. Liu et al used similar arguments describing the synthesis of CdS nanowires using solution of sulfur powders in organic diamines [34]. Therefore, we suggest that the red colored precursor solution of MEA chelated complex of tungstate and Sdissolved, when exposed to microwave irradiation, triggered off instantaneous generation of thiotungstate ions along with trace amounts of oxythiotungstate ions in the solution. The above observations could be summarized by the following reaction:
 |
Figure 1.

UV-vis absorption spectra of (a) the microwave-irradiated precursor solution; (b) ethanol dispersion of the WS2 powders annealed at 750 °C.
The S2− ions (or H2S) required for the above reaction were probably generated through disproportionation of sulfur in the alkaline solution [35, 36] of MEA. The hydroxyl (OH−) ions required for the reaction were provided by water impurity of MEA and by the aqueous solution of tungstic acid present in the reaction mixture. The alkaline solution of sulfide, bisulfide, or polysulfide is thermodynamically unstable, and the dissolved sulfur (Sdissolved) probably decomposed (at temperatures higher than 85 °C) to thiosulfate (S2O32−) and HS−, in accord with the net consumption of dissolved sulfur [35]. Consequently, microwave irradiation should have accelerated the decomposition of dissolved sulfur in MEA and, hence, augmented the generation of HS− ion in the solution. The generated HS− ions remained in equilibrium with the S2− ions in solution. Drawing similarities with the available literature [35, 36], the probable chemical reactions for the generation of S2− ions through microwave-induced disproportionation of sulfur (Sdissolved) in alkaline solution of MEA, in presence of OH− ions in the reaction medium, can be summarized by the following set of equations:
The formation of S2− in the microwave heated alkaline solution of MEA and sulfur (i.e. Sdissolved) was tested through blackening of lead acetate soaked paper. The presence of S2O32− ions in the solution was tested through formation of white precipitate with aqueous solution of BaCl2. The formation of S2O32−, HS− and S2− ions in the microwave irradiated solution of MEA and sulfur (i.e. Sdissolved) was also validated through UV-vis spectroscopy. The absorbance bands at 232, 360 and 305 nm in the UV-vis spectra may be respectively assigned to HS−, S2− ions [37, 38] and zero-oxidation sulfur (S0) in soluble state [36, 39]. The band at 232 nm can be ascribed to S2O32− ions. This assignment was confirmed by comparing the observed band with those for a known solution of MEA and sodium thiosulphate. It could thus be inferred that the microwave irradiated solution of sulfur and MEA was composed of S2O32−, HS−, S2− ions and neutral sulfur species in the soluble state (S0).
Acidification of the microwave irradiated, red colored precursor solution, containing thiotungstate ions and sulfur dissolved in MEA with concentrated HCl, generated brown precipitates, which turned to yellowish brown on drying in vacuum. Formation of brown amorphous precipitates of WS3 through acidification of thiotungstate precursors is well documented [40] and is given by the following equation:
Corroborating with the literature and also from the fact that WS3 and WS2 are the only two known sulfides of tungsten, the obtained precipitate was identified as WS3. However, to gain more credence, the precipitate was characterized by FTIR spectroscopy and its solubility was qualitatively tested in concentrated nitric acid and MEA. In contrast to WS2, the yellowish brown precipitate dissolved in concentrated nitric acid leaving a minor yellow residue. The yellowish brown precipitate was, however, completely soluble in MEA without any residue. The yellow solid, that was obtained as a residue when the yellowish brown precipitate was treated with nitric acid but did not appear when treated with MEA, was possibly of sulfur. Sulfur is insoluble in concentrated nitric acid but is soluble in MEA. Thus, the yellowish brown precipitate, obtained on acidification of the microwave irradiated precursor solution, was possibly a mixture of WS3 and sulfur.
FTIR spectroscopy studies of the yellowish brown precipitate showed a medium and broad absorption band centered around 515 cm−1, which could be assigned to the doubly degenerate W–S bond stretching mode for WS3 [41]. In addition, a weak but distinct absorption band was observed at 468 cm−1. This band matched with the characteristic S–S stretching mode for elemental sulfur (S8) [42], thereby, confirming the presence of sulfur in the obtained precipitate. Furthermore, the absence of the characteristic absorption bands corresponding to the symmetric and asymmetric S–W–S stretching modes for WS2 molecule at 533 cm−1 and 528 cm−1 [41], respectively, confirms that the precipitate is a mixture of WS3 and S. The above observation was further corroborated by EDX studies of the precipitate, which revealed the higher W : S atomic ratio than the expected ratio of 1 : 3.
It can therefore be ascertained that apart from WS3, elemental sulfur also precipitates out of the microwave irradiated precursor solution of MEA chelated complex of tungstate and Sdissolved on acidification, through decomposition of S2O32− ions in the solution. The simultaneous precipitation of WS3 and elemental sulfur, through acidification of microwave irradiated precursor solution, can be represented by the following equations:
 |
 |
The dried yellowish brown precipitate, containing WS3 and elemental sulfur, when annealed at 750 °C yields black powder of WS2 with volatization of sulfur. The generation of WS2 can thus be given as follows:
Solubility tests were carried out for the black annealed powders, to qualitatively assess the composition of the final product. Unlike the as-prepared yellowish brown precipitates, the annealed powder was insoluble in nitric acid and MEA. As expected for WS2, the black powders dissolved in HF-aqua regia mixture. This qualitatively established the absence of WS3 and elemental sulfur in the annealed black powders. The observation was corroborated by the FTIR spectra for the powders, which was also marked by the absence of the characteristic absorption bands for S and WS3. Furthermore, EDX studies of the annealed powders revealed the atomic percentage of S and W in the final sample as ∼64.8% and 35.1% (within the instrumental error of ±5%), respectively, and the S : W elemental ratio of 1.84. EDX measurements, therefore, indicated that the sulfur content and the W : S atomic ratio in the annealed powders was within the acceptable range of the WS2 stoichiometry [19].
Thermal decomposition behavior of the yellowish brown precursors, obtained after acidification of precursor solution, is shown in figure 2. The TG curve showed a gradual weight loss of ∼71% up to 500 °C corresponding to two endothermic heat effects observed in the DTA and a broad peak in the DTG curve in the same temperature range.
Figure 2.

Thermograms of the decomposition of the precipitates obtained on acidification of precursor solution.
The first endothermic peak observed below 200 °C in the DTA curve, which was manifested by a weight loss of ∼2.5% in the TG curve, was assigned to the loss of adsorbed moisture and the escape of trapped gases (such as, H2S, SO2 and HCl) from the powder matrix. Since the composition of the precursor was inferred as a mixture of WS3 and S, the major weight loss of ∼68.5% (as reflected in the TG curve) occurring between 200 and 500 °C could be cumulatively assigned to the loss of interlayer water molecules, the conversion of WS3 to WS2 through loss of sulfur and the volatilization of free elemental sulfur contained in the sample. The entire heat loss process was manifested by a small endothermic effect in the DTA curve in the corresponding temperature range. However, according to [43, 44], WS3 is stable up to ∼340 °C and it decomposes exothermally to S and WS2 (as indicated by equation (8)) above 340 °C with W6+ reduced to W4+ and S2− oxidized to S0, simultaneously. In figure 2, the DTA plot for the present sample showed a small endothermic peak instead of the expected exothermic event in the corresponding temperature range. This was possibly because the expected exothermal event in the conversion of WS3 to WS2 got overly compensated by endothermic heat effect generated through vaporization of the sulfur contained in the sample. The exothermic peak at about 630 °C in the DTA curve, which shows no significant weight loss in TG, can be attributed to the phase change from amorphous to crystalline WS2.
X-ray powder diffraction (XRD) of the as-prepared, yellowish brown precipitate showed broad modulations without any peak formation, indicating the material to be amorphous. XRD pattern of the powders obtained on subsequent annealing of the precursors at 750 °C for 1.5 h revealed broad peaks (as shown in figure 3), which were indexed to the 2H WS2 phase according to JCPDS card no. 08-0237. The XRD pattern of the powders showed hexagonal unit cell dimensions in agreement with the literature values. However, the major diffraction peak at 2θ ≈ 14.04°, corresponding to the 002 reflection, slightly shifted towards lower angles in comparison to the bulk 2H WS2 system (2θ ≈ 14.32°). The shift may be attributed to the strain relaxation in the 2H WS2 crystal lattice due to the formation of curved nanostructures [13, 14]. This observation was supported by the calculated cell parameters of the same a (0.31 nm) and somewhat longer c (1.26 nm) compared with those of the 2H WS2 system (a=0.31 and c=1.236 nm; JCPDS card no. 08-0237).
Figure 3.
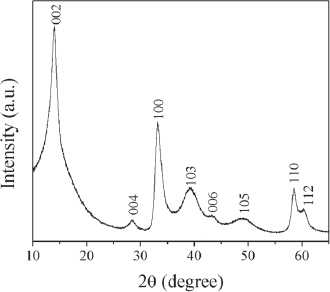
XRD pattern of the nanostructured WS2 powders obtained through heat treatment of the precursors at 750 °C for 1.5 h under argon atmosphere.
Room temperature UV-vis spectrum of the annealed (at 750 °C for 1.5 h) precursors is depicted in figure 1(b). It shows two weak absorption bands centered at 646 and 533 nm, which can be assigned to the excitons A and B of WS2, respectively. The observed positions of the excitonic bands are shifted to higher wavelength in comparison to the respective excitons at 635.8 and 525.3 nm, reported for the bulk 2H WS2 system [45]. The shift is however lower than reported for the IF-WS2 nanostructures (i.e. at 649.1 and 551 nm, respectively) [14, 45]. Our spectra thus indicate formation of WS2 nanostructures with intermediate exciton wavelength between the bulk 2H WS2 and IF-like WS2 nanostructures.
Bright-field TEM micrographs for the annealed powders of WS2 are shown in figures 4(a) and (b). The figures show the formation of WS2 nanowire-like structures, with thickness ∼5–10 nm. Li et al reported similar morphology for hydrothermally synthesized MoS2 nanowires [46]. HRTEM image of the same sample clearly identifies a single nanowire of WS2 with well-defined lattice fringes (figure 4(b)). Figure 4(c) illustrates the selected area electron diffraction (SAED) patterns for the sample. It shows distinct rings, characteristic of an assembly of nanocrystallites, which can be indexed to the 002, 103, 105 and 112 reflections of the 2H WS2 phase.
Figure 4.

(a) Low-resolution TEM image of WS2 nanowires; (b) HRTEM image of a single nanowire with visible lattice fringes; (c) selected area electron diffraction pattern for the WS2 nanowires.
The inter-planar distance along the c-axis, corresponding to the d002 plane, was measured from the average distance between two adjacent lattice fringes and was found to be approximately 0.64 nm, which is consistent with the XRD result. The value of the corresponding d002 spacing for the bulk 2H-WS2 is 0.618 nm (from JCPDS file), which is slightly smaller than the measured value, supporting our inference on the formation of folded nanostructures of WS2. The exact mechanism of formation of WS2 nanowires through microwave-induced localized temperature and pressure effects at the reaction sites requires further investigations.
Summary
A simple and rapid method has been developed for the synthesis of WS2 nanowires, with thickness ∼5–10 nm, through microwave irradiation induced reaction of tungstic acid with elemental sulfur in a medium of dilute monoethanolamine (MEA) via formation of tetrathiotungstate ions as intermediates. Amorphous precipitates, obtained on acidification of the precursor solution of tetrathiotungstate ions, when annealed at 750 °C, yield the WS2 nanowires. In the process, microwave induced reactions facilitate the solubilization of tungstic acid in MEA through formation of MEA chelated complex of tungstate and also aid in the in-situ formation of tetrathiotungstate ions via the generation of S2− ions from monoethanolaminium polysulfide complex. Thus, MEA not only acts as a solvent but also as a coordinating agent. Structural studies indicate strain relaxation in 2H-WS2 crystal lattices along the c-axis, suggesting the formation of curved nanostructures. Optical absorption studies support the findings through the red shift of the exciton bands.
Acknowledgments
The authors highly acknowledge MHRD, Govt of India and DST, Govt of India for the financial support of the present research work.
References
- Iijima S. Nature. 1991;354:56. doi: 10.1038/354056a0. [DOI] [Google Scholar]
- Cheng F Y. and Chen J. J. Mater. Res. 2006;21:2744. doi: 10.1557/jmr.2006.0337. [DOI] [Google Scholar]
- Wu F. 2004. Appl. Phys. A 78 989 10.1007/s00339-002-1992-5 [DOI] [Google Scholar]
- Chhowalla M. and Amaratunga G A J. Nature. 2000;407:164. doi: 10.1038/35025020. [DOI] [PubMed] [Google Scholar]
- Xu B H, Lin B Z, Sun D Y. and Ding C. Electrochim. Acta. 2007;52:3028. doi: 10.1016/j.electacta.2006.09.046. [DOI] [Google Scholar]
- Alonso G, Valle M D, Cruz J, Claverie A L, Petranovskii V. and Fuentes S. Catal. Lett. 1998;52:55. doi: 10.1023/A:1019067319305. [DOI] [Google Scholar]
- Harris S. and Chianelli R R. J. Catal. 1984;86:400. doi: 10.1016/0021-9517(84)90385-3. [DOI] [Google Scholar]
- Rapoport L, Fleischer N. and Tenne R. J. Mater. Chem. 2005;15:1782. doi: 10.1039/b417488g. [DOI] [Google Scholar]
- Zhou Z R. and Vincent L. Wear. 1999;229:962. doi: 10.1016/S0043-1648(99)00038-1. [DOI] [Google Scholar]
- Genut M, Margulis L, Hodes G. and Tenne R. Thin Solid Films. 1992;217:91. doi: 10.1016/0040-6090(92)90611-E. [DOI] [Google Scholar]
- Tenne R, Margulis L, Genut M. and Hodes G. Nature. 1992;360:444. doi: 10.1038/360444a0. [DOI] [Google Scholar]
- Margulis L, Salitra G, Tenne R. and Talianker M. Nature. 1993;365:113. doi: 10.1038/365113b0. [DOI] [Google Scholar]
- Feldman Y, Wasserman E, Srolovitz D J. and Tenne R. Science. 1995;267:222. doi: 10.1126/science.267.5195.222. [DOI] [PubMed] [Google Scholar]
- Feldman Y, Frey G L, Homyonfer M, Lyakhovitskaya V, Margulis L, Cohen H, Hodes G, Hutchison J L. and Tenne R. J. Am. Chem. Soc. 1996;118:5362. doi: 10.1021/ja9602408. [DOI] [Google Scholar]
- Rosentsveig R, Margolin A, Feldman Y, Popovitz-Biro R. and Tenne R. Chem. Mater. 2002;14:471. doi: 10.1021/cm010630f. [DOI] [Google Scholar]
- Zhu Y Q, Hsu W K, Grobert N, Chang B H, Terrones M, Terrones H, Kroto H W. and Walton D R M. Chem. Mater. 2000;12:1190. doi: 10.1021/cm991189k. [DOI] [Google Scholar]
- Wu J. and Fu X. Mater. Lett. 2007;61:4332. doi: 10.1016/j.matlet.2007.01.099. [DOI] [Google Scholar]
- Therese H A, Li J, Kolb U. and Tremel W. Solid State Sci. 2005;7:67. doi: 10.1016/j.solidstatesciences.2004.10.006. [DOI] [Google Scholar]
- Shang Y, Xia J, Xu Z. and Chen W. J. Dispersion Sci. Technol. 2005;26:635. doi: 10.1081/DIS-200057684. [DOI] [Google Scholar]
- Nath M, Govindaraj A. and Rao C N R. Adv. Mater. 2001;13:283. doi: 10.1002/1521-4095(200102)13:4<283::AID-ADMA283>3.0.CO;2-H. [DOI] [Google Scholar]
- Alonso G, Yang J, Siadati M H. and Chianelli R R. Inorg. Chim. Acta. 2001;325:193. doi: 10.1016/S0020-1693(01)00648-X. [DOI] [Google Scholar]
- Li Y D, Li X L, He R R, Zhu J. and Deng Z X. J. Am. Chem. Soc. 2002;124:1411. doi: 10.1021/ja012055m. [DOI] [PubMed] [Google Scholar]
- Remskar M, Skraba Z, Regula M, Ballif C, Sanjines R. and Levy F. Adv. Mater. 1998;10:246. doi: 10.1002/(SICI)1521-4095(199802)10:3<246::AID-ADMA246>3.0.CO;2-6. [DOI] [Google Scholar]
- Sergei I N, Koltypin Y, Mastai Y, Koltypin M. and Gedanken A. J. Mater. Chem. 2002;12:1450. doi: 10.1039/b110867k. [DOI] [Google Scholar]
- Rao K J, Vaidhyanathan B, Ganguli M. and Ramakrishnan P A. Chem. Mater. 1999;11:882. doi: 10.1021/cm9803859. [DOI] [Google Scholar]
- Tompsett G A, Conner W C. and Yngvesson K S. Chem. Phys. Chem. 2006;7:296. doi: 10.1002/cphc.200500449. [DOI] [PubMed] [Google Scholar]
- Patra C R, Odani A, Pol V G, Aurbach D. and Gedanken A. J. Solid State Electrochem. 2007;11:186. doi: 10.1007/s10008-005-0086-7. [DOI] [Google Scholar]
- Panda A B, Glaspell G. and El-Shall M S. J. Am. Chem. Soc. 2006;128:2790. doi: 10.1021/ja058148b. [DOI] [PubMed] [Google Scholar]
- Chen D, Tang K, Shen G, Sheng J, Fang Z, Liu X, Zheng H. and Qian Y. Mater. Chem. Phys. 2003;82:206. doi: 10.1016/S0254-0584(03)00206-2. [DOI] [Google Scholar]
- McDonald J W, Friesen G D, Rsenhein L D. and Newton W E. Inorg. Chim. Acta. 1983;72:205. doi: 10.1016/S0020-1693(00)81720-X. [DOI] [Google Scholar]
- Stirling D. 2000. The Sulfur Problem: Cleaning up Industrial Feedstocks J H Clark. Cambridge, UK: Royal Society of Chemistry; p 17 [Google Scholar]
- Roof G L and Go T S. 2003. Polysulfide solutions and hydroxalkylaminium ions for stabilizing elemental sulfur US Patent No. 6605234 [Google Scholar]
- Hou H W, Xin X Q. and Shi S. Coord. Chem. Rev. 1996;153:25. doi: 10.1016/0010-8545(95)01224-9. [DOI] [Google Scholar]
- Liu H B, Li Y L, Luo H Y, Fang H J, Li H M, Xiao S Q, Shi Z Q, Xiao S X and Zhu D B. 2003. Eur. Phys. J. D 24 405 10.1140/epjd/e2003-00188-3 [DOI] [Google Scholar]
- Licht S and Davis J. 1997. J. Phys. Chem. B 101 2540 10.1021/jp962661h [DOI] [Google Scholar]
- Dubois P, Lelieur J P. and Lepoutre G. Inorg. Chem. 1989;28:195. doi: 10.1021/ic00301a008. [DOI] [Google Scholar]
- Blandamerj M J, Grossan J M. and Symons M C R. Trans. Faraday Soc. 1964;60:494. doi: 10.1039/tf9646000494. [DOI] [Google Scholar]
- Fischer H, Schulz-Ekloff G. and Wohrle D. Chem. Eng. Technol. 1997;20:462. doi: 10.1002/ceat.270200705. [DOI] [Google Scholar]
- Dubois P, Lelieur J P. and Lepoutre G. Inorg. Chem. 1988;27:3032. doi: 10.1021/ic00290a027. [DOI] [Google Scholar]
- Lassner E and Schubert W D. 1999. Tungsten: Properties, Chemistry, Technology of the Element, Alloys, and Chemical Compound Heidelberg: Springer; p 166 [Google Scholar]
- Liang B and Andrews L. 2002. J. Phys. Chem. A 106 6945 10.1021/jp025915+ [DOI] [Google Scholar]
- Coates J. 2000. Interpretation of Infrared Spectra, A Practical Approach in Encyclopedia of Analytical Chemistry Meyers R A. Chichester: Wiley; p 10829 [Google Scholar]
- Müller A, Diemann E, Jostes R. and Bögge H. Angew. Chem., Int. Ed. Engl. 1981;93:957. doi: 10.1002/ange.19810931106. [DOI] [Google Scholar]
- Müller A, Diemann E, Jostes R. and Bögge H. Angew. Chem., Int. Ed. Engl. 1981;20:934. doi: 10.1002/anie.198109341. [DOI] [Google Scholar]
- Frey G L, Elani S, Homyonfer M, Feldman Y and Tenne R. 1998. Phys. Rev. B 57 6666 10.1103/PhysRevB.57.6666 [DOI] [Google Scholar]
- Li W J, Shi E W, Ko J M, Chen Z, Ogino H. and Fukuda T. J. Cryst. Growth. 2003;250:418. doi: 10.1016/S0022-0248(02)02412-0. [DOI] [Google Scholar]