

Abstract
Metal ion coordination in discrete or extended metallo-supramolecular assemblies offers ample opportunity to fabricate and study devices and materials that are equally important for fundamental research and new technologies. Metal ions embedded in a specific ligand field offer diverse thermodynamic, kinetic, chemical, physical and structural properties that make these systems promising candidates for active components in functional materials. A key challenge is to improve and develop methodologies for placing these active modules in suitable device architectures, such as thin films or mesophases. This review highlights recent developments in extended, polymeric metallo-supramolecular systems and discrete polyoxometalates with an emphasis on materials science.
Keywords: metallo-supramolecular chemistry, metal ion coordination, polymers, polyelectrolyte amphiphile complex, polyoxometalate, surfactant-encapsulated cluster, mesophase, nanostructure, nanotechnology
Introduction
Nature continually demonstrates delicate control over the intramolecular forces that underlie the very existence of life, from the molecular level, as in DNA, to the microscopic level, as in viruses, up to the macroscopic scale, as in bone or wood. Therefore, natural materials often have an intrinsic structural hierarchy, which is responsible for properties such as mechanical strength and toughness. In addition, natural materials often have several functions. For example, the cytoskeleton determines cell and tissue shape but also influences a wide range of fundamental cellular functions, including migration, movement of organelles and cell division [1]. The intricate relationship between structure and properties reveals itself in photosynthesis and respiratory reactions, where metal ions organized in a protein matrix play a key role in the reaction mechanisms. When the metal ions are arranged precisely in three-dimensional (3D) space, highly efficient electron transfer and energy conversion occurs. Moreover, biological systems are responsive to external stimuli, that is, they can adapt their structure and function as a result of the weak and competitive interactions that keep the components in place. Responsiveness is also needed for information signaling and transduction. One of the most intriguing aspects of biological systems is their ability to self-repair and self-replicate. Inspired by nature, the introduction of specific and selective molecular interactions to guide the association of functional modules is an overarching theme in current materials science and device design [2]. The technological and functional advantages of using molecular-based devices have been recognized for some time [3]. However, the formation of functional nanoscaled or molecular devices from non-biological components or modules has begun only recently [4, 5].
There are essentially two methodologies for creating devices at the nanometer scale, namely, those based on lithography (top down) and those based on self-assembly (bottom up). While lithography allows the creation of ever smaller structures, self-assembly is claimed to be an important avenue toward the manufacture of next-generation molecular materials [5]. The advantages of self-assembly include parallel fabrication, (molecular) dimension control, component alignment and repair mechanisms. The current challenge in the field of self-assembly is, therefore, to develop strategies to combine, orient and order structural and functional modules in predictable ways because material and device performance is critically dependent on the spatial arrangement of the functional modules. In order to support, handle, manipulate and operate devices at the nanoscopic scale, it will also be advantageous to collect and arrange the components in surface-confined structures, however, there are no generic methodologies available for achieving this goal [6]. In the last few years, the development of new methodologies for constructing molecular objects with nanoscopic dimensions capable of mesoscopic pattern formation has been the focus of supramolecular science [7]. With the remarkable progress in the self-assembly of molecular components into specific arrays and polymers, many supramolecular species have now been proposed as potential components for nanoscopic devices [8].
Nanotechnology attempts to establish molecular-based devices and machinery of nanoscopic dimensions, i.e. functional supramolecular architectures that process specific elementary steps within a coherent sequence of matter/signal transformations. The potential fields of application for molecular devices are diverse; currently investigated ‘hot topics’ include (arrays of) chemosensors to derive electric signals from molecular recognition events (nanosensors) and molecular switches and logical elements for highly integrated circuits (molecular electronics). Self-assembling supramolecular systems represent a feasible and efficient approach to fabricating the required quantities of nanosized supramolecular architectures. The final supramolecular entity spontaneously evolves from a chemical library of suitably shaped molecular building blocks through a sequence of recognition, growth and termination steps [9]. Discrete nanosized as well as extended supramolecular assemblies have thus been synthesized exploiting ligand–metal ion interactions, π–π interactions, or hydrogen-bonding-mediated recognition processes.
The combination of supramolecular modules as functional components and amphiphiles as structural components adds another dimension to self-assembly. The interaction of amphiphiles with supramolecular modules occurs spontaneously and is driven by the release of counter ions, as well as electrostatic and hydrophobic interactions thus giving rise to a large array of structures, as well as lyo- and thermotropic mesophases. Amphiphilic self-assembly is by far the most fundamental mechanism in the construction of soft matter materials, leading to materials with significant technological value, such as microfluidic devices, electrical nanodevices, gene therapeutics, prostheses, cellular engineering and medical devices, as well as sensing, catalysis and signal transduction [4, 10, 11]. Complexes of surfactants and lipids with polyelectrolytes have a well-defined nanostructure as well as interesting mechanical and dielectric properties. The resulting composites, containing biological or natural polyelectrolytes, have been explored in drug delivery, pH-switchable systems and templates for directing the structure of polymer architectures. Amphiphilic self-assembly is a simple yet efficient way of producing melt-processable, soluble materials with properties that range from thermoplastic through elastomeric to thermosetting [12]. In addition, the self-assembly of amphiphiles and supramolecular modules offers a promising solution to a prevailing problem in materials chemistry. The modularity of this approach provides extensive control of the structure and function from the molecular to the macroscopic scale. In addition, the use of modules provides an unsurpassed degree of synthetic simplicity, diversity and flexibility. The ability to control the spatial arrangement of functional and structural components is of critical importance with respect to the encoding of new (collective) properties and the full exploitation of a material’s potential. However, the potential lack of symmetrical invariance (e.g. crystallinity) in such devices and materials poses a serious challenge to structure determination by traditional diffraction methods, which is of critical importance for the accurate understanding of structure–property relationships. Further progress in this area will therefore call for interdisciplinary research programs that go beyond the classical approaches in chemistry, physics and engineering.
Discrete or extended metallo-supramolecular modules made by metal-ion-directed self-assembly are of particular interest for the construction of technological devices [13]. They provide a set of well-defined coordination geometries, a range of binding strengths and ligand exchange kinetics that allow the reversible assembly–disassembly of supramolecular architectures that include switchable interaction sites. In addition, they possess a variety of photochemical, electrochemical and reactive properties that are relevant to functional devices. Metal ions collect and spatially direct the assembly of ligands according to predetermined coordination algorithms. The final properties can, therefore, be tailored through the judicious choice of steric and electronic ligand–metal ion interactions. The occurrence of semioccupied d-orbitals gives rise to some of the most prominent properties including strong absorption, high quantum yields, suitable excited state lifetimes, luminescence and tunable redox states. These features can be explored to achieve optical nonlinearity [14], as well as photomediated charge separation [15] for artificial photosynthesis. The splitting of the d-orbitals in a ligand field of appropriate symmetry and strength can give rise to thermally or photoinduced spin transition and spin crossover phenomena, the most intriguing ones being light-induced excited spin state trapping (LIESST), reverse and low-spin LIESST [16]. While photomediated charge separation in polynuclear complexes has been studied with the aim of realizing artificial photosynthesis, such systems are also interesting for information-processing devices [17].
Polyoxometalates (POMs) are an intriguing class of discrete metallo-supramolecular systems, well-defined, discrete nanoscopic clusters with a wide variety of applications in fundamental and applied science including catalysis, electrochemistry, electrooptics and medicine [18]. A bewildering variety of POM structures have been characterized, which recently culminated in the discovery of giant nanosized polymolybdate clusters [19]. In contrast to most semiconductor nanoparticles and quantum dots, POMs are transition-metal oxide clusters that are uniform at the atomic level. POMs form in a series of steps through the sequential self-assembly of metal-oxygen building blocks. An attractive feature of POM clusters is the ability of the metal-oxygen framework to accommodate excess electrons [20]. This reduction is generally reversible and occurs with marginal structural rearrangement. The reduced POMs frequently display a deep blue color and are active electrocatalysts for the hydrogen evolution reaction and oxygen reduction. The ability to accept electrons under the alteration of their light-absorbing properties is an interesting effect for the construction of functional devices and materials. The extinction coefficient of the colored POM state is comparable to that of organic dyes; however, the photochemical stability of POMs is far superior to that of organic molecules. Photochromic properties arise in the presence of certain counter ions, such as alkylammonium, anilinium and pyridinium, which can undergo photoinduced proton transfer to the POM framework. The reduction of the POM cluster results in the accumulation of negative charge, which increases the basicity of the POM anion. The reduction process may, therefore, be accompanied by concomitant protonation and, as a result, the redox properties of POMs are markedly pH-sensitive. Although through crystal engineering, extended 2D and 3D solid-state arrays may be constructed, the exploitation and encoding of the value-adding properties of POMs as functional components in advanced materials remain elusive, mainly due to the fact that these materials are obtained as crystalline solids that are hard to process. Due to the high lattice energies associated with crystallization, the occurrence of distinct supramolecular architectures, such as liquid crystalline phases, is rarely observed, and methodologies have only recently been developed to explore such new POM phases [21]. To support, handle, manipulate and operate devices that contain POMs as functional components, it will be advantageous to arrange the clusters in surface-confined structures; however, until recently there have been no generic methodologies available for achieving this goal [6].
Another interesting class of materials is macromolecular assemblies based on inorganic elements because they add another dimension to the field of polymer and materials science. Coordination numbers and geometries become extra variables, and metal ions provide redox, magnetic, optical or reactive properties that are not available in carbon-based polymers [22, 23]. Their potential impact on the technology of inorganic–organic composite materials may be gleaned from the importance of metal ions in biological systems as cofactors in enzymes and metalloproteins for oxygen transport in respiration or for electron transfer in photosynthesis. Over 40 elements embracing main group metal elements (Si, Ge), transition metals or rare-earth elements in addition to the 10 elements found in organic polymers (C, H, N, O, B, P, halides) are available for organometallic polymers. Therefore, the variations of organometallic polymers are endless. Several types of organometallic polymers can be distinguished on the basis of how the metal is integrated in the polymer, as outlined by Rehahn (figure 1) [24]. Metals can be pendant to or they can be part of the polymer backbone. In coordination polymers the coordinative bond between the metal ion and the ligand is an integral part of the backbone. The polymer backbone can also act as a polymeric chelate, which embeds the metal ion; breaking the coordinative bond does not affect the polymer backbone. Finally, metal ions can be part of network structures with a continuous 3D topology.
Figure 1.
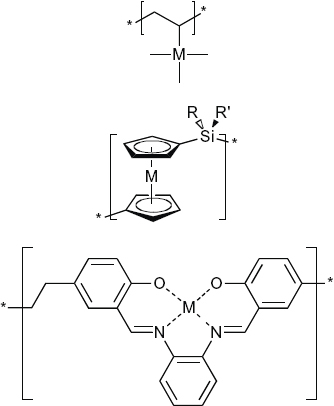
A metal, M, can be pendant to (top) or part of the polymer backbone (center), or the metal is embedded in a polymeric chelate (bottom). In the coordination polymer (center), the coordinative bond is an integral part of the polymer backbone (metallo-supramolecular polymer). Depending on the strength of the interaction between the metal ion and the coordinating ligand, coordination polymers are dynamic equilibrium systems or kinetically inert polymers.
A central challenge in utilizing supramolecular modules as molecular devices and advanced materials is to control the surface chemical properties in order to incorporate the components in suitable device architectures. Modifying the surface chemical properties is a central step in mediating the contact with common technical substrates and junctions, in tailoring the compatibility with organic materials and biological tissue, in manipulating the solubility and in engineering novel nano- and mesoscopic supramolecular architectures. Many possible applications involve thin films or layered materials, such as displays, sensors and protective coatings. The chemical routes that govern self-assembly have been extensively investigated and the self-organization of functional modules into extended, highly ordered ensembles represents the next milestone towards functional hierarchically structured materials. To exploit the novel collective properties of ensembles, it will be important to gain more precise control of the spatial arrangement of modules in their supramolecular self-organized environment. To achieve these goals it is, therefore, necessary to develop new methods and to improve existing methods of fabricating thin films and other device architectures of well-defined composition and dimensions with supramolecular modules as functional components.
This review focuses on methods to introduce various functional supramolecular modules into different device architectures including mesophases, thin films, nanostructures, etc. These methods have in common that weak non-covalent interactions and self-assembly are employed in order to combine, position, and orient the functional modules in the final device architecture. Results on extended metallo-supramolecular polymers and discrete POM clusters will be presented. These systems have in common that they contain transition metal ions that are both important for structure formation and the properties of the systems.
Metallo-supramolecular polymers
Here, we are interested in soluble dynamic coordination or metallo-supramolecular polymers, respectively, assembled from molecular constituents [25]. The topic of coordination polymers with macromolecular ligands has been reviewed recently [26]. If the binding constants between ligands and metal ions are very large, kinetically inert polymers are formed that are similar to conventional, covalent polymers in terms of their polymer physics and properties. If the binding constants are small, polymeric assemblies do not form in solution but form only in the solid state. On the other hand, if the binding constants, that is, the interactions between the metal ions and the ligands, are of intermediate strength, macromolecular assemblies form in solution. This is not necessarily a limitation because the binding constant depends on the pH and temperature as well as on the ligand design. By carefully choosing the boundary conditions of self-assembly, it is, therefore, possible to find a window of opportunity where soluble metallo-supramolecular polymers form and can be studied. Coordination polymers can be assembled directly from metal ions and ligands. Although polymers based on kinetically inert transition–metal complexes are readily characterized in solution by standard analytical means, polymers formed by kinetically labile transition–metal complexes in solution have only very recently been successfully characterized in detail. The overwhelming majority of the resulting networks or metal-organic frameworks (MOFs) are isolated and characterized as crystalline solids [27, 28]. Here, the binding constants are generally so small that no polymeric assemblies are formed in solution. The coordination network exists only in the solid state. The diversity of the resulting framework architectures is remarkable and has been extensively discussed in previous reviews [29–31]. Due to their large apparent surface area, perhaps the most promising application of MOFs is the specific uptake of gases through the control of functional group chemistry and the windows, pores and channels of the architecture [32]. The technological needs regarding fuel gases such as methane and hydrogen have fostered research in this area [33, 34]. The ability to characterize these crystalline materials by crystallography provides a good perspective on establishing accurate structure–property relationships.
The above-mentioned examples underline the prospects of using metal ion–ligand interactions for constructing metallo-supramolecular materials with various structures, properties, and applications. Recent developments indicate a new trend in which kinetically labile metal–ligand interactions are employed to form macromolecular assemblies in solution. Kinetically labile interactions enable the construction of materials with unprecedented properties: materials built up through weak interactions can assemble, disassemble and reconstruct in a dynamic fashion under ambient conditions, thus establishing optimization routines and error correction in the assembly algorithm! Since the interactions are often on the order of kT and are comparable to entropic forces, such materials can be adaptive and responsive. Moreover, different interactions, like hydrogen bonding, electrostatics or coordinative bonds, can compete within a complex molecular architecture so that structure and properties are dynamic, that is, they depend on external parameters, such as temperature, pH, the solvent, ionic strength or external fields. In addition, such materials have the ability to self-repair, self-anneal and self-correct under ambient conditions (on account of the weak interactions). In their pioneering work on metallohelicates Lehn and co-workers demonstrated the possibility of this principle [35].
Metallo-supramolecular chemistry, which is producing increasingly sophisticated polymetallic architectures based on various intercomponent interactions, has transformed the field of inorganic coordination chemistry. The resulting complicated and aesthetically appealing structures have provided the driving force to rationalize their formation, culminating in ‘the principle of maximum site occupancy’ [36, 37]. Here, we discuss the important aspects of macromolecular assemblies that are formed by the interaction between metal ions and ditopic ligands [38].
We make the following assumptions: the ligand, L, or the monomer consists of two identical metal ion receptors linked through an arbitrary but rigid spacer (figure 2). We will consider only the case of extended linear macromolecular assemblies. Discrete ring-type structures are not considered here because they are not expected to exhibit interesting dynamic properties. The analysis is based on thermodynamics following the law of mass action. Several species exist in solution: nn ML complex in which a metal ion is bound to one receptor, and an ML2 complex in which a metal ion is coordinated to two receptors. The ML2 complex constitutes the link in the polymer backbone. The end groups are either free receptor sites or ML complexes.
Figure 2.
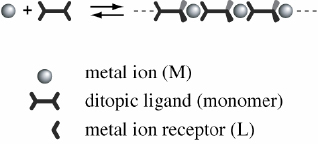
Metal-ion-induced self-assembly of ditopic ligands resulting in macromolecular assemblies. In the case of an intermediate binding strength between metal ions and ligands, dynamic equilibrium polymers form in solution. The ditopic ligand consists of two metal ion receptors linked by a spacer. Here only linear, extended structures are considered, e.g. through rigid ligands.
In the simplest case, the metal ions bind independently of each other and the equilibrium concentrations of all species can be described by the stability constants of the two types of complexes:
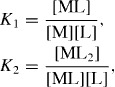 |
where [L], [M], [ML] and [ML2] are the concentrations of free receptors, free metal ions and metal ions coordinated by one and two receptors, respectively. At higher concentrations we can, of course, anticipate deviations from these assumptions. As outlined by van der Gucht and co-workers the concentrations of the various species are given by [38]
where p is the fraction of receptors involved in ML complexes and q is the fraction of receptors involved in ML2 complexes. The concentration of the ditopic ligand is given by [Mon] and the ratio of the concentration of the metal ion to that of the monomer is given by the coefficient y. These equations completely describe the equilibrium concentration of each species. The following graph illustrates the average number of monomers per assembly 〈n〉 as a function of concentration for different values of y (figure 3). In this example, the stability constants of the Fe(II) terpyridine (tpy= terpyridine) complex are used (log[K1]=7, log[K2]=14) [39]. Notably, the molecular weight depends on the concentration and stoichiometry of the constituents. In the case of an exact stoichiometry of the metal ion to the ligand, we observe exponential polymer growth with increasing concentration similar to that observed in classical condensation polymerization. It can be seen that terpyridine ligands and Fe(II) form assemblies with very high molecular weight. In the case of a different stoichiometry (y≠1) polymer growth stops when the deficient component is consumed, which corresponds to the plateau region observed at higher concentrations. It is, therefore, possible to control the molecular weight of the assemblies through the stoichiometry.
Figure 3.
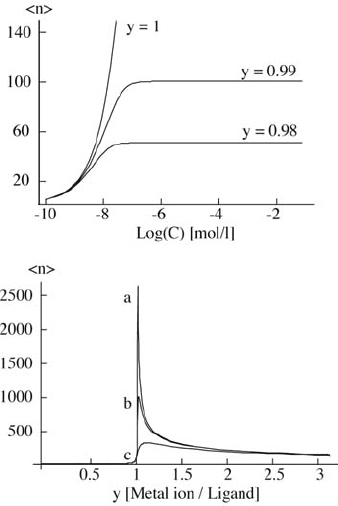
Top: average number of monomers (ditopic ligands) per assembly 〈n〉 as a function of concentration for different stoichiometries y. Here, the stability constants of Fe(II) and terpyridine are used (log[K1]≪log[K2]). Bottom: average number of monomers per assembly 〈n〉 as a function of stoichiometry y for different monomer concentrations ((a) 10−3 mol l−1, (b) 10−4 mol l−1 and (c) 10−5 mol l−1).
The dependence of the average number of monomers in an assembly as a function of stoichiometry y for different concentrations is also shown in figure 3. The average assembly length depends strongly on the stoichiometry. Note that the curves are asymmetric with respect to y=1. An excess of metal ions results in longer assemblies than an excess of monomers. Experimentally, it is generally difficult to achieve an exact 1:1 stoichiometry. It is, therefore, advisable to use a slight excess of metal ions in the synthesis. For the synthesis of metallo-supramolecular coordination polymers, the ditopic terpyridine–Fe(II) system is, therefore, an excellent model system because it forms large macromolecular assemblies. Similarly, the first-row transition elements such as Ni(II), Co(II) and Fe(II) are kinetically labile, thus, the assembly process can be carried out at room temperature in aqueous media [40].
We can draw several important conclusions from this analysis. Metallo-supramolecular coordination polymers can form very high molar masses comparable to those of conventional polymers if the metal ion, the ligand, and the concentration are chosen appropriately. However, this analysis does not take into account additional effects that can occur during assembly, such as the accumulation of charge or the formation of lyotropic phases [41]. If the assemblies carry charge, then additional effects may arise through the polyelectrolyte effect such as ionic-strength-dependent properties. Methods that interfere with the equilibrium, such as chromatography or viscosity, which are commonly used in polymer analysis, cannot be applied to these systems. However, it is possible to choose a stoichiometry such that the length of the assembly becomes independent of concentration for higher concentrations. This effect can be used, for instance to maximize signal intensity, e.g. in scattering experiments, by increasing the concentration without affecting the length distribution. Another way of controlling the length is to use a monotopic ligand, which acts as a capping unit.
Owing to the weak interactions between the ligands and the metal ions we can anticipate the response of these materials to mechanical shear. Rupturing the metal ion–ligand interactions results in smaller polymer segments, which is associated with a reduction in viscosity (vide infra). The choice of metal ions and the design of ligands provide additional variables for tailoring the properties of the resulting assemblies. The ligands are generally Lewis bases and, therefore, protons and metal ions compete for the ligands; thus, the equilibrium is affected by the pH of the solution. The pH can be used to shift the equilibrium, e.g. to move the length into a specific range independently of the ligand and metal ion concentrations, or the polymer can be considered to be pH-responsive. The same is true for coordinating solvent molecules. The simultaneous equilibria of protons, solvent molecules, metal ions and ligands are also temperature-dependent [42]. In summary, there are a considerable number of variables that affect the architecture and the polymer properties of these systems, which adds to the richness of these systems. Systematic studies on how molecular weight correlates with polymer-like properties (enhanced viscosity, glassy solid state, etc) for such systems have yet to be established.
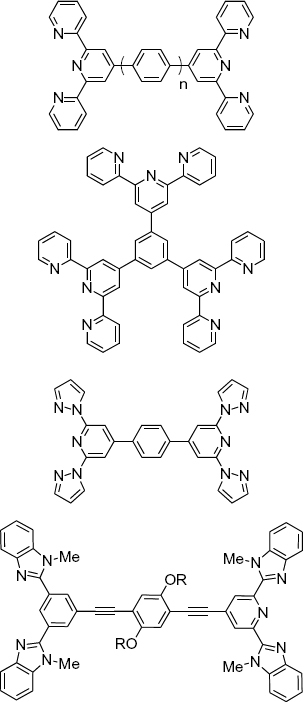
One of the first systematic studies of well-defined coordination polymers based on kinetically labile complexes was presented by Rehahn and co-workers. Using phenanthroline-based ditopic ligands, they prepared a variety of polymers with Ag(I) and Cu(I) (figure 4) [43, 44]. The ligand design essentially prevents the formation of discrete species, thus giving rise to extended macromolecular assemblies. These coordination polymers are sensitive to the type of solvent molecules and readily decompose via the displacement of metal ions by coordinating solvent molecules. Decomposition is avoided if the polymer is dissolved in nonpolar solvents. In an appropriate solvent, ligand exchange is reduced to such an extent that the polymer can be characterized. The metal ions render the polymers highly charged, resulting in the need for polar solvents. Long alkyl chains, attached to the ligand, were used as a means of tailoring the solubility. In nonpolar solvents like 1,1,2,2-tetrachloroethane (TCE), the resulting polymers behave as well-defined macromolecules. In the presence of coordinating solvents, like acetonitrile or pyridine, the species behave more similarly to low-molecular-weight aggregates. NMR titrations indicate a degree of polymerization of 6–8 if 0.8 equivalents of Cu(I) is used. At a 1 : 1 stoichiometry, the degree of polymerization increases to above 20. In contrast to Cu(I), the Ag(I) polymer shows rapid ligand exchange in a TCE/CH3CN mixture (4 : 1). In the absence of coordinating species there is practically no ligand exchange, as documented by mixing Cu(I) and Ag(I) polymers. To avoid experimental errors, capillary viscosimetry was used to adjust the stoichiometry to 1 : 1 to obtain high-molar-mass polymers. The analysis of the viscosity yielded an apparent intrinsic viscosity of [η]≈30 ml g−1 for a high-molar-mass polymer based on Cu(I) in 0.01 M NH4PF6 acetone−1. The salt is added to suppress polyelectrolyte effects. A similar value is found for a stable, random-coil Ru(II) coordination polymer of molar mass Mn≈40.000 g mol−1 [45]. However, the Huggins constant (kH≈9) of the kinetically labile polymer is very large, which is a result of the equilibration. A lowering of the concentration causes the depolymerization of the coordination polymer, thus enhancing the reduction of the viscosity. Time-dependent measurements support this hypothesis. The addition of a solvent causes the re-equilibration of the system, which is manifested as the time dependent-decay of the viscosity to a final value. The authors point out that even a small amount of coordinating species in the solution, such as water, causes depolymerization.
Figure 4.
Dynamic coordination polymers based on the ditopic phenanthroline ligand and Cu(I) and Ag(I) ions, M.
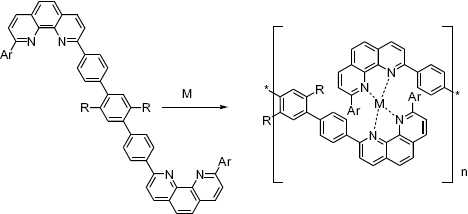
Constable introduced the concept of polytopic ligands based on terpyridines to construct metallo-supramolecular coordination compounds (figure 5) [46]. This early study focused on complexes with kinetically inert Ru(II), which can be characterized by standard techniques. Since then, 2,2′:6′,2″-terpyridine has attracted great interest for the construction of metallo-supramolecular polymers because it binds many transition-metal ions, generally possesses high-binding constants due to the chelate effect and forms stereochemically well-defined complexes. Terpyridine forms octahedral complexes with most transition-metal ions, thus it is ideally suited for constructing linear supramolecular polymers through substitution at the 4-position of the central pyridine ring. The high-binding constants allow us to also study metal-ion-induced self-assembly in aqueous media, which is highly attractive for putting such systems into practical use. In addition, terpyridine transition–metal complexes possess a variety of attractive properties that make them interesting for photophysical, electrochemical and magnetic studies [47].
Figure 5.
Structures of polytopic ligands based on terpyridines, pyrazolylpyridine and 2,6-bis(methylbenzimidazolyl)pyridine for the construction of metallo-supramolecular coordination polymers [46, 48].
Owing to their charge, the solubility of these so-called metallo-supramolecular polyelectrolytes (MEPEs) in aqueous solutions critically depends on the counter ions, particularly because terpyridine ligands are generally not very soluble in water. The first water-soluble MEPEs based on 1,4-bis(2,2′:6′,2″-terpyridin-4-yl)benzene and Fe(OAc)2 were reported more than five years after Constable’s work and finally opened an avenue to study and employ these systems in many ways (figure 6) [49]. Although the solution structure and the solution properties such as the rheological and polyelectrolyte properties require further studies, the solid-state structure of the MEPE based on FeOAc2 and 1,4-bis(2,2′:6′,2″-terpyridin-4-yl)benzene was recently determined by electron diffraction (figure 6). As pointed out above it is generally not possible to grow crystals of equilibrium polymers by supersaturation, which is commonly employed for crystal growth, because under these conditions the assemblies become very large and form amorphous precipitates. However, we can use the following principles to grow nanoscopic crystals of MEPEs directly on a surface under dilute conditions. Theoretically it has been predicted that in the absence of strong polymer–surface interactions long chains experience a larger entropy loss than shorter ones upon adsorption. Therefore, the surface region is predominantly occupied by short chains. At low temperatures and under dilute conditions the formation of extended assemblies is further inhibited. Finally, if the stoichiometry of the ligand to the metal ion is larger than 1, the degree of polymerization remains constant if the concentration is increased. This approach may be of general use for characterizing the first, nanocrystalline particulates in coordination polymers and frameworks to address fundamental questions on how such systems grow. Due to the low concentration, only nanoscopic crystals that are not suitable for x-ray diffraction are obtained. Therefore, we decided to attempt an analysis by electron diffraction. The analysis revealed a primitive monoclinic unit cell, in which the MEPE forms linear rods, which are organized into sheets (figure 6). Four sheets intersect the unit cell, while the adjacent sheets are rotated by 90° with respect to each other. Mössbauer spectroscopy of bulk samples confirmed the pseudo-octahedral coordination geometry of the MEPE and indicated an average length of approximately 8 repeat units in the solid state. The formation of the Fe(II)-tpy2-complexes has also been confirmed by the characteristic MLCT band observed in UV-vis spectroscopy. Recently, it was shown that MEPE can also assemble in the pores of zeolite-like materials [50].
Figure 6.
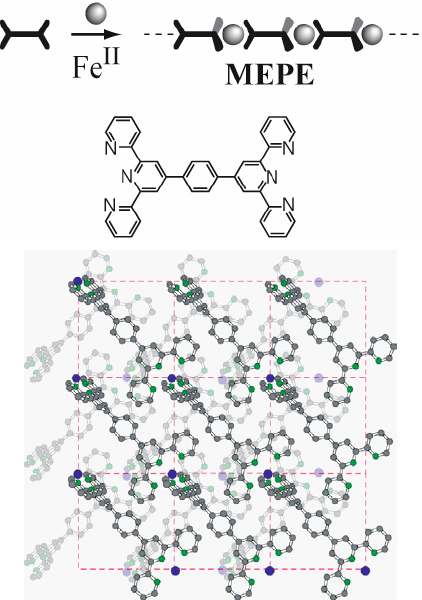
Top: MEPE self-assembled from FeOAc2 and 1,4-bis(2,2′:6′,2″ -terpyridin-4-yl)benzene. Bottom: solid-state structure determined by electron diffraction and molecular modeling. The MEPE forms linear rods that are organized into sheets. The unit cell consists of four sheets, while each sheet is rotated by 90° with respect to each other. The coordination geometry is pseudo-octahedral [51].
In addition to terpyridine, other ligands can be used for coordination polymers. For instance, coordination polymers based on Schiff base rare-earth complexes were reported in 1994 by Archer and co-workers (figure 7) [52]. In polar solvents such as DMSO or NMP, molar masses of 30 000 have been achieved. These polymers exhibit good stability and high glass-transition temperatures. The concept has been extended to other well-characterized rare-earth coordination polymers [53, 54]. The incorporation of rare-earth metal ions makes these systems interesting for novel photophysical studies and applications. Embedding metal ions in liquid crystal polymers is a step towards the realization of mesophases that combine the properties of metal ions and mesophases. For instance, Serrano and co-workers explored this concept to synthesize nematic polyesters based on metallomesogenic metal moieties (figure 7) [55, 56]. The polymer shown in figure 7 displays an enantiotropic nematic mesophase. Cooling the melt results in the formation of a nematic glass. Macroscopic ordering is induced by drawing fibers from the nematic melt. X-ray scattering indicates that the orientation of the mesogenic cores is along the stretching direction, which demonstrates that such polymers are readily processable. Owing to the Cu(II) centers, these polymers are paramagnetic and a weak exchange interaction of antiferromagnetic character between the copper centers was detected. Although many other systems have been reported in the literature [57], strong coupling between the metal centers, and, therefore, cooperative phenomena are generally not observed because the distance between the metal centers is too large [58]. The rich chemistry of terpyridines has afforded a number of other interesting ligands with photoluminescent properties, as reported by Würthner and co-workers [59, 60], as well as chiral coordination polymers [61, 62].
Figure 7.
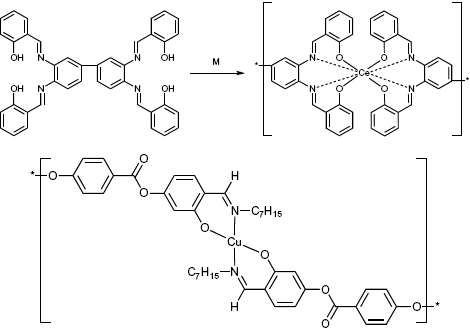
Top: example of a linear cerium(IV) Schiff-base coordination polymer. Bottom: coordination polymers as part of a mesophase described by Serrano and co-workers [63].
The rich photo- and electrochemical properties of porphyrins make the corresponding polymers interesting materials from the point of view of charge storage and transport, solar energy conversion and nonlinear optics. Michelsen and Hunter reported an approach to forming soluble porphyrin polymers through the coordination of the central metal ion and the pendant, covalently attached, pyridine rings (figure 8) [64]. Solubility is ensured by introducing 2-ethylhexyl side chains to the porphyrin core. Metallation with Co(II) produced a monomer that self-assembles to form a porphyrin polymer. At concentrations greater than 10 mM the polymer precipitates and can only be redissolved in coordinating solvents such as pyridine. This observation indicates the formation of high-molar-weight polymers. Unfortunately, the authors have not investigated the resulting precipitates, which may also be interesting colloids (vide supra). In solution, the occurrence of polymerization was shown by pulsed-gradient spin-echo NMR diffusion experiments, which provide insight into the relative size of the assemblies. By adding a monomer to the polymer solution, which induces chain shortening, the authors further demonstrated the polymeric nature of the assembly by size exclusion chromatography. The association constant was determined to be approximately 106 M−1 and the polydispersity index was between 1.5 and 2.5, as expected for equilibrium polymers. The maximum molar weight observed was 136 kDa, corresponding to approximately 100 repeat units. Precipitation occurred at higher concentrations indicating even higher molar masses.
Figure 8.
Self-assembly of a Co(II)-porphyrin polymer as described by Michelsen and Hunter [64].
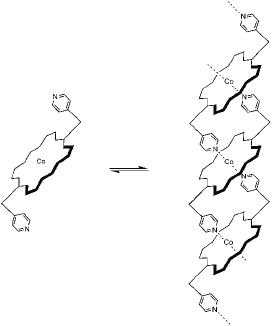
Another area of interest for which supramolecular chemistry holds great promise is that of stimuli–responsive materials, because environmental variables can have a large effect on the degree of interactions between the individual components of the materials. This is particularly true for metallo-supramolecular polymers. An alteration of the strength of the noncovalent part of the intermolecular bonds can result in a dramatic modification of the supermolecular structure and thus cause significant changes in the properties. Rowan and Beck have introduced multistimulus, multiresponsive metallo-supramolecular polymers from lanthanides and 2,6-bis(benzimidazolyl)-pyridine (BIP) ligands (figure 9) [65, 66]. This ligand can bind transition metal ions in a 2 : 1 ratio as well as lanthanides in a 3 : 1 ratio. Therefore, assembling ligands, transition-metal ions and lanthanides in the appropriate stoichiometry results in the formation of metallo-supramolecular gels.
Figure 9.
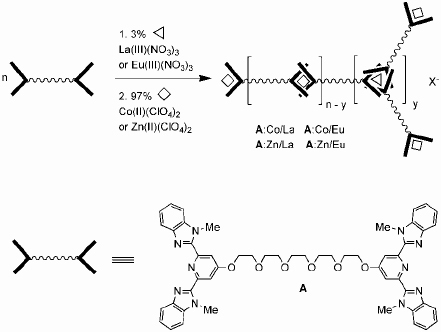
Representative scheme showing the formation of metallo-supramolecular gels by assembling BIP, transition-metal ions and lanthanides in the appropriate ratio.
Transition metals such as Zn(II) or Co(II) give rise to linear polymers due to the preferred octahedral coordination geometry of the ligand and these metal ions. Adding 3 mol.% of the lanthanide (Eu(III) or La(III)) with respect to the ligand spontaneously results in the formation of a gel in a CHCl3/CH3CN mixture. A total of four different gels were prepared and characterized (Co/La, Zn/La, Co/Eu and Zn/Eu). All four gels are thermoresponsive and show a reversible gel–sol transition upon heating (figure 10). The optical changes indicate that upon heating, the La/ligand interaction is thermally broken. It is not surprising that these gels are also thixotropic, that is, they exhibit shear-thinning under stress. Shaking the Zn/La gel results in a free-flowing liquid, which, upon standing, thickens again to a gel-like material.
Figure 10.
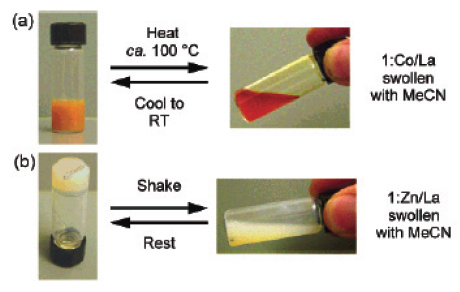
(a) Thermoresponsive nature of the Co/La system and (b) thixotropic response of the Zn/La system (with permission from [65]).
The Eu(III)-containing polymer exhibits intense metal centered luminescence, which is facilitated by the antenna effect of the ligand. This is effectively a light conversion process, which occurs by the absorption of radiation by the ligand, followed by a ligand-to-metal energy transfer process finally resulting in a metal-centered emission. The photochemical properties make these materials interesting for various applications but they also offer a method of investigating the assembly–disassembly process. Heating the Zn/Eu polymer results in a substantial reduction in the lanthanide-based emission, providing further evidence that the lanthanide-ligand bond is thermally broken. Lanthanides are oxophilic and bind well to carboxylic acids. Therefore, it is not surprising that the polymers are also chemoresponsive. The addition of 0.85 wt.% formic acid to the Zn/Eu polymer results in the loss of mechanical stability and quenches the Eu(III) emission. This result is consistent with that of formic acid displacing the ligand on the Eu(III) cation resulting in a switching-off of the antenna effect. The process is reversed upon drying the material in vacuum to remove the formic acid followed by reswelling in acetonitrile. The rheological behavior has been studied in more detail by Rowan and co-workers [67, 68]. The susceptibility of coordination polymers based on diphosphanes and Pd(II) to sheer has been elegantly demonstrated by Sijbesma. In toluene this polymer is sufficiently kinetically inert that an analysis of the molecular weight is possible by standard chromatographic techniques. Also, the assembly kinetics is sufficiently slow for it not to interfere with chromatography; equilibration requires several days. Exposing the solution to ultrasound results in a significant reduction of the molecular weight and re-equilibration restores the original molecular weight. Notably, ultrasonic chain scission is a nonrandom process that acts preferentially on longer chains. A possible application of ultrasonic chain scission may lie in the area of transition–metal catalysis by the creation of highly reactive species under controlled conditions [69].
With the aim of developing organic/inorganic hybrid materials that are stable at high temperatures but yet processable, Rowan and co-workers employed the ditopic ligand shown in figure 5, which combines the BIP unit (vide supra) for dynamic polymerization and 1,4-diethynylbenzene as a functional unit. Owing to the conjugation, this new ligand is more emissive than similar BIP ligands that lack conjugation [70–72]. The optical properties of this compound allow following the self-assembly process in solution. Upon the addition of Zn(II), three isosbestic points are observed, suggesting the equilibrium of a finite number of spectroscopically distinct species. The authors assign the bands in the UV-vis spectra to free ligand, 1 : 1 Zn-ligand and 1 : 2 Zn-ligand complexes. Metal binding also has a pronounced effect on the emission characteristics. The formation of polymers is further demonstrated by an increase in the viscosity of the solutions at higher concentrations. This approach to assemble conjugated polymers from smaller building blocks has high potential for the utilization of these materials; because high-molecular-weight conjugated polymers are generally difficult to process due to their high transition temperatures, limited solubility and high solution viscosities. In addition, structural defects and impurities in such polymers are not uncommon, making their processing often intricate and time-consuming [72]. Using Zn(II) and Fe(II) the authors readily prepared fibers and thin films amendable to further characterization.
The nature of the core in the ditopic BIP ligand has a profound impact on the material’s properties. While rigid ligands are expected to form rigid-rod-type polymers, the introduction of flexible linkers allows the formation of rings [73]. A short core, e.g. penta(ethylene glycol), in the ligands encourages the formation of macrocyclic species, and the resulting material does not show polymer-like properties. On the other hand, macromonomers based on poly(tetrahydrofuran) telechelic units give rise to polymeric materials with improved mechanical strength. This work also hints to the existence of another problem in the formation of high-molecular-weight materials. If polydisperse macromonomers are used, it is intrinsically not possible to achieve the desired 1 : 1 ratio of ligand to metal by simply weighing the components. The authors used NMR titration to assess the maximum degree of polymerization. Here it becomes clear that (simple) standard procedures have to be developed to control the stoichiometry precisely in order to achieve comparable and reproducible results.
Recently, Ruben and co-workers prepared an MEPE from 1,4-bis(1,2′:6′,1″-bis-pyrazolylpyridin-4′-yl) benzene and Fe(II) (figure 5) [74]. Although the authors do not present data on the molecular weight of this new MEPE, they report a reversible spin transition above room temperature that occurs at approximately 323 K with a thermal hysteresis of 10 K. In contrast to the terpyridine-based ligands, the bite angle in the bis-pyrazolylpyridin is wider, which reduces the ligand field and, therefore, opens an avenue towards spin-crossover at ambient temperatures [74]. Interestingly, the authors use a twofold excess of metal ions to limit the molecular weight (vide infra figure 3) and to keep the formed MEPE in solution.
Craig and co-workers have demonstrated an elegant method of manipulating the dynamics of metal–ligand interactions by employing the equivalent of a macromolecular ‘kinetic isotope effect’ (figure 11) [75]. The independent control of the dynamic versus thermodynamic properties is achieved by a simple steric effect at the metal center of square-planar Pd(II) and Pt(II) complexes. Ligand exchange at metal centers generally occurs through an associative mechanism, in which the attacking nucleophile associates with the metal center prior to the departure of the original ligand. The pentacoordinate transition state is sterically crowded relative to the reactant and product complex. Steric effects in the spectator ligands of the complex, therefore, profoundly affect the rate of ligand exchange while having little effect on the stability of the complex. Pincer motifs have proven to be attractive in this regard due to the ease of steric manipulation of the spectator ligands and the added advantage of increased stability relative to the moderate stability of aryl Pd(II) complexes [76, 77]. Using this approach, the authors studied supramolecular networks with a continuous 3D topology based on polypyridine and a ditopic pincer ligand (crosslinker) in DMSO. They showed that the mechanical properties of the material are linked to solvent-mediated ligand displacement reactions and that the dynamical properties are governed by the dissociation of the crosslinks in the network. The mechanical properties are, therefore, determined by the relaxation that occurs when the crosslinks are dissociated from the polymer backbone. The dynamic properties are scaled through the dissociation rates of the crosslinks independently of their rate of formation; thus, dissociation is effectively equivalent to equilibration. Note that this work shows how dynamics and mechanism of metal–ligand exchange reactions are not only a qualitative but also a quantitative representation of the viscoelastic properties of bulk materials [78]. Although thermodynamics are a primary design consideration in supramolecular chemistry, the dynamics of the interactions are particularly important under nonequilibrium conditions, such as those imposed under mechanical stress [76, 77, 79]. Recently, Craig and co-workers used single-molecule force spectroscopy to study the mechanical activation of the ligand substitution reaction. By measuring the rupture force as a function of loading rate, the authors could determine the dissociation rate constants in these systems [80].
Figure 11.
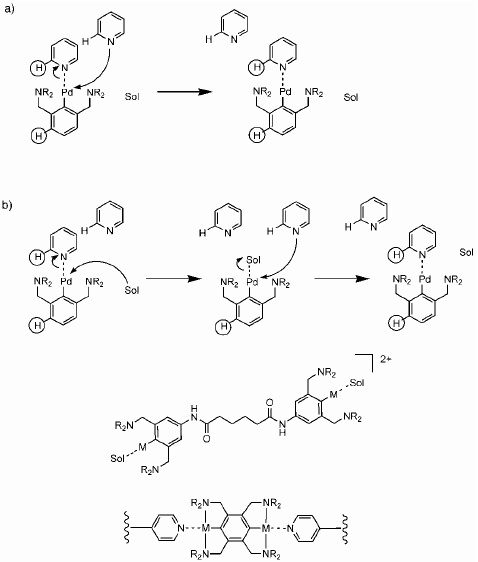
Exchange mechanism for pincer–pyridine complexes. The dynamics of ligand exchange can be controlled through steric manipulation of the spectator ligand, R, around the metal center. (a) Direct displacement of one pyridine by another pyridine. (b) Solvent-assisted ligand exchange. Bottom: ditopic pincer crosslinkers [75].
The concept of dynamic 3D topologies has been taken to a higher degree of complexity by Weck and co-workers by combining metal ion coordination and hydrogen bonding as crosslinks (figure 12) [81]. By balancing the thermodynamic and kinetic variables of the constituents, the authors envisage systems allowing rapid prototyping and facile modification of the physical properties of the crosslinked materials. These types of orthogonal functionalization and crosslinking offer new options for overcoming the functional group incompatibility encountered in many polymerization techniques [82].
Figure 12.
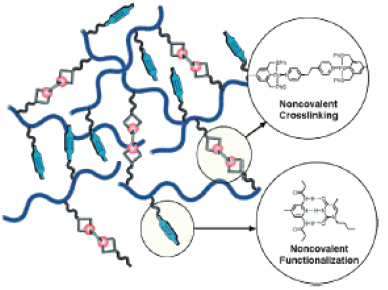
Scheme illustrating the approach of Weck and co-workers using metal ion coordination and hydrogen bonding to generate reversible crosslinks to realize new materials with tailored thermodynamic and kinetic properties (with permission from [81]).
A system of great practical use is presented by Stuart and co-workers [83]. They employed water-soluble 2,6-dicarboxypyridine ligands that bind transition-metal ions and lanthanides. Recently, this group reported on using the hierarchical self-assembly of MEPEs and diblock copolymers to make coacervate core micelles (C3Ms) as shown in figure 13 [84]. Here, the negatively charged MEPE interacts with the positively charged part of the diblock copolymer, which results in the formation of a complex in the interior of the micelle. The assembly is stabilized by the hydrophilic poly(ethylene oxide) part of the coblock copolymer, which forms a shell around the interior complex.
Figure 13.
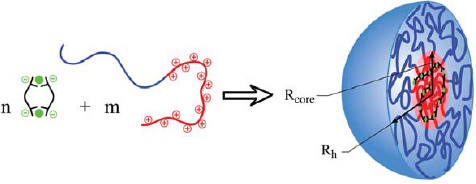
Self-assembly of diblock copolymers and MEPE resulting in the formation of a micellar structure (complex coacervate core micelles) (with permission from [84]).
As mentioned above, some metallo-supramolecular polymers tend to precipitate from their solution if the concentration of the constituents exceeds a critical limit. Because of the use of organic ligands, solubility is an issue that needs careful attention in metallo-supramolecular polymer research. Oh and Mirkin have used this property to form colloidal particles from coordination polymers (figure 14) [85]. A homochiral carboxylate-functionalized binaphtyl bis-metallo-tridentate Schiff base (BMSB) is used as a building block. The particles form via coordination to the carboxylate groups on the BMSB periphery. The process of particle formation is completely reversible, as evidenced by the formation of the starting materials upon the addition of excess pyridine. The choice of the BMSB ligand, the type of metallation and the ancillary ligands make it possible to tailor the chemical and physical properties of the resulting polymers and particles. It is interesting to note that the particles are stable in both water and common organic solvents. The inclusion of binaphtyl in the ligand affords fluorescence from the complex as well as from the particles. The ancillary ligands (L in figure 14) also allow the manipulation of the electronic nature of the metal ions. For instance, increasing the σ-donor capability of the ancillary ligand induces a redshift of the absorption maximum. The ability to produce these materials in enantiopure form makes them attractive in catalysis and separation. This concept has recently been used by Maeda to obtain nanoscale architectures from dipyrrin ligands (figure 15) [86].
Figure 14.
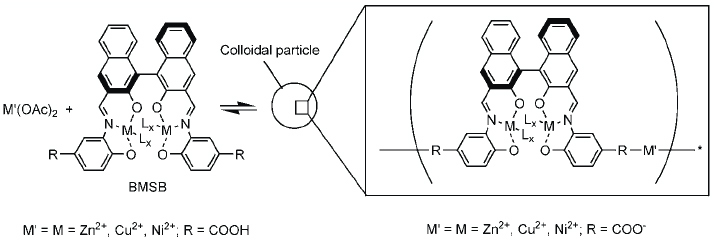
Preparation of colloidal particles from metal ions and BMSB. Adding diethyl ether to the reaction mixture results in particle formation and precipitation. A coordinating solvent such as pyridine dissolves the particles (adapted from [85]).
Figure 15.
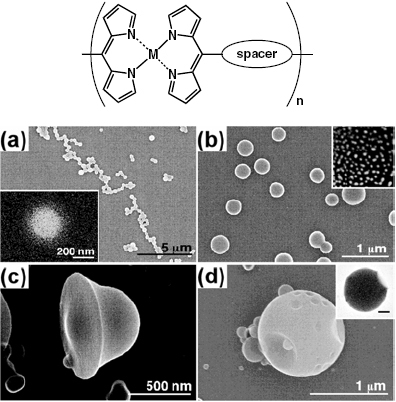
Ditopic phenylethynyl-bridged dipyrrins give coordination polymers with Zn(II), which produce fluorescent colloidal objects in solution (a, b). Using a mixture of THF and water, unique morphologies, such as bell-shaped (c) and ‘golf ball’-like (d) architectures, are observed [86].
Thin films
The positive charge can be used to deposit MEPEs on an oppositely charged surface using electrostatic layer-by-layer deposition. The alternating deposition of MEPEs and suitable polyelectrolytes such as poly(styrene-sulfonate) gives rise to well-defined multilayers on flat substrates as well as colloidal templates (figure 16) [87, 88]. It is likely that the release of counter ions drives the irreversible deposition of the polyelectrolytes on the surface. An alternative route to incorporating metal complexes in thin films involves using discrete metallounits that interact through weak bonds. Pyrenyl tails attached to the terpyridine moiety cause assembly of the discrete units into linear arrays, particularly in water where the π–π interactions are strongest. The formation of π-stacking is evident from the crystal structure and fluorescence properties of the thin films [89]. The applications of such systems currently include electrochromic devices [90–92]. Although the LbL procedure offers many advantages, such as deposition from solution, the use of simple equipment and the formation of coatings on arbitrary surfaces, the resulting layers do not have the same degree of order as the Langmuir–Blodgett (LB) films of PACs (vide infra) [49, 93].
Figure 16.
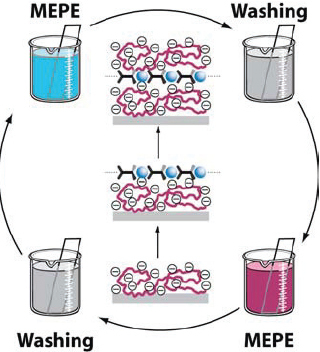
Electrostatic layer-by-layer assembly of MEPEs affording well-defined multilayers. The method can be applied to any type of surface, provides excellent thickness control, is readily automated and a large number of different components can be assembled into the layers.
Mesophases
The hierarchical self-assembly of amphiphiles and MEPEs gives rise to metallo-supramolecular mesophases. The amphiphiles add another variable to the modularity of these assemblies. Through the choice of ligands, metal ions, amphiphiles and their respective stoichiometry, the structure and properties of the resulting assemblies can be controlled at every length scale. For instance, positively charged MEPEs based on metal ions and ditopic bis-terpyridines can be used to decorate metallounits with negatively charged amphiphiles [94–97]. The amphiphilic self-assembly of an MEPE and dihexadecyl phosphate (DHP) affords the corresponding PAC. This colloid-chemical approach is based on counter-ion exchange, and the association of MEPE and DHP has a profound impact on the properties of the final material. PACs dissolve in common organic solvents indicating that the amphiphiles effectively shield the hydrophilic portion of the MEPE. We, therefore, assume that, in solution, the amphiphiles are predominately located around the hydrophilic metal-ion centers as depicted in figure 17. The PAC forms nanostructures on graphite and spreads at the air–water interface. The resulting Langmuir monolayer can be transferred onto solid supports by the LB technique [98]. In addition, PACs exhibit thermotropic polymorphism and spin-crossover [99].
Figure 17.
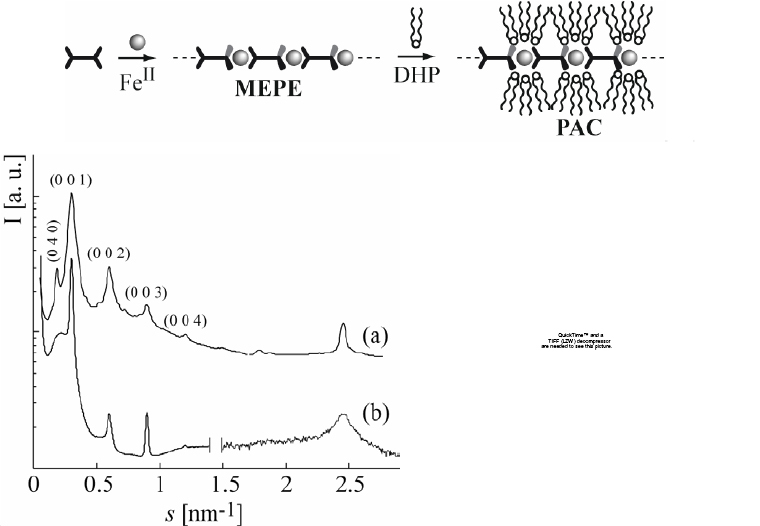
Top: sequential self-assembly of ditopic bis-terpyridines, metal ions and amphiphiles resulting in polyelectrolyte-amphiphile complexes (PAC) (DHP = dihexadecyl phosphate). The PAC readily dissolves in organic solvents, forms nanostructures on graphite, as well as Langmuir and LB layers, and exhibits spin-crossover with Fe(II) as the central metal ion. Through the choice of the ligands, metal ions, amphiphiles and their respective stoichiometry, it is possible to tailor the structure and properties of the PACs. Bottom left: calculated (a) and experimental (b) scattering curves of the PAC. Right: structural model on which the computed scattering pattern is based showing the lamellar architecture of the PAC with interdigitated amphiphiles. The hexagonal alkyl chain packing of the orthorhombic DHP unit cell comprises 12 molecules. This unit cell satisfies the steric requirements of the MEPE repeat unit. Phosphate groups are randomly oriented toward the top or bottom of the strata.
The combination of rigid-rod-shaped polymers and flexible surfactants gives rise to polymorphism. The structure of the room-temperature phase of the PAC based on DHP, 1,4-bis(2,2′:6′,2″-terpyridin-4-yl)benzene, and Fe(II) is shown in (figure 17) [97]. A combination of x-ray scattering and molecular modeling was used to reveal details of the architecture. Notably, DHP forms an interdigitated layer in this structure in contrast to the solid-state structure of DHP and the typical packing motifs encountered in amphiphilic architectures [100]. The PAC structure is a good example of a multicomponent hierarchical architecture. At the molecular level, the structure is determined by the design of the ligands and the metal coordination algorithm. At the mesoscopic scale, the structure is formed through the interaction of the MEPE rods and the amphiphilic molecules. Finally, at the macroscopic level, the structure arises through the packing of the PAC rods into the final architecture.
Investigations of the temperature dependence of the structure and properties revealed that the Fe(II)-PAC show a spin-crossover from a diamagnetic low spin state to a paramagnetic high-spin state. This phase transition in the amphiphilic matrix induces mechanical strain in the assembly of the tightly coupled metal ion coordination centers, which in turn results in a distortion of the coordination geometry. As a result, the crystal field splitting of the d-orbital subsets decreases resulting in a spin-crossover (figure 18). Liquid crystalline materials are readily processed into various device architectures, and the concept can be expanded to virtually all metallo-supramolecular polymers with suitable electronic configurations [101]. It will be interesting to see if, in the future, it will be possible to affect the thermal hysteresis behavior of these materials, which is an important requirement for switching applications.
Figure 18.
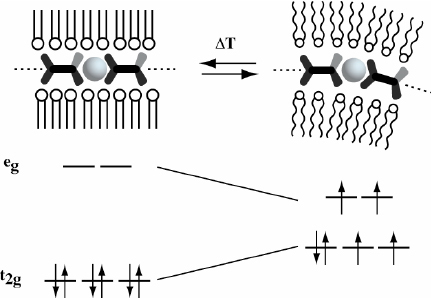
Melting of the amphiphilic matrix in the PAC results in a distortion of the coordination geometry of the tightly coupled metal centers, giving rise to a spin-crossover from a diamagnetic low-spin state to a paramagnetic high-spin state (schematic representation).
An unusual system showing ferromagnetic exchange interactions was reported by Haase and co-workers [102]. Here, the Schiff base mesogenic units containing Ni(II) metal ions are located in the side chain; however, the units form a ladder-like structure (figure 19). Presumably, the metal centers and the counter ions form a linear chain so that the metal centers come in close contact enabling cooperativity. In this respect, we can also consider this material to be a polymer with labile metal–ligand interactions. However, the material does not show liquid crystalline properties presumably due to the crosslinking of polymers through the metallomesogenic units. In fact, the metallopolymers are not soluble; during the synthesis they form a colloidal solution and are precipitated as a solid. The ferromagnetic interaction can be described by that of either that of ferromagnetically coupled dimers or linear chains. So far, no detailed structural data is available for this material due to its insolubility. The presence of cooperative magnetic properties such as remnant magnetization and coercivity indicates a strong superparamagnetism. Introducing Co(II) into these methacrylate polymers results in mesophases leading to the formation of 1D Heisenberg antiferromagnetic chain structures [103]. However, the presence of monomeric Co centers that obey Curie–Weiss behavior and dimeric Co ions, which are best described by an isotropic Heisenberg exchange operator, constitute limiting factors preventing a true 1D magnetic arrangement. The question of which type of magnetic ordering can be achieved in mesophases, therefore, remains of strong interest [16].
Figure 19.
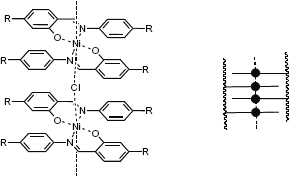
Structural model for the nickel metallopolymer. Left: Schiff base Ni complex (R denotes the polymer backbone). Right: schematic ladder structure showing the stacking in a linear chain [102].
Valkama et al described a concept in which a block copolymer, polystyrene-block-poly(4-vinylpyridine) (PS-block-P4VP), is complexed with dodecylbenzenesulfonate (DBS) as amphiphilic component and zinc dodecylbenzenesulfonate (Zn(DBS)2), leading to the polymeric supramolecular PS-block-P4VP[Zn(DBS)2]y (figure 20) [104]. The mesoporous material made from this complex has a lamellar structure, and even indicates the existence of a structural hierarchy for high-molecular-weight block copolymers. Moreover, Zn(DBS)2 can be extracted from the template with a selective solvent such as methanol, leading to a porous structure. Using P4VP, 2, 6-bis(octylaminomethyl)pyridine and Zn(DBS)2, Valkama et al further described a concept for self-organizing tricomponent mixtures [105]. According to this concept, the Zn2+ centers coordinate to the pyridine groups of PVP and 2,6-bis(octylaminomethyl)pyridine. The amphiphilic counter ions, DBS, bind to the positive metal center through electrostatic interactions, and, therefore, also contribute to the resulting architecture. The structure is denoted as a multicomb architecture with the format poly[(4VP)Zn(2,6-bis(octylaminomethyl)pyridine) Zn(DBS)2]. Because of the enhanced side-chain crowding, a cylindrical organization is obtained. Other hierarchical polyelectrolyte-amphiphile structures include molecular wires formed by a halogen-bridged platinum complex [Pt(en)2][PtCl2(en)2]4- (en=1,2-diaminoethane) with anionic amphiphiles [106], and a rigid metallohost obtained from a ruthenium–bipyridine complex and a glycoluril-based receptor cavity [107].
Figure 20.
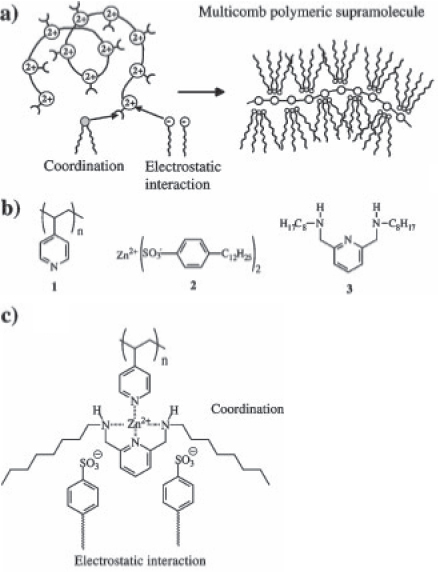
(a) Multicomb polymeric superstructures derived from a polymeric backbone (1), and the metal ion coordination of counter ions (2) and amphiphilic ligands (3). (b) Modules: P4VP (1), Zn (DBS)2 (2), and 2,6-bis(octylaminomethyl)pyridine (3). (c) Suggested complexation with a 1 : 1 : 1 molar mixture of 1, 2 and 3 to form poly[(4VP)Zn(2,6-bis(octylamino-methyl)pyridine)(DBS)2] (adapted from [105]).
Langmuir monolayer and LB multilayers
The LB technique was one of the first methods used to fabricate thin films with long-range order and precise thickness control, and played a key role in the development of molecular electronics [108]. The defined conditions of the air–water interface permit us to explore the surface activity and provide molecular-level control to construct layered materials by LB film transfer. The general concept of Langmuir monolayer preparation and LB transfer is depicted as follows. A known amount of the sample is dissolved in a water-immiscible volatile solvent, and the solution is spread on the water surface contained within a Langmuir trough. Upon evaporation of the solvent, a monolayer remains at the air–water interface, which is subsequently compressed with a moving barrier, while the surface pressure, π, is measured and displayed as a function of the corresponding area, A, revealing details of the phase behavior of the monolayer as well as information about the size of the molecules. Langmuir monolayers can be transferred onto solid substrates by dipping an appropriate substrate through the interface at constant pressure. Repeated monolayer transfer generally yields highly ordered lamellar multilayers.
One of the first reports of polymer–metal complexes at the air–water interface and the corresponding LB multilayers was by Nagel and Oertel [109]. In this approach, a metal ion complex from the subphase binded to recognition sites in the Langmuir-monolayer at the air–water interface. In another approach, the preassembled water-insoluble metallo-supramolecular assembly is directly spread on the water surface. For instance, it is possible to spread the above-mentioned PAC, based on Fe(II), 1,4-bis(2,2′:6′, 2″-terpyridin-4-yl)benzene and DHP, directly at the air–water interface because owing to the amphiphilic molecules, the assembly is insoluble in water (figure 21) [98, 110]. As a result of the surface tension across the interface, the PAC self-reorganizes at the air–water interface. The DHP molecules assemble at the top of the interface, while the hydrophilic MEPE is located in the subphase. Both strata are coupled to each other through electrostatic interactions. The interaction between DHP and MEPE produces an untilted hexagonal liquid-condensed phase of aliphatic alkyl chains even at low surface pressure. The electrostatic coupling is thought to be responsible for the stability of the Langmuir monolayer and its high collapse pressure. In contrast to the Langmuir monolayer of neat DHP, the PAC isotherm shows no distinct phase transition between the tilted and untilted liquid-condensed phase, which is ascribed to the polycrystalline nature of the PAC Langmuir monolayer. The architecture of the PAC Langmuir monolayer is rationalized as stratified bilayer, as schematically depicted in figure 21. The structural flexibility required to form this stratified architecture is provided by the noncovalent interactions within the PAC assembly.
Figure 21.
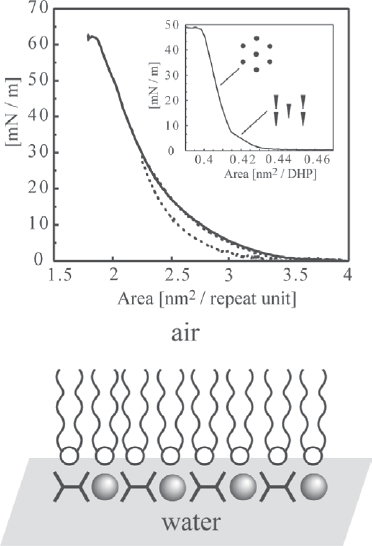
Top: compression isotherm (solid line) and expansion hysteresis (dotted line) of the PAC-1 monolayer at the air/water interface. The monolayer shows no distinct phase transitions. The area per molecule at the collapse pressure is ∼2.2 nm2 per molecule corresponding to six DHPs. The collapse pressure of 60 mN/m−1 is remarkably high. The monolayer of neat DHP (inset) shows a distinct phase transition. The structure of the alkyl chains in both phases is schematically depicted. Bottom: schematic bilayer architecture of the PAC-1 monolayer at the air–water interface. The amphiphilic component forms a (partially) charged template layer, onto which MEPE is adsorbed from underneath.
PAC monolayers are readily transferred onto solid substrates using the LB technique [94]. The average thickness per layer is 2.8±0.2 nm, corresponding to that of a monolayer. Repeated transfer leads to the realization of a LB multilayer. X-ray reflectometry (XRR) curves show both Kiessig fringes and Bragg peaks indicating the high order of the LB layers. Notably, the XRR curves of a film made of l layers exhibits only l/2 Kiessig fringes before the first-order Bragg peak, indicating that the LB films are Y-type and consist of amphiphile/MEPE sandwich layers. The LB layers are also oriented within the plane of the layers. The transfer of monolayers on the solid support causes the macroscopic alignment of the MEPE rods along the transfer direction. This architecture is in marked contrast to that of the volume phase discussed above. The DHP molecules in LB films are not interdigitated due to the pre-organization at the air–water interface. This comparison demonstrates how the modular nature allows the control of the architecture from molecular to macroscopic scales through the boundary conditions during self-assembly.
Nanostructures
Macromolecules often get kinetically trapped at interfaces during adsorption due to their great size, which effectively prohibits reversible self-assembly processes and the (long-range) ordering of macromolecules. Even shape-persistent macromolecules such as DNA, or proteins generally exhibit only short-range ordering in surface-confined architectures [111]. On the other hand, numerous molecules self-assemble on suitable interfaces, such as long chain alkanes on graphite, giving rise to highly ordered structures [112]. We, therefore, wondered if it was possible to utilize molecular self-assembly to organize PACs at interfaces in predictable ways, so that the amphiphilic component induces self-assembly on a suitable template layer.
Again, the PAC based on ditopic bis-terpyridines, Fe(II) and DHP is considered here. Spin-coating a dilute solution of a mixture of the PAC and a long-chain alkane at a mass ratio of approximately around 1:10 on graphite leads to the formation of perfectly straight nanostructures, with lengths of up to 200 nm under these experimental conditions, as indicated by scanning force microscopy (SFM) (see figure 22) [96]. Notably, no crossing molecules or kinks are observed. The distance between the PAC rods is always 5.5±0.3 nm, irrespective of the length of the alkane utilized, which indicates that the alkane layer orients and straightens the PAC, but does not determine the distances between adjacent PACs. This multicomponent self-assembly process consists of several steps. First, the alkanes are adsorbed epitaxially on the lattice of the basal plane of graphite, a long-known phenomenon, giving rise to a lamellar monolayer. The alkane wets the graphite preferentially because it is used in excess and because of the commensurability of the alkane and the graphite lattice. This monolayer represents a corrugated surface potential and, therefore, acts as a template for the adsorption of the PACs. The entire process evolves through multistep self-assembly with interactions at several levels and length scales involving metal ion coordination, electrostatics, and van der Waals forces. The molecular structure encoded in the design of the ligand and the coordination geometry is, therefore, amplified from the nanoscopic to the macroscopic scales. The modularity of this approach provides a means of encoding the value-adding physicochemical properties of metallo-supramolecules devices into nanoscopic architectures that can be addressed and manipulated individually by ‘scanning probe techniques’.
Figure 22.

Adsorption of PACs in predictable ways on the basal plane of graphite. Top: representative SFM image of PAC adsorbed in the presence of C32H66 on the basal plane of graphite. Middle: SFM image of a sample prepared from about twice the total concentration. The bottom layer consists of regularly ordered lamellae of the self-assembled alkane monolayer (inset at the bottom left shows a filtered image of part of the surface covered with the pure alkane monolayer). Perfectly straight PAC rods are observed on top of the alkane template layer. Bottom: proposed model for the resulting nanostructures (simplified representation, not to scale). The alkanes (thick rods) are adsorbed epitaxially on the lattice of the basal plane of the underlying graphite surface. This alkane monolayer serves as a template for orienting the rigid rods of the PAC.
POM clusters
Ultra thin films
The self-assembly of alternating layers of POM anions and oppositely charged species by LbL self-assembly (figure 16) is deceptively simple [113]. Starting with a negatively charged substrate, such as a layer of polystyrene-sulfonate (PSS) or a charged monolayer of an alkyl-silane [114] or alkyl-thiol [115], its immersion in a polycation solution leads to irreversible adsorption of the polycations, thereby recharging the surface. A few minutes are typically sufficient to form a complete layer [116]. After rinsing, immersion in a POM solution results in the adsorption of the next layer, and repeated deposition leads to the build-up of a multilayer. The only requirement is that the components are sufficiently charged to be irreversibly adsorbed at the interface. Several POMs of different shapes, sizes and charge have been successfully incorporated in multilayers (figure 23) [117–120].
Figure 23.
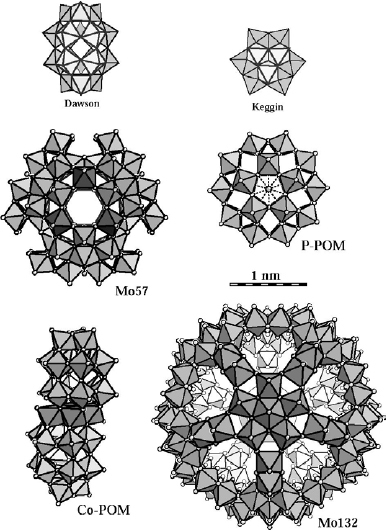
Structures of some of the POMs used for the fabrication of LbL multilayers. Keggin: [XM12O40]3−/4− (X=P, Si, M=W, Mo). Dawson: [P2M18O62]6− (M=W, Mo). Preyssler (P-POM): [M(H2O)P5W30O112]14/12− (M=Na, Eu). The P-POM has the shape of a flattened ellipsoid with dimensions 1.3×1.7 nm. Co-POM: [CoII4(H2O)2P4W30O112]16−. The ‘sandwich’ type Co(II)-substituted Co-POM is approximately 2.3 nm in length and 1.4 nm in width. Mo57: [H3Mo57V6(NO)6O183(H2O)18]21−. The external form of Mo57 is similar to that P-POM with dimensions of 1.6×2.4 nm. Mo132: giant Keplerate Mo132O372(CH3COO)30H2O)72]42−. The nearly spherical Mo132 with a diameter of approximately 3 nm shows promise for host–guest chemistry and size-selective catalysis because it possesses an open Mo–O framework architecture that encloses a central cavity with an approximate inner diameter of 1.7 nm. The Mo132 framework spans (Mo–O) 9-ring openings with an average ring aperture of 0.43 nm, which is comparable in size to pores in zeolitic architectures.
For all POM-based multilayers, as with many other systems prepared by this technique, the total thickness of multilayer assemblies increases linearly with the number of adsorbed layers, indicating a stepwise and regular growth process. The final coverage does not depend on the dipping time; however, it depends on the experimental parameters, such as ionic strength. Generally, up to two molecular layers of POMs are deposited per deposition step. The occurrence of regular growth is consistent with a build-up of excess interfacial charge by the POM anions, which the next layer that positively charged PE can bind to.
The deposition process is governed by a delicate balance of adsorption and desorption equilibria. While the immersion step should lead to the irreversible adsorption of a controlled amount of material onto the interface, the rinsing process is used for the desorption of excess, loosely bound species. The optimization of LbL in terms of controlling the amount deposited as a result of two competing processes requires the judicious selection of the pH and ionic strength of the dipping solutions as well as the careful control of the kinetics of the process. For instance, the layering of smaller POM anions is critically affected by the pH of the solutions. The presence of protons in the dipping solutions is known to significantly enhance the quantity of POM anions irreversibly adsorbed.
For smaller POMs, the surface coverage can be increased from sub-monolayer to multilayer coverage simply by adjusting the ionic strength of the dipping solutions. This is demonstrated in figure 24 for the deposition of [Eu(III)(H2O)P5W30O110]12− and PAH [121]. If no salt is added to the POM solution, residual electrostatic and dipolar repulsion within the interfacial layer keep the surface-confined POM anions separated, thereby resulting in sub-monolayer coverage, compared to the packing density of the POMs in the crystalline solid. In the presence of salt in the POM dipping solutions, the surface coverage increases to approximately one monolayer. The high ionic strength screens repulsive interactions; thus, the anions can become more closely packed. It is interesting to note that for very high ionic strength of the dipping solution, regular film growth may not occur, which is in marked contrast to the adsorption of polyelectrolytes [122]. If, in addition, salt is added to the PAH dipping solution, the surface coverage increases to approximately two monolayers, which is attributed to the penetration of POM anions into the top polyelectrolyte layer.
Figure 24.
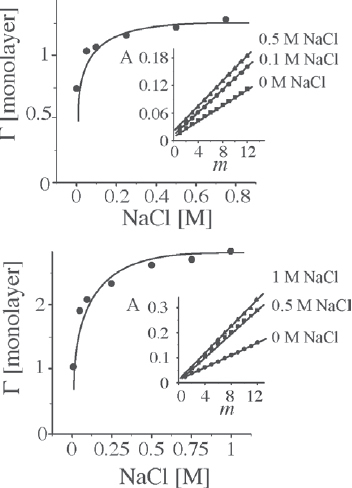
Surface coverage per layer, Γ, of Eu-POM in (Eu-POM/PAH)m multilayers, as functions of NaCl concentration of the Eu-POM (Top) and PAH solution (Bottom). Top: PAH is deposited from aqueous solution containing no NaCl. Bottom: Eu-POM is deposited from aqueous solution containing 0.1 M NaCl (adapted from [121]).
This method, therefore, provides control of the film growth and thickness at the nanometer scale. It does not require specialized equipment or substrates, and it can be used without adaption for automated fabrication. Furthermore, it is economical and readily amenable to scaling-up for the fabrication of large-area, defect-free devices on any type and shape of surface. The deposited films are mechanically robust and generally permeable for small molecules.
UV-vis spectroscopy is probably the easiest method of monitoring multilayer buildup because of the strong characteristic adsorption band of POM anions in the range of 200–400 nm [117]. Equivalent to measuring the optical absorbance, one can also determine the film thickness by ellipsometry or XRR. Furthermore, XRR provides information for the interior multilayer architecture and structural differences depending on the POM anion. Well-resolved Kiessig fringes occur in films of Mo132/PAH and Mo57/PAH. These multilayers are, therefore, homogeneous at the macroscopic scale. The total interfacial roughness is typically below 1 nm. No Bragg peaks are observed in these films, indicating the lack of internal structure (stratification), which is attributed to the sub-monolayer coverage. However, in general, these multilayers are not strictly stratified because adjacent polyelectrolyte layers partially interpenetrate each other and are also less ordered than self-assembled monolayers or LB films [123]. However, for the smaller POMs, XRR does not resolve Kiessig fringes because the interfacial roughness is too large to allow coherent interference of the x-rays from the top and bottom of the LbL layer. The smaller POMs are probably more dispersed across the interface and penetrate the polyelectrolyte matrix more easily, thus, reducing the degree of striation. However, Kiessig fringes become apparent if an additional PAH/PSS layer pair is placed on top of the multilayer. These complementary methods are simple to use and provide elementary information on deposition and structure. Many other analytical tools are available to study adsorption kinetics, layer growth and structure. Most POM anions can undergo a rapid reversible reduction. Therefore, cyclic voltammetry (CV) is another frequently used tool in POM multilayer research.
In terms of possible components for electrochromic devices, POMs are promising candidates due to their ability to undergo a rapid reversible reduction accompanied by coloration. Some of the first electrochromic coatings were obtained from poly(diallyldimethylammonium) chloride and [W10O32]4− [124] or [P2Mo18O62]6− [119]. As a step towards realistic technological implementation, we selected the europium derivative of the Preyssler anion, (NH4) 11.5K0.5[Eu(H2O)P5W30O110]·34H2O, (Eu-POM), as an active component because it exhibits reversible electrochemical behavior accompanied by a large electrochromic response [125]. As can be seen in figure 25, the oxidized form of Eu-POM is completely transparent in the visible region, whereas the reduced Eu-POM exhibits a broad absorption band at approximately 700 nm. Each redox step is accompanied by increasing coloration of the solution.
Figure 25.
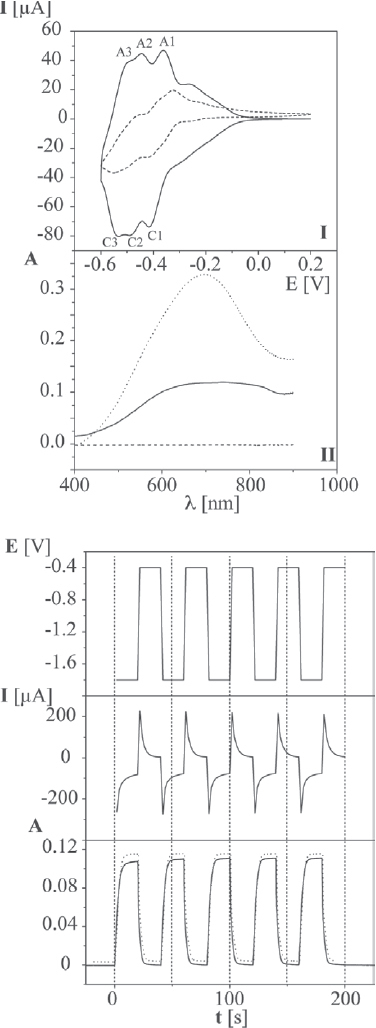
Top: (I) CV characteristics of Eu-POM in solution (dashed line, ITO electrode, 0.25 mM, ordinate magnified 5×) and a (PSS/PAH/Eu-POM/PAH)20 multilayer (solid line, ITO electrode). (II) UV-vis spectra of the oxidized (dashed line) and reduced (dotted line) Eu-POM in solution and the reduced (PSS/PAH/Eu-POM/PAH)20 multilayer (solid line). Bottom: potential, E, current, I, and absorbance, A, at 700 nm response of the (PSS/PAH/Eu-POM/PAH)20-coated ITO electrode to subsequent double potential steps between −0.4 and −1.8 V. Bottom: 1st cycle (solid line) and 500th (dashed line) cycle; traces are offset for clarity. The response times for coloration and bleaching are 4.2 and 4.4 s, respectively.
Device fabrication rests on the LbL deposition of Eu-POM, PAH and PSS on indium tin oxide (ITO)-coated glass slides. A visually noticeable optical contrast (transparent to dark blue) during potential scanning shows that the film is electrochromic. A high optical contrast is readily achieved even for thin films. The UV/vis spectrum of the (PSS/PAH/Eu-POM/PAH)20-modified ITO electrode shown in figure 25 was recorded during CV. The absorbance at 700 nm A700 nm, for a (PSS/PAH/Eu-POM/PAH)20 modified ITO electrode increases from 0 to 0.12. For a (PSS/PAH/Eu-POM/PAH)100-modified ITO electrode, A700 nm becomes as large as 0.7. A single PSS/PAH/Eu-POM/PAH layer generates an optical density of 0.006 in the reduced state. A film less than 1 μm thick would create an optical density of approximately 1, which is sufficient for many practical device applications.
A typical time dependence of the absorbance of a (PSS/PAH/Eu-POM/PAH)20 multilayer at 700 nm during potential-step chronoamperometry is illustrated in figure 25 (right). Both current and absorbance respond within seconds during coloration and bleaching. The multilayer has good stability and reversibility as the CV characteristics, the response times for coloration and bleaching, and the absorbance do not change noticeably even after 500 cycles. The multilayers also show considerable optical memory, that is, the layer remains in the colored state after reduction even if the potential source is disconnected. Therefore, it is possible to display information using this device without power consumption. The low operation voltage and low power consumption are additional advantages of the device. Power is required only for switching, and leakage currents can be minimized by operating the device under open-circuit conditions. The method can be readily extended for use with patterned substrates, thus films can be prepared on predefined areas. Moreover, the devices can be prepared under ambient conditions by a robot. Recently, a dual-mode device was introduced that combines photochromic and electrochromic properties [126]. Several other systems have also been studied recently [127–130].
A large number of POMs undergo a series of reversible one- and two-electron reductions, which facilitates their use as redox catalysts [131]. By selecting suitable cationic motives, the POM-based multilayer can be explored as a two-component, bifunctional electrocatalyst that can catalyze both reduction and oxidation. Cox et al immobilized the ruthenium metallodendrimer (RuDen) on the electrode-adsorbed Dawson-type POM anion, [P2W18O62]6− [132, 133]. The obtained electrodes are highly organized, bifunctional catalytical systems, which are electrocatalytically active toward the reduction of iodate and the oxidation of arsenite (as shown in figure 26).
Figure 26.
Left: reduction of iodate at a bare glassy carbon electrode (A) and a (P2W18O626−/RuDen)10-coated electrode (B). The mediated reduction of the iodate by the multilayer is demonstrated by the amplification of the P2W18O626− reduction peak current. Right: oxidation of arsenite at a bare glassy carbon electrode (C) and a (P2W18O626−/RuDen)10-coated electrode (D). The mediated oxidation of arsenite by the multilayer is demonstrated by the amplification of the reduction peak current for the oxidation of RuII (adapted from [132]).
The reduction of POM increases the negative charge density of the metal-oxide framework and thus its basicity. As a consequence, the reduction is generally pH-dependent. This feature allows the use of POMs as electrochemical probes to survey microenvironmental effects. We selected Co-POM because of its good pH stability (pH 2–9). It is interesting to note that the details of the electrochemical response of CoPOM-containing multilayers depend on the layer architecture [134]. The CV characteristics of (PSS/PAH/Co-POM/PAH)10 and (Co-POM/PAH)10-modified electrodes are shown in figure 27. The (PSS/PAH/Co-POM/PAH)10-modified ITO electrode assembled from salt-free PAH solution (solid line) shows redox waves with peaks at −0.574 V (C1), 0.73 V (C2) and 0.52 V (A1). A different electrochemical response is observed if the film is assembled with a PAH solution containing NaCl (0.5 M). During the cathodic sweep, only one reduction peak appears at a potential of −0.744 V (C3), and during the anodic sweep two peaks appear at −0.563 V (A1) and −0.687 V (A2). The two two-electron reduction steps appear to merge into one four-electron process. However, a different response is observed if the layer sequence is altered. The CV characteristic of a (Co-POM/PAH)10 multilayer (dotted line) assembled with PAH containing NaCl (0.5 M) exhibits two peaks at potentials of −0.643 V (C1) and −0.731 V (C2) during the cathodic sweep, and two peaks at −0.554 V (A1) and −0.69 V (A2) V during the anodic sweep. These examples demonstrate that the redox properties of the immobilized Co-POM can reveal subtle variations in the film structure and composition. This property suggests that POMs can be used as electroactive probes to study microenvironmental effects in LbL multilayers.
Figure 27.
Left: CV characteristics of (PSS/PAH/Co-POM/PAH)10 (solid) assembled with salt-free solutions, (PSS/PAH/Co-POM/PAH)10 (dashed) and (PSS/PAH/Co-POM/PAH)10 (dotted) assembled with PAH solution containing NaCl (0.5 M). (ITO electrode, supporting electrolyte: 0.2 M PBS pH 5.0, scan rate: 10 mV s−). Right: CV characteristics of PSS/PAH(Co-POM/PAH)10 assembled with PAH solution containing NaCl (0.5 M). Solid line: pH 5.0, dotted line: pH 6.0, dashed line: pH 7.0. The inset shows the dependence of the redox potential on pH (ITO electrode, supporting electrolyte: 0.2 M PBS, scan rate: 10 mV s−1) (adapted from [134]).
The voltammetric response of the LbL-based Co-POM multilayer is also pH-dependent. Representative CV characteristics as a function of pH are shown in figure 27. The relationship between the redox potential and pH is shown in the inset. With decreasing pH, the redox potential shifts to a positive potential. This feature makes Co-POM-based multilayers potential candidates for pH microsensors, e.g. in physiological media. This approach provides a simple and inexpensive means of fabrication, and the device operates reversibly, the electrodes have long-term stability and, in addition, the electrochemical response characteristics, such as sensitivity and response time, can be adjusted through the structure and composition of the sensing layer. Other applications of Co-POM-based multilayers include various sensors, for instance for detecting of nitric oxide [135, 136].
Surfactant-encapsulated POM cluster
A strategy used to modify the surface chemical properties of POM clusters relies on a ligand-exchange process in the second coordination sphere; the counter cations from the hydration sphere of the POM anions are replaced by suitable amphiphiles, resulting in discrete supramolecular modules. Due to the close packing of the long-chain amphiphiles on the surface of the POM, we coined the term ‘surfactant-encapsulated clusters’ (SECs) to emphasize the particular architecture of these core-shell assemblies (figure 28) [137]. This colloid chemical approach has been successfully used to stabilize a variety of semiconductor [138] and precious metal nanoparticles [139], but has not previously been used in POM chemistry to purpose discrete supramolecular assemblies [18, 140].
Figure 28.
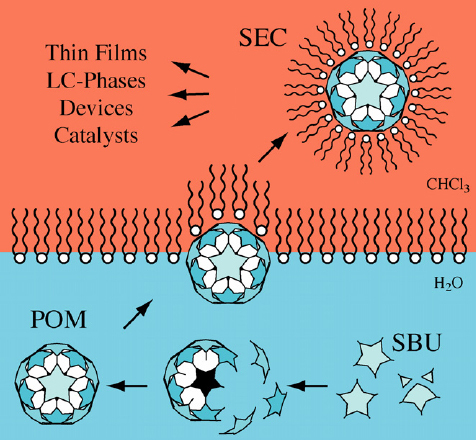
Sequential self-assembly of POMs from secondary building units (SBU) and concomitant amphiphilic self-assembly followed by phase transfer of SECs into the organic phase.
Most SECs are prepared by a straightforward two-step procedure. First, a water-soluble salt of the POM is prepared and its structure is characterized by standard analytical methods (e.g. single-crystal x-ray structure analysis). In the second step, an aqueous solution of the POM salt is treated with a water-immiscible organic solvent containing an appropriate amount of a cationic surfactant. This preparation scheme permits the characterization of the materials used in each step. This approach has also been successfully used to encapsulate POMs with dendritically branching amphiphiles [141–144]. These results show that surfactant encapsulation is widely applicable to different POM clusters and amphiphilic systems. These SECs form intriguing supramolecular architectures that are comparable to the structures of naturally occurring capsule-forming proteins such as the iron storage protein ferritin.
In contrast to this approach, Moriguchi et al prepared a decatungstate-containing bilayer composite, DODA/[W10O32]4− (DODA=dioctadecyldimethylammonium) [145], by a one-phase reaction. An aqueous solution of [W10O32]4− is added to a quaternary aqueous alkylammonium compound under vigorous stirring, which immediately results in the formation of precipitates. After filtration, washing and drying in vacuum, the resulting DODA/[W10O32]4− complex does not dissolve in water, ethanol or 2-propanol, but only in chloroform. Chambers et al. reported another more sophisticated strategy for synthesizing the first bis(alkyl)-substituted amphiphilic, asymmetrical POM species {[CH3(CH2)3]4N}4{[CH3(CH2)11Si]2OSiW11O39} by adding two equivalents of dodecyltrichlorosilane in CH3CN to an aqueous solution of the Keggin lacunary POM precursor at 50 °C [146]. This synthesis also yields an LB monolayer at the air-water interface that is stable and forms reversibly, as evident by the lack of hysteresis in compression–expansion cycles.
The analytical characterization of such large assemblies constitutes an enormous challenge, particularly because single-crystal x-ray diffraction cannot be used due to the lack of long-range order of liquid–crystal-like SEC materials. A host of complementary techniques has been used to probe the structural integrity of the POM in the SEC. The presence of the POM anion in the SEC has been proven by Raman, IR and UV/vis spectroscopy due to the characteristic vibrational and electronic transitions of the cluster anions. The surfactant shell can be investigated by 1H-NMR spectroscopy, which reveals that the positively charged head groups of the surfactants point towards the negatively charged cluster surface. Both, neutron and x-ray scattering can be used to examine the outer sphere and particle core of the SEC and confirm that a single cluster anion resides within the SEC. The scattering data is in agreement with the dimensions of the POM anions, as determined by single-crystal x-ray structure analysis. These measurements answer the important question of whether or not the structural integrity of the POM cluster is preserved in aqueous solution and in the SECs. In addition, molecular dynamics (MD) simulations are also a promising tool for refining the dynamic structure models of SECs down to atomic resolution.
Experiments with different surfactant/POM combinations indicate that the steric requirements for the packing of the surfactant alkyl chains and the molecular balance of the hydrophobic/hydrophilic properties play an important role in stabilizing the structure of the SECs. For example, aqueous solutions of the partially reduced POM [H3MO57V6(NO)6O83(H2O)18)]21−, [Mo57], were identically treated with stoichiometric quantities of the commercially available surfactants DODA•Br, octadecyl trimethylammonium bromide ((CH3)3(C18H37)3)N•Br), or trioctadecylmethylammonium bromide (CH3(C18H37)3)N•Br); the transport of the Mo57 cluster into the organic phase was only achieved using the DODA surfactant, while the phase transfer of the cluster failed or was incomplete in the case of the other surfactants. A compound corresponding to the empirical formula (DODA)20(NH4) [Mo57] was isolated from the organic solution, and its physicochemical properties were studied in great detail. Results from analytical ultracentrifugation, small angle x-ray scattering and Langmuir compression isotherms are consistent with the presence of a single Mo57 core encapsulated within a shell of 20 DODA molecules. A single ammonium cation was introduced into the molecular formula to compensate the 21 negative charges of the cluster anion. Although the applied methods do not allow the unambiguous detection of a single ammonium cation in such a large assembly, the following is a reasonable postulate. The center of the cluster anion contains a cavity suitable for binding an ammonium cation. In fact, the cavity with its preorganized oxygen electron pairs resembles the binding site of ammonium-binding crown ethers. In contrast to the water-soluble starting material (NH4)21[Mo57], the SEC (DODA)20(NH4) [Mo57] dissolves readily in organic solvents such as benzene, toluene, and chloroform. The solubility properties suggest that the alkyl chains form a compact hydrophobic shell, which shields the enclosed cluster anion. To demonstrate the high stability of this compound, an aqueous dispersion of the SEC was refluxed and sonicated for several minutes, after which there were no signs that decomposition had occurred.
Another SEC with an intricate supramolecular architecture, which appears promising in terms of host–guest chemistry and homogeneous size-selective catalysis, is (DODA)40(NH4)2[(H2O)n⊂Mo132O372(CH3COO)30(H2O)72] (DODA)40(NH4)2 [Mo132]) (figure 29) [142, 144]. On the basis of the results of the elemental analysis, 40 DODA molecules encapsulate the cluster, leading to a discrete, nearly spherical particle with a molecular mass of approximately 43 9000 g mol−1. The solvent-accessible surface (SAS) of the Mo132 cluster calculated for a probe radius of 0.28 nm is approximately 33.4 nm2, which yields an average surfactant area of 0.84 nm2. Within the given range of uncertainty, this corresponds reasonably well to the empirical value of 32(±8) nm2 for the total surface area of 40 DODA molecules.
Figure 29.
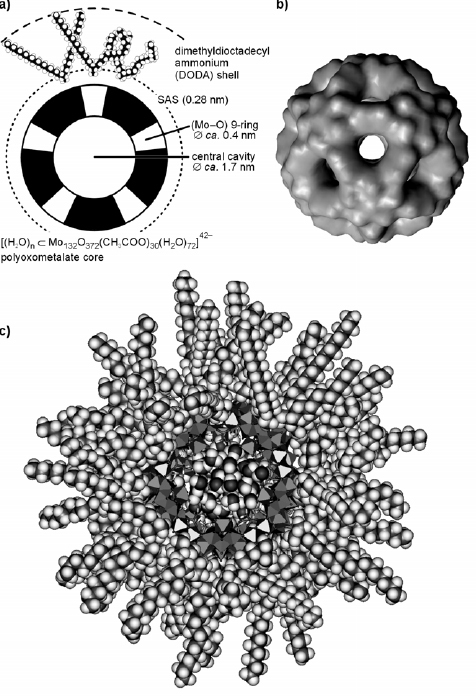
(a) Supramolecular architecture of (DODA)40 (NH4)2 [Mo132]. (b) Solid rendered representation of the Connolly surface of the Keplerate cluster Mo132. (c) The cross-section through a SEC model shows the surfactant shell and the clathrated H2O molecules in space-filling representations, while the Mo–O framework is displayed as a polyhedral model (adapted from [144]).
The geometric matching of the two juxtaposed ionic surfaces constituting the POM anion and the surfactants may be of critical importance for driving the self-encapsulation process to completion. The surface charge density of Mo132 is such that all DODA molecules have sufficient space to form a single layer at a van der Waals distance from the cluster surface. The covered surface area of a 0.84 nm2 per DODA molecule furthermore suggests the tight packing of the amphiphile at the cluster surface. The SAS of Mo132 for a 0.28 nm probe displays a continuous spherical surface, which indicates that the DODA cations cannot penetrate the large central cavity of the Keplerate. In contrast, the SAS of Mo132 for a 0.14 nm probe, which is often referred to as the water-accessible (Connolly) surface, extends into the central cavity through each of the 20 noncircular (Mo-O) 9-ring openings (figure 29). The whole assembly of the cluster and surfactant at this level of structural organization resembles a reversed micelle in which the hydrophilic cavity is completely filled by the large cluster anion. An MD simulation of the SEC gives an indication of the packing of the surfactant molecules around the nanosized cluster. The simulation in figure 29 indicates that the cluster is completely encapsulated and shielded by the long hydrophobic octadecyl chains, which explains the high solubility of the SECs in nonpolar organic solvents.
Other surfactant-encapsulated POM complexes include (DODA)16As4W30Cu4O112·114H2O [147, 148], C16TA/[EuW10O36]9− (C16TA = hexadecyltrimethylammonium cation) [149], obtained by employing a similar method and the complexes of the ‘giant Ferris wheel’ [(MoO3)176(H2O)80H32]32− with a variety of surfactants [150]. The (DODA)16As4W30Cu4O112·114 H2O SEC can form a stable monolayer at the air/water interface. However, the ellipsoid is significantly distorted at the air–water interface, although it is believed that the general core-shell structure remains intact. The complexation of wheel-shaped clusters with surfactants delivers supramolecular organic–inorganic hybrid aggregates with an extended disk-like shape, consisting of a rigid and hydrophilic core and a flexible hydrophobic shell. Therefore, SECs undergo self-assembly into a columnar liquid crystalline similar to organic liquid crystals [150, 151].
The latest development in this field is the design of artificial enzymes, so-called dendrizymes, which mimic the structure and function of naturally occurring catalytically active metalloproteins, an example being the dendron-encapsulated cluster (C52H60NO12)12[Mn(H2O)3(SbW9O33)2] [143]. Here, the catalytic activity of POMs is combined with the steric properties of tailored dendritic surfactants towards size-selective catalytic systems. The SECs thereby bear a resemblance to natural metalloproteins (dendrizymes). Elemental analysis indicates that the POM anion is encapsulated in a densely packed shell of 12 dendrons, thus yielding a discrete, hydrophobic supramolecular architecture with a molar mass of 15 778 g mol−1.
The reasons for spontaneously occurring SEC assembly are not well understood yet, since accurate values for the contributing enthalpy and entropy terms are difficult to determine. Currently, we assume that the process is mainly driven by an increase in Coulomb interactions: placing the cationic head groups in close vicinity to the POM surface efficiently screens the electrostatic charge of the encapsulated anion. Hydrophobic interactions between the alkyl chains of the close-packed surfactant shell may further stabilize the SEC. Finally, the gain of hydration enthalpy upon the release of counter anions (e.g. Cl− or Br−) into the aqueous phase, and the entropically favorable liberation of a large number of ammonium cations and water molecules from the cluster surface may equally contribute to the free energy of SEC formation. Recently, many other studies concerning surfactant-encapsulated POMs have been published [152–155].
LB films of SECs
Compared with ‘naked’ POM clusters, SECs have the following feature: the surfactant shell improves the stability of the encapsulated cluster against fragmentation; enhances the solubility of the encapsulated cluster in nonpolar, aprotic organic solvents; neutralizes the charge of the anionic POM, thereby leading to discrete, electrostatically neutral assemblies; and alters the surface chemical properties of the POM (e.g. self-aggregation, surface adhesion, wetting behavior) in a predictable manner. Of these features, the hydrophobic nature of SECs is particularly interesting because it enables the production of well-defined thin films by the LB technique [156, 157].
The SECs can be directly spread at the air–water interface to yield a homogeneous SEC monolayer. From Langmuir isotherms, the surface area of a single (DODA)20(NH4)[Mo57] species is determined to be 10.4 nm2, which corresponds to an object with a diameter of 3.6 nm (figure 30). The (DODA)40(NH4)2[Mo132] species occupies an area of 15 nm2), which corresponds to a diameter of 4.4 nm. Both values are in excellent agreement with the proposed structural model in which a single cluster anion resides within a close shell of surfactant molecules. Analytical characterization of SEC Langmuir films by Brewster angle microscopy, optical ellipsometry and grazing-angle x-ray diffraction confirm monolayer coverage.
Figure 30.
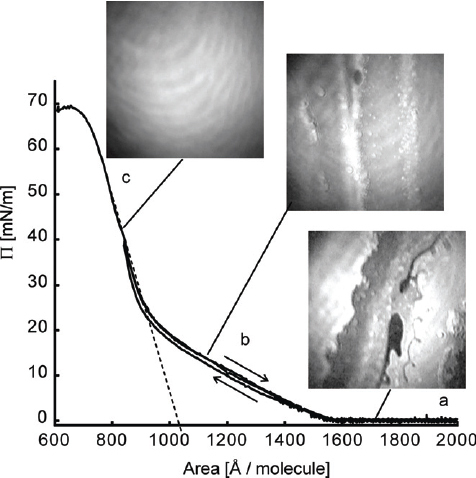
Representative surface-pressure (π–A) diagram of a (DODA)20(NH)4 [Mo57] monolayer at the air–water interface. The inserted photographs were obtained from Brewster angle microscopic images at different surface pressures. (a) π=0 mN, (b) π=20 mN and (c) y=40 mN.
The LB transfer of SEC monolayers is achieved by the following procedure. SECs are spread from a chloroform solution on the water surface, the resulting SEC monolayer is compressed and the film is transferred at constant surface pressure onto a solid substrate. The substrates on which SECs have been deposited include silicon, quartz and gold-sputtered glass slides. The substrate is immersed in the subphase before spreading, and multilayers are formed by repeated LB transfer. The transfer ratio is close to unity in all cases. Investigations by optical ellipsometry and UV/vis-spectroscopy demonstrate that LB transfer is highly reproducible and that film growth is essentially linear, that is, in each dipping cycle equivalent amounts of SECs are transferred onto the substrate. The XRR of SEC LB multilayers on silicon substrates shows well-resolved Kiessig fringes, indicating a uniform film thickness. In addition, several Bragg reflections are discernible, which implies that these LB-deposited films have an internal layered structure in contrast with the structure of the corresponding LbL multilayers.
The static contact angles, γ, of (DODA)20(NH4)[Mo57] LB multilayers are 102° for water and 31° for hexadecane. Similarly, the water contact angle of (DODA)40(NH4)2[Mo132] LB multilayers amounts to 97°. These values clearly indicate the hydrophobic nature of the SEC-coated substrate surface and demonstrate how efficiently the DODA surfactant shell screens the underlying hydrophilic cluster. The absolute values of the contact angles suggest that the SEC alkyl chains are somewhat disordered. For comparison, the methyl-terminated surface of a self-assembled monolayer typically has a contact angle in the range of 110–115° [158].
It is not yet clear how SECs are stabilized at the air–water interface because hydrophobic compounds typically aggregate and float as lenses on the water surface. Since SECs have finite water contact angles, partial wetting may cause the partial immersion of the spherical SEC into the aqueous subphase [159]. This may trap the SEC at the air–water interface and prevent aggregation into droplets.
In addition, we found that SECs spontaneously assemble into highly ordered 2D arrays. TEM studies on SEC thin films deposited on solid substrates either by LB transfer or by the simple evaporation of dilute SEC solutions, reveal that SECs have a strong tendency to self-assemble into extended, well-ordered 2D arrays. figure 31, as an example, shows a TEM micrograph of a thin film of DODA40(NH4)2[Mo132]. The inorganic cores of the SECs appear as dark spots embedded in a bright matrix of surfactant molecules. Monolayer regions, darker regions representing a bilayer and the brighter uncovered substrate are clearly distinguishable. Small domains exhibit hexagonal arrays of SECs corresponding to the 2D close packing of spherical particles (arrows). Both, the diameter of the dark objects (approximately 3 nm) as well as the average distance between them (approximately 4.5 nm) are consistent with the proposed SEC model. In thicker films, the order improves and becomes 3D. An ordered region is shown in figure 31(b); the related electron diffraction pattern (insert) clearly reveals long-range order (spots rather than rings) and a threefold symmetry of the pattern of reflections corresponding to a spacing of 4.2 nm. The packing of the SECs may be tentatively described as that of an fcc lattice with a cubic unit cell axis of approximately 6 nm. The observed diffraction pattern thus corresponds to the close packing of SECs in a (111) plane normal to the electron beam. It is noteworthy that upon heating thick SEC films, dewetting and terracing are observed, as in the case of block copolymers.
Figure 31.

(a) TEM micrograph of a thin film of DODA40(NH4) [Mo132] (scale bar 50 nm). Extended monolayers, interspersed with holes and double layer regions are visible. Arrows mark some regions with apparent hexagonal order. (b) TEM micrograph of an ordered region of a thicker film at high magnification (scale bar 20 nm). The inset shows a low-angle electron diffraction pattern recorded from a larger area (5 Mm2) containing the region shown.
In a few cases it has been shown that SECs exhibit liquid crystalline behavior. In these cases, the liquid crystallinity is provided by the surfactant component, whereas the inorganic component effectively follows the direction of the surfactant phase [134, 151, 155].
The highly ordered and assembled surfactant shells of SECs control the amount and the spatial arrangement of POM ions. Therefore, the hydrophilic interlayers of SECs are expected to act as a spatially and chemically constrained reaction field for the size- and shape-controlled synthesis of inorganic nanoparticles and in catalytic reactions. Moriguchi et al used a DODA/[W10O32]4− composite as the photocatalyst for the conversion of 2-propanol to acetone [145]. The DODA/[W10O32]4− composite powder dispersed in 2-propanol photocatalyzes the oxidative dehydrogenation of 2-propanol into acetone in the presence of oxygen and under UV irradiation. The DODA/[W10O32]4− exhibited a unique temperature dependence; the photocatalytic activity is markedly enhanced above the phase-transition temperature of the amphiphilic bilayer. When the DODA/[W10O32]4− composite was combined with Pt nanoparticles, the photocatalytic dehydrogenation of 2-propanol into acetone and H2 proceeds in the absence of oxygen because Pt promotes the reoxidation of protonated [H2W10O32]4− to the original [W10O32]4− (figure 32).
Figure 32.
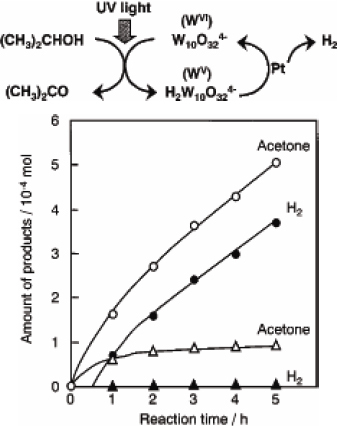
Top: catalytic cycle of the dehydrogenation of 2-propanol into acetone using DODA/[W10O32]4−/Pt[x]. Bottom: time courses of photocatalytic reactions of DODA/[W10O32]4−/Pt[0.3] (circles) and DODA/[W10O32]4− (triangles) for the dehydrogenation of 2-propanol in a nitrogen gas atmosphere at 35 °C. The catalysts (1×10−4 mol) are dispersed in 200 ml of 2-propanol.
Moreover, SECs are promising candidates for diverse applications including electrocatalysis, display devices, and photonic materials. The results observed so far are still limited and further studies are necessary to elucidate the full potential of these materials.
Conclusions and outlook
Metal ion coordination in discrete or extended assemblies enables the reparation of functional components that are of high potential for the development of functional materials and nanotechnological devices. A central challenge is to characterize these mostly complex soft materials lacking crystalline order, to establish accurate structure–property relationships and effectively control their structure and functionality. Although an increasing number of materials has been reported in the last few years, methods for incorporating these nanoscaled self-assembled materials or objects into devices on a production (economically feasible) scale have lagged significantly behind. It has been recognized that these self-assembled systems will have to subsequently or concomitantly self-organize into hierarchical structures that may also form interconnects to the macroscopic world. Therefore, for many of these devices based on organic or hybrid inorganic–organic compounds, in analogy to biological systems, the structural organization of supramolecular assemblies must be controlled. Moreover, the potential usefulness and functionality of these materials and systems have largely been demonstrated by proof-of-principle experiments, such as those using scanning-probe tips to construct and evaluate the devices. The future developments in the preparation and functionalization of suitable assemblies, the advancements in controlling the structural organization, and the progress in developing new tools to construct devices are expected to further promote the developments outlined in this review.
Footnotes
Invited paper.
References
- Jamora C and Fuchs E. 2002. Nat. Cell Biol. E 4 101 10.1038/ncb0402-e101 [DOI] [PubMed] [Google Scholar]
- Sarikaya M, Tamerler C, Jen A K Y, Schulten K. and Baneyx F. Nat. Mater. 2003;2:577. doi: 10.1038/nmat964. [DOI] [PubMed] [Google Scholar]
- Alivisatos A P. et al Adv. Mater. 1998;10:1297. doi: 10.1002/(SICI)1521-4095(199811)10:16<1297::AID-ADMA1297>3.0.CO;2-7. [DOI] [Google Scholar]
- Hamley I W. Angew. Chem. Int. Edn. Engl. 2003;42:1692. doi: 10.1002/anie.200200546. [DOI] [PubMed] [Google Scholar]
- Lehn J M. et al Science. 2002;295:2400. doi: 10.1126/science.1071063. [DOI] [PubMed] [Google Scholar]
- Swalen J D. et al Langmuir. 1987;3:932. doi: 10.1021/la00078a011. [DOI] [Google Scholar]
- Shimomura M. and Sawadaishi T. Curr. Opin. Colloid Interface Sci. 2001;6:11. doi: 10.1016/S1359-0294(00)00081-9. [DOI] [Google Scholar]
- Stoddart J F. Acc. Chem. Res. 2001;34:410. doi: 10.1021/ar010084w. [DOI] [PubMed] [Google Scholar]
- Lehn J M. Proc. Natl. Acad. Sci. USA. 2002;99:4763. doi: 10.1073/pnas.072065599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thunemann A F. Prog. Polym. Sci. 2002;27:1473. doi: 10.1016/S0079-6700(02)00017-5. [DOI] [Google Scholar]
- Thunemann A F, Muller M, Dautzenberg H, Joanny J F O. and Lowne H. vol 166. Berlin: Springer; 2004. Polyelectrolytes with Defined Molecular Architecture II; p. p 113. [Google Scholar]
- Ober C K. and Wegner G. Adv. Mater. 1997;9:17. doi: 10.1002/adma.19970090104. [DOI] [Google Scholar]
- Holliday B J. and Mirkin C A. Angew. Chem. Int. Edn. Engl. 2001;40:2022. doi: 10.1002/1521-3773(20010601)40:11<2022::AID-ANIE2022>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Asselberghs I, Therien M J, Coe B J, McCleverty J A. and Clays K. Vol 928. Washington, DC: American Chemical Society; 2006. Metal-Containing and Metallosupra-molecular Polymers and Materials; p. p 527. [Google Scholar]
- Scandola F, Chiorboli C, Prodi A, Iengo E. and Alessio E. Coord. Chem. Rev. 2006;250:1471. doi: 10.1016/j.ccr.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Gaspar A B, Ksenofontov V, Seredyuk M. and Gutlich P. Coord. Chem. Rev. 2005;249:2661. doi: 10.1016/j.ccr.2005.04.028. [DOI] [Google Scholar]
- Alam M S, Stromsdorfer S, Dremov V, Muller P, Kortus J, Ruben M. and Lehn J M. Angew. Chem. Int. Edn. Engl. 2005;44:7896. doi: 10.1002/anie.200502743. [DOI] [PubMed] [Google Scholar]
- Long D L, Burkholder E. and Cronin L. Chem. Soc. Rev. 2007;36:105. doi: 10.1039/b502666k. [DOI] [PubMed] [Google Scholar]
- Muller A, Kogerler P. and Dress A W M. Coord. Chem. Rev. 2001;222:193. doi: 10.1016/S0010-8545(01)00391-5. [DOI] [Google Scholar]
- Sadakane M. and Steckhan E. Chem. Rev. 1998;98:219. doi: 10.1021/cr960403a. [DOI] [PubMed] [Google Scholar]
- Davidson P. and Gabriel J C P. Curr. Opin. Colloid Interface Sci. 2005;9:377. doi: 10.1016/j.cocis.2004.12.001. [DOI] [Google Scholar]
- Sato O. Acc. Chem. Res. 2003;36:692. doi: 10.1021/ar020242z. [DOI] [PubMed] [Google Scholar]
- Holder E, Langeveld B M W. and Schubert U S. Adv. Mater. 2005;17:1109. doi: 10.1002/adma.200400284. [DOI] [Google Scholar]
- Rehahn M. Acta Polym. 1998;49:201. doi: 10.1002/(SICI)1521-4044(199805)49:5<201::AID-APOL201>3.0.CO;2-0. [DOI] [Google Scholar]
- Kurth D G. and Higuchi M. Soft Matter. 2006;2:915. doi: 10.1039/b607485e. [DOI] [PubMed] [Google Scholar]
- Andres P R. and Schubert U S. Adv. Mater. 2004;16:1043. doi: 10.1002/adma.200306518. [DOI] [Google Scholar]
- Hosseini M W. Acc. Chem. Res. 2005;38:313. doi: 10.1021/ar0401799. [DOI] [PubMed] [Google Scholar]
- Brammer L. Chem. Soc. Rev. 2004;33:476. doi: 10.1039/b313412c. [DOI] [PubMed] [Google Scholar]
- O'Keeffe M. and Yaghi O M. J. Solid State Chem. 2005;178:V. doi: 10.1016/S0022-4596(05)00368-3. [DOI] [Google Scholar]
- Yaghi O M, O'Keeffe M, Ockwig N W, Chae H K, Eddaoudi M. and Kim J. Nature. 2003;423:705. doi: 10.1038/nature01650. [DOI] [PubMed] [Google Scholar]
- Eddaoudi M, Moler D B, Li H L, Chen B L, Reineke T M, O'Keeffe M. and Yaghi O M. Acc. Chem. Res. 2001;34:319. doi: 10.1021/ar000034b. [DOI] [PubMed] [Google Scholar]
- Fletcher A J, Thomas K M. and Rosseinsky M J. J. Solid State Chem. 2005;178:2491. doi: 10.1016/j.jssc.2005.05.019. [DOI] [Google Scholar]
- Rosi N L, Eckert J, Eddaoudi M, Vodak D T, Kim J, O'Keeffe M. and Yaghi O M. Science. 2003;300:1127. doi: 10.1126/science.1083440. [DOI] [PubMed] [Google Scholar]
- Eddaoudi M, Kim J, Rosi N, Vodak D, Wachter J, O'Keefe M. and Yaghi O M. Science. 2002;295:469. doi: 10.1126/science.1067208. [DOI] [PubMed] [Google Scholar]
- Hasenknopf B, Lehn J M, Baum G. and Fenske D. Proc. Natl. Acad. Sci. USA. 1996;93:1397. doi: 10.1073/pnas.93.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamacek J, Borkovec M. and Piguet C. Chem.—Eur. J. 2005;11:5217. doi: 10.1002/chem.200500290. [DOI] [PubMed] [Google Scholar]
- Hamacek J, Borkovec M. and Piguet C. Chem.—Eur. J. 2005;11:5227. doi: 10.1002/chem.200500289. [DOI] [PubMed] [Google Scholar]
- Vermonden T, van der Gucht J, de Waard P, Marcelis A T M, Besseling N A M, Sudholter E J R, Fleer G J. and Stuart M A C. Macromolecules. 2003;36:7035. doi: 10.1021/ma030353t. [DOI] [Google Scholar]
- Holyer R H, Hubbard C D, Kettle S F A. and Wilkins R G. Inorg. Chem. 1966;5:622. doi: 10.1021/ic50038a027. [DOI] [Google Scholar]
- Hogg R. and Wilkins R G. J. Chem. Soc. 1962;341 [Google Scholar]
- Ciferri A. 2003. J. Macromol. Sci.—Polym. Rev. C 43 271 [Google Scholar]
- Bullock J I. and Simpson P W G. J. Chem. Soc., Faraday Trans. I. 1981;77:1991. doi: 10.1039/f19817701991. [DOI] [Google Scholar]
- Velten U. and Rehahn M. Chem. Commun. 1996:2639. [Google Scholar]
- Loeb S J. and Shimizu G K H. J. Chem. Soc., Chem. Commun. 1993:1395. [Google Scholar]
- Kelch S. and Rehahn M. Macromolecules. 1997;30:6185. doi: 10.1021/ma9702814. [DOI] [Google Scholar]
- Constable E C. and Thompson A. J. Chem. Soc., Dalton Trans. 1992:3467. [Google Scholar]
- Han F S, Higuchi M. and Kurth D G. Organic. Lett. 2007;9:559. doi: 10.1021/ol062788h. [DOI] [PubMed] [Google Scholar]
- Barigelletti F, Flamigni L, Balzani V, Collin J P, Sauvage J P, Sour A, Constable E C. and Thompson A. J. Am. Chem. Soc. 1994;116:7692. [Google Scholar]
- Schutte M, Kurth D G, Linford M R, Colfen H. and Mohwald H. Angew. Chem. Int. Edn. Engl. 1998;37:2891. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2891::AID-ANIE2891>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Akcakayiran D, Kurth D G, Rohrs S, Rupprechter G. and Findenegg G H. Langmuir. 2005;21:7501. doi: 10.1021/la050230x. [DOI] [PubMed] [Google Scholar]
- Kolb U, Büscher K, Helm C A, Lindner A, Thünemann A F, Menzel M, Higuchi M. and Kurth D G. Proc. Natl. Acad. Sci. USA at press. 2006 doi: 10.1073/pnas.0601092103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H Y, Cronin J A. and Archer R D. Macromolecules. 1994;27:2174. [Google Scholar]
- Chen H Y. and Archer R D. Macromolecules. 1995;28:1609. [Google Scholar]
- Chen H Y, Cronin J A. and Archer R D. Inorg. Chem. 1995;34:2306. [Google Scholar]
- Alonso P J, Puertolas J A, Davidson P, Martinez B, Martinez J I, Oriol L. and Serrano J L. Macromolecules. 1993;26:4304. [Google Scholar]
- Marcos M, Oriol L, Serrano J L, Alonso P J. and Puertolas J A. Macromolecules. 1990;23:5187. [Google Scholar]
- Oriol L. and Serrano J L. Adv. Mater. 1995;7:348. [Google Scholar]
- Palacio F, Ramos J. and Castro C. Mol. Cryst. Liq. Cryst. 1993;232:173. [Google Scholar]
- Dobrawa R, Lysetska M, Ballester P, Grune M. and Würthner F. Macromolecules. 2005;38:1315. [Google Scholar]
- Dobrawa R. and Würthner F. Chem. Commun. 2002:1878. doi: 10.1039/b205478g. [DOI] [PubMed] [Google Scholar]
- Barron J A, Glazier S, Bernhard S, Takada K, Houston P L. and Abruna H D. Inorg. Chem. 2003;42:1448. doi: 10.1021/ic020691v. [DOI] [PubMed] [Google Scholar]
- Okamura T, Iwamura T, Seno S, Yamamoto H. and Ueyama N. J. Am. Chem. Soc. 2004;126:15972. doi: 10.1021/ja044310j. [DOI] [PubMed] [Google Scholar]
- Valdebenito N, Oriol L, Barbera J, Diaz F. and Serrano J L. Macromol. Chem. Phys. 2000;201:2573. doi: 10.1002/1521-3935(20001101)201:17<2573::AID-MACP2573>3.0.CO;2-Z. [DOI] [Google Scholar]
- Michelsen U. and Hunter C A. Angew. Chem. Int. Edn. Engl. 2000;39:764. doi: 10.1002/(SICI)1521-3773(20000218)39:4<764::AID-ANIE764>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Beck J B. and Rowan S J. J. Am. Chem. Soc. 2003;125:13922. doi: 10.1021/ja038521k. [DOI] [PubMed] [Google Scholar]
- Rowan S J. and Beck J B. Faraday Discuss. 2005;128:43. doi: 10.1039/b403135k. [DOI] [PubMed] [Google Scholar]
- Zhao Y Q, Beck J B, Rowan S J. and Jamieson A M. Macromolecules. 2004;37:3529. doi: 10.1021/ma0497005. [DOI] [Google Scholar]
- Weng W G, Beck J B, Jamieson A M. and Rowan S J. J. Am. Chem. Soc. 2006;128:11663. doi: 10.1021/ja063408q. [DOI] [PubMed] [Google Scholar]
- Paulusse J M J. and Sijbesma R P. Angew. Chem. Int. Edn. Engl. 2004;43:4460. doi: 10.1002/anie.200460040. [DOI] [PubMed] [Google Scholar]
- Iyer P K, Beck J B, Weder C. and Rowan S J. Chem. Commun. 2005;319 doi: 10.1039/b410734a. [DOI] [PubMed] [Google Scholar]
- Knapton D, Iyer P K, Rowan S J. and Weder C. Macromolecules. 2006;39:4069. doi: 10.1021/ma060500y. [DOI] [Google Scholar]
- Knapton D, Rowan S J. and Weder C. Macromolecules. 2006;39:651. doi: 10.1021/ma0520747. [DOI] [Google Scholar]
- Beck J B, Ineman J M. and Rowan S J. Macromolecules. 2005;38:5060. doi: 10.1021/ma050369e. [DOI] [Google Scholar]
- Rajadurai C, Fuhr O, Kruk R, Ghafari M, Hahn H. and Ruben M. Chem. Commun. 2007:2636. doi: 10.1039/b702468a. [DOI] [PubMed] [Google Scholar]
- Yount W C, Loveless D M. and Craig S L. J. Am. Chem. Soc. 2005;127:14488. doi: 10.1021/ja054298a. [DOI] [PubMed] [Google Scholar]
- Yount W C, Juwarker H. and Craig S L. J. Am. Chem. Soc. 2003;125:15302. doi: 10.1021/ja036709y. [DOI] [PubMed] [Google Scholar]
- Yount W C, Loveless D M. and Craig S L. Angew. Chem. Int. Edn. Engl. 2005;44:2746. doi: 10.1002/anie.200500026. [DOI] [PubMed] [Google Scholar]
- Loveless D M, Jeon S L. and Craig S L. Macromolecules. 2005;38:10171. doi: 10.1021/ma0518611. [DOI] [Google Scholar]
- Kersey F R, Lee G, Marszalek P. and Craig S L. J. Am. Chem. Soc. 2004;126:3038. doi: 10.1021/ja0499501. [DOI] [PubMed] [Google Scholar]
- Kersey F R, Yount W C. and Craig S L. J. Am. Chem. Soc. 2006;128:3886. doi: 10.1021/ja058516b. [DOI] [PubMed] [Google Scholar]
- Pollino J M, Nair K P, Stubbs L P, Adams J. and Weck M. Tetrahedron. 2004;60:7205. doi: 10.1016/j.tet.2004.05.055. [DOI] [Google Scholar]
- Pollino J M. and Weck M. Chem. Soc. Rev. 2005;34:193. doi: 10.1039/b311285n. [DOI] [PubMed] [Google Scholar]
- Vermonden T, van Steenbergen M J, Besseling N A M, Marcelis A T M, Hennink W E, Sudholter E J R. and Stuart M A C. J. Am. Chem. Soc. 2004;126:15802. doi: 10.1021/ja0458928. [DOI] [PubMed] [Google Scholar]
- Yan Y, Besseling N A M, de Keizer A, Marcelis A T M, Drechsler M. and Stuart M A C. Angew. Chem. Int. Edn. Engl. 2007;46:1807. doi: 10.1002/anie.200604039. [DOI] [PubMed] [Google Scholar]
- Oh M. and Mirkin C A. Nature. 2005;438:651. doi: 10.1038/nature04191. [DOI] [PubMed] [Google Scholar]
- Maeda H, Hasegawa M, Hashimoto T, Kakimoto T, Nishio S. and Nakanishi T. J. Am. Chem. Soc. 2006;128:10024. doi: 10.1021/ja0637301. [DOI] [PubMed] [Google Scholar]
- Caruso F, Schuler C. and Kurth D G. Chem. Mater. 1999;11:3394. doi: 10.1021/cm9911058. [DOI] [Google Scholar]
- Kurth D G, Caruso F. and Schuler C. Chem. Commun. 1999:1579. doi: 10.1039/a905115e. [DOI] [Google Scholar]
- Krass H, Plummer E A, Haider J M, Barker P R, Alcock N W, Pikramenou Z, Hannon M J. and Kurth D G. Angew. Chem. Int. Edn. Engl. 2001;40:3862. doi: 10.1002/1521-3773(20011015)40:20<3862::AID-ANIE3862>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Kurth D G, Lopez J P. and Dong W F. Chem. Commun. 2005:2119. doi: 10.1039/b500994d. [DOI] [PubMed] [Google Scholar]
- Bernhard S, Goldsmith J I, Takada K. and Abruna H D. Inorg. Chem. 2003;42:4389. doi: 10.1021/ic0207146. [DOI] [PubMed] [Google Scholar]
- Hayashi A, Higuchi M, Ohtsuka Y. and Kurth D G. Chem. Mater. at press 2008 [Google Scholar]
- Schutte M, Stolle C. and Kurth D G. Supramol. Chem. 2003;15:549. [Google Scholar]
- Kurth D G, Lehmann P. and Schutte M. Proc. Natl. Acad. Sci. USA. 2000;97:5704. doi: 10.1073/pnas.97.11.5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattari Z, Hatta E, Kurth D G. and Fischer T M. J. Chem. Phys. 2001;115:9923. [Google Scholar]
- Kurth D G, Severin N. and Rabe J P. Angew. Chem. Int. Edn. Engl. 2002;41:3681. doi: 10.1002/1521-3773(20021004)41:19<3681::AID-ANIE3681>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Meister A, Forster G, Thunemann A F. and Kurth D G. Chem. Phys. Chem. 2003;4:1095. doi: 10.1002/cphc.200300702. [DOI] [PubMed] [Google Scholar]
- Lehmann P, Symietz C, Brezesinski G, Krass H. and Kurth D G. Langmuir. 2005;21:5901. doi: 10.1021/la050841p. [DOI] [PubMed] [Google Scholar]
- Bodenthin Y, Pietsch U, Grenzer J, Geue T, Mohwald H and Kurth D G. 2005. J. Phys. Chem. B 109 12795 10.1021/jp0458001 [DOI] [PubMed] [Google Scholar]
- Kurth D G. and Bein T. Langmuir. 1995;11:578. doi: 10.1021/la00002a037. [DOI] [Google Scholar]
- Bodenthin Y, Pietsch U, Mohwald H. and Kurth D G. J. Am. Chem. Soc. 2005;127:3110. doi: 10.1021/ja0447210. [DOI] [PubMed] [Google Scholar]
- Werner R, Falk K, Ostrovsky S. and Haase W. Macromol. Chem. Phys. 2001;202:2813. doi: 10.1002/1521-3935(20011001)202:14<2813::AID-MACP2813>3.0.CO;2-T. [DOI] [Google Scholar]
- Bustamante E A S, Galyametdinov Y G, Griesar K, Schuhmacher E. and Haase W. Macromol. Chem. Phys. 1998;199:1337. [Google Scholar]
- Valkama S, Ruotsalainen T, Kosonen H, Ruokolainen J, Torkkeli M, Serimaa R, ten Brinke G. and Ikkala O. Macromolecules. 2003;36:3986. doi: 10.1021/ma020538k. [DOI] [PubMed] [Google Scholar]
- Valkama S. et al Macromol. Rapid Commun. 2003;24:556. doi: 10.1002/marc.200390084. [DOI] [Google Scholar]
- Kimizuka N, Oda N. and Kunitake T. Inorg. Chem. 2000;39:2684. doi: 10.1021/ic000189f. [DOI] [PubMed] [Google Scholar]
- Elemans J, Rowan A E. and Nolte R J M. J. Am. Chem. Soc. 2002;124:1532. doi: 10.1021/ja012061i. [DOI] [PubMed] [Google Scholar]
- Mobius D. and Kuhn H. J. Appl. Phys. 1988;64:5138. doi: 10.1063/1.342421. [DOI] [Google Scholar]
- Nagel J. and Oertel U. Polymer. 1995;36:381. doi: 10.1016/0032-3861(95)91329-6. [DOI] [Google Scholar]
- Lehmann P. and Brezesinski C. Chem. Eur. J. 2001;7:1646. doi: 10.1002/1521-3765(20010417)7:8<1646::AID-CHEM16460>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Sheiko S S. and Moller M. Chem. Rev. 2001;101:4099. doi: 10.1021/cr990129v. [DOI] [PubMed] [Google Scholar]
- Rabe S. and Buchholz S. Science. 1991;253:424. doi: 10.1126/science.253.5018.424. [DOI] [PubMed] [Google Scholar]
- Caruso F, Kurth D G, Volkmer D, Koop M J. and Muller A. Langmuir. 1998;14:3462. doi: 10.1021/la980177v. [DOI] [Google Scholar]
- Kurth D G. and Bein T. Langmuir. 1995;11:3061. doi: 10.1021/la00008a035. [DOI] [Google Scholar]
- Schwartz D K. Annu. Rev. Phys. Chem. 2001;52:107. doi: 10.1146/annurev.physchem.52.1.107. [DOI] [PubMed] [Google Scholar]
- Kurth D G. and Osterhout R. Langmuir. 1999;15:4842. doi: 10.1021/la981760e. [DOI] [Google Scholar]
- Kurth D G, Volkmer D, Ruttorf M, Richter B. and Muller A. Chem. Mater. 2000;12:2829. doi: 10.1021/cm001032o. [DOI] [Google Scholar]
- Ingersoll D, Kulesza P J. and Faulkner L R. J. Electrochem. Soc. 1994;141:140. doi: 10.1149/1.2054673. [DOI] [Google Scholar]
- Kuhn A. and Anson F C. Langmuir. 1996;12:5481. doi: 10.1021/la960461r. [DOI] [Google Scholar]
- Ichinose I, Tagawa H, Mizuki S, Lvov Y. and Kunitake T. Langmuir. 1998;14:187. doi: 10.1021/la970797g. [DOI] [Google Scholar]
- Liu S Q, Kurth D G, Bredenkotter B. and Volkmer D. J. Am. Chem. Soc. 2002;124:12279. doi: 10.1021/ja026946l. [DOI] [PubMed] [Google Scholar]
- Losche M, Schmitt J, Decher G, Bouwman W G. and Kjaer K. Macromolecules. 1998;31:8893. doi: 10.1021/ma980910p. [DOI] [Google Scholar]
- Schmitt J, Grunewald T, Decher G, Pershan P S, Kjaer K. and Losche M. Macromolecules. 1993;26:7058. doi: 10.1021/ma00077a052. [DOI] [Google Scholar]
- Moriguchi I. and Fendler J H. Chem. Mater. 1998;10:2205. doi: 10.1021/cm980127b. [DOI] [Google Scholar]
- Liu S Q, Kurth D G, Mohwald H. and Volkmer D. Adv. Mater. 2002;14:225. doi: 10.1002/1521-4095(20020205)14:3<225::AID-ADMA225>3.0.CO;2-F. [DOI] [Google Scholar]
- Liu S Q, Mohwald H, Volkmer D. and Kurth D G. Langmuir. 2006;22:1949. doi: 10.1021/la0523863. [DOI] [PubMed] [Google Scholar]
- Xue B, Peng J, Xin Z F, Kong Y M, Li L. and Li B. J. Mater. Chem. 2005;15:4793. doi: 10.1039/b511659g. [DOI] [Google Scholar]
- Gao G G, Xu L, Wang W J, Wang Z Q, Qiu Y F. and Wang E B. Electrochim. Acta. 2005;50:1101. doi: 10.1016/j.electacta.2004.08.006. [DOI] [Google Scholar]
- Gao G G, Xu L, Wang W J, An W J, Qiu Y F, Wang Z Q and Wang E B. 2005. J. Phys. Chem. B 109 8948. [DOI] [PubMed] [Google Scholar]
- Gao G G, Xu L, Wang W J, An W J. and Qiu Y F. J. Mater. Chem. 2004;14:2024. [Google Scholar]
- Keita B. and Nadjo L. J. Mol. Catal. A: Chem. 2007;262:190. [Google Scholar]
- Cheng L. and Cox J A. Chem. Mater. 2002;14:6. [Google Scholar]
- Zynek M, Serantoni M, Beloshapkin S, Dempsey E. and McCormac T. Electroanalysis. 2007;19:681. [Google Scholar]
- Liu S Q, Kurth D G. and Volkmer D. Chem. Commun. 2002:976. doi: 10.1039/b200942k. [DOI] [PubMed] [Google Scholar]
- Liu S Q, Volkmer D. and Kurth D G. Anal. Chem. 2004;76:4579. doi: 10.1021/ac0495283. [DOI] [PubMed] [Google Scholar]
- Jin Y N, Xu L, Zhu L D, An W J. and Gao G G. Thin Solid Films. 2007;515:5490. [Google Scholar]
- Nyman M, Ingersoll D, Singh S, Bonhomme F, Alam T M, Brinker C J. and Rodriguez M A. Chem. Mater. 2005;17:2885. [Google Scholar]
- Kurth D G, Lehmann P. and Lesser C. Chem. Commun. 2000:949. [Google Scholar]
- Fendler J H. and Meldrum F C. Adv. Mater. 1995;7:607. [Google Scholar]
- Clemente-Leon M, Coronado E, Gomez-Garcia C L, Mingotaud C, Ravaine S, Romualdo-Torres G. and Delhaes P. Chem. Eur. J. 2005;11:3979. doi: 10.1002/chem.200401063. [DOI] [PubMed] [Google Scholar]
- Kurth D G, Lehmann P, Volkmer D, Colfen H, Koop M J, Muller A. and Du Chesne A. Chem. Eur. J. 2000;6:385. doi: 10.1002/(SICI)1521-3765(20000117)6:2<385::AID-CHEM385>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Kurth D G, Lehmann P, Volkmer D, Muller A. and Schwahn D. J. Chem. Soc., Dalton Trans. 2000:3989. [Google Scholar]
- Volkmer D, Bredenkotter B, Tellenbroker J, Kogerler P, Kurth D G, Lehmann P, Schnablegger H, Schwahn D, Piepenbrink M. and Krebs B. J. Am. Chem. Soc. 2002;124:10489. doi: 10.1021/ja017613b. [DOI] [PubMed] [Google Scholar]
- Volkmer D, Du Chesne A, Kurth D G, Schnablegger H, Lehmann P, Koop M J. and Muller A. J. Am. Chem. Soc. 2000;122:1995. [Google Scholar]
- Moriguchi I, Orishikida K, Tokuyama Y, Watabe H, Kagawa S. and Teraoka Y. Chem. Mater. 2001;13:2430. [Google Scholar]
- Chambers R C, Atkinson E J O, McAdams D, Hayden E J. and Brown D J A. Chem. Commun. 2003:2456. doi: 10.1039/b307808f. [DOI] [PubMed] [Google Scholar]
- Bu W F, Fan H L, Wu L X, Hou X L, Hu C W, Zhang G. and Zhang X. Langmuir. 2002;18:6398. doi: 10.1021/la020085c. [DOI] [Google Scholar]
- Bu W F, Wu L X, Hou X L, Fan H L, Hu C W. and Zhang X. Colloid Interface Sci. 2002;251:120. doi: 10.1006/jcis.2002.8380. [DOI] [PubMed] [Google Scholar]
- Zhang T R, Lu R, Zhang H Y, Xue P C, Feng W, Liu X L, Zhao B, Zhao Y Y, Li T J. and Yao J N. J. Mater. Chem. 2003;13:580. doi: 10.1039/b210965d. [DOI] [Google Scholar]
- Polarz S, Smarsly B. and Antonietti M. ChemPhysChem. 2001;2:457. doi: 10.1002/1439-7641(20010716)2:7<457::AID-CPHC457>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Li W, Yi S Y, Wu Y Q and Wu L X. 2006. J. Phys. Chem. B 110 16961 10.1021/jp062074o [DOI] [PubMed] [Google Scholar]
- Li H L, Sun H, Qi W, Xu M. and Wu L X. Angew. Chem. Int. Edn. Engl. 2007;46:1300. doi: 10.1002/anie.200603934. [DOI] [PubMed] [Google Scholar]
- Jiang M, Zhai X D. and Liu M H. J. Mater. Chem. 2007;17:193. doi: 10.1039/b610029e. [DOI] [Google Scholar]
- Fan D W, Jia X F, Tang P Q, Hao J C. and Liu T B. Angew. Chem. Int. Edn. Engl. 2007;46:3342. doi: 10.1002/anie.200604180. [DOI] [PubMed] [Google Scholar]
- Zhang T R, Spitz C, Antonietti M. and Faul C F J. J. Chem. Eur. J. 2005;11:1001. doi: 10.1002/chem.200400796. [DOI] [PubMed] [Google Scholar]
- Clemente-Leon M, Ito T, Yashiro H, Yamase T. and Coronado E. Langmuir. 2007;23:4042. doi: 10.1021/la0635146. [DOI] [PubMed] [Google Scholar]
- Clemente-Leon M, Ito T, Yashiro H. and Yamase T. Chem. Mater. 2007;19:2589. doi: 10.1021/cm070436e. [DOI] [Google Scholar]
- Bain C D, Troughton E B, Tao Y T, Evall J, Whitesides G M. and Nuzzo R G. J. Am. Chem. Soc. 1989;111:321. doi: 10.1021/ja00183a049. [DOI] [Google Scholar]
- Horvolgyi Z, Nemeth S. and Fendler J H. Colloids Surf. A: Physicochem. Eng. Aspects. 1993;71:327. doi: 10.1016/0927-7757(93)80048-J. [DOI] [Google Scholar]