

Abstract
Theranostic nanoparticles currently have been regarded as an emerging concept of ‘personalized medicine’ with diagnostic and therapeutic dual-functions. Eu3+ doped hydroxyapatite (HAp) has been regarded as a promising fluorescent probe for in vivo imaging applications. Additionally, substitution of Ca2+ with Fe3+ in HAp crystal may endow the capability of producing heat upon exposure to a magnetic field. Here we report a preliminary study of doping mechanism and photoluminescence of Eu3+ and Fe3+ doped HAp nanoparticles (Eu/Fe:HAp). HAp with varied concentration of Eu3+ and Fe3+ doping are presented as Eu(10 mol%):HAp, Eu(7 mol%)-Fe(3 mol%):HAp, Eu(5 mol%)-Fe(5 mol%):HAp, Eu(3 mol%)-Fe(7 mol%):HAp, and Fe(10 mol%):HAp in the study. The results showed that the HAp particles, in nano-size with rod-like morphology, were successfully doped with Eu3+ and Fe3+, and the particles can be well suspended in cell culture medium. Photoluminescence analysis revealed that particles have prominent emissions at 536 nm, 590 nm, 615 nm, 650 nm and 695 nm upon excitation at a wavelength of 397 nm. Moreover, these Eu/Fe:HAp nanoparticles belonged to B-type carbonated HAp, which has been considered an effective biodegradable and biocompatible drug/gene carrier in biological applications.
Keywords: Eu3+/Fe3+ doped-hydroxyapatite, theranostic nanoparticles and photoluminescence
1. Introduction
Nanotechnology has been seen as the way to the future in medical care. Recently, lanthanide-doped nanoparticles have been developed as a new class of luminescent optical labels that have become potential alternatives to organic fluorophores and quantum dots for applications in biology due to their high quantum yield, large Stokes shift, long lifetime and high stability [1, 2]. However, there are still some major challenges waiting to be overcome, such as having an ideal biocompatible and biodegradable nanoparticle-imaging probe for clinical use [3, 4].
The use of hydroxyapatite (HAp, Ca10(PO4)6(OH)2) in cell imaging is now an emerging topic because of its excellent biocompatibility, bioactivity, biodegradability and convenience for surface modification. Hydroxyapatite is also the major inorganic component in bones and teeth, and it plays an important role in tissue engineering, drug and gene delivery, and other biomedical areas [5, 6]. Due to its biocompatible and biodegradable properties, HAp has been regarded as one of the best host materials for metallic ions doping, such as Ag, Zn, Fe, Gd and Mn ions; the metallic ions doping could effectively regulate the properties of HAp and reduce the cytotoxicity from metallic ions [7–11]. Additionally, HAp could be a good host material for lanthanide doping by substitution of Ca2+ with lanthanide ions into the HAp crystal lattice [7, 12]. The lanthanide doped HAp may endow the particles with fluorescent properties, which can overcome low fluorescence intensity and low photostability of traditional fluorescence dyes, and avoid toxicity and disposal issues of quantum dot probes [13]. It has been reported that lanthanide ions, such as europium and terbium, exhibited the typical fluorescence for cell imaging under the specific excitation wavelength [7, 12, 13]. In particular, doping of europium, which has 4 f-4 f intraorbital electronic transitions spanning the visible and near-infrared ranges, leads to an important material for biomedical applications in the near-infrared spectral range [11, 14].
Theranostic agents are an emerging concept of ‘personalized medicine’ with diagnostic and therapeutic functions [15, 16]. Chen et al [11] reported that the presence of Eu3+/Gd3+ ions doped in calcium phosphate nanospheres could lead to the nanoparticles with photoluminescent and magnetic properties. Although HAp doped with Gd3+ could also be expected for use in tumor therapy via magnetic fluid hyperthermia, the heat generation is mild from Gd3+ compared to the use of Fe ions [11, 17]. In considering an ideal theranostic agent for clinical use, we select Eu3+ and Fe3+ as the doping ions in HAp nanocrystal in this study, named Eu/Fe:HAp.
Substitution of Ca2+ with Fe2+ or Fe3+ in HAp crystal has showed the capability of producing heat upon exposure to a magnetic field, so that HAp doped with Fe ions can also be considered thermal seeds in hyperthermia treatment [18]. Over the past few years, our lab has developed several Fe-doped ceramics as thermal seeds for cancer hyperthermia [19–22]. Among those, Fe-doped HAp, as so-called mHAp, yielded a great possibility for heat generation upon exposure to the magnetic field; that may selectively killed cancer cells and keep normal cells alive in the range of 43–46 °C. Although iron oxide-based phases (maghemite or magnetite) have also been regarded as potential magnetic nanoparticles showing the ability to generate the heat under the magnetic field, the long-term side effects on the human body have been a concern [18, 23]. Compared to iron-oxide base materials, mHAp nanoparticles could reveal more valid bioactive, bioresorbable and low-toxic properties; recently magnetic apatite has been offered as a new alternative to the iron oxide-based phases for hyperthermia treatment [18, 24, 25].
Until now, few papers have discussed the concept of calcium phosphate-based theranostic agents via metal ions doping, and in particular, the use of hydroxyapatite as a host material with Eu3+ and Fe3+ dual-ions doping has not been reported yet. Therefore, the objective of this preliminary study is the doping mechanism and photoluminescence properties of Eu/Fe:HAp nanoparticles. The synthesized Eu/Fe:HAp nanoparticles were prepared by wet chemical precipitation with total doping concentration of Eu3+ and Fe3+ fixed at 10 mol% relative to Ca2+ in HAp host material. X-ray diffraction (XRD), scanning electron microscopy (SEM), inductively coupled plasma atomic emission spectrometry (ICP-AES), Fourier transform infrared spectroscopy (FTIR) and photoluminescence spectroscopy (PL) were used for the analysis of crystal structure, morphology, composition, functional groups, and luminescence properties, respectively.
2. Experimental procedures
2.1. Synthesis of Eu/Fe:HAp nanoparticles
Preparation of Eu/Fe:HAp, Ca10 − x − yEuxFey(PO4)6(OH)2 with x and y between 0 and 1 was conducted by a wet chemical precipitation method. Pure Ca(OH)2 was first prepared from hydration of CaO, which was obtained from heat decomposition of CaCO3 [26]. The 0.3 M H3PO4 solution was prepared with various concentrations of Eu(NO3)3 and Fe(NO3)3. Then, the complex solution of H3PO4 was added dropwise (1.3 ml min−1) into the 0.5 M Ca(OH)2 solution and the mixed solution was vigorously stirred for 2 h at pH 8.0 with temperature maintained at 80 °C. The total doping concentration of Eu3+ and Fe3+ was fixed at 0.05 M, which was equal to 10 mol% relative to Ca2+, and themolar ratios of Eu3+ to Fe3+ were 1 : 0, 2 : 1, 1 : 1, 1 : 2 and 0 : 1, which are representatives of Eu(10 mol%):HAp, Eu(7 mol%)-Fe(3 mol%):HAp, Eu(5 mol%)-Fe(5 mol%):HAp, Eu(3 mol%)-Fe(7 mol%):HAp and Fe(10 mol%):HAp respectively. The ultimate mixed solution was left to age at 37 °C for 24 h. After aging, the solution was centrifuged and washed with de-ionized water three times and freeze-dried for further analyses by XRD, ICP-AES, FE-SEM, FTIR and PL.
2.2. Characterization
The physical and chemical characteristics of the Eu/Fe:HAp were examined by several methods. The crystal structure and crystallinity were characterized by XRD (X’Pert, Philips) using Cu Kα radiation (λ = 0.15406 nm) at 40 kV and 30 mA. The crystallite size (D) of HAP and Eu/Fe:HAp particles was averaged from the major reflection peaks, (002), (202), (310), (222) and (213), using the equation of Debye-Scherrer [27]. Where λ is the wavelength of the monochromatic x-ray beam, B is the full width at half-maximum (FWHM) of peak at the maximum intensity and θ is the peak diffraction angle that satisfies Bragg’s law for the (hkl) plane.
The lattice parameters (a and c) were calculated with the following equation, where d is the interplanar spacing, hkl are Miller indices [28].
ICP-AES (ICPE-9000) was used for the quick and accurate analysis of Ca, P, Fe and Eu elements in samples by prior dilution, dissolution or microwave assisted closed vessel digestion according to the regulations [29]. The amount of detected ions was further calculated and illustrated in ionic ratio (table 1 and figure 3). The doping efficiency was calculated by the amount of actual doping concentration divided to the theoretical doping concentration.
Table 1.
Ionic composition ratio and P deficiency of HAp with various Eu3+ and Fe3+ doping ratios.
| Ca/P (molar ratio) | (Eu+Fe)/Ca (molar ratio) | P deficiency (%) | |
|---|---|---|---|
| HAp | 1.91 | — | 14.37 |
| Eu(10 mol%):HAp | 1.65 | 5.96 | 5.27 |
| Eu(7 mol%)-Fe(3 mol%):HAp | 1.72 | 7.53 | 11.6 |
| Eu(5 mol%)-Fe(5 mol%):HAp | 1.71 | 8.75 | 12.44 |
| Eu(3 mol%)-Fe(7 mol%):HAp | 1.73 | 9.03 | 14.1 |
| Fe(10 mol%):HAp | 1.78 | 9.34 | 17.8 |
Figure 3.
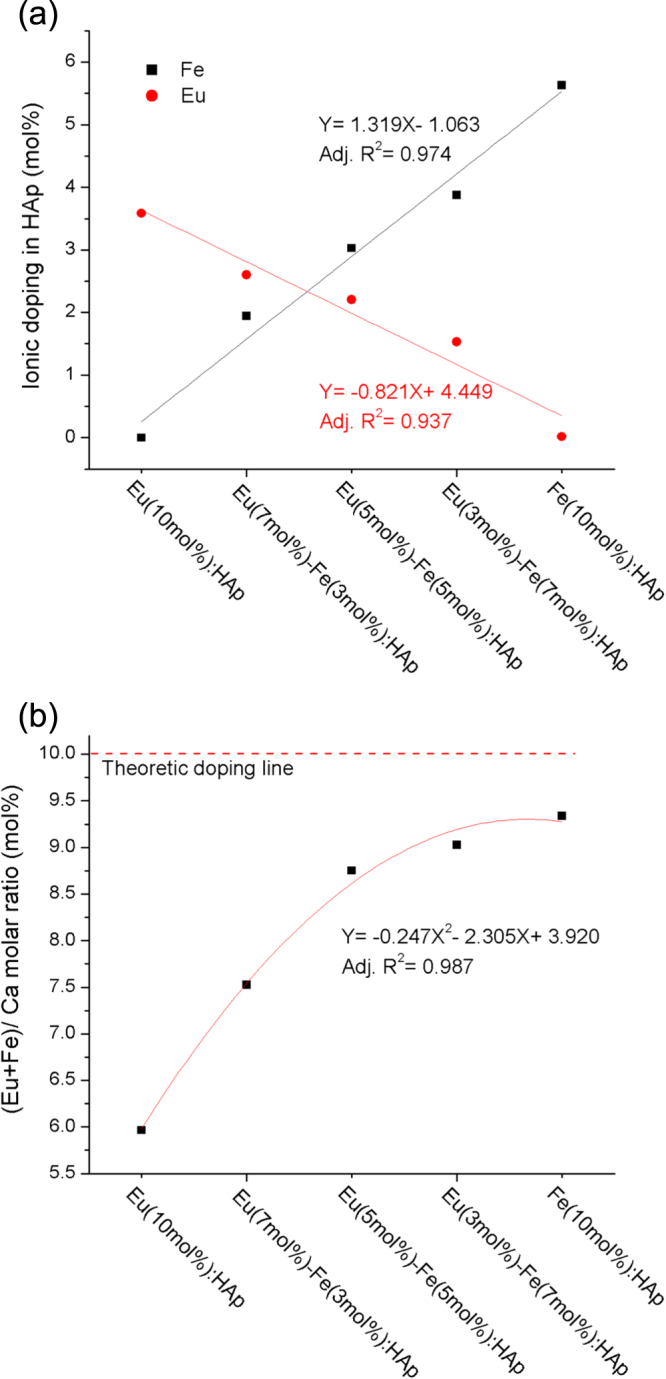
The ICP-AES results of (a) Eu3+ and Fe3+ doping amount and (b) (Eu+Fe)/ Ca molar ratio of HAp with varied dopant concentration.
Particles mixed with KBr were characterized by FTIR spectroscopy (FT/IR-4100N, JASCO) using a diffuse reflection technique to characterize surface functional groups of particles. All FTIR spectra were recorded from 500 cm−1 to 4000 cm−1 in steps of 4 cm−1.
To study the suspension ability of HAp and Eu/Fe:HAp nanoparticles in aqueous solution, turbidity of 1.5 mg ml−1 of particles suspended in culture medium with 10% fetal bovine serum (FBS) was measured by UV spectrophotometer at 320 nm as an indicator of the suspended concentration at each standing time [30].
For imaging of morphology, particles were coated with a thin gold layer by sputtering. The particle size and morphology of Eu(5 mol%)-Fe(5 mol%):HAp were evaluated from SEM images (S-4700 microscope, Hitachi, 20 kV).
For further imaging the particles in cell culture environment, human HeLa cell line was seeded in a 35 mm glass bottom Petri dish with a density of 5 × 104 cells/well in minimum essential media (MEM) supplemented with 10% FBS and maintained at 37 °C in a humidified air containing 5% CO2. After incubation of cells for 24 h, Eu(5 mol%)-Fe(5 mol%):HAp particles were added in the dish at a finial concentration of 1 μg ml−1. After incubation for 5 h, the cells were fixed with 4% paraformaldehyde, labeled with a Hoechst stain, and observed in an optical microscope (CLSM, SP5, Leica).
A spectrofluorometer (FP-6300, JASCO) equipped with a cut-off filter Y-52 (JASCO, Japan) was used to characterize the photoluminescence properties of the Eu/Fe:HAp particles. Emission spectra were recorded at room temperature under 397 nm excitation, and the excitation spectra were measured for the 615 nm emission.
3. Results and discussion
3.1. Phase, composition, morphology and doping mechanism
Figure 1 shows the XRD patterns of the Eu/Fe:HAp obtained. The calculated values of grain size and lattice parameters are presented in figure 2. The XRD patterns reveal that the synthesized samples can well be indexed to hydroxyapatite, which belongs to a hexagonal system with P63/m space group (ICDD file No. 00-009-0432). The major peaks in the XRD patterns are consistent to the (211), (300), (002), (202), (222) and (213) reflections of the Ca10(PO4)6(OH)2 lattice; none of the patterns displayed additional crystallized phase, indicating that Eu3+ and Fe3+ have been successfully doped into the crystal lattice of HAp. The effect of Eu3+ and Fe3+ doping appears in the XRD patterns of Eu/Fe:HAp as a change in peaks position, intensity and broadening. The lower intensity and a slight shift to lager angles are attributed to the smaller particles size and greater structural strain in Eu/Fe:HAp relative to pure HAp [31]. Increasing the concentration of Fe3+ into the materials apparently leads to the decreasing of the peak intensity, which indicates Fe3+ can reduce the crystallinity of HAp formation. It is assumed that Fe3+ with ionic radius 69 pm, which is smaller than the radius of Ca2+ (114 pm), could inhibit the growth of HAp crystals by shrinking the lattice as Fe3+ substitutes into the Ca2+ sites [10, 31, 32]. In contrast to the Fe3+, the ionic radius of Eu3+ is 108.7 pm, which is closer to Ca2+, and therefore the peak intensity seems unobviously suppressed by the substitution of Eu3+ in Ca2+ site. Based on the equation of Debye Scherrer [28], the broadening of peaks (increase of the FWHM) is related to the Eu3+ and Fe3+ substitution in HAp that creates lattice defects and suppresses the grain growth [31]. The average grain size decreases gradually from 18 nm to 13 nm with the increasing Fe3+ concentration (figure 2(a)).
Figure 1.
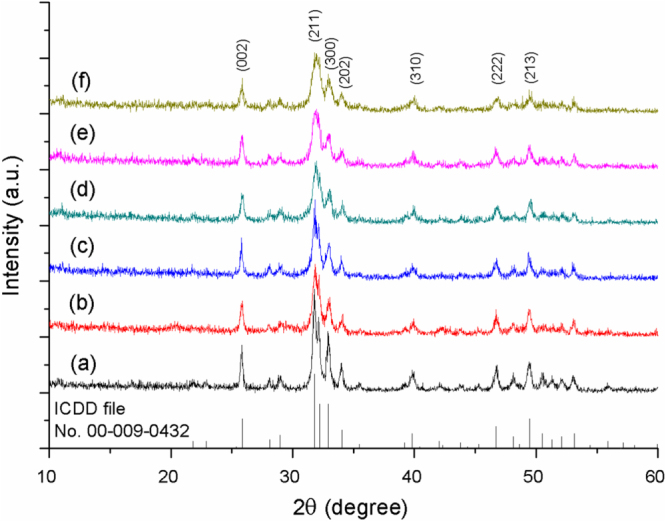
XRD patterns of (a) HAp and (b)–(f) Eu/Fe:HAp with varied Eu3+ and Fe3+ doping concentrations. The doping concentrations of Eu3+/ Fe3+ are 10 mol% relative to Ca2+, and the molar ratios of Eu3+ to Fe3+ from (b) to (f) are 1:0, 2:1, 1:1, 1:2 and 0:1 respectively.
Figure 2.

The calculated (a) grain size and (b), (c) lattice parameters of HAp and Eu/Fe:HAp with various Eu3+ and Fe3+ doping concentrations, based on XRD data (n = 5).
The results of lattice parameters reveals that doping of Eu3+ and Fe3+ would cause the a-axis lattice parameter to decrease (figure 2(b)). For hexagonal lattice system of HAp, a-axis lattice is equal to the b-axis lattice. As the amount of Fe3+ doping increased to 10 mol%, the a-axis lattice parameter approached the minimum logically due to the substitution of Fe3+ with smaller ionic radius, which will decrease the lattice parameters [31]. Both the reducing intensity of XRD peaks (figure 1) and the decrease of a-axis lattice have demonstrated the substitution of Fe3+ in HAp lattice crystal. Generally, c-axis lattice parameter should also be expected to decrease like a-axis as a result of the smaller radius of Fe3+ ions. However, there is no any significant decrease in c-axis with Fe3+ ions doping (figure 2(c)), for the substitution of carbonate anion for the phosphate anion provokes the expansion along the c-axis [33]. The carbonate anion comes from atmospheric CO2 or CaCO3 impurities in the reactant [8]. However, the substitution of carbonate anion for phosphate anion does not cause appreciable change in XRD patterns (figure 1), for phosphate and carbonates anions have similar x-ray scattering factors and the amount of phosphate substitution by carbonates is quite low [8], less than 15% carbonate anion doped in Eu/Fe:HAp according to the ICP-AES results (table 1).
The doping concentrations of Eu3+ and Fe3+ in HAp measured by ICP-AES were close to the theoretical values (figure 3(a)). The doping efficiency is related to the ionic radius [20], so that the smaller radius of Fe3+ results in a higher concentration (5.63 mol%) compared to Eu3+ (3.58 mol%) in a host material. The strongest effect of Eu3+ and Fe3+ substitution in HAp lattice is observed in sample Fe(10 mol%):HAp where it approaches 9.34 mol% of relative doping amount of Fe3+ to Ca2+ (figure 3(b)), also indicating Fe3+ has high doping efficiency of around 93% than Eu3+ (60%). The results also point out the Ca/P molar ratio is higher than expected molar ratio of HAp (Ca/P = 1.67) and Eu/Fe:HAp (Ca/P = 1.5) according to the stoichiometry of pure HAp (table 1). The increase of Ca/P molar ratio indicates that the substitution of carbonate anion for the phosphate groups in the HAp has taken place. These substitutions reduce the amount of phosphate group (P deficiency), thus leading the Ca/P ratio to increase [8]. It can be seen that Eu/Fe:HAp generally has lower P deficiency than HAp, for the doping ions possibly interfere with the substitution of carbonate anions for phosphate sites. Particularly, P deficiency is reduced with the increasing Eu3+ concentration, which shows Eu3+ has higher tendency than Fe3+ to inhibit carbonate anions substitution.
Moreover, FTIR spectra (figure 4) of Eu/Fe:HAp indicate that all samples possess fundamental apatite structure with PO43− and OH− absorption bands [34]. The CO32− and H2O adsorption bands are also detected in all spectra. The carbonate groups also proves partial PO43− groups were replaced by CO32− in the HAp lattice and lead to the formation of particles, which belonged to B-type carbonated HAp [10, 35]. The carbonated HAp has been reported as the major inorganic component of vertebrate bones [36] and has shown to have an improved bioactivity and biodegradability compared with pure HAp [37]. In contrast, carbonated HAp has a dissolution rate 5 times higher than pure HAp due to the substitution of carbonate ions, which will increase the bioactivity of HAp particles by increasing the number of triple-junction grain boundaries [37, 38]. Due to the substitution of carbonate, this Eu/Fe:HAp nanoparticle has been considered a biodegradable material and expected to be a next generation biomaterial.
Figure 4.
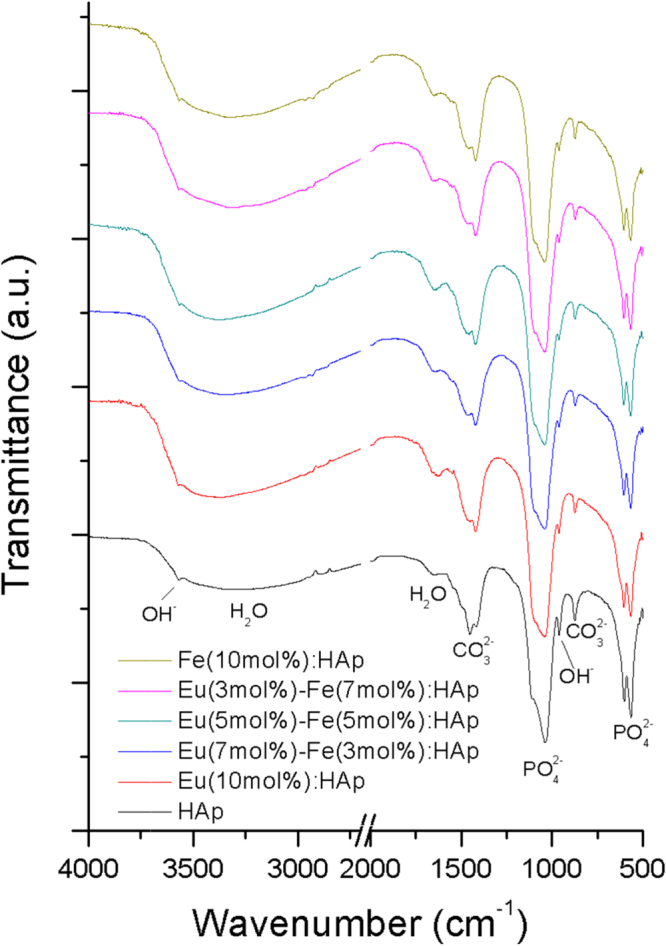
FTIR spectra of the synthesized HAp and Eu/Fe:HAp nanoparticles developed with various Eu3+ and Fe3+ doping concentrations.
HAp nanoparticles can be produced by different techniques, such as wet chemical precipitation, sol-gel and solid state synthesis [39]. However, the finial particles always aggregate easily in aqueous based solvent due to its high surface energy when the size of particles is in nanometer range [40]. In order to have better application in biomedical field, it is an important issue to take the fabrication of water-dispersible nanoparticles into consideration [41]. In this study, we have successfully synthesized Eu/Fe:HAp with various doping concentrations in water without addition of any surfactant, and those nanoparticles could be well re-suspended in culture medium (with 10% FBS) for a relatively long period of time (figure 5(a)). Generally, the suspension ability of HAp nanoparticles highly depended on the use of aqueous solution, with complete aggregation observed within several minutes in an aqueous solution [42]. However, they could be homogenously suspended in citrate solution due to the excellent chelating ability within citrate ions and Ca2+ ions, which forms the repulsion among particles and increases the dispersibility of nanoparticles [43]. To more clearly understand the particles in biological application, the synthesized particles were incubated in culture medium; turbidity was measured by UV spectrophotometer at 320 nm as an indicator of HAp suspension ability at each standing time [30]. The results reveal that after 1 h of incubation, more than 90% of Eu/Fe:HAp particles were still suspended in culture medium (figure 5(b)). However, after 3 h of standing time, HAp without ionic doping shows lower suspended concentration than Eu/Fe:HAp, which is reasonable because the lower crystallinity and grain size of Eu/Fe:HAp particles may result in a higher dispersibility in aqueous solution compared to HAp. The SEM image of Eu(5 mol%)-Fe(5 mol%):HAp is shown in figure 5(c), which is representative of all Eu/Fe:HAp nanoparticles. All Eu/Fe:HAp samples have similar crystal morphology, which resembles that of HAp particles without Eu3+ and Fe3+ doping. The image clearly shows the particles are homogeneously dispersed in the loose agglomeration (polydispersity Index around 0.20–0.28 by dynamic light scattering analysis) having rod-like shapes with a length of about 100 nm. Optical imaging further demonstrated that Eu/Fe:HAp particles can be well dispersed in culture medium and homogenously bound on cell membrane (figure 5(d)). According to the literature reported by Barua and Mitragotri [44], the nanoparticles could enhance tissue penetration but those smaller than 10 nm are easily cleared by renal excretion and phagocytosis. Nanoparticles around 100 nm exhibit prolongation in blood circulation system and cause relative low rate of mononuclear phagocyte system uptake compared to smaller or larger particles. Furthermore, HAp-based nanomaterials are considered easily internalized into the cells through cell endocytosis, and facilitate to endosomal escape due to its biodegradable properties in response to endosomal acidic pH [30, 45, 46]. Overall, the results have demonstrated that Eu/Fe-doped HAp nanoparticles could find biological applications as drug and gene vehicles.
Figure 5.

(a) The photograph of re-suspended HAp and Eu/Fe:HAp particles in culture medium with 10% FBS. The symbol A represents the sample HAp; the molar ratios of Eu3+ to Fe3+ from B to F are 1:0, 2:1, 1:1, 1:2 and 0:1 respectively. (b) Assessment of particle suspension in culture medium by turbidity measurement at 320 nm. (c) SEM image of synthesized Eu(5 mol%)-Fe(5 mol%):HAp nanoparticles. (d) Imaging of cells incubated with Eu(5 mol%)-Fe(5 mol%):HAp nanoparticles; cell nucleus was labeled with a Hoechest stain.
3.2. Photoluminescence properties
Photoluminescence properties have provided important information about Eu3+ substitution in HAp. PL spectra show the Eu/Fe:HAp with different Eu3+ dopant concentration have maximum emission at 615 nm under 397 nm excitation (figure 6); the excitation wavelength was chosen according to the excitation spectrum in figure 6(a) and all spectra corresponded to the typical 5D0 →︀ 7FJ emissions from Eu3+ transition. In the PL emission spectra (figure 6(b)), three intense emission peaks appear at about 590, 615 and 695 nm and two weak peaks at about 536 and 650 nm, and they can be attributed to the 5D0 →︀ 7F1, 5D0 →︀ 7F2, 5D0 →︀ 7F3, 5D0 →︀ 7F0 and 5D0 →︀ 7F3, transitions of Eu3+ ions respectively [13, 47, 48]. These luminescence transitions demonstrate the result of the spin–orbit coupling of six electrons in the f-subshell and the transitions are attributed to the mixing of odd terms due to the crystal field. The non-degenerate 5D0 →︀ 7F0 transition clearly indicates the presence of Eu3+ in the host of HAp [49]. According to the literature, the ideal crystalline HAp has two different calcium sites: Ca(I) and Ca(II), and 5D0 →︀ 7F1 transition is related to Eu3+ substitution in Ca(I) while 5D0 →︀ 7F2 transition is due to the substitution of Eu3+ in Ca(II) [50]. Our results show that the peak from 603–640 nm has higher intensity than that from 570–603 nm, indicating that Eu3+ ions have higher substitution percentage in Ca(II) sites than in Ca(I) sites of HAp host matrix. The intensity of emission increases with an increase in the concentration of Eu3+ from 0 mol% to 10 mol%. However, the luminescence is quenched with the Fe3+ doped in HAp crystal lattice (figure 7). The suppressed intensity may be due to either cross relaxation or crystal defects in the host material [51]. Chen and Liu [52] reported that the intensity of emission is related to the electronic transition from 5D0 to 7FJ, which occurred in Eu3+-O2− bond; however, the electronic transition may be perturbed either by an impurity ion such as O2− or by a structural distortion, which could result in emission suppression. For trivalent Fe3+ substitution at a Ca2+ site, charge compensation is considered to induce defects and nonstoichiometry in the HAp lattice. At the Ca(I) site, a nonstoichiometric system is introduced, while Fe3+ substitution at Ca(II) sites might generate vacancies at hydroxyl and Ca(II) sites [53]. Hence, it may be the reason why the substitution of Fe3+ for Ca2+, which causes more vacancies and defects in crystal, will disrupt the mechanism of charge transfer of Eu3+-O2− from the 2p orbital of O2− to the 4f orbital of the Eu3+ ion [13, 48] and will lead to the decrease of emission intensity. Additionally, ICP-AES (table 1) also suggests the carbonate substitution is increasing with the increasing concentration of Fe3+; and the amount of carbonate substitution is also inversely proportional to the emission intensity. Thus, we may not exclude the possibility of carbonate substitution in Eu/Fe:HAp, which will obstruct charge transfer between Eu3+ and O2−, and quench the emission intensity.
Figure 6.
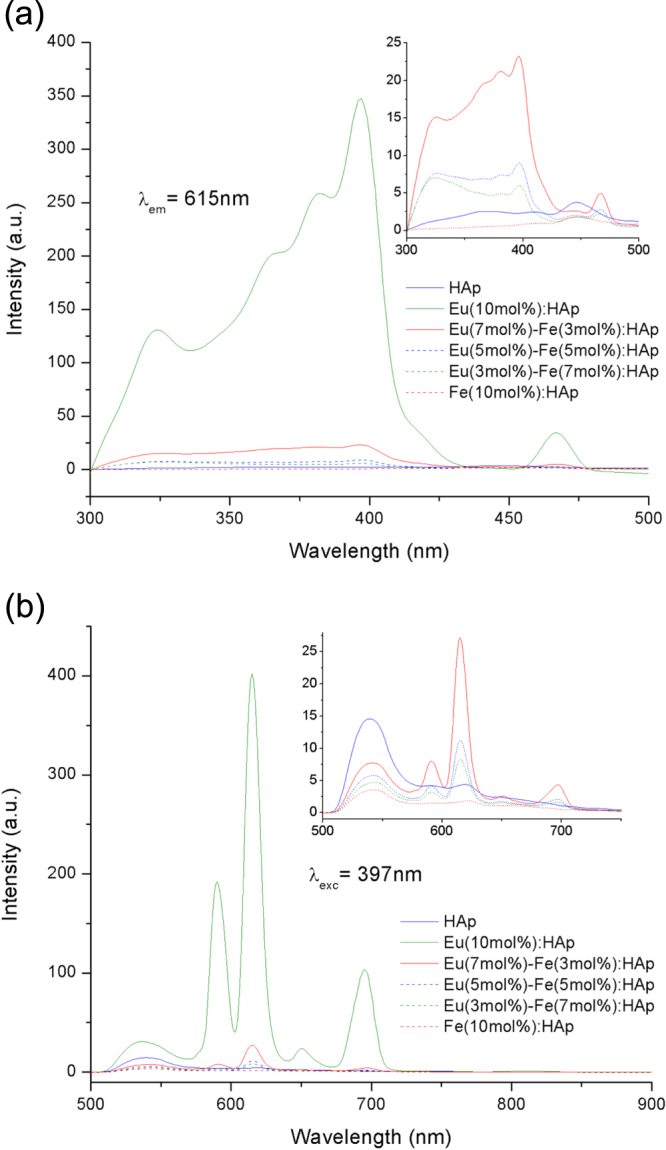
(a) The PL excitation spectra and (b) PL emission spectra of HAp and Eu/Fe:HAp with different dopant concentrations. Inset shows the magnified spectra of samples except the sample of Eu(10 mol%):HAp.
Figure 7.
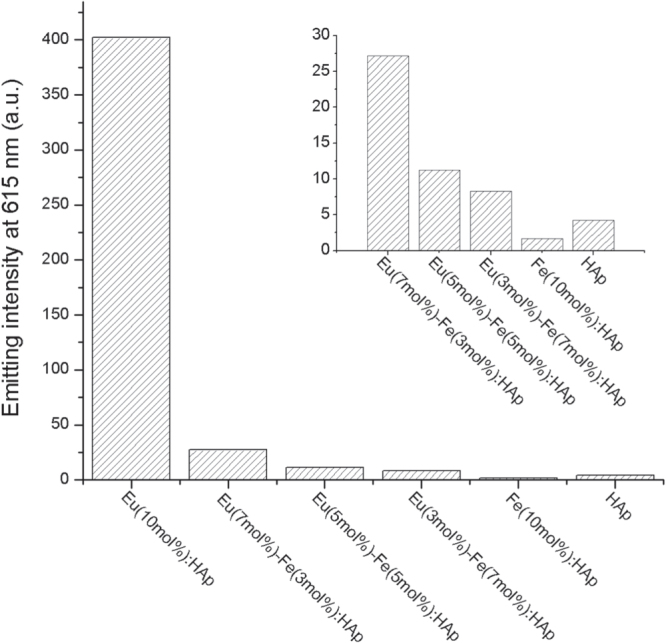
Luminescence intensity (615 nm) of Eu/Fe:HAp with varied atomic doping molar ratio under the excitation at a wavelength of 397 nm. Inset shows data for the last five samples.
Our preliminary results indicated that luminescent Eu/Fe:HAp nanoparticle may find biological applications. The second part would be to evaluate the cytotoxicity, cell imaging, heat generation, and possibility of using the developed nanoparticle as thermal seeds for cancer hyperthermia in rat model; that would be shown in the other article.
4. Conclusions
We have reported a preliminary study on the doping mechanism and photoluminscence of Eu/Fe:HAp nanoparticles. Herein, nano-sized, single-phase, biodegradable and luminescent Eu/Fe:HAp nanoparticles were synthesized through the wet chemical precipitation in water without addition of any surfactant, showing rod-like particles with a length of about 100 nm that can be suspended in culture medium. The nanoparticles were synthesized as hexagonal single crystals without the appearance of second phase, and the nanoparticles belonged to the biodegradable B type carbonated HAp. These nanoparticles emitted luminescence with prominent peaks at 536, 590, 615, 650 and 695 nm under 397 nm excitation. Our results indicate that Eu/Fe:HAp nanoparticles could be an ideal candidate for biological applications. With these encouraging results, future work will involve the confirmation of magnetic and hyperthermia properties of these Eu/Fe:HAp nanoparticles as an in vitro and in vivo model.
Acknowledgments
This work is an international cooperation supported by The Asia-Oceania Top University League on Engineering (AOTULE) program. M-H Chen thanks all members from Profs. Tanaka and Ikoma laboratory in Tokyo Tech for their supports. The authors are particularly grateful to Nanotechnology Innovation Station in NIMS, for UV spectrophotometer, DLS and cell imaging supports.
References
- Tagaya M, Ikoma T, Yoshioka T, Motozuka S, Minami F. and Tanaka J. Mater. Lett. 2011;65:2287. doi: 10.1016/j.matlet.2011.04.011. [DOI] [PubMed] [Google Scholar]
- Gnach A. and Bednarkiewicz A. Nano Today. 2012;7:532. doi: 10.1016/j.nantod.2012.10.006. [DOI] [Google Scholar]
- Chapman S. Nano Today. 2013;8:454. doi: 10.1016/j.nantod.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y C, Huang X C, Luo Y L, Chang Y C, Hsieh Y Z. and Hsu H Y. Sci. Technol. Adv. Mat. 2013;14(044407) doi: 10.1088/1468-6996/14/4/044407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung R J. Biomed. Eng. Appl. Basis Commun. 2011;23:107. doi: 10.4015/s1016237211002451. [DOI] [Google Scholar]
- Okada M. and Furuzono T. Sci. Technol. Adv. Mat. 2012;13(064103) doi: 10.1088/1468-6996/13/6/064103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui J, Zhang X, Zhang Z, Wang S, Tao L, Wei Y. and Wang X. Nanoscale. 2012;4:6967. doi: 10.1039/c2nr32404k. [DOI] [PubMed] [Google Scholar]
- Devanand Venkatasubbu G, Ramasamy S, Ramakrishnan V. and Kumar J. 3 Biotech. 2011;1:173. doi: 10.1007/s13205-011-0021-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciobanu C S, Iconaru S L, Pasuk I, Vasile B S, Lupu A R, Hermenean A, Dinischiotu A. and Predoi D. Mater. Sci. Eng. C Mater. Biol. Appl. 2013;33:1395. doi: 10.1016/j.msec.2012.12.042. [DOI] [PubMed] [Google Scholar]
- Li Y, Nam C T. and Ooi C P. J. Phys. Conf. Ser.; 2009. [Google Scholar]
- Chen F, Huang P, Zhu Y J, Wu J. and Cui D X. Biomaterials. 2012;33:6447. doi: 10.1016/j.biomaterials.2012.05.059. [DOI] [PubMed] [Google Scholar]
- Ciobanu C S, Iconaru S L, Massuyeau F, Constantin L V, Costescu A. and Predoi D. J. Nanomater. 2012;2012:1. doi: 10.1155/2012/942801. [DOI] [Google Scholar]
- Hasna K, Kumar S S, Komath M, Varma M R, Jayaraj M K. and Kumar K R. Phys. Chem. Chem. Phys. 2013;15:8106. doi: 10.1039/c3cp42648c. [DOI] [PubMed] [Google Scholar]
- Petit L, Griffin J, Carlie N, Jubera V, García M, Hernández F E. and Richardson K. Mater. Lett. 2007;61:2879. doi: 10.1016/j.matlet.2007.01.072. [DOI] [Google Scholar]
- Terreno E, Uggeri F. and Aime S. J. Control. Release. 2012;161:328. doi: 10.1016/j.jconrel.2012.05.028. [DOI] [PubMed] [Google Scholar]
- Park Y I. Adv. Mater. 2012;24:5755. doi: 10.1002/adma.201202433. [DOI] [PubMed] [Google Scholar]
- Drake P, Cho H J, Shih P S, Kao C H, Lee K F, Kuo C H, Linb X Z. and Lin Y J. J. Mater. Chem. 2007;17:4914. doi: 10.1039/b711962c. [DOI] [Google Scholar]
- Tampieri A. Acta Biomater. 2012;8:843. doi: 10.1016/j.actbio.2011.09.032. [DOI] [PubMed] [Google Scholar]
- Wu H C, Wang T W, Sun J S, Wang W H. and Lin F H. Nanotechnology. 2007;18(165601) doi: 10.1088/0957-4484/18/16/165601. [DOI] [Google Scholar]
- Hou C H, Chen C W, Hou S M, Li Y T. and Lin F H. Biomaterials. 2009;30:4700. doi: 10.1016/j.biomaterials.2009.05.018. [DOI] [PubMed] [Google Scholar]
- Wang T W, Wu H C, Wang W R, Lin F H, Lou P J, Shieh M J. and Young T H. J. Biomed. Mater. Res. A. 2007;83:828. doi: 10.1002/jbm.a.31411. [DOI] [PubMed] [Google Scholar]
- Hou C H, Hou S M, Hsueh Y S, Lin J, Wu H C. and Lin F H. Biomaterials. 2009;30:3956. doi: 10.1016/j.biomaterials.2009.04.020. [DOI] [PubMed] [Google Scholar]
- Zhao S, Lin X, Zhang L, Sun L, Li J, Yang W. and Sun Z. Biomed. Eng. Appl. Basis Commun. 2012;24:229. doi: 10.4015/s1016237212500056. [DOI] [Google Scholar]
- Iafisco M, Sandri M, Panseri S, Delgado-López J M, Gómez-Morales J. and Tampieri A. Chem. Mater. 2013;25:2610. doi: 10.1021/cm4007298. [DOI] [Google Scholar]
- Panseri S, Cunha C, D’alessandro T, Sandri M, Giavaresi G, Marcacci M, Hung C T. and Tampieri A. J. Nanobiotechnology. 2012;10:32. doi: 10.1186/1477-3155-10-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi M, Matsumoto H N, Yamada T, Koyama Y, Takakuda K. and Tanaka J. Biomaterials. 2004;25:63. doi: 10.1016/S0142-9612(03)00472-1. [DOI] [PubMed] [Google Scholar]
- Brundavanam R K, Poinern G E J. and Fawcett D. Am. J. Mater. Sci. 2013;3:84. [Google Scholar]
- Webster T J, Massa-Schlueter E A, Smith J L. and Slamovich E B. Biomaterials. 2004;25:2111. doi: 10.1016/j.biomaterials.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Fischer L, Zipfel B, Koellensperger G, Kovac C, Bilz S, Kunkel A, Venzago C. and Hann S. J. Pharm. Biomed. Anal. 2014;95:121. doi: 10.1016/j.jpba.2014.02.016. [DOI] [PubMed] [Google Scholar]
- Chowdhury E H, Maruyama A, Kano A, Nagaoka M, Kotaka M, Hirose S, Kunou M. and Akaike T. Gene. 2006;376:87. doi: 10.1016/j.gene.2006.02.028. [DOI] [PubMed] [Google Scholar]
- Gamal G A, Al-Mufadi F A. and Said A H. Eng. Technol. Appl. Sci. Res. 2013;3:532. [Google Scholar]
- Miyaji F, Kono Y. and Suyama Y. Mater. Res. Bull. 2005;40:209. doi: 10.1016/j.materresbull.2004.10.020. [DOI] [Google Scholar]
- Borum-Nicholas L, Wilson O C., Jr Biomaterials. 2003;24:3671. doi: 10.1016/S0142-9612(03)00239-4. [DOI] [PubMed] [Google Scholar]
- Kumar R, Prakash K H, Cheang P. and Khor K A. Langmuir. 2004;20:5196. doi: 10.1021/la049304f. [DOI] [PubMed] [Google Scholar]
- Uskokovic V. and Uskokovic D P. J. Biomed. Mater. Res. B Appl. Biomater. 2011;96:152. doi: 10.1002/jbm.b.31746. [DOI] [PubMed] [Google Scholar]
- Nagai A, Tanaka K, Tanaka Y, Nakamura M, Hashimoto K. and Yamashita K. J. Biomed. Mater. Res. A. 2011;99:116. doi: 10.1002/jbm.a.33131. [DOI] [PubMed] [Google Scholar]
- Porter A, Patel N, Brooks R, Best S, Rushton N. and Bonfield W. J. Mater. Sci. Mater. Med. 2005;16:899. doi: 10.1007/s10856-005-4424-1. [DOI] [PubMed] [Google Scholar]
- Barralet J, Akao M, Aoki H. and Aoki H. J. Biomed. Mater. Res. 2000;49:176. doi: 10.1002/(sici)1097-4636(200002)49:2<176::aid-jbm4>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Hadeel A, Jeffrey L E, Ramin R, Hans C. and Fariba D. Trends Biomater. Artif. Organs. 2011;25:12. [Google Scholar]
- Abidi S S A. and Murtaza Q. U.P.B. Sci. Bull. Ser. B. 2013;75:3. doi: 10.1007/s12262-013-0839-y. [DOI] [Google Scholar]
- Zhou R, Si S. and Zhang Q. Appl. Surf. Sci. 2012;258:3578. doi: 10.1016/j.apsusc.2011.11.119. [DOI] [Google Scholar]
- Tan J, Chen M. and Xia J. Appl. Surf. Sci. 2009;255:8774. doi: 10.1016/j.apsusc.2009.06.044. [DOI] [Google Scholar]
- Martins M A, Santos C, Almeida M M. and Costa M E. J. Colloid Interface Sci. 2008;318:210. doi: 10.1016/j.jcis.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Barua S. and Mitragotri S. Nano Today. 2014;9:223. doi: 10.1016/j.nantod.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M, Rhodes C J, Moncol J, Izakovic M. and Mazur M. Chem. Biol. Interact. 2006;160:1. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Kimura T, Nibe Y, Funamoto S, Okada M, Furuzono T, Ono T, Yoshizawa H, Fujisato T, Nam K. and Kishida A. J. Drug Deliv. 2011;2011(962743) doi: 10.1155/2011/962743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Zhu J. and Zhou K. Mater. Res. Bull. 2012;47:24. doi: 10.1016/j.materresbull.2011.10.013. [DOI] [Google Scholar]
- Graeve O A, Kanakala R, Madadi A, Williams B C. and Glass K C. Biomaterials. 2010;31:4259. doi: 10.1016/j.biomaterials.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Jagannathan R. and Kottaisamy M. J. Phys.: Condens. Matter. 1995;7:8453. doi: 10.1088/0953-8984/7/44/014. [DOI] [Google Scholar]
- André R S, Paris E C, Gurgel M F C, Rosa I L V, Paiva-Santos C O, Li M S, Varela J A. and Longo E. J. Alloy. Compd. 2012;531:50. doi: 10.1016/j.jallcom.2012.02.053. [DOI] [Google Scholar]
- Yang P, PD P. and Yin Z. J. Lumin. 2002;97:51. doi: 10.1016/s0022-2313(01)00426-4. [DOI] [Google Scholar]
- Chen X Y. and Liu G K. J. Solid State Chem. 2005;178:419. doi: 10.1016/j.jssc.2004.09.002. [DOI] [Google Scholar]
- Jiang M, Terra J, Rossi A, Morales M, Baggio Saitovitch E. and Ellis D. Phys. Rev. B. 2002;66(224107) doi: 10.1103/physrevb.66.224107. [DOI] [Google Scholar]