

Abstract
Surface-mediated gene transfer systems using apatite (Ap)-based composite layers have received increased attention in tissue engineering applications owing to their safety, biocompatibility and relatively high efficiency. In this study, DNA-antibody–apatite composite layers (DA–Ap layers), in which DNA and antibody molecules are immobilized within a matrix of apatite nanocrystals, were fabricated using a biomimetic coating process. They were then assayed for their gene transfer capability for application in a specific cell-targeted gene transfer. A DA–Ap layer that was fabricated with an anti-CD49f antibody showed a higher gene transfer capability to the CD49f-positive CHO-K1 cells than a DNA–apatite composite layer (D–Ap layer). The antibody facilitated the gene transfer capability of the DA–Ap layer only to the specific cells that were expressing corresponding antigens. When the DA–Ap layer was fabricated with an anti-N-cadherin antibody, a higher gene transfer capability compared with the D–Ap layer was found in the N-cadherin-positive P19CL6 cells, but not in the N-cadherin-negative UV♀2 cells or in the P19CL6 cells that were pre-blocked with anti-N-cadherin. Therefore, the antigen–antibody binding that takes place at the cell–layer interface should be responsible for the higher gene transfer capability of the DA–Ap than D–Ap layer. These results suggest that the DA–Ap layer works as a mediator in a specific cell-targeted gene transfer system.
Keywords: transfection, antibody, hydroxyapatite, composite, coating, cell targeting
1. Introduction
The gene transfer technique is an effective stimulation method for altering cellular characteristics, functions and behaviors that include proliferation and differentiation. It has emerged as an important tool even in tissue engineering applications. Among conventional gene transfer systems, a calcium phosphate system [1–6] that is mediated by nano- and microscale particles of DNA–apatite composites is superior in safety and biocompatibility to other alternatives, such as viral [7–9] and lipid [10–12] systems. However, the gene transfer efficiency of the calcium phosphate system is relatively low. It was improved by developing a non-particulate and surface-mediated gene transfer system using a DNA–apatite composite layer (D–Ap layer), in which DNA molecules are immobilized within a matrix of apatite nanocrystals [13, 14]. Recently, we have further improved the efficiency of gene transfer by co-immobilizing a cell adhesion protein, in this case either laminin [15, 16] or fibronectin [17, 18], within the D–Ap layer. Although the mechanisms are not yet fully clarified, we believe that enhanced endocytosis activity and/or elevated DNA concentration in the microenvironment at the interface between the cell and the layer could contribute to the increase in gene transfer efficiency of the cell adhesion protein-immobilized D–Ap layer [15–17].
We hypothesized that the immobilization of the antibody within the D–Ap layer would increase the gene transfer efficiency in a cell-specific manner through antigen–antibody binding at the cell–layer interface. Antibodies are Y-shaped glycoproteins that are categorized as part of the immunoglobulin superfamily, and they bind to antigens on the cell membrane with a high specificity. Therefore, a targeted gene transfer to a specific cell type is feasible if an antibody is co-immobilized within the D–Ap layer and the resulting DNA–antibody–apatite composite layer (DA–Ap layer) is used for gene transfer. Cell-targeted gene transfer utilizing an antibody has been extensively studied in the past, with attention to gene therapy applications [19–25]. However, these previous studies were carried out on conventional gene transfer systems using particulate complexes of antibody and plasmid DNA [19–21] or those further combined with viruses [22], cationic polymers [23], lipids [24] or calcium phosphates [25]. To the best of our knowledge, no attempts have been made to create a cell-targeted gene transfer system using apatite-based composite layers.
The objective of this study was to fabricate DA–Ap layers, i.e. apatite layers immobilizing both DNA and antibody molecules within them, and to evaluate their gene transfer efficiency for application in cell-targeted gene transfer. The DA–Ap layers were fabricated on an ethylene-vinyl alcohol copolymer (EVOH) substrate utilizing an amorphous calcium phosphate (ACP)-assisted biomimetic coating process [26]. In this process, the substrate was pre-coated with ACP nanoparticles and then immersed in a coating solution, i.e., a metastable supersaturated calcium phosphate solution (CP solution [27]) supplemented with DNA and an antibody. The content of anti-CD49f antibody in the resulting DA–Ap layers was varied by changing its concentration in the coating solution. The layers were analyzed for their surface morphology, structure and capability to transfer a reporter gene to the CD49f-positive CHO-K1 cells. Then, a DA–Ap layer was fabricated under optimal conditions using an anti-N-cadherin antibody and used in the cell-targeted gene transfer feasibility study. In this study, the gene transfer capabilities of a D–Ap layer (control) and the DA–Ap layer were evaluated using the N-cadherin-positive P19CL6 cells, N-cadherin-negative UV♀2 cells, and P19CL6 cells pre-blocked with an anti-N-cadherin antibody.
2. Materials and methods
2.1. Materials
We used the following antibodies: anti-CD49f rat immunoglobulin G (IgG) (GoH3, Becton, Dickinson and Company, USA) and anti-N-cadherin mouse IgG, (D-4, Santa Cruz Biotechnology, USA). DNA from the pGL3 control vector including the complementary DNA of luciferase (Promega Co., USA) was propagated and purified to obtain 0.7–1.2 mg ml−1 of the plasmid. Upon translation, the pGL3 control vector functions as a genetic reporter by expressing luciferase within the cells. All the chemical reagents used to prepare the coating solutions were purchased from Nacalai Tesque Inc., Japan.
The substrate material was EVOH with a nominal ethylene content of 32 mol% (generously supplied by Kuraray Co. Ltd, Japan, for other experiments). Substrates of EVOH were prepared by hot-pressing EVOH pellets and then cutting the resulting plate (1 mm thickness) into 5 mm × 10 mm rectangles. They were then abraded on the top surface with SiC paper (average grain size 7.6 μm), ultrasonically washed with acetone and ethanol, and dried in a vacuum for 24 h.
In the gene transfer experiments we used CHO-K1 ovary cells (RIKEN BioResource Center, Japan), P19CL6 embryonal carcinoma cells (Riken BioResource Center, Japan) and UV♀2 vascular endothelial cells (Riken BioResource Center, Japan), together with their respective culture media: RPMI1640 (Invitrogen Corporation, USA), minimum essential medium (MEM, Invitrogen Corporation, USA) and Dulbecco's modified Eagle medium (DMEM, Invitrogen Corporation, USA). All cell culture media were supplemented with fetal bovine serum (FBS, Life Technologies) at a concentration of 10%.
2.2. Conformation of antigens expressed on the cell surface by fluorescent immunostaining
The CHO-K1, P19CL6 and UV♀2 cells were seeded in each well of a 24-well cell culture plate at a density of 5 × 104 cells/0.5 ml per well. After incubating for 24 h, the cells were washed twice with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde (Kanto Kagaku, Japan) at 4 °C for 45 min. After washing twice with PBS, immunostaining for the CHO-K1 cells was carried out by incubating the cells with anti-CD49f rat IgG, GoH3 (primary antibody, dilution rate = 200) at 4 °C overnight, and then with FITC-conjugated anti-rat IgG (secondary antibody, dilution rate = 320, Sigma-Aldrich, USA) at 37 °C for 2 h. Immunostaining for the P19CL6 and UV♀2 cells was conducted by incubating the cells with anti-N-cadherin mouse IgG, D-4 (primary antibody, dilution rate = 50) at 4 °C overnight, and then with FITC-conjugated anti-mouse IgG (secondary antibody, dilution rate = 100, Sigma-Aldrich, USA) at 37 °C for 2 h. After washing twice with PBS, the cells were observed under a fluorescence microscope (IX71, Olympus, Japan).
2.3. Alternate dipping treatment for pre-coating with ACP
The EVOH substrate was subjected to an alternate dipping treatment [26, 28, 30, 31] that was simplified from the Taguchi's method [29]. First, the substrate was dipped in 20 ml of 200 mM aqueous solution of CaCl2 (Nacalai Tesque, Japan) for 10 s, then dipped in 20 ml of ultrapure water for 1 s, and dried in air for a few minutes. The substrate was subsequently dipped in 20 ml of 200 mM aqueous solution of K2HPO4·3H2O (Nacalai Tesque) for 10 s, dipped again in 20 ml of ultrapure water for 1 s, and dried in air for a few minutes. The alternate dipping in the CaCl2 and the K2HPO4·3H2O solutions was performed three times. As a result of this treatment, nanoparticulates of ACP, which is a precursor of apatite [32], were pre-coated onto the EVOH surface [33]. The EVOH substrate pre-coated with ACP was sterilized with ethylene oxide gas.
2.4. Biomimetic coating process for coating with DA–Ap layers
The CP solution (142 mM NaCl, 1.50 mM K2HPO4·3H2O, 3.75 mM CaCl2, 50 mM tris(hydroxymethyl)aminomethane (Tris) [27]) was prepared by dissolving NaCl, K2HPO4·3H2O, HCl (40 mM), and CaCl2 in ultrapure water and then buffering the solution at pH = 7.40 at 25.0 °C with Tris and HCl [16–18]. Coating solutions were prepared by adding DNA at a concentration of 40 μg ml−1 and an antibody at concentrations of 0, 5, 10 or 20 μg ml−1 to the CP solution. The resulting coating solutions were sterilized by filtration.
The EVOH substrate that was pre-coated with ACP as described in section 2.3 was aseptically immersed in 1.5 ml of each coating solution at 25 °C for 24 h using a polystyrene bottle with a screw cap. After removal from the coating solution, the substrate was gently washed with ultrapure water before the surface analysis (section 2.5), or washed with PBS before the gene transfer experiments (sections 2.6 and 2.7). The coated substrates were denoted as samples DA0, DA5, DA10 and DA20 according to the respective antibody concentration in the coating solution: 0, 5, 10 and 20 μg ml−1.
2.5. Analysis of sample surfaces and coating solutions
The surface morphologies and structures of samples prepared with the anti-CD49f antibody were examined using a scanning electron microscope (SEM, XL30, FEI Company Ltd., USA) and a thin-film x-ray diffractometer (TF-XRD, RINT Ultima X, Rigaku Co., Japan) employing CuKα x-rays.
The coating solutions were clear and induced no spontaneous precipitation during the coating process. The DNA and antibody concentrations in the coating solutions were measured before and after the coating process using an ultraviolet–visible (UV–Vis) spectrophotometer (Model V-550, Jasco Corporation, Japan). The amount of DNA and antibody immobilized on the sample surfaces was estimated by subtracting the final concentrations from the initial concentrations of DNA and antibody in the coating solution. The DNA concentration was evaluated via absorbance at 260 nm. The antibody concentration was calculated from the absorbance at 595 nm, using the Bradford method and a protein assay kit (Bio-Rad Laboratories Inc., USA). The DNA standard solutions were prepared by adding DNA at concentrations of 0, 20 or 40 μg ml−1 to the CP solution (for sample DA0) or the CP solution supplemented with 5 (for sample DA5), 10 (for sample DA10) or 20 (for sample DA20) μg ml−1 of anti-CD49f antibody. The antibody standard solutions were prepared by adding anti-CD49f at concentrations of 0, 5, 10 or 20 μg ml−1 to the CP solution supplemented with 40 μg ml−1 of DNA. Before the measurement, all the freshly prepared standard solutions were kept under the same condition as in the biomimetic coating process (section 2.4), i.e. they were sealed in the polystyrene bottle with a screw cap after filtration and then incubated at 25 °C for 24 h.
2.6. Confirmation of effect of antibody in the DA–Ap layer on gene transfer capability
The gene transfer capability of each sample prepared with the anti-CD49f antibody was evaluated by luciferase assay using the CHO-K1 cells. Each sample was placed in one well of a 24-well cell culture plate, and the CHO-K1 cells were seeded at a cell concentration of 5 × 104 cells/0.5 ml per well. After incubation for 72 h, the cells were washed three times with PBS and lysed in 200 μl of cell culture lysis reagent (Promega Corporation, USA). After vortexing, the cell lysate was centrifuged at 12 000 g for 2 min and the supernatant was retained. The supernatant was analyzed using a luciferase assay kit (Promega) to quantify the luciferase activity of the cultured cells according to the manufacturer's instruction. Briefly, the relative amount of luciferase (luminescent enzyme) in the supernatant was quantified by measuring its luminescence intensity with a luminometer (Gene Light 55, Microtec Co. Ltd, Japan) after the enzymatic reaction with luciferin (luminescent substrate). The thus quantified luciferase activity of the cells represents the gene transfer capability of the sample, since luciferase is produced by the cells as a result of expression of luciferase gene delivered from the sample.
2.7. Feasibility study on cell-targeted gene transfer
The gene transfer capability of samples D and DA10, which were prepared with the anti-N-cadherin antibody, was evaluated according to the protocol described in section 2.6 using the P19CL6 and UV♀2 cells. Some of the P19CL6 cells were pre-blocked with N-cadherin prior to the gene transfer experiment. The N-cadherin blocking was performed by incubating the P19CL6 cells (1 × 106 cells ml−1) at 37 °C for 1 h in Opti-MEM (Invitrogen Corporation, USA) supplemented with the anti-N-cadherin antibody (10 μg ml−1) and then washing the cells twice in MEM supplemented with 10% FBS.
3. Results
3.1. Fluorescent immunostaining
Fluorescent immunostaining study showed that the CHO-K1, P19CL6 and UV♀2 cells were CD49f-positive, N-cadherin-positive and N-cadherin-negative, respectively. The CHO-K1 cells were clearly stained with anti-CD49f antibody (figure 1(left)), showing the presence of CD49f antigens on the cell surface. On the other hand, the P19CL6 cells were clearly stained with anti-N-cadherin antibody (figure 1(center)), but the UV♀2 cells were not stained with anti-N-cadherin antibody (figure 1(right)). This result shows that N-cadherin antigens were present at the P19CL6 cell surface, whereas they were vanishingly scarce at the UV♀2 cell surface.
Figure 1.
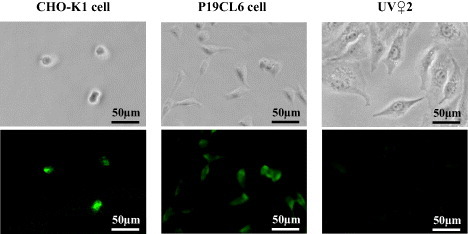
Phase-contrast (top row) and fluorescence (bottom row) images of the CHO-K1 cells immunostained with anti-CD49f antibody (left), the P19CL6 cells immunostained with anti-N-cadherin antibody (center), and the UV♀2 cells immunostained with anti-N-cadherin antibody (right).
3.2. Surface structures of samples
According to the SEM, TF-XRD and UV–Vis results, a D–Ap layer formed on the surface of sample DA0, and DA–Ap layers formed on the surfaces of samples DA5, DA10 and DA20. Uniform layers with a microscale cavernous and lumpy structure, as shown in the SEM images in figure 2(a), were observed over the entire surface of all samples that were prepared with the anti-CD49f antibody. The layers were composed of apatite [34] of poor crystallinity, as judged from the broad TF-XRD peaks (figure 2(b)). The apatite layers on samples DA5, DA10 and DA20 immobilized both the DNA and the antibody, whereas the apatite layer on sample DA0 immobilized only the DNA, according to the UV–Vis results (figure 3). As shown in figure 3(a), the DNA content of sample DA20 was significantly lower than that of samples DA5 and DA10. As shown in figure 3(b), the mean antibody content of the samples increased with the initial antibody concentration in the coating solution in the following order: DA0 < DA5 < DA10 < DA20.
Figure 2.
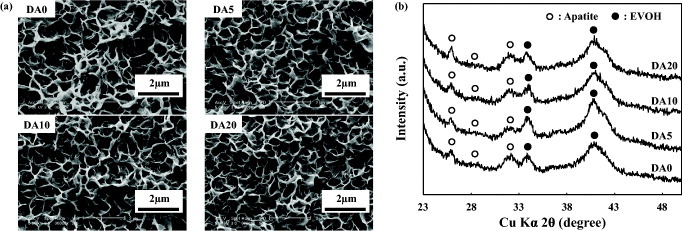
SEM images (a) and TF-XRD patterns (b) of the surfaces of samples DA0, DA5, DA10, and DA20, which were prepared with the anti-CD49f antibody.
Figure 3.
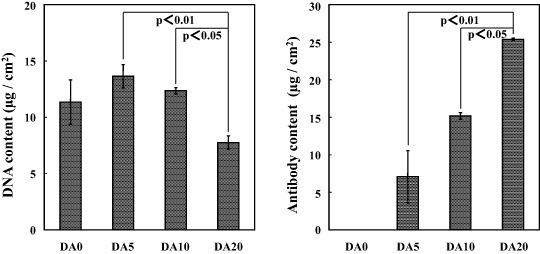
Contents of DNA (a) and antibody (b) on the surfaces of samples DA0, DA5, DA10 and DA20, which were prepared with the anti-CD49f antibody (mean ± SD).
3.3. Effect of antibody in the DA–Ap layer on gene transfer capability of samples
The immobilization of anti-CD49f antibody within a D–Ap layer was effective in facilitating gene transfer to the CD49f-positive CHO-K1 cells. Figure 4 shows the luciferase activity of CHO-K1 cells cultured on samples DA0, DA5, DA10 and DA20 prepared with the anti-CD49f antibody. The mean luciferase activity of the CHO-K1 cells cultured on samples DA5, DA10 and DA20, which were coated with the DA–Ap layers, was higher than that of the cells cultured on sample DA0, which was coated with the D–Ap layer, although the difference between samples DA0 and DA20 was statistically insignificant. There was no significant difference in luciferase activity among samples DA5, DA10 and DA20, which were coated with the DA–Ap layers. However, sample DA10 showed a relatively high and uniform luciferase activity, and therefore its fabrication conditions were used in the following experiments.
Figure 4.
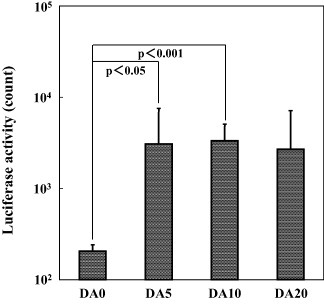
Luciferase activity of the CHO-K1 cells after being cultured for 72 h on samples DA0, DA5, DA10 and DA20, which were prepared with the anti-CD49f antibody (mean + SD).
3.4. Feasibility study of cell-targeted gene transfer
The immobilization of anti-N-cadherin antibody within the D–Ap layer was effective in facilitating gene transfer to the N-cadherin-positive P19CL6 cells, but not to the N-cadherin-negative UV♀2 cells. Figure 5 shows the luciferase activity of the P19CL6 (a) and UV♀2 (b) cells cultured on samples DA0 and DA10 prepared with the anti-N-cadherin antibody. As shown in figure 5(a), the luciferase activity of the P19CL6 cells cultured on sample DA10, which was coated with the DA–Ap layer, was significantly higher than that of the cells cultured on sample DA0, which was coated with the D–Ap layer. On the other hand, as shown in figure 5(b), the luciferase activities were comparable for the N-cadherin-negative UV♀2 cells cultured on sample DA0 and those cultured on sample DA10.
Figure 5.
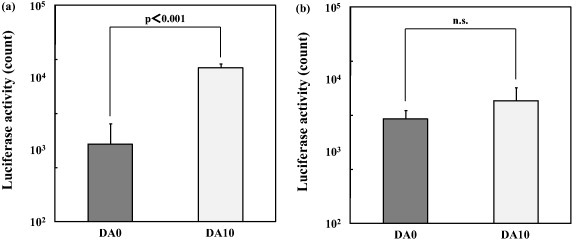
Luciferase activity of the P19CL6 cells (a) and the UV♀2 cells (b), after being cultured for 72 h on samples DA0 and DA10, which were prepared with the anti-N-cadherin antibody (mean + SD, n.s.: no statistical difference).
The increase in the gene transfer capability of the DA–Ap layer due to the immobilization of the anti-N-cadherin antibody was depleted by pre-blocking the P19CL6 cells with anti-N-cadherin antibody. As shown in figure 6, the luciferase activity of the P19CL6 cells cultured on sample DA10 was decreased to a level comparable to that of the cells cultured on sample DA0 when the cells were pre-blocked with anti-N-cadherin antibody.
Figure 6.
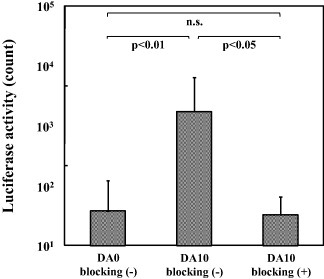
Luciferase activity of the P19CL6 cells, that have and have not been pre-blocked with anti-N-cadherin antibody, after being cultured for 72 h on samples DA0 and DA10, which were prepared with the anti-N-cadherin antibody (mean + SD, n.s.: no statistical difference).
4. Discussion
DA–Ap layers were successfully fabricated with the ACP-assisted biomimetic coating process using metastable coating solutions, i.e., the CP solution supplemented with DNA and antibody at various concentrations. According to the SEM (figure 2(a)) and TF-XRD (figure 2(b)) results, these layers were similar in their surface morphology and crystalline phase irrespective of the initial antibody concentration in the coating solution. The significant differences among these layers lie in the DNA and antibody contents of the DA–Ap layers, which depend on the initial antibody concentration in the coating solution (figure 3). The DA–Ap layers are thought to form in the coating solutions via the growth of an apatite layer on the EVOH surface and the simultaneous immobilization of DNA and antibody, on the basis of our previous report on the ACP-assisted biomimetic coating process [35]. During this coating process, a crucial step affecting the DNA and antibody contents of the DA–Ap layer could be the adsorption of DNA and antibody molecules through electrostatic interactions on apatite crystals or their precursors including an amorphous phase [36–39]. The increase in the initial antibody concentration in the coating solution resulted in the higher mean antibody content of the resulting DA–Ap layer (figure 3(b)), probably by accelerating the antibody adsorption rate. Sample DA20 had a lower DNA content than samples DA5 and DA10 (figure 3(a)), although the initial DNA concentration in the coating solutions was the same. The competitive adsorption of DNA and antibody molecules might lead to a reduced DNA content on the surface of sample DA20.
The DNA and antibody molecules are most likely to be immobilized inside the DA–Ap layer as well as on the layer's outermost surface according to our previous transmission electron microscopy results [35]. However, surface-sensitive x-ray photoelectron spectroscopy (XPS) results reveal higher contents of DNA and antibody on the outermost surface than inside of the layer. The N/Ca atomic ratio of the DA–Ap layer determined by XPS was comparable for all the samples (0.80 ± 0.05 for DA10), and was considerably higher than that (0.07 ± 0.00 for DA10) determined by chemical analysis of the coating solutions. The chemical analysis was carried out by UV–Vis for DNA and antibody (figure 3) and by inductively coupled plasma atomic emission spectroscopy for calcium (data not shown); the N/Ca atomic ratio of the DA–Ap layer was determined using the nitrogen concentration of the DNA (16.8 wt% according to the base sequence of pGL3 control vector) and antibody (16 wt% according to Jones factor [40]) molecules. Note that XPS probed only the outermost surface (<10 nm) whereas chemical analysis averaged over the whole thickness of the DA–Ap layer. Hence, the higher N/Ca atomic ratio determined by XPS suggests that the DNA and antibody molecules are not homogeneously distributed through the thickness of the DA–Ap layer, but are concentrated on the layer's outermost surface.
The biological activity of DNA was retained even after the ACP-assisted biomimetic coating process (see figures 4–6), as reported previously [15–18]. This retention of activity is due to the mild coating conditions: neutral pH, normal pressure and temperature, and enzyme-free acellular environment. It was suggested from the luciferase assay results that the antigen-specific binding sites of the antibody also remained active even after the present coating process. According to the luciferase assay results, the DA–Ap layers fabricated with the anti-CD49f antibody showed a significantly higher gene transfer capability to the CD49f-positive CHO-K1 cells in comparison with the D–Ap layer (figure 4). With the exception of the antibody content, the observed surface properties of these samples were comparable (figures 2 and 3). Therefore, the anti-CD49f antibody that was immobilized within the DA–Ap layer played a critical role in facilitating gene transfer to the CHO-K1 cells. The role of the immobilized antibody in gene transfer has not yet been fully clarified, but we believe that antigen–antibody binding at the cell–layer interface could be responsible for the increased gene transfer capability of the DA–Ap layer compared with that of the D–Ap layer. Antigen–antibody binding can facilitate gene transfer by enhancing the adhesion of cells to the DA–Ap layer and/or by promoting endocytosis by the cell [20, 41].
Antibody's facilitating effects on gene transfer have also been observed for conventional gene transfer systems using particulate complexes of an antibody and DNA [19–21] or those further combined with viruses [22], cationic polymers [23], lipids [24] and/or calcium phosphates [25]. For example, Kozlova et al showed that the covalent attachment of a CD11c antibody onto calcium phosphate-based nanoparticles increased gene transfer efficiency for the CD11c-positive cells [25]. It should be emphasized that all these conventional systems including Kozlova's system are rather different from our system using the DA–Ap layers in terms of the configuration of transfection reagents. The conventional systems are particle-mediated systems, in which particulate transfection reagents (DNA complexes) are either added onto precultured cells or injected into intended sites of the body by parenteral administration. On the other hand, our system is a surface-mediated system, in which a DA–Ap layer serves as both a cell culture substrate and a transfection reagent. This feature is advantageous in tissue engineering applications because apatite-based composite layers can be coated on various types of base materials including bioresorbable poly(ε-caprolactone) [30, 31] and poly(l-lactide) [42]. These base materials can take forms of not only tabular substrates but three-dimensional porous bodies [30, 31, 43], fibers [44] and sponges [45], which are suitable as scaffold structures. Thus-coated composite layers provide biocompatible surface to support cell adhesion and growth [13, 15–17], and can stimulate the cell effectively via the area-specific gene transfer on their surfaces [17, 46]. For example, a DNA-fibronectin-apatite composite layer coated on a ceramic scaffold induced specific cell differentiation and tissue regeneration around the layer in vivo [43, 47]. These facts demonstrate that the apatite-based composite layers are truly useful as a surface component of tissue engineering scaffolds.
There was no significant difference in the gene transfer capabilities among samples DA5, DA10 and DA20 (figure 4), although the mean antibody content in the DA–Ap layer increased in the following order: DA5 < DA10 < DA20 (figure 3(b)). This lack of a difference may occur because the binding sites of antigens on the CHO-K1 cells were saturated even in sample DA5. Sample DA20 had a lower DNA content than samples DA5 and DA10 (figure 3(a)) in spite of having the highest antibody content, and this may also explain the similar gene transfer capability among these samples. This is because a lower DNA content in an apatite-based composite layer may have an adverse effect on gene transfer capability of the layer [16]. If the DNA content in the DA–Ap layer is increased by increasing DNA concentration in the coating solution then the gene transfer capability of the composite layer could be further improved, although this is yet to be clarified.
It should be noted that the antibody's facilitating effect on the gene transfer capability of the DA–Ap layer was only observed for the specific cells that were expressing the corresponding antigens on their surfaces. This finding was supported by previous reports on conventional gene transfer systems using particulate complexes including an antibody [19–25]. As shown in figure 5, the anti-N-cadherin antibody immobilized within the DA–Ap layer could facilitate gene transfer to the N-cadherin-positive P19CL6 cells (a) but not to the N-cadherin-negative UV♀2 cells (b). In addition, the anti-N-cadherin antibody's facilitating effect on gene transfer to the P19CL6 cells was depleted when the cells were pre-blocked with anti-N-cadherin antibody (figure 6). These results support our assertion that the antigen-antibody binding at the cell–layer interface could be responsible for the higher gene transfer capability of the DA–Ap layer relative to the D–Ap layer. These results also suggest the possibility that the DA–Ap layer works as a mediator in a specific cell-targeted gene transfer system. By selecting a suitable antibody for immobilization within a DA–Ap layer, preferential gene transfer to target cells within multiclonal cell systems, including an in vivo system, might be feasible on the layer. Such a cell-targeted gene transfer system using a DA–Ap layer would be highly useful in tissue engineering applications, as it could reduce the risk of unfavorable gene transfer to unintended cells and improve therapeutic effects [19–25], though this is a challenge for a future study.
5. Conclusions
DA–Ap layers were successfully fabricated using the ACP-assisted biomimetic coating process. The gene transfer capability was higher in the DA–Ap layer fabricated with the anti-CD49f antibody than in the D–Ap layer when both were tested with the CD49f-positive CHO-K1 cells. A similar phenomenon was observed for the DA–Ap layer that was fabricated with the anti-N-cadherin antibody and tested with the N-cadherin-positive P19CL6 cells, but not with the N-cadherin-negative UV♀2 cells or P19CL6 cells that were pre-blocked with anti-N-cadherin. Therefore, the antigen–antibody binding that occurs at the cell–layer interface could be responsible for the higher gene transfer capability of the DA–Ap than D–Ap layer. The gene transfer system using a DA–Ap layer can target specific cells and therefore should be useful in tissue engineering applications.
Acknowledgments
This work was supported by the grant-in-aid for young scientists (B) no. 22700499 from the Ministry of Education, Culture, Sport, Science and Technology of Japan. A part of this work was conducted at the Nano-Processing Facility, which was supported by the IBEC Innovation Platform of the National Institute of Advanced Industrial Science and Technology (AIST), Japan.
References
- Graham F L. and van der Eb A J. Virology. 1973;52:456. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- Batard P, Jordan M. and Wurm F. Gene. 2001;270:61. doi: 10.1016/S0378-1119(01)00467-X. [DOI] [PubMed] [Google Scholar]
- Roy I, Mitra S, Maitra A. and Mozumdar S. Int. J. Pharm. 2003;250:25. doi: 10.1016/S0378-5173(02)00452-0. [DOI] [PubMed] [Google Scholar]
- Chowdhury E H, Kunou M, Harada I, Kundu A K. and Akaike T. Anal. Biochem. 2004;328:96. doi: 10.1016/j.ab.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Kakizawa Y, Furukawa S. and Kataoka K. J. Control. Release. 2004;97:345. doi: 10.1016/j.jconrel.2004.03.031. [DOI] [PubMed] [Google Scholar]
- Kutsuzawa K, Chowdhury E H, Nagaoka M, Maruyama K, Akiyama Y. and Akaike T. Biochem. Biophys. Res. Commun. 2006;350:514. doi: 10.1016/j.bbrc.2006.09.081. [DOI] [PubMed] [Google Scholar]
- Moolten F L. and Wells J M. J. Natl Cancer Inst. 1990;82:297. doi: 10.1093/jnci/82.4.297. [DOI] [PubMed] [Google Scholar]
- Culver K W, Ram Z, Walbridge S, Ishii H, Oldfield E H. and Blaese R M. Science. 1992;256:1550. doi: 10.1126/science.1317968. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, Chen C, Li J. and Xiao X. Nature Biotechnol. 2005;23:321. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- Bennett M J, Aberle A M, Balasubramaniam R P, Malone J G, Malone R W. and Nantz M H. J. Med. Chem. 1997;40:4069. doi: 10.1021/jm970155q. [DOI] [PubMed] [Google Scholar]
- Templeton N S, Lasic D D, Frederik P M, Strey H H, Roberts D D. and Pavlakis G N. Nature Biotechnol. 1997;15:647. doi: 10.1038/nbt0797-647. [DOI] [PubMed] [Google Scholar]
- Uduehi A N, Moss S H, Nuttall J. and Pouton C W. Pharm. Res. 1999;16:1805. doi: 10.1023/A:1018986922710. [DOI] [PubMed] [Google Scholar]
- Shen H, Tan J. and Saltzman W M. Nature Mater. 2004;3:569. doi: 10.1038/nmat1179. [DOI] [PubMed] [Google Scholar]
- Sun B, Tran K K. and Shen H. Biomaterials. 2009;30:6386. doi: 10.1016/j.biomaterials.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyane A, Tsurushima H. and Ito A. Gene Ther. 2007;14:1750. doi: 10.1038/sj.gt.3303041. [DOI] [PubMed] [Google Scholar]
- Oyane A, Tsurushima H. and Ito A. J. Gene Med. 2010;12:194. doi: 10.1002/jgm.1425. [DOI] [PubMed] [Google Scholar]
- Oyane A, Murayama M, Yamazaki A, Sogo Y, Ito A. and Tsurushima H. J. Biomed. Mater. Res. A. 2010;92:1038. doi: 10.1002/jbm.a.32449. [DOI] [PubMed] [Google Scholar]
- Yazaki Y, Oyane A, Tsurushima H, Sogo Y, Ito A. and Yamazaki A. Biomaterials. 2011;32:4896. doi: 10.1016/j.biomaterials.2011.03.021. [DOI] [PubMed] [Google Scholar]
- Poncet P, Panczak A, Goupy C, Gustafsson K, Blanpied C, Chavanel G, Hirsch R. and Hirsch F. Gene Ther. 1996;3:731. [PubMed] [Google Scholar]
- Durrbach A. Cancer Gene Ther. 1999;6:564. doi: 10.1038/sj.cgt.7700085. [DOI] [PubMed] [Google Scholar]
- Deas O, Angevin E, Cherbonnier C, Senik A, Charpentier B, Levillain J P, Oosterwijk E, Hirsch F. and Dürrbach A. Hum. Gene Ther. 2002;13:1101. doi: 10.1089/104303402753812502. [DOI] [PubMed] [Google Scholar]
- Haisma H J, Grill J, Curiel D T, Hoogeland S, van Beusechem V W, Pinedo H M. and Gerritsen W R. Cancer Gene Ther. 2000;7:901. doi: 10.1038/sj.cgt.7700198. [DOI] [PubMed] [Google Scholar]
- Coll J L, Wagner E, Combaret V, Metchler K, Amstutz H, Iacono-Di-Cacito I, Simon N. and Favrot M C. Gene Ther. 1997;4:156. doi: 10.1038/sj.gt.3300375. [DOI] [PubMed] [Google Scholar]
- Bartsch M, Weeke-Klimp A H, Meijer D K, Scherphof G L. and Kamps J A. J. Liposome Res. 2005;15:59. doi: 10.1081/lpr-64961. [DOI] [PubMed] [Google Scholar]
- Kozlova D, Chernousova S, Knuschke T, Buer J, Westendorf A M. and Epple M. J. Mater. Chem. 2012;22:396. doi: 10.1039/c1jm14683a. [DOI] [Google Scholar]
- Oyane A. J. Ceram. Soc. Japan. 2010;118:77. doi: 10.2109/jcersj2.118.77. [DOI] [Google Scholar]
- Uchida M, Oyane A, Kim H M, Kokubo T. and Ito A. Adv. Mater. 2004;16:1071. doi: 10.1002/adma.200400152. [DOI] [Google Scholar]
- Oyane A, Tsurushima H. and Ito A. Chem. Lett. 2006;35:1300. doi: 10.1246/cl.2006.1300. [DOI] [Google Scholar]
- Taguchi T, Kishida A. and Akashi M. Chem. Lett. 1998;8:711. doi: 10.1246/cl.1998.711. [DOI] [Google Scholar]
- Oyane A, Uchida M, Choong C, Triffitt J, Jones J. and Ito A. Biomaterials. 2005;26:2407. doi: 10.1016/j.biomaterials.2004.07.048. [DOI] [PubMed] [Google Scholar]
- Oyane A, Uchida M, Yokoyama Y, Choong C, Triffitt J. and Ito A. J. Biomed. Mater. Res. A. 2005;75:138. doi: 10.1002/jbm.a.30397. [DOI] [PubMed] [Google Scholar]
- Suzuki O. Acta Biomater. 2010;6:3379. doi: 10.1016/j.actbio.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Oyane A, Uchida M, Ishihara Y. and Ito A. Key Eng. Mater. 2005;284–286:227. doi: 10.4028/www.scientific.net/KEM.284-286.227. [DOI] [Google Scholar]
- Kay M I, Young R A. and Posner A S. Nature. 1964;204:1050. doi: 10.1038/2041050a0. [DOI] [PubMed] [Google Scholar]
- Oyane A, Uchida M, Onuma K. and Ito A. Biomaterials. 2006;27:167. doi: 10.1016/j.biomaterials.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Daculsi G, Pilet P, Cottrel M. and Guicheux G. J. Biomed. Mater. Res. 1999;47:228. doi: 10.1002/(SICI)1097-4636(199911)47:2%3C228::AID-JBM13%3E3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Oyane A, Yokoyama Y, Uchida M. and Ito A. Biomaterials. 2006;27:3295. doi: 10.1016/j.biomaterials.2006.01.029. [DOI] [PubMed] [Google Scholar]
- Oyane A, Wang X, Sogo Y, Ito A. and Tsurushima H. Acta Biomater. 2012;8:2034. doi: 10.1016/j.actbio.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Oyane A, Ootsuka T, Hayama K, Sogo Y. and Ito A. Acta Biomater. 2011;7:2969. doi: 10.1016/j.actbio.2011.04.012. [DOI] [PubMed] [Google Scholar]
- Jones D B. USDA Circ. 1931;183:1. [Google Scholar]
- Raghavan M. and Bjorkman P J. Annu. Rev. Cell Dev. Biol. 1996;12:181. doi: 10.1146/annurev.cellbio.12.1.181. [DOI] [PubMed] [Google Scholar]
- Yokoyama Y, Oyane A. and Ito A. J. Mater. Sci.: Mater. Med. 2007;18:1727. doi: 10.1007/s10856-007-3024-7. [DOI] [PubMed] [Google Scholar]
- Wang X, Oyane A, Tsurushima H, Sogo Y, Li X. and Ito A. Biomed. Mater. 2011;6:064204. doi: 10.1088/1748-6041/6/4/045004. [DOI] [PubMed] [Google Scholar]
- Yokoyama Y, Oyane A. and Ito A. J. Biomed. Mater. Res. B. 2008;86:341. doi: 10.1002/jbm.b.31025. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Oyane A, Ikeno F, Lyons J K. and Yeung A C. Catheter Cardiovasc. Interv. 2009;74:468. doi: 10.1002/ccd.22024. [DOI] [PubMed] [Google Scholar]
- Oyane A, Yazaki Y, Araki H, Sogo Y, Ito A, Yamazaki A. and Tsurushima H. J. Mater. Sci.: Mater. Med. 2012;23:1011. doi: 10.1007/s10856-012-4581-y. [DOI] [PubMed] [Google Scholar]
- Zhang W, Tsurushima H, Oyane A, Yazaki Y, Sogo Y, Ito A. and Matsumura A. J. Biomed. Sci. 2011;18:62. doi: 10.1186/1423-0127-18-62. [DOI] [PMC free article] [PubMed] [Google Scholar]