

Abstract
In this work, the relationship between the unsaturated chain end group content and the thermal oxidative degradation rate was systematically studied with binary polymer blends of isotactic polypropylene (iPP) with and without the unsaturated chain end group. The iPPs with and without the unsaturated chain end group were synthesized by a metallocene catalyst in the absence of hydrogen and by a Ziegler catalyst in the presence of one, respectively. The thermal oxidative degradation rate of the binary iPP blends was estimated from the molecular weight and the apparent activation energy (ΔE), which were obtained through size exclusion chromatography (SEC) and thermogravimetric analysis (TGA) measurements, respectively. These values exhibited a negative correlation against the mole content of the unsaturated chain end group. The thermal oxidative degradation rate apparently depends on the content of the unsaturated chain end group. This tendency suggests that the unsaturated chain end acts as a radical initiator of the iPP degradation reaction.
Keywords: polypropylene, unsaturated chain end groups, activation energy, radical initiator
Introduction
Isotactic polypropylene (iPP) has been widely used as materials in the household appliance, medical ware, automotive and other industrial sections. However, iPP easily undergoes oxidative degradation under the influence of elevated temperature and sunlight, and the degradation leads to a deterioration of the mechanical properties and yellowing [1–5].
The reaction mechanism of the degradation has been recognized as a free-radical chain reaction, leading to polymer chain scission [1–5]. This radical reaction is a cyclic reaction and is called ‘autoxidation’. Here, it is noted that the presence of a radical species is indispensable for the initiation of autoxidation. Since iPP is a saturated hydrocarbon, it rarely becomes a radical initiator by itself under general degradation conditions. The production of a radical species and its spread are an important factor in oxidative degradation.
Based on the detailed kinetic studies of iPP degradation using chemiluiminescence (CL), Celina and co-workers proposed ‘a spreading model’ for the progress of the thermal oxidative reaction [6–8]. This model was constructed assuming that the unoxidized polymer fraction was infected by the spatial expansion from the initial oxidized zone. The validity of the model has been confirmed experimentally [9–12]. Low-molecular-weight materials, such as dialkyl peroxides, hydroperoxides and macroradical species, have been considered as initiators [6, 9, 11, 12].
We have reported that unsaturated chain end groups influence the rate of iPP degradation [13]. Considering the higher resonance stability of the allyl radical, the unsaturated chain end group is expected to function as an initiator of degradation. In addition, iPP certainly contains the unsaturated chain end group, which is produced by a chain transfer reaction during polymerization [14, 15] and by physical iPP chain scission during molding [16]. Its role in the initial stage of degradation should be clarified in detail since the unsaturated end group may act as not only the radical initiator but the capture agent. The double bond can easily react with radical species, including the allyl radical in another chain. In this case, the unsaturated chain end group would be the degradation inhibitor such as antioxidant. It is important to examine which characteristics of the unsaturated chain end group are dominant during the initial stage of degradation. The characteristics naturally depend on various conditions such as the content and the molecular mobility of the unsaturated chain end group. The elucidation requires a systematic iPP series with various contents of the unsaturated chain end group. It is difficult, however, to directly synthesize such an iPP series. The unsaturated chain end group is produced by chain transfer reactions such as monomer transfer and hydrogen elimination [14]. The change in content requires various polymerization conditions, leading to changes in the molecular characteristics such as molecular weight and tacticity. A better method is to blend iPPs without and with the unsaturated chain end group to generate such an iPP series. Recently, many studies of PPs synthesized by metallocene catalyst have been reported subsequent to the discovery made by Kaminsky et al [17]. The feature of the metallocene catalyst is that it enables the production of tailor-made PP through a ligand design at the metal center [14, 17, 18], and this catalyst is naturally able to yield iPP with unsaturated chain end groups.
In this study, the iPP series with varying unsaturated end group content was prepared using the binary iPP blend of Ziegler-catalyzed iPP (ZiPP) without unsaturated chain end groups and metallocene-catalyzed iPP (MiPP) with 0.7 mol.% of unsaturated chain end groups, and the effect of the content of the unsaturated group on thermal oxidative degradation was studied by thermogravimetric analysis (TGA) and size exclusion chromatography (SEC).
Experimental
Materials
Propylene of research grade (donated by Chisso Co.), nitrogen (purchased from Uno Sanso Co.), meso-ethylenebis (1-indenyl)zirconium dichloride (purchased from Wako Pure Chemical Industry), and methylaluminoxane (MAO; purchased from Tosoh Finechem Co. in the toluene solution) were used without further purification. Toluene and xylene solvent was purified by passing them through a molecular sieve 13 X column.
Ziegler-catalyzed isotactic polypropylene (ZiPP)
ZiPP was supplied by Toho Catalyst Co., Ltd. Since the polymer chain transfer was carried out with hydrogen, its chain end group was a saturated hydrocarbon. In order to remove unfavorable catalyst residues, ZiPP was stirred with distilled water containing a small amount of ethanol at room temperature overnight, dried, and then was reprecipitated from a boiling xylene solution into methanol under nitrogen atmosphere.
Metallocene-catalyzed iPP (MiPP)
The meso-ethylenebis(1-indenyl)zirconium dichloride and MAO catalyst system simultaneously yielded a slightly low isotactic iPP and a considerably low one. The lower iPP (MiPP) contained a number of unsaturated chain end groups [13]. In this work, the polymerization was performed under a propylene pressure of the 5 kgf cm−2 with an Al/Zr molar ratio of 3000 at 50 °C for 3 h in toluene as solvent. In order to remove catalyst residues, the whole iPP was reprecipitated from a boiling xylene solution into methanol under nitrogen atmosphere and then dried in vacuo. This whole iPP was fractionated by boiling n-hexane in a soxhlet apparatus for 6 h, and the obtained soluble part was used as MiPP after drying it in vacuo.
Polymer blend preparation
The ZiPP and the MiPP were blended in a boiling xylene solution in pure nitrogen at atmospheric pressure. The compositions of the components in blends of ZiPP/MiPP were 99/1, 95/5, and 90/10 (w/w). All of polymer blends were precipitated in methyl alcohol and then dried in a vacuum oven at 60 °C for 6 h.
1C- and 13C -NMR measurements
The contents of the unsaturated chain end group and meso pentad fraction (mmmm) were determined by 1H- and 13C-NMR spectroscopy using a Varian Gemini-300 spectrometer at 120 °C on 20% (w/v) solution in hexachloro- 1,3-butadiene. 1,1,2,2-Tetrachloroethane-d2 was added as an internal lock and used as an internal chemical shift reference.
SEC analysis
The sample in the small vial was dissolved in 5 ml of o-dichlorobenzene containing 2,6-di-t-butyl-p-cresol as an antioxidant, and the obtained sample solution was directly measured by SEC. The molecular weight was determined by SEC (Senshu, SSC-7100) with styrene-divinylbenzene gel columns (SHODEX, HT-806M) at 140 °C using o-dichlorobenzene as a solvent.
Thermogravimetric analysis (TGA)
The decrease in the weight of the sample was investigated by thermogravimetric analysis (TGA, METTER TG50) at several constant temperatures (130, 135, 140 and 145 °C) in air.
Results and discussion
Figure 1 shows the 1H-NMR spectrum of MiPP. The existence of vinylidene, vinyl and vinylene unsaturated chain end groups can be confirmed. The total content of unsaturated chain end groups was estimated to be 0.7 mol.% from the area of each corresponding peak. ZiPP contained no unsaturated chain end group because of the polymer chain transfer by hydrogen. The polymer blend (ZiPP/MiPP) provided an iPP series with various contents of the unsaturated chain end group. The blend ratios (wt.%/wt.%) of ZiPP/MiPP were 99/1, 95/5 and 90/10. The molecular characteristics of ZiPP, MiPP and ZiPP/MiPP are summarized in table 1.
Figure 1.
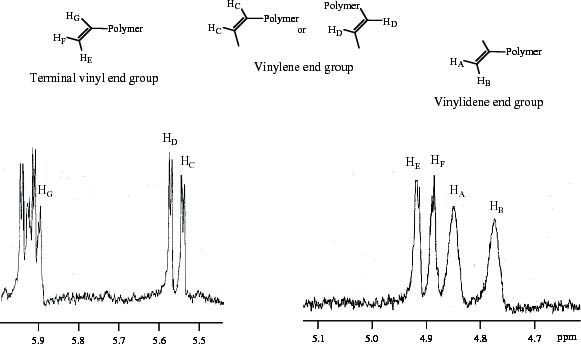
Region of unsaturated chain end groups in 1H-MR spectrum of MiPP.
Table 1.
Molecular characteristics of ZiPP, MiPP and ZiPP/MiPP polymer blends.
| Sample | Meso pentad fraction mmmm (mol.%) | Mn (g mol.−1) | MW/Mn | Chain end group content∗∗(mol.%) |
|---|---|---|---|---|
| ZiPP | 97 | 9.4 104 | 4.9 | ≈0 |
| MiPP | 72 | 3.0 103 | 2.6 | 0.7 |
| ZiPP/MiPP (99/1)∗ | – | – | – | 0.007 |
| ZiPP/MiPP (95/5)∗ | – | – | – | 0.035 |
| ZiPP/MiPP (90/10)∗ | – | – | – | 0.07 |
∗Blend ratio: (wt.%/wt.%). ∗∗Calculation from dilution factor.
The content dependence of thermal oxidative degradation was investigated by SEC measurement. Figure 2 shows the changes in the ratio of molecular weights after and before thermal oxidative degradation. The molecular weight decreases for all amounts of additional unsaturated chain end groups. The rate of change apparently depends on the content of unsaturated chain end groups, indicating that the existence of unsaturated groups accelerates the degradation reaction.
Figure 2.
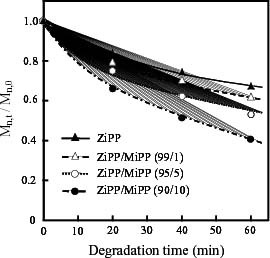
Changes in ratio of molecular weights (Mn, t/Mn, 0) of ZiPP and ZiPP/MiPP during thermal oxidative degradation at 130 °C; Mn, t and Mn, 0 are number average molecular weights after and before thermal oxidative degradation, respectively.
In our previous works [13, 19], we have reported that TGA measurement is a useful method of studying the rate of thermal oxidative degradation. The weight change curves obtained by TGA are shown in figure 3. The curves exhibit multiple stages composing the three periods of constant, increasing and decreasing weight. Here the increase and decrease of weight are attributed to the incorporation of oxygen and the volatilization of highly degraded parts, respectively. The start of the increase in weight means the end of the induction period of thermal oxidative degradation. In order to evaluate the degradation rate, in several samples, the durations of the induction period (tdi) were determined using the weight change curves obtained from the measurements at several constant temperatures (130, 135, 140 and 145 °C). The tdi has been defined as the time (second) at the intersection of the tangent at the inflection point with the plateau line of the induction period [13, 19]. The apparent activation energy (ΔE) was calculated from the Arrhenius plot of ln tdi versus the reciprocal of temperature.
Figure 3.
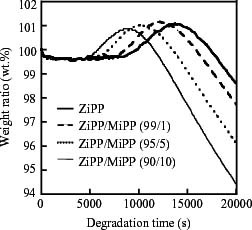
Weight change curves of ZiPP and ZiPP/MiPP at 130 °C in air.
Figure 4 shows the Arrhenius plots of tdi of ZiPP and ZiPP/MiPP. tdi apparently obeys the Arrhenius law within the chosen temperature limit. The values of tdi and ΔE are summarized in table 2. It is noted here that the ΔE value depends on the content of the unsaturated chain end group. As shown in figure 5, ΔE exhibits a negative correlation with the mole content of unsaturated chain end groups. The fall of ΔE suggests that the existence of unsaturated chain end groups affects the reaction mechanism of iPP degradation.
Figure 4.
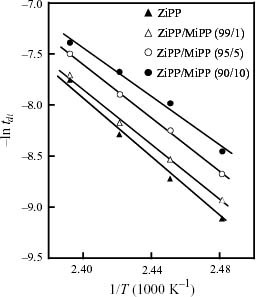
Arrhenius plots of duration of induction period (tdi) for thermal oxidative degradation of ZiPP and ZiPP/MiPP.
Table 2.
Value of tdi at various temperatures and apparent activation energies (ΔE).
| Sample | tdi(×103 s) | ΔE(kJ mol.−1) | |||
|---|---|---|---|---|---|
| 130 °C | 135 °C | 140 °C | 145 °C | ||
| ZiPP | 8.4 | 6.1 | 4.0 | 2.3 | 119 |
| ZiPP/MiPP (99/1) | 7.6 | 5.1 | 3.6 | 2.2 | 113 |
| ZiPP/MiPP (95/5) | 5.8 | 3.8 | 2.7 | 1.8 | 108 |
| ZiPP/MiPP (90/10) | 4.7 | 2.9 | 2.1 | 1.6 | 101 |
Figure 5.
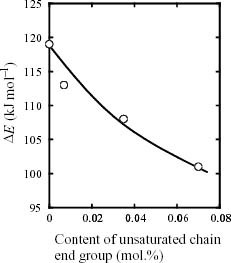
Apparent activation energy (ΔE) versus content of unsaturated chain end group.
There is much literature on the mechanisms of the iPP autoxidation process [1–5, 20]. Many researchers agree fundamentally with the following mechanistic scheme.
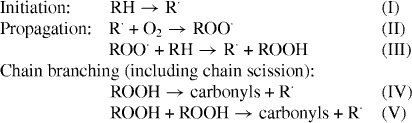 |
Among these reaction steps, the hydroperoxide decompositions (IV) and/or (V) are the rate-determining steps [4, 20, 21]. The ΔEs obtained here corresponds to those of the hydroperoxide decomposition [20]. In addition, the ΔE of bimolecular hydroperoxide decomposition (V) is considerably lower than that of the unimolecular one (IV) [4, 20]. The existence of adjacent hydroperoxides leads to the bimolecular decomposition reaction (V) path [4, 20] and, thus, apparently lowers the ΔE observed in the degradation reaction. If the formation rate of adjacent hydroperoxides increases with the increase in the content of the unsaturated chain end group, the observed ΔE changes can be explained by the competition between the bimolecular decomposition with a lower ΔE and unimolecular one with a higher ΔE. The behavior of the lower ΔE is thought to be closely related to the increasing population of adjacent hydroperoxides.
As materials that accelerate the thermal oxidative degradation of iPP, benzoyl peroxide (BPO) [11], formaldehyde [11, 22] and organic acids [11, 23] have been reported. The acceleration effect of BPO is due to its radical-initiating ability [11], whereas the formaldehyde and organic acid additives are considered to catalyze the hydroperoxide decomposition as prodegradants [11, 22, 23]. Considering the chemical structure, the acceleration effect of unsaturated chain end groups is likely due to not only the catalysis of the hydroperoxide decomposition, but also their action as degradation initiators. The unsaturated chain end group should be the initiator because the allyl radical to be produced has higher resonance stability. In fact, in the case of polyethylene, Allen and colleagues have already pointed out that the unsaturated group is the initiator [24, 25]. The added MiPP would be expected to function as the degradation initiator. The amount of hydroperoxide increases with the addition of MiPP, and the ratio of adjacent hydroperoxides becomes higher. The decrease of ΔE originates from this mechanism.
These results demonstrate that the unsaturated chain end group becomes the radical initiator, not the capture agent. It is natural that this behavior is also associated with the chemical structure and molecular weight of the added polymer containing unsaturated chain end groups. The MiPP has the iPP structure and a considerably low molecular weight (Mn=3000 g mol.−1). Thus, the added MiPP is uniformly dispersed in the ZiPP and have such a slow molecular chain motion that the possibility of encountering other radicals is considerably low. The degradation appears to proceed uniformly and efficiently upon the addition.
Conclusion
In order to clarify the function mechanism of the unsaturated chain end group in iPP thermal oxidative degradation, the correlation between the unsaturated chain end group content and degradation rate was studied in a ZiPP/MiPP polymer blend. This blend allowed the unsaturated chain end group content to be varied in order to clarify a role of the group as a degradation initiator. The changes in the molecular weight and ΔE showed that the increase of the content led to the acceleration of the iPP degradation reaction. These results suggested that the unsaturated chain end group acted as a radical initiator.
Acknowledgment
This work was supported by a Grant-in-Aid for Scientific Research, No. 17550194 from the Japan Society for the Promotion of Science.
References
- Kato Y, Carlsson D J. and Wiles D M. J. Appl. Polym. Sci. 1969;13:1447. doi: 10.1002/app.1969.070130708. [DOI] [Google Scholar]
- Carlsson D J. and Wiles D M. Macromolecules. 1969;6:597. doi: 10.1021/ma60012a007. [DOI] [Google Scholar]
- Billingham N C. Makromol. Chem. Macromol. Symp. 1989;28:145. [Google Scholar]
- Audouin L, Gueguen V, Tcharkhtchi A. and Verdu J. J. Polym. Sci. A: Polym. Chem. 1995;33:921. doi: 10.1002/pola.1995.080330605. [DOI] [Google Scholar]
- Alam M S, Nakatani H, Ichiki T, Goss Ben G S, Liu B. and Terano M. J. Appl. Polym. Sci. 2002;86:1863. doi: 10.1002/app.11108. [DOI] [Google Scholar]
- Celina M. and George G A. Polym. Degrad. Stab. 1993;40:323. doi: 10.1016/0141-3910(93)90138-9. [DOI] [Google Scholar]
- Celina M, George G A. and Billingham N C. Polym. Degrad. Stab. 1993;42:335. doi: 10.1016/0141-3910(93)90229-C. [DOI] [Google Scholar]
- Celina M, George G A, Lacey D J. and Billingham N C. Polym. Degrad. Stab. 1995;47:311. doi: 10.1016/0141-3910(94)00134-T. [DOI] [Google Scholar]
- Celina M. and George G A. Polym. Degrad. Stab. 1995;50:89. doi: 10.1016/0141-3910(95)00136-A. [DOI] [Google Scholar]
- Goss B G S, Nakatani H, George G A. and Terano M. Polym. Degrad. Stab. 2003;82:119. doi: 10.1016/S0141-3910(03)00172-1. [DOI] [Google Scholar]
- Celina M, Clough R L. and Jones G D. Polymer. 2005;46:5161. doi: 10.1016/j.polymer.2005.04.033. [DOI] [Google Scholar]
- Celina M, Clough R L. and Jones G D. Polym. Degrad. Stab. 2006;91:1036. doi: 10.1016/j.polymdegradstab.2005.07.022. [DOI] [Google Scholar]
- Nakatani H, Suzuki S, Tanaka T. and Terano M. Polym. Int. 2007;56:1147. doi: 10.1002/pi.2252. [DOI] [Google Scholar]
- Busico V. and Cipullo R. Prog. Polym. Sci. 2001;26:443. doi: 10.1016/S0079-6700(00)00046-0. [DOI] [Google Scholar]
- Janiak C, Lange K C H, Marquardt P, Krüger R P. and Hanselmann R. Macromol. Chem. Phys. 2002;203:129. doi: 10.1002/1521-3935(20020101)203:1<129::AID-MACP129>3.0.CO;2-C. [DOI] [Google Scholar]
- Sebastião V C. and Babetto A C. Adv. Polym. Technol. 2002;21:243. doi: 10.1002/adv.10028. [DOI] [Google Scholar]
- Kaminsky W, Külper K, Brintzinger H H. and Wild F R W P. Angew. Chem. Int. Edn. Engl. 1985;24:507. doi: 10.1002/anie.198505071. [DOI] [Google Scholar]
- Brintzinger H H, Fischer D, Mülhaupt R, Rieger B. and Waymouth R M. Angew. Chem. Int. Edn. Engl. 1995;34:1143. doi: 10.1002/anie.199511431. [DOI] [Google Scholar]
- Nakatani H, Suzuki S, Tanaka T. and Terano M. Polymer. 2005;46:12366. doi: 10.1016/j.polymer.2005.10.131. [DOI] [Google Scholar]
- Achimsky L, Audouin L, Verdu J, Rychly J. and Matisova-Rychla L. Polym. Degrad. Stab. 1997;58:283. doi: 10.1016/S0141-3910(97)00059-1. [DOI] [Google Scholar]
- Korcek S, Chenier J H B, Howard J A. and Ingold K U. Can. J. Chem. 1972;50:2285. doi: 10.1139/v72-365. [DOI] [Google Scholar]
- Eriksson P, Reitberger T. and Stenberg B. Polym. Degrad. Stab. 2002;78:183. doi: 10.1016/S0141-3910(02)00132-5. [DOI] [Google Scholar]
- Broska R, Rychlý J. and Csomorová K. Polym. Degrad. Stab. 1999;63:231. doi: 10.1016/S0141-3910(98)00097-4. [DOI] [Google Scholar]
- Chirinos-Padrón A J, Hernández P H, Chávez E, Allen N S, Vasiliou C. and DePoortere M. Eur. Polym. J. 1987;23:935. doi: 10.1016/0014-3057(87)90036-X. [DOI] [Google Scholar]
- Allen N S, Edge M, Holdsworth D, Rahman A, Catalina F, Fontan E, Escalona A M. and Sibon F F. Polym. Degrad. Stab. 2000;67:57. doi: 10.1016/S0141-3910(99)00121-4. [DOI] [Google Scholar]