

Abstract
Porous coordination polymers (PCPs), which are microporous materials, have been given much attention from both scientific and commercial aspects regarding their application to gas storage, gas separation and catalytic reaction because of the regularity of their pore shape and pore size, accompanied with the functionality. Moreover, in recent years, flexible PCPs, which are structurally transformable depending upon external stimuli, have been attractive because they provide unique properties, dissimilar to those of zeolites. In this review, the chemistry and application of flexible crystalline PCPs are summarized and discussed.
Keywords: porous coordination polymer, storage, separation, flexible framework, porosity
Introduction
In recent years, porous coordination polymers (PCPs), the properties of which are promising in chemistry and for applications in catalysis [1–6], storage [7–12], ion exchange [13, 14] and separation [15–22], and polymerization [23–25], as well as zeolites have been widely studied. Furthermore, PCPs possess structural regularity, high porosity, high surface area, and designability resulting in a greater potential for application than zeolite or activated carbon. The word polymer is used because they extend an infinite structure constructed of coordination bonds of metal ions and organic ligands [26–28]. PCPs have not only coordination bonds but also other weak interactions or noncovalent bonds (H-bonds, π-electron stacking or van der Waals interaction), resulting in the structural transformation ability. The transition metal ions (d and f blocks) and electron acceptors (Lewis acid) are usually used as joints. Because of the variety of the coordination numbers of metal ions, one can obtain various coordination geometries (e.g. linear, T-shape, trigonal-planar, tetrahedral, octahedral), and consequently, various structural architectures. To elucidate the structures of PCPs, x-ray measurements and computational interpretation techniques have been improved. Not only have the structures of PCPs been resolved, but the specific alignment and properties of guest molecules, as determined by gas adsorption, have been investigated [29]. The linkers are electron donors (Lewis bases), in particular, the highly symmetrical multidentate ligands with N, O and S donor atoms [30]. These ligands are the key to forming infinite extended structures. PCPs possess a regularity of the framework, which provides void structures of desirable pore size, pore shape and functional pore surfaces, which are advantageous in separation applications [31]. Yaghi's group have created a series of PCPs, so-called MOFs (metal organic frameworks) based on Zn4O, which act as joints, while the dicarboxylate linker controls the pore size and pore function —these compound series are good representatives of the concept of reticular design [11, 32, 33]. Since high void space originates from long bridging ligands, however, that brings about catenation (interpenetration and interweaving) resulting in decrease of void space. To solve this problem, the author's group has proposed pillared layer construction for the synthesis of PCPs, so-called CPL-n (coordination polymer n with pillared layer structures) [34, 35], to avoid catenation. These CPL-n compounds show some structural transformation ability after guest accommodation. This ability suggests that PCPs can exhibit a structural flexibility different from the rigid zeolites or activated carbon. Moreover, the compounds from Ferey's group, namely MIL-53 and MIL-88 (materials of institute Lavoisier), produce the distinct structural transformation induced by guest molecules [36–38]. Thus, the PCPs can be subcategorized into robust and flexible PCPs.
Synthetic method
The PCPs are generally synthesized in the liquid phase by using solvent as a medium to induce the self-assembly of a regular framework. The reaction can be carried out by mixing the solution of metal ions with the solution of ligands at room temperature, or under hydrothermal/solvothermal conditions [39]. To obtain the desired PCPs, metal ion and ligand, solvent and counterions must be involved. Generally, rigid or semi-rigid ligands are usually used because product complexes from flexible ligands tend to be amorphous. Aromatic ligands are usually employed because an aromatic part is more rigid than an aliphatic one; three kinds of ligands are often used: neutral, cation and anion ligands. The as-synthesized PCPs initially have no void space because all the cavities are filled with guests, which may be solvent molecules, excess ligands or counterions. Solvent can be used to remove such guest molecules, to make the void space usable. In such a case, the solvent should be sufficiently volatile or exchangeable to create vacant space. However, some PCPs cannot exist without the guest molecule, they collapse after the removal of solvents. The solvent is likely to be unimportant in the use of PCPs because it will be removed after synthesis. However, solvents play a significant role in the formation of PCPs; the size and shape of pore in PCPs can be controlled by employing different solvents. Moreover, the solvent can function as a template for constructing the framework and also prohibits catenation, enabling the formation of an open structure. For example, the compounds { [Co3 (ndc)3 (bipyen)1.5 ]⋅H2O} n and { [Co2 (ndc)2 (bipyen)] ⋅ H2O ⋅ C6H6 } n (see figure 1), are good examples of the effect of solvent on the synthesis of PCPs [40]. Hydrothermal reactions of both compounds were carried out under similar conditions except that a benzene molecule was added to the mother liquor of the latter one. Both crystallographic structures show 3-fold interpenetration with different pore size and shape. The pore size of a compound that has only the lattice water molecule is 4.3 × 4.3 Å. Another compound has both water and benzene lattices occupying pores of different sizes (6.6 × 6.2 Å for benzene and 4.4 × 3.5 Å for water). On the basis of this point, the key factors for achieving intentional PCPs are the selection and combination of metal joints, ligand linkers and solvent or counterions.
Figure 1.
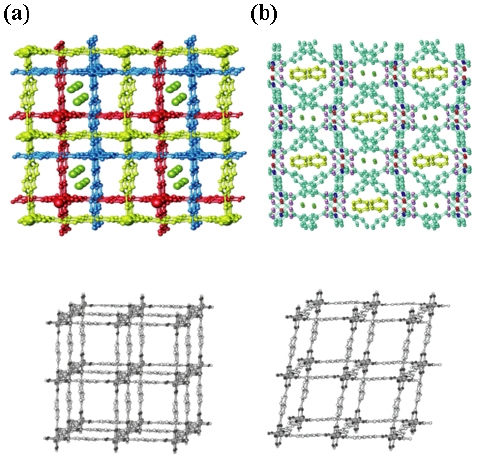
(a) Structure of {[Co3(ndc)(bipyen)1.5]⋅H2O}n. The oxygen atoms of guest water molecules are shown as green spheres. (b) Structure of {[Co3(ndc)(bipyen)1.5]⋅H2O⋅C6H6}n. Guest benzene and water molecules are yellow and green, respectively. Single structure of (a) and (b) are shown by the lower gray frameworks. © 2004, Wiley-VCH [40], with permission.
Robustness and flexibility
The PCPs can be categorized into robust and flexible classes on the basis of the structural transformation ability. The robust PCPs do not show any structural transformations upon applying external stimuli, e.g. guest molecule, heat, or magnetic or electric field, while the flexible PCPs do. High porosity and high surface area, together with permanent pore size and pore shape, are the merits of robust PCPs for gas storage application.
Application of robust PCPs: gas storage
Fuel gases such as H2 and CH4 have been realized as vehicle fuels, with environmental, economical and trade balance benefits. These gases have attracted much interest as replacements of petroleum and diesel power sources [41]. However, inefficient storage, that is, the low mileage owing to low volumetric density, is a demerit of fuel gas for automobile use. Recently, porous materials have been attracting attention as gas storage vessels [42, 43]; the gas will be stored in the adsorbate phase, which enables an increase in volumetric density. The experiment on CH4 adsorption was carried out at room temperature. Fuel gas-storage material is one of the possible uses of PCPs and will reduce the cost and safety problems, thanks to their high porosity and surface area. The first demonstration of CH4 sorption/desorption by PCP at room temperature at pressure of 0–30 atm was carried out in 1997, and opened up the field of porous chemistry relevant to gas storage [44].
The intermolecular distance of H2 in 3-D PCPs is about 3.0 Å, which is shorter than 3.6 Å, the intermolecular distance in pure solid cluster hydrogen. This result supports the idea that PCPs may be applicable as hydrogen storage materials. The Zn4O-based MOF compounds, which have high porosity and large surface area, are well known for their application to fuel gas storage. These series of MOFs were synthesized by varying the bridging ligands, as shown in figure 2. In general, pore sizes of MOFs are too large for the effective adsorption of hydrogen. The spherical pore size of MOF-5 constructed of 1,4-bdc acid is about 15 Å in diameter, while the kinetic size of hydrogen gas is about 2.89 Å in diameter [9]. Unused space or dead volume increases the volumetric density, and hence smaller pores are favorable. It is believed that the interaction between the aromatic ring of the ligand and the fuel gas gives rise to the storage ability [45]. To modify the pore size in order to achieve effective adsorption, nonvolatile guests are intercalated in the PCP space to decrease the pore size. Active surface guests also contribute to adsorption. For example, the large-pore MOF-177, 11.8 Å in diameter, can be doped by C60 molecules (6.83 Å in diameter) or polymers with a highly conjugated system (because of the adsorption ability of the graphene sheet of activated carbon) in order to increase the surface area for H2 adsorption [32].
Figure 2.
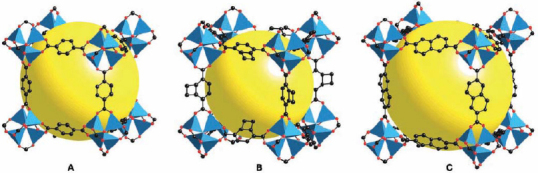
Examples of metal organic framework (MOFs) by Yaghi's group. © 2002, AAAS [11], with permission.
In the case of flexible PCPs, frameworks can response to external stimuli [46]. This flexibility is a unique characteristic of PCPs, making it superior to the other porous materials (zeolite and activated carbon). We believe that because of this special feature, flexible PCPs could provide more applicable and efficient functions.
Classification of porous coordination polymers [26]
One of the authors predicted the simultaneous existence of two concepts, softness and regularity, in a certain material in 1998, and categorized coordination polymers into three generations. The first generation shows frameworks that remain stable only under the guest inclusion condition. After guest removal, the framework will collapse irreversibly to form a framework. The second-generation PCPs are so robust that their frameworks are maintained even after the removal of guest molecules. In addition, non-conformationally changeable framework can be realized with the construction of the rigid bridging ligands. The third-generation PCPs exhibit flexible frameworks and dynamic functionalities [47–49].
Flexible porous coordination polymers
A unique property of third-generation PCPs is the flexibility of crystals. As described previously, such flexibility is an advantage solely of PCPs. This kind of PCP responds to external stimuli by reversible structural transformation. Generally, guest molecules act as the stimuli for the transformation of flexible PCPs, and the guest-induced distortion phenomena can be classified into the following categories.
Guest induced crystal-to-amorphous transformation (type 3rd -1)
After the removal of guest molecules, type 3rd-1 PCPs collapse and become amorphous. However, crystallinity is regained under the initial conditions. For instance, the lanthanide (III) coordination polymers { [M2 (BDOA)3 (H2O)4 ]⋅6H2O} n (M = Tb, Gd and Sm; BDOA=benzene -1,4-dioxylacetate) show the crystal-to-amorphous transformation property [50]. The aliphatic–OCH2–group on the ligand causes flexibility of the ligand, and can form H-bonds with guest molecules. The three compounds contain both coordinated and guest water molecules. The as-synthesized structures possess crystallinity. However, after the removal of eight water molecules per formula unit, four of which are guest water and two of which are coordinated water molecules, by heating to 120 °C, the compound formulae became [M2 (BDOA)3 (H2O)2 ]n and crystallinity disappeared, as confirmed from the x-ray powder diffraction (XRPD) pattern. However, the reversible formation of the crystalline phase can be achieved by soaking in water.
In comparison with the extensively investigated rigid ligand, 1,3,5-benzenetricarboxylic acid (BTC), a tripodal ligand, 4, 4′, 4″-tris(carboxymethyl)-1,3,5-benzenetricarboxamide, in which the amide groups are introduced, increases the flexibility of the entire structure owing to the presence of free rotation sites. The four novel 3-D compact frameworks { [ML(H2O)3 ]2 [M(H2O)6 ](H2O)3 } n (M = Zn, Mn, Ni, and Co, L = 4,4,4′,4″-tris(carboxymethyl)-1,3,5-benzenetricarboxamide) are composed of 2-D non-interpenetrating double anionic layers that are separated by layers of hexaqua metal cations { [M(H2O)6 ]2 + } n via H-bonds between the carboxyl groups (–CONH–) and the coordinated water molecules [51]. The amide groups in the ligand play an important role in the organization of these interesting supramolecular solids and make them dynamic molecular solids that exhibit a water-induced reversible crystal-to-amorphous transformation, which were further confirmed from their dehydration and rehydration behaviors by thermogravimetric and XRPD analysis.
Guest induced crystal-to-crystal transformation (type 3rd -2)
Frameworks of PCPs in this category are transformed to other networks with the retention of crystallinity, after the removal or exchange of guest molecules. [Zn2 (1,4–bdc)2 (dabco)]n (1,4–bdc = 1, 4 benzenedicarboxylate and dabco =1,4-diazabicyclo[2.2.2]octane), which has a 3-D framework of a jungle-gym structure, has structural transformability [52]. From the XRPD patterns, the as-synthesized compound, { [Zn2 (1,4–bdc)2 (dabco)]⋅4DMF⋅1/2H2O} n, the guest-free compound { Zn2 (1,4–bdc)2 (dabco)} n and the benzene-included compound { [Zn2 (1,4–bdc)2 (dabco)]⋅2C6H6 }n are found to have different structures. The framework shrinks upon guest inclusion because of the induced fit ability of flexible PCPs, as shown in figure 3. Because of this phenomenon, the O–Zn bond became elastic. The coordination geometry of Zn2 + can be distorted by guest inclusion without a change in the coordination number—reversible structural transformation occurs without the loss of crystallinity.
Figure 3.
(a) View along 4-fold axis of the structure in [Zn2 (1,4-bdc)2 (dabco)]n. One {Zn2(1,4-bdc)2} 2-D layer is colored orange to emphasize the alternation of the stacking layer. Hydrogen atoms and guest molecules are omitted. (b) Space-filling representation of evacuated framework, in which the open square channels are emphasized; view along fourfold axis. (c) Side view of evacuated framework, showing the windows interconnecting the channels. (d) Space-filling representation of the metal organic framework structure in [Zn2 (1,4–bdc)2 (dabco)]n C6H6, showing rhombic-grid motif of [Zn2 (1,4–bdc)2 ] layers. The guest molecules and dabco hydrogens are not shown. Legend: Zn green, N blue, O red, C grey and H white. © 2004, Wiley-VCH [52], with permission.
Structural transformation of flexible PCPs by guest molecule (see figure 4)
Figure 4.
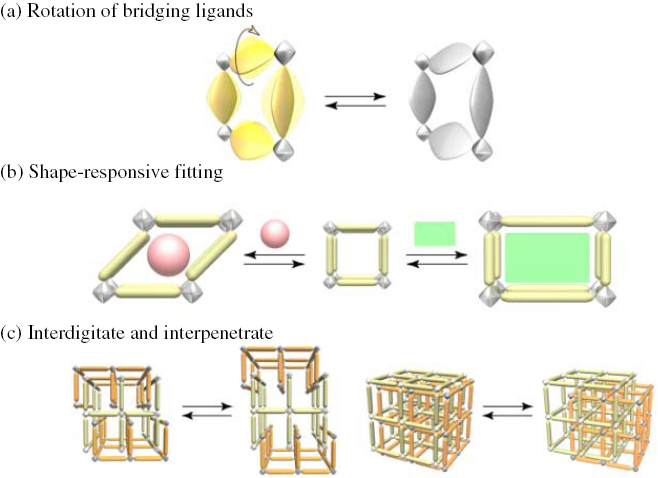
Schematic representation of flexible property of PCPs.
Rotation of bridging ligands
The PCPs with high void space exhibit a more dynamic ability than other nonporous solids. Ample space allows ligand molecules to undergo a partial motion such as rotation. For instance, a 3-D PCP, { [Zn3 (ntb)2 (EtOH)2 ]⋅4EtOH} n (ntb = NN,N′,N″-nitrilotrisbenzoate), see figure 5, shows reversible structural transformation through the coordination geometry of Zn2 +, changing from a trigonal bipyramid ({ [Zn3 (ntb)2 (EtOH)2 } n) to a tetrahedron ([Zn3 (ntb)2 ]n) after the removal of coordinating EtOH molecules without the collapse of the framework [53]. On the other hand, the rotation of the O–C–O plane of the carboxylate part results in the geometrical change.
Figure 5.
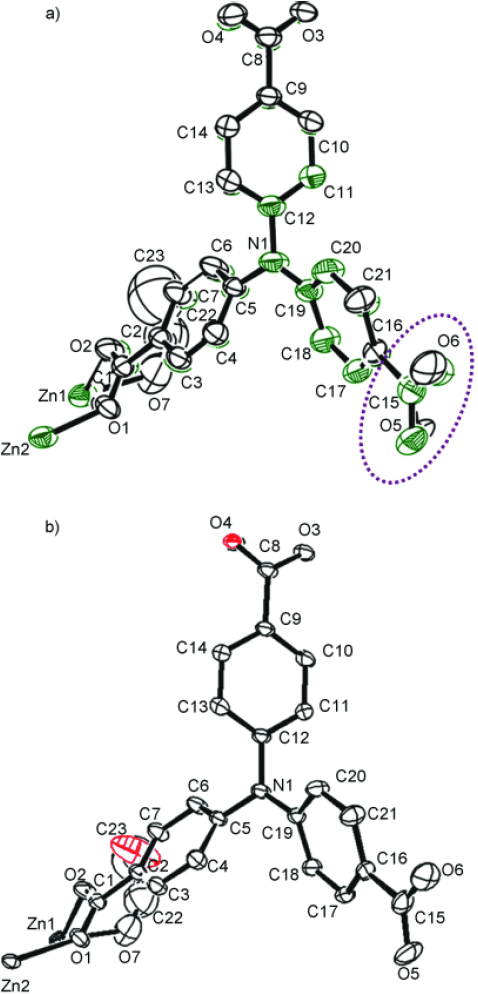
Superimposition of the crystallographic asymmetric units. (a) Original crystal [Zn3 (ntb)2 (EtOH)2 ]n⋅4nEtOH (black) and desolvated crystal (green). Rotation of O5-C15-O6 plane is depicted within dotted circle. (b) Original crystal [Zn3(ntb)2(EtOH)2]n⋅4nEtOH (black) and resolvated crystal (red). © 2007, Wiley-VCH [53], with permission.
The x-ray crystal structure of { Co(IN)2 ⋅0.5EtOH} n (IN = isonicotinate) shows channels containing ethanol guest molecules H-bonded to the carboxylate oxygen of the framework [54]. Its structure may be regarded as a 1-D cobalt-carboxylate chain aligned along the crystallographic a-direction, with the pyridyl group pointing out from the chains, as shown in figure 6. These chains cross-link via their pyridyl groups and trans coordination sites at the metals. There are channels in the structure along the a-axis; their width and shape vary dramatically. The variation in cross section is caused by the alternating open and closed rotational positions of the pyridine rings. At the open pyridines, the cavities are large enough to accommodate ethanol guest molecules. The structure of the desolvated compound ([Co(IN)2 ]n) has connectivity identical to that of the as-synthesized one. However, some adjustments such as reduction of the bond length occur after guest removal. Interestingly, some rotations of pyridine rings also occur. In particular, rings that are mutually trans across metal centers change from being mutually twisted in the solvated phase to being coplanar in the desolvated phase and still retains the alternation of open and closed rings along each channel. However, the registry of open and closed positions between the channels changes. The different rotational positions of the rings in the two phases suggest that pyridyl rotation may occur within the crystal during desolvation, and help to explain how oversized ethanol guests are able to move in and out of the host framework.
Figure 6.
Crystal structures of (a) [Co(IN)2]⋅0.5EtOH and (b) [Co(IN)2 ] (Co = green, O = red, N = blue and C = gray) with some H-atoms omitted for clarity. Segments of one carboxylate-bridged chain of each compound are shown in (c) and (d), respectively. The chain is shown by bold lines and axial ligation by pyridine from other carboxylate chains is shown by faint line. The mutual rotation of trans-pyridyl groups upon changing from solvated to desolvated states is shown in (d). © 2004, RSC [54], with permission.
Solid-state nuclear magnetic resonance (NMR) spectroscopy has been used to characterize structures and dynamic behaviors. The relaxation time is useful for characterizing the overall tumbling and internal dynamics of molecules. Because solid-state NMR spectra are sensitive to not only the rate of dynamic motion but also the geometry, this technique has recently been applied to study the dynamic properties of PCPs. The ligand rotation of the diamagnetic compound [Zn2 (1,4–ndc)2 (dabco)]n (1,4–ndc = 1,4–naphthalenedicarboxylate, dabco = 1,4-diazabicyclo[2,2,2]octane) was studied by 2H-solid state NMR [55]. 2H is a spin I= 1 nucleus and thus it possesses a quadrupole moment (quadrupole coupling is about 200 kHz). This coupling provides information about the motional mechanism, and dominates other anisotropic interactions in diamagnetic compounds, thus the dynamic motion can be analyzed. In this study, 1-D spectra are measured at various temperatures, and NMR data reveals that under the guest-free condition, the rotation rate constant of naphthalene rings around their C1–C4 axes (see figure 7) is in the range of 3.0 × 106 s−1 at 203 K to 5 × 107 s−1 at 223 K. In contrast, under the benzene guest-inclusion condition, the rotation of the ligand was inhibited. The rotational behavior is reversible by guest adsorption/desorption.
Figure 7.
View of guest-induced reversible rotation in [Zn2 (1,4–ndc)2 (dabco)]n. Benzene guests are blue; rotationally free naphthalene rings are yellow. © 2006, Wiley-VCH [55], with permission.
The dynamic rotation depends on thermal energy and does not cause a geometrical transformation, thus this dynamic property cannot be investigated by the XRD technique. However, because the rotation of the ligand is associated with the dynamic pore size and pore shape, it should affect the adsorption/desorption properties of PCPs.
Shrinking-swelling and shape-responsive fitting
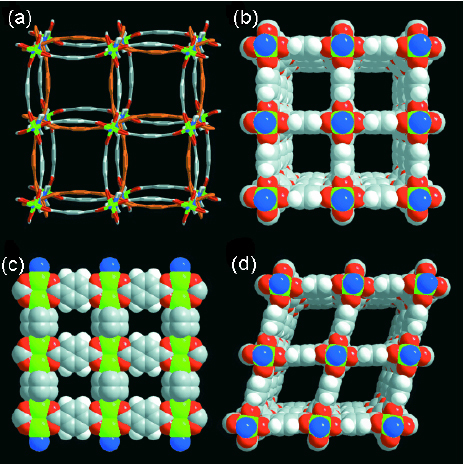
The flexibility of PCPs is attributed to coordination bonds, H-bonds, and other weak nonbonding interactions. Structural adjustments, such as shrinkage or expansion occur to accommodate the guest molecules. The 3-D compounds { M(μ 2–OH)(1,4–bdc)} n (M = Al3 +, Cr3 +), denoted MIL-53, are good examples of the shrinking-expanding framework depending on guest molecules [7, 56]. The as-synthesized MIL-53 framework encapsulates the excess 1,4-bdc ligands that form H-bond with the oxygen atom of the carboxylic and μ 2-OH groups. After the removal of the unreacted ligand, the framework turns expands, resulting in a larger pore space, as shown in figure 8. However, a more contracted form can be produced in the hydrated form. Similar to the 1,4-bdc excess ligand, water guest molecules form H-bonds with oxygen atoms. Since the water molecule is smaller than that of 1,4-bdc, the hydrated MIL-53 possesses smaller pores than the ligand-accommodated MIL-53; water and 1,4-bdc act as bridges that connect one corner of the framework with the opposite corner. Moreover, with the combination of flexibility and functionality, this compound also shows a good selectivity to guest sorption. There is no evidence of guest exchange in hydrated MIL-53 upon dispersing into acetone or ethanol, whereas the encapsulated water can be replaced by DMF. This is because of the weaker interaction between the cage of MIL-53 and the acetone and ethanol molecules. In contrast, the formation of H-bonds in DMF is more favorable than in water with the-N+= C–O− resonance structure.
Figure 8.
View of the pore systems of (a) MIL-53as (1,4-bdc- accommodated phase), (b) MIL-54ht (degassed phase), and MIL-53lt (water-accommodated phase).
The 3-D structure of { Cu(IN)2H2O} n(IN = isonicotinate) consists of square pyramidal copper atoms coordinated by two pyridyl groups and two carboxylate groups of four IN units in a monodentate fashion at the equatorial positions [57]. Another IN moiety occupies the remaining apical site of the square pyramidal geometry as a result of carboxylate monodentate coordination. The square pyramidal copper atoms are linked by five two-connected tridentate IN units in such a way that a novel five-connected three-dimensional network is formed with spiral open-framework channels running along the a-axis. After water molecules were removed, the stable open-framework [Cu(IN)]n is obtained. By soaking the guest-free crystals in methanol and 1-propanol, methanol- and 1-propanol-inclusion complexes, respectively, were obtained. In the mixed solvents ethanol-pentane and ethanol-1-propanol, the adsorption processes are very selective and only ethanol molecules are included in the structure. The three new inclusion polymers{ [Cu(IN)2 ]⋅CH3OH}, { [Cu(IN)2 ]⋅CH3CH2OH} and { [Cu(IN)2]⋅0.75CH3 (CH2)2OH} were characterized by single-crystal XRD. Surprisingly, these inclusion complexes revealed that the spiral open-framework is not only highly stable in its original dimensions and capable of selective adsorption but also expandable. The symmetry of the original spiral open-framework structure was maintained in the expanded structures. Moreover, in the mixtures of alkane and alcohol, only hydrophilic alcohol molecules were found in the channels. The alkane molecules are hydrophobic and cannot enter into the structure. In the case of mixed ethanol-1-propanol, ethanol molecules took the positions in the channels, which revealed that the adsorption property of this complex is size-selective.
To achieve the guest recognition ability, PCPs should be both shrinkable and expandable or either of these process alone. The deformation of the host such that it is effectively fitted with the guest is so-called shape-responsive fitting. The pillar layer compound [Cu2 (pzdc)2 (bpy)]n (bpy =4,4′-bipyridine), namely, CPL-2, can shrink and expand in response to the shape and size of the benzene guest molecule [26]. The channel shape of apohost is closed into a rectangular shape with the dimensions of 5.6 × 7.2 Å. An anisotropic benzene molecule (3.3 × 6.6 × 7.3 Å) sits periodically in CPL-2 in the commensurate fashion. The pore shape of CPL-2⊃benzene is similar to the letter ‘Z' and is well suitable for accommodating benzene. The contraction volume of CPL-2⊃benzene is about 4.9% compared with apohost CPL-2.
Interdigitation and interpenetration
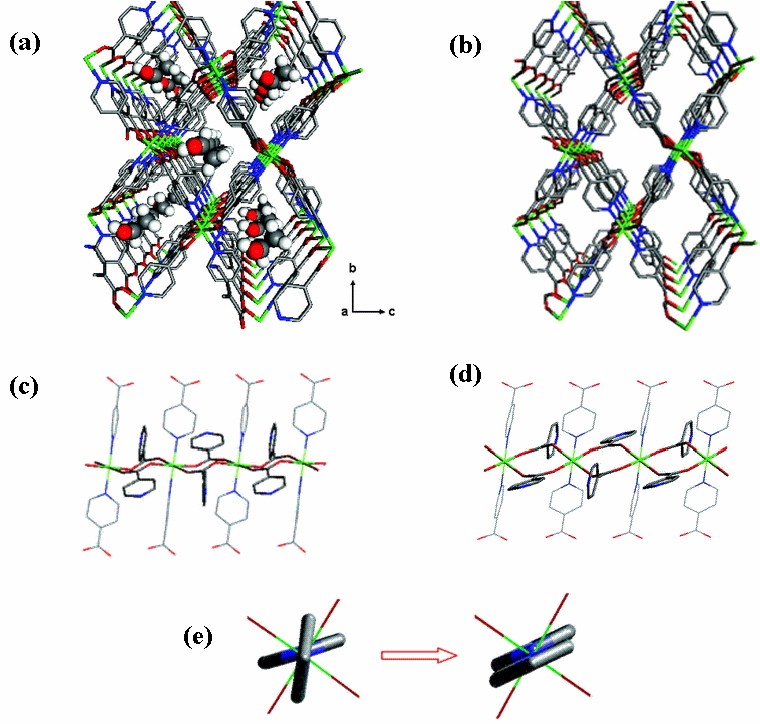
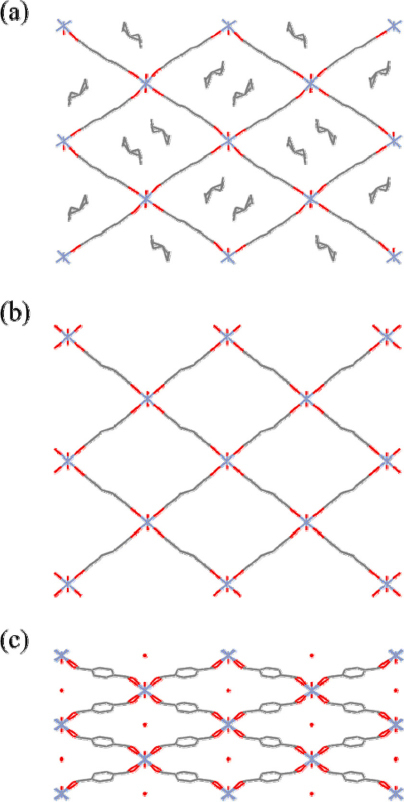
Nature abhors a vacuum [58]. This is proven in crystalline materials. The catenation of two or more framework motifs occurs to fill the voids in PCPs when using large ligands. A large number of PCPs exhibit mutual aggregation of frameworks, which results in densely packed, interdigitating or interpenetrating frameworks. Although close packing of these entangled networks hinders ligand rotation or individual framework expansion or shrinkage, the cooperative motion between frameworks brings about mutual sliding. The compound { [Cd2 (bpea)(1,4–bdc)(phen)2 ][Cd(1, 4–bdc)(phen)2 ⋅H2O]} n (bpea = biphenylethene - 4,4′-dicarboxylate, phen = 1,10-phenanethroline) shows an interdigitating architecture, as shown in figure 9 [59]. The 1-D zigzag chains tangle to form a puckered sheet. The phen groups are perpendicular to the propagation direction and point out in the opposite direction; this is suitable for forming interdigitating structure by π–π stacking. In addition, this 2-D sheet still entangles with the upper and lower sheets in the ABAB sequence to form a 3-D array. In the compound { [Zn2 (ip)2 (bpy)2 ⋅DMF]} n (ip = isophathalate and bpy = 4,4′-bpy), namely CID-1 (CID: coordination polymer with interdigitated structures), the ip ligands are perpendicular to the 2-D layer sheets that are mutually interdigitated to create stable 3-D frameworks [60]. The as-synthesized CID-1, in which DMF solvent molecules are accommodated, will be packed in the dehydrated state. CID-1 shows guest-dependent adsorption properties as well as structural transformation. It also exhibits good affinity for alcohol sorption without any stress, while it does not adsorb benzene vapor at 298 K. The 3-D structural compound { Zn4 (μ 4-OH)(L1)3 (dmf) × 4DMF × 3CH3OH× 2H2O} n (L1 = 6,6′-dichloro-2,2′-diethoxy-1,1′-binaphthyl-4,4′-di benzoate) possesses a large cubic cavity of 19 × 19 × 19 Å [43]. Because of this large void space, a 4-fold interpenetrating network is formed. The size of the cavity becomes smaller after interpenetration (4.1 × 5.0 Å along a-axis, 3.2×4.0 and 2 × 2.5 Å along b-axis, and 4.7×5.4 and 1.8 × 3.4 Å along c-axis), resulting in effective H2 adsorption. The extent of sorption is 1.12 wt.% at 48 bar, which is comparable to that of purified single-wall carbon nanotubes.
Figure 9.
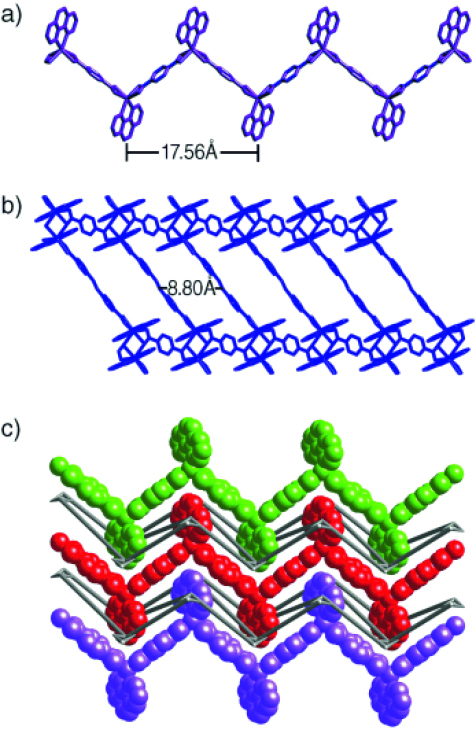
Crystal structure of {[Cd2(bpea)(1,4–bdc)(phen)2][Cd(1, 4–bdc)(phen)2⋅H2O]}n. View of (a) zigzag chain, (b) puckered sheet and (c) entanglement of the 1-D chains and 2-D sheets in the ABAB mode. The interdigitation of the phen groups is clearly visible. © 2004, Wiley-VCH [59], with permission.
Although interpenetration reduces the pore diameter, one can use this phenomenon to improve the framework properties, such as the sorption property. Because the interactions between individual motifs of interpenetrating or interdigitating frameworks are weak, the sliding of motifs influenced by guest molecules is possible, resulting in flexible PCPs. On the other hand, interdigitation and interpenetration are strategies of modifying the properties of PCPs by the entrapment mechanism. Effective adsorption depends on the appropriate size and shape of pores in the host—they should be well matched with the size and shape of guests in the sense of host–guest interaction. Therefore, the self-structural adjustment of interdigitating and interpenetrating frameworks is a useful contrivance for selective adsorption.
Adsorption characteristics of flexible porous coordination polymers
Sorption/desorption isotherms
Adsorption is defined as the accumulation of molecules on solid surfaces, and can be classified into physical adsorption and chemical adsorption depending on the interaction between the adsorbent and adsorbate. Adsorption observed on PCPs usually is physisorption, which is a reversible process and depends upon pore size and shape. The International Union of Pure and Applied Chemistry (IUPAC) classifies the gas/vapor adsorption isotherms based on BDDT classification (Brunauer and co-workers) into six classes. These isotherms describe the interactions between guest molecules and the surfaces and the pore sizes and shapes of adsorbents without structural transformation during the sorption process. Although the pore size and shape of PCPs have very uniform distributions, the flexible PCPs show dynamic properties. PCPs are microporous materials with a pore diameters less than 2 nm, and adsorption in such a small pore is usually reversible without hysteresis. Because of a functional surface and flexibility, the adsorption of PCPs does not simply follow one of the common six isotherms but is qualitatively distinct from that of rigid microporous materials. The isotherms of flexible PCPs usually indicate properties that follow a combination of isotherms or a new isotherm (see figure 10).
Figure 10.
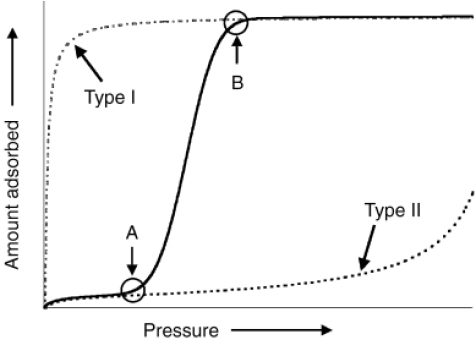
Typical adsorption isotherm for flexible PCPs that underwent a structural transformation. Dashed lines represent the type I and type II isotherms. Points A and B indicate the gate-opening and gate-closing pressures which accompany the start and end of structural transformation, respectively. © 2004, Wiley-VCH [58], with permission.
Thermal property of flexible PCPs
To understand and develop adsorption/desorption properties of PCPs in relation to their flexible properties, the thermal conductivity of PCPs must be investigated. Because sorption processes are sensitive to ambient temperature fluctuation, the stored gases must be protected from it. However, few studies on the thermal conductivity of PCPs have been reported so far. Generally, the PCPs have rather low thermal conductivity because of their low density. Since PCPs are dielectric materials, their energy carriers are associated with the vibrational modes of crystal lattices, called phonon phenomena [61, 62]. The thermal conductivity of PCPs depends on the mode of phonon scattering: phonon–grain boundary scattering defect–phonon scattering or phonon–phonon scattering. Recently, the thermal conductivity of single-crystal MOF-5 has been reported from both experimental and theoretical points of view [63, 64]. At temperatures below 35 K, thermal conductivity increases with increasing temperature and defect-phonon scattering is the dominate mechanism. In the temperature range from 35 to 100 K, the thermal conductivity of MOF-5 decreases rapidly with increasing temperature and the scattering is mainly controlled by phonon–phonon scattering. At temperatures above 100 K, thermal conductivity is mostly temperature-independent (0.32 W m K−1 at temperature 300 K), indicating the minimum phonon mean free path. From the mean free path analysis, the MOF-5 mean free path is minimized into the cage size. This heteroatomic crystal shows a much shorter phonons mean free path than a homoatomic crystal such as diamond. Hence, the scattering of phonons should occur on the joints of heteroatomic sites which are the carboxylate carbon group in the case of MOF-5. This study reveals the thermal conductivity mechanism of PCPs.
In the gas storage applications, the correlation of heat transfer between host and guest molecules is to be elucidated. Adsorption on a porous material is an exothermic process; the temperature tends to increase during adsorption. The storage capacity is dependent on the removal of the heat of adsorption. The thermal property of a system depends not only on the thermal conductivity of adsorbent, but also on the adsorbate properties (size, thermal conductivity, geometry, etc). The factors that may be useful in designing adsorption properties of flexible PCPs are still unclear and must elucidate in order to develop new types of PCPs.
Gate opening process
The role of a guest inclusion in a rigid porous framework is limited by the kinetic size of the guest molecule and the diameter of the pore windows of the host framework. The flexibility of PCPs allows a certain guest molecule to be recognized preferentially, different from classical rigid frameworks. Guest molecules act as an external chemical stimulus on structural transformation; this has not been observed in the case of rigid porous materials. For instance, the 2-D framework [Cu(dhbc)2 (4,4′–bpy)]n (dhbc=2,5–dihydroxybenzoicacid) has a mutually interdigitated form as a result of the π stacking interaction that gives rise to the 1-D channel structure [65]. This compound shows no N2 diffusion into the framework. However, at 298 K, an unusual N2 adsorption isotherm is obtained; the isotherm profile suddenly increases from the pressure of 50 atm and the specific surface area becomes 320 m2 g−1. Even though the adsorption process is exothermic, referred to as Le Chatelier's principle, the amount of adsorption should decrease with increasing temperature; this flexible PCP exhibits the opposite adsorption behavior. The higher temperature supplies the kinetic energy of framework movement, and accompanied with a sufficiently strong pressure to open the gate, the structural transformation from nonporous to porous phases occurs. This onset pressure is referred to the ‘gate-opening pressure' (figure 11). Interestingly, in the desorption process, the abrupt decrease of the isotherm is at 30 atm, which is called the ‘gate-closing pressure'. The difference in the pressure for the gate opening and closing causes a hysteretic behavior. Even though the hysteretic profile occurs in this adsorption/desorption isotherm during the expansion and shrinkage of this PCP, the crystallinity of the two phases still remains over the transformation.
Figure 11.
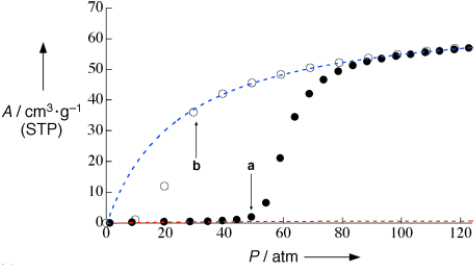
Nitrogen adsorption (filled circles) and desorption isotherms (open circles) for 1 b at 298 K. Blue and red dashed lines were determined by fitting the linear parts of the Langmuir plots in the higher (from 83 to 122 atm) and lower (from 8 to 34 atm) pressure ranges, respectively. © 2003, Wiley-VCH [65], with permission.
An unusual guest response is found in the compound [Ni2 (4,4′–bipy)3 (NO3)4]n [66]. The cavity in this PCP shows a much smaller window of 2.5 × 4.9 Å. This material is, however, able to reversibly adsorb toluene of a large size (4 × 6.6 Å), which is opposite of what is expected for rigid porous materials. In order to accommodate guest molecules with a larger size than the size of pore window of the host, the framework must be opened up. The competitive π–π stacking interaction of toluene and the framework opens the windows of the closed framework.
The compound { [Cu(bpy)(BF4)2 (H2 O)2 ]⋅(bpy)} n with a non-interpenetrated structure is a 2-D sheet where the upper and lower sheets are stacked with H-bonds and π stacking of bpy [67]. Therefore, pore space is not observed in any direction and consequently, guest molecules should not be able to be accommodated in this compound. The adsorption of gas on this complex suddenly begins at a definite relative pressure regardless of almost nil adsorption below the gate-opening pressure. Such a unique adsorption phenomenon is attributed to the H-bond-regulated microporous nature of this complex. H-bonds in pores block adsorption below the gate pressure. Additionally, the gate pressure depends on both the adsorbate and the pretreatment temperature of the Cu complex solid.
Applications of flexible crystal in adsorption
Storage/trapping
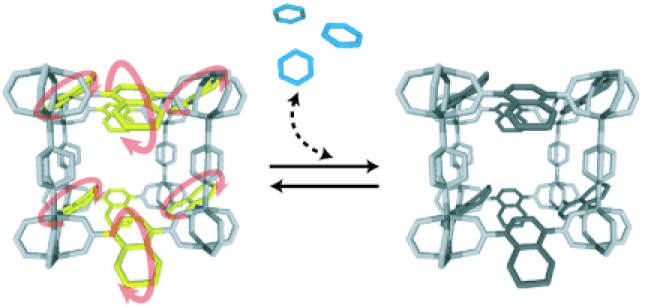
The strategy of the adsorption of robust PCPs is to tune the pore size to that of the adsorbed gas. On the other hand, because flexible PCPs are structurally transformable, the interaction between host and guest could have self-tuning mechanisms to adjust the pore shape and size toward the most stable state with guests. From this characteristic, flexible PCPs are effective for stabilizing the explosive gases such as acetylene. As mentioned above, the structure of CPL series provides unique functionalities, for instance, CPL-2 changes via shape-responsive fitting [68, 69]. When this compound adsorbs benzene as a guest molecule, the 2-D sheets mutually slide to fit the shape of the host and the guest (figure 12). Moreover, CPL-1 [68, 69] can be used as template to construct a stable acetylene array aggregate with a milder intermolecular interaction than that of pure acetylene. Acetylene molecules are trapped individually by forming the double H-bonds with an uncoordinated oxygen atom on pzdc, resulting in structural change. In pure dense acetylene gas, the strong intermolecular interaction causes an explosion. When acetylene is placed in PCP, the interaction between individual acetylene molecule and the wall of PCPs occurs, and then the interaction between acetylene molecules is reduced; the system is stabilized.
Figure 12.
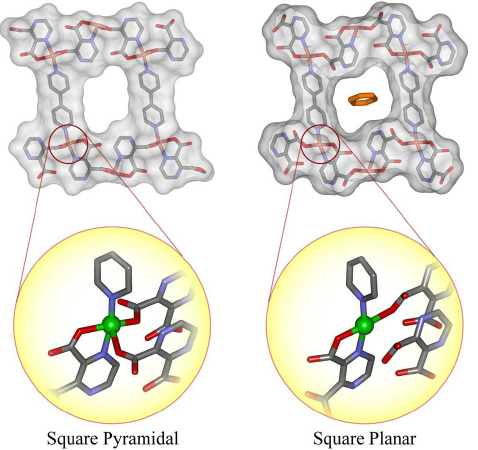
Schematic representation of the structural transformation with ‘shape-responding fitting' triggered by benzene adsorption. The mutual sliding of the 2-D sheets and pore shrinkage are observed and are accompanied by the change of the Cu coordination geometry from a square pyramid to a square plane.
A large surface area for adsorption, together with a dynamic property, is the merit of flexible PCPs in storage applications. Therefore, flexible PCPs are excellent materials in practical applications. Rational synthesis of porous frameworks is often thwarted by interpenetration and architectural frailty, which arise from the presence of large linkers that extend into the space between the nodes. Therefore, the regulation of this interpenetration in order to remove any obstacles to porous functionalities is an important challenge in crystal engineering. Recently, there was an intriguing claim that even an interpenetrating framework tends to have a certain porous functionality [70–72]. Stiff interpenetrating frameworks maintain their structural integrity in guest molecule storage, whereas structural flexibility, such as mutual framework sliding, gives rise to a new type of porous functionality, the controllable gate-opening property.
The 2-fold interpenetrating { Cu3 (BTB)2 (H2O)3⋅(DMF)9 (H2O)2 } n, (BTB = 4,4′,4″–benzene–1,3,5–tribenzate), so-called MOF-14, shows a high H2 uptake because of the large void space even with the interpenetrating fashion [73]. By the above examples, it was demonstrated that effective adsorption requires the proper pore size and shape. The flexibility of PCPs will lead to promising new adsorption properties.
Selective adsorption and separation
Selective sorption and separation are another promising application of PCPs. Generally, porous materials can separate a mixture of gases by size selection methods. In the case of flexible PCPs, the combination of flexibility and a functional surface enable effective selectivity. A 2-fold interpenetrating framework { [Ni(bpe)2 (N(CN)2)](N(CN)2 } n (bpe = 1,2–bis(4–pyridyl)ethane, N(CN)2−= dicyanamide) exhibits the selective gas adsorption and anion exchange property [74]. Even though the interpenetration results in the reduction of porosity, this PCP shows highly selective guest sorption. In the evacuated state, the compound has no adsorptivity to small-size gases compared with the size of cage windows, such as N2, O2 and Xe, whereas it selectively adsorbs the same-size CO gas or the larger MeOH and Me2CO gases. The reason why this compound does not adsorb N2, O2 or Xe is probably because the strong interaction between gases and pore windows results in the closed-window phase; hence, the framework no longer has open channels. For CO2 adsorption, an electric field produced by dipole-induced dipole interaction between the host and the guest effectively enhances CO2 adsorption. In the case of a selective anion exchange, the framework provides one free N(CN)2− per unit as a counterion. The shape of the ion is almost linear. The experiments of anion exchange with N3−, NCO−, N3− and BF4− were carried out. Only N3− shows the irreversible exchange reaction. The reason is the different symmetries, shapes and sizes of NCO−, NO3− and BF4− to N(CN)2−.
The surface properties of the framework for adsorption can be modified by tuning the properties of the organic ligands. The modifiable active site and dynamic ability are the advantages of flexible PCPs. The selective adsorption arises from the functional surface accompanied by the guest-induced structural transformation ability; effective and stable adsorption is expected. In the case of rigid porous frameworks, the adsorption sites give rise to effective adsorption via the interaction between adsorbent and adsorbate. However, the adsorptivity must be linked with the shape fitting that can be found in biological molecules, such as in the case of the high recognition of enzyme and the substrate binding process. Therefore, the elastic structures of flexible PCPs provide one strategy for novel selective adsorption.
Nanoreactors for polymerization
When one-dimensional nanochannels of crystalline porous compounds are filled with guest molecules with polymerizable groups, the monomers can be polymerized there. Confined nanospaces accompanied by interactions between the host framework and the monomer affect the behavior, regularity and reactivity of monomers, resulting in precisely controlled polymerization and polymer arrangement [24]. Because flexible dynamic frameworks respond to guest molecules, a selective linear radical polymerization of divinylbenzene (DVBs) in the channels of [M2 (1,4–bdc)2 (dabco)]n (bdc = benzenedicarboxylate, dabco = 1,4–diazabicyclo[2.2.2]octane, M = Zn2+, Cu2+) has been reported [23]. The pore sizes of the two frameworks are identical (7.8 × 7.8 Å2 along their c axes). However, the polymerization occurs only in [Zn2 (1,4–bdc)2 (dabco)]n. This is because, even though the introduction of p-DVB into the channel does not affect the porous nature of the framework, it induces a small amount of lattice expansion compared with the original host. Only the Zn complex is able to show dynamic and flexible properties, while the Cu complex does not. No trace of a polymeric product was observed in the space of the rigid framework, [Cu2 (1,4–bdc)2 (dabco)]n. This is because the flexibility of the host allows the adsorption of relatively large p-DVB molecules by expansion of the host structure such that the monomer molecules are arranged adjacent to each other, which is essential for polymerization.
Conclusion
Flexible porous coordination polymers behave differently from rigid ones. Because of the flexibility, structural adaption to external stimuli is possible in the flexible porous compounds. In particular, flexible PCPs provide new and versatile gas storage, gas separation and guest exchange applications because of their better recognition and structural transformation abilities than those of rigid porous materials.
Footnotes
Invited paper.
References
- Fujita M, Kwon Y J, Washizu S. and Ogura K. J. Am. Chem. Soc. 1994;116:1151. doi: 10.1021/ja00082a055. [DOI] [Google Scholar]
- Seo J S, Whang D, Lee H, Jun S I, Oh J, Jeon Y J. and Kim K. Nature. 2000;404:982. doi: 10.1038/35010088. [DOI] [PubMed] [Google Scholar]
- Hasegawa S, Horike S, Matsuda R, Furukawa S, Mochizuki K, Kinoshita Y. and Kitagawa S. J. Am. Chem. Soc. 2007;129:2607. doi: 10.1021/ja067374y. [DOI] [PubMed] [Google Scholar]
- Gomez-Lor B, Gutierrez-Puebla E, Iglesias M, Monge M A, Ruiz-Valero C. and Snejko N. Chem. Mater. 2005;17:2568. doi: 10.1021/cm047748r. [DOI] [PubMed] [Google Scholar]
- Evans O R, Ngo H L. and Lin W. J. Am. Chem. Soc. 2001;123:10395. doi: 10.1021/ja0163772. [DOI] [PubMed] [Google Scholar]
- Tada M. and Iwasawa Y. Coor. Chem. Rev. 2007;251:2702. doi: 10.1016/j.ccr.2007.06.008. [DOI] [Google Scholar]
- Ferey G, Latroche M, Serre C, Millange F, Loiseau T. and Percheron-Guegan A. Chem. Commun. 2003:2976. doi: 10.1039/b308903g. [DOI] [PubMed] [Google Scholar]
- Rosi N L, Eckert J, Eddaoudi M, Vodak D T, Kim J, O'Keeffe M. and Yaghi O M. Science. 2003;300:1127. doi: 10.1126/science.1083440. [DOI] [PubMed] [Google Scholar]
- Rowsell J L. and Yaghi O M. Angew. Chem. Int. Edn. 2005;44:4670. doi: 10.1002/anie.200462786. [DOI] [PubMed] [Google Scholar]
- Noro S, Kitagawa S, Kondo M. and Seki K. Angew. Chem. Int. Edn. 2000;39:2081. doi: 10.1002/1521-3773(20000616)39:12<2081::AID-ANIE2081>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Eddaoudi M, Kim J, Rosi N, Vodak D, Wachter J, O'Keeffe M. and Yaghi O M. Science. 2002;295:469. doi: 10.1126/science.1067208. [DOI] [PubMed] [Google Scholar]
- Seki K and Mori W. 2002. J. Phys. Chem. B 106 1380 10.1021/jp0130416 [DOI] [Google Scholar]
- Min K S. and Suh M P. J. Am. Chem. Soc. 2000;122:6834. doi: 10.1021/ja000642m. [DOI] [Google Scholar]
- Yaghi O M. and Li H. J. Am. Chem. Soc. 1996;118:295. doi: 10.1021/ja953438l. [DOI] [Google Scholar]
- Pan L, Olson D H, Ciemnolonski L R, Heddy R. and Li J. Angew. Chem. Int. Edn. 2006;45:616. doi: 10.1002/anie.200503503. [DOI] [PubMed] [Google Scholar]
- Rood J A, Noll B C. and Henderson K W. Inorg. Chem. 2006;45:5521. doi: 10.1021/ic060543v. [DOI] [PubMed] [Google Scholar]
- Ma S, Wang X S, Manis E S, Collier C D. and Zhou H C. Inorg. Chem. 2007;46:3432. doi: 10.1021/ic070338v. [DOI] [PubMed] [Google Scholar]
- Snurr R Q, Hupp J T. and Nguyen S T. AIChE J. 2004;50:1090. doi: 10.1002/aic.10101. [DOI] [Google Scholar]
- Uemura K, Kitagawa S, Kondo M, Fukui K, Kitaura R, Chang H-C. and Mizutani T. Chem. Eur. J. 2002;8:3586. doi: 10.1002/1521-3765(20020816)8:16<3586::AID-CHEM3586>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Min K S. and Suh M P. Chem. Eur. J. 2001;7:303. doi: 10.1002/1521-3765(20010105)7:1<303::AID-CHEM303>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Dybtsev D N, Chun H, Yoon S H, Kim D. and Kim K. J. Am. Chem. Soc. 2004;126:32. doi: 10.1021/ja038678c. [DOI] [PubMed] [Google Scholar]
- Bradshaw D, Prior T J, Cussen E J, Claridge J B. and Rosseinsky M J. J. Am. Chem. Soc. 2004;126:6106. doi: 10.1021/ja0316420. [DOI] [PubMed] [Google Scholar]
- Uemura T, Hiramatsu D, Kubota Y, Takata M. and Kitagawa S. Angew. Chem. Int. Edn. 2007;46:4987. doi: 10.1002/anie.200700242. [DOI] [PubMed] [Google Scholar]
- Uemura T, Horike S. and Kitagawa S. Chem. Asian J. 2006;1:36. doi: 10.1002/asia.200600074. [DOI] [PubMed] [Google Scholar]
- Uemura T, Kitaura R, Ohta Y, Nagaoka M. and Kitagawa S. Angew. Chem. Int. Edn. 2006;45:4112. doi: 10.1002/anie.200600333. [DOI] [PubMed] [Google Scholar]
- Matsuda R, Kitaura R, Kitagawa S, Kubota Y, Kobayashi T C, Horike S. and Takata M. J. Am. Chem. Soc. 2004;126:14063. doi: 10.1021/ja046925m. [DOI] [PubMed] [Google Scholar]
- James S L. Chem. Soc. Rev. 2003;32:276. doi: 10.1039/b200393g. [DOI] [PubMed] [Google Scholar]
- Rowsell J L C. and Yaghi O M. Micro. Meso. Mater. 2004;73:3. doi: 10.1016/j.micromeso.2004.03.034. [DOI] [Google Scholar]
- Kubota Y, Tanaka M, Kobayashi T C. and Kitagawa S. Coor. Chem. Rev. 2007;251:2510. doi: 10.1016/j.ccr.2007.07.025. [DOI] [Google Scholar]
- Robin A Y. and Fromm K M. Coord. Chem. Rev. 2006;250:2127. [Google Scholar]
- Ariga A V K, Hill J P. and Mori T. Coord. Chem. Rev. 2007;251:2562. doi: 10.1016/j.ccr.2007.02.024. [DOI] [Google Scholar]
- Chae H K, Siberio-Perez D Y, Kim J, Go Y, Eddaoudi M, Matzger A J, O'Keeffe M. and Yaghi O M. Nature. 2004;427:523. doi: 10.1038/nature02311. [DOI] [PubMed] [Google Scholar]
- Li H, Eddaoudi M, O'Keeffe M. and Yaghi O M. Nature. 1999;402:276. doi: 10.1038/46248. [DOI] [Google Scholar]
- Kondo M, Okubo T, Asami A, Noro S-I, Yoshitomi T, Kitagawa S, Ishii T, Matsuzaka H. and Seki K. Angew. Chem. Int. Edn. 1999;38:140. doi: 10.1002/(SICI)1521-3773(19990115)38:1/2<140::AID-ANIE140>3.0.CO;2-9. [DOI] [Google Scholar]
- Kitagawa S. and Matsuda R. Coord. Chem. Rev. 2007;251:2490. doi: 10.1016/j.ccr.2007.07.009. [DOI] [Google Scholar]
- Loiseau T, Serre C, Huguenard C, Francis G F, Henry T M, Bataille T. and Ferey G. Chem. Eur. J. 2004;10:1373. doi: 10.1002/chem.200305413. [DOI] [PubMed] [Google Scholar]
- Barthelet K, Marrot J, Riou D. and Férey G. Angew. Chem. Int. Edn. 2002;41:281. doi: 10.1002/1521-3773(20020118)41:2<281::AID-ANIE281>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Serre C, Millange F, Thouvenot C, Nogues M, Marsolier G, Louer D. and Ferey G. J. Am. Chem. Soc. 2002;124:13519. doi: 10.1021/ja0276974. [DOI] [PubMed] [Google Scholar]
- Chen X M. and Tong M L. Acc. Chem. Res. 2007;40:162. doi: 10.1021/ar068084p. [DOI] [PubMed] [Google Scholar]
- Choi E-Y, Park K, Yang C-M, Kim H, Son J-H, Lee S W, Lee Y H, Min D. and Kwon Y-U. Chem. Eur. J. 2004;10:5535. doi: 10.1002/chem.200400178. [DOI] [PubMed] [Google Scholar]
- Menon V C. and Komarneni S. J. Porous Mater. 1998;5:43. doi: 10.1023/A:1009673830619. [DOI] [Google Scholar]
- Mueller U, Schubert M, Teich F, Puetter H, Schierle-Arndt K. and Pastre J. J. Mater. Chem. 2006;16:626. doi: 10.1039/b511962f. [DOI] [Google Scholar]
- Kesanli B, Cui Y, Smith M R, Bittner E W, Bockrath B C. and Lin W. Angew. Chem. Int. Edn. 2005;44:72. doi: 10.1002/anie.200461214. [DOI] [PubMed] [Google Scholar]
- Kondo M, Yoshitomi T, Matsuzaka H, Kitagawa S. and Seki K. Angew. Chem. Int. Edn. 1997;36:1725. doi: 10.1002/anie.199717251. [DOI] [Google Scholar]
- Nechaev Y S. and Alexeeva O K. Int. J. Hydrogen Energy. 2003;28:1433. doi: 10.1016/S0360-3199(03)00004-1. [DOI] [Google Scholar]
- Kawano M. and Fujita M. Coord. Chem. Rev. 2007;251:259. doi: 10.1016/j.ccr.2007.07.022. [DOI] [Google Scholar]
- Kitagawa S. and Uemura K. Chem. Soc. Rev. 2005;34:109. doi: 10.1039/b313997m. [DOI] [PubMed] [Google Scholar]
- Bradshaw D, Claridge J B, Cussen E J, Prior T J. and Rosseinsky M J. Acc. Chem. Res. 2005;38:273. doi: 10.1021/ar0401606. [DOI] [PubMed] [Google Scholar]
- Cussen E J, Claridge J B, Rosseinsky M J. and Kepert C J. J. Am. Chem. Soc. 2002;124:9574. doi: 10.1021/ja0262737. [DOI] [PubMed] [Google Scholar]
- Hong X L, Li Y Z, Hu H, Pan Y, Bai J. and You X Z. Cryst. Growth Des. 2006;6:1221. doi: 10.1021/cg0506323. [DOI] [Google Scholar]
- Sun R, Li Y Z, Bai J. and Pan Y. Cryst. Growth Des. 2007;7:890. doi: 10.1021/cg060626m. [DOI] [Google Scholar]
- Dybtsev D N, Chun H. and Kim K. Angew. Chem. Int. Edn. 2004;43:5033. doi: 10.1002/anie.200460712. [DOI] [PubMed] [Google Scholar]
- Suh M P, Cheon Y E. and Lee E Y. Chem. Eur. J. 2007;13:4208. doi: 10.1002/chem.200601534. [DOI] [PubMed] [Google Scholar]
- Wei Q, Nieuwenhuyzen M, Meunier F, Hardacre C. and James S L. Dalton Trans. 2004:1807. doi: 10.1039/b404485a. [DOI] [PubMed] [Google Scholar]
- Horike S, Matsuda R, Tanaka D, Matsubara S, Mizuno M, Endo K. and Kitagawa S. Angew. Chem. Int. Edn. 2006;45:7226. doi: 10.1002/anie.200603196. [DOI] [PubMed] [Google Scholar]
- Ferey G, Mellot-Draznieks C, Serre C, Millange F, Dutour J, Surble S. and Margiolaki I. Science. 2005;309:2040. doi: 10.1126/science.1116275. [DOI] [PubMed] [Google Scholar]
- Lu J Y. and Babb A M. Chem. Commun. 2002:1340. doi: 10.1039/b200213b. [DOI] [PubMed] [Google Scholar]
- Kitagawa S, Kitaura R. and Noro S. Angew. Chem. Int. Edn. 2004;43:2334. doi: 10.1002/anie.200300610. [DOI] [PubMed] [Google Scholar]
- Wang X-L, Qin C, Wang E-B, Xu L, Su Z-M. and Hu C-W. Angew. Chem. Int. Edn. 2004;43:5036. doi: 10.1002/anie.200460758. [DOI] [PubMed] [Google Scholar]
- Horike S, Tanaka D, Nakagawa K. and Kitagawa S. Chem. Commun. 2007:3395. doi: 10.1039/b703502k. [DOI] [PubMed] [Google Scholar]
- Kittel C. New York: Wiley; 1995. Introduction to Solid State Physics. [Google Scholar]
- Tritt T M. Berlin: Springer; 2005. Thermal Conductivity: Theory, Properties, and Applications. [Google Scholar]
- Huang B L, McGaughey A J H. and Kaviany M. Int. J. Heat Mass Transfer. 2007;50:393. doi: 10.1016/j.ijheatmasstransfer.2006.10.002. [DOI] [Google Scholar]
- Huang B L, Ni Z, Millward A, McGaughey A J H, Uher C, Kaviany M. and Yaghi O. Int. J. Heat Mass Transfer. 2007;50:405. doi: 10.1016/j.ijheatmasstransfer.2006.10.001. [DOI] [Google Scholar]
- Kitaura R, Seki K, Akiyama G. and Kitagawa S. Angew. Chem. Int. Edn. 2003;42:428. doi: 10.1002/anie.200390130. [DOI] [PubMed] [Google Scholar]
- Fletcher A J, Cussen E J, Bradshaw D, Rosseinsky M J. and Thomas K M. J. Am. Chem. Soc. 2004;126:9750. doi: 10.1021/ja0490267. [DOI] [PubMed] [Google Scholar]
- Li D. and Kaneko K. Chem. Phys. Lett. 2001;335:50. doi: 10.1016/S0009-2614(00)01419-6. [DOI] [Google Scholar]
- Kubota Y, Takata M, Matsuda R, Kitaura R, Kitagawa S. and Kobayashi T C. Angew. Chem. Int. Edn. 2006;45:4932. doi: 10.1002/anie.200600976. [DOI] [PubMed] [Google Scholar]
- Matsuda R. et al Nature. 2005;436:238. doi: 10.1038/nature03852. [DOI] [PubMed] [Google Scholar]
- Halder G J, Kepert C J, Moubaraki B, Murray K S. and Cashion J D. Science. 2002;298:1762. doi: 10.1126/science.1075948. [DOI] [PubMed] [Google Scholar]
- Seki K. Phys. Chem. Chem. Phys. 2002;4:1968. doi: 10.1039/b110899a. [DOI] [Google Scholar]
- Reineke T M, Eddaoudi M, Moler D, O'Keeffe M. and Yaghi O M. J. Am. Chem. Soc. 2000;122:4843. doi: 10.1021/ja000363z. [DOI] [PubMed] [Google Scholar]
- Chen B, Eddaoudi M, Hyde S T, O'Keeffe M. and Yaghi O M. Science. 2001;291:1021. doi: 10.1126/science.1056598. [DOI] [PubMed] [Google Scholar]
- Maji T K, Matsuda R. and Kitagawa S. Nat. Mater. 2007;6:142. doi: 10.1038/nmat1827. [DOI] [PubMed] [Google Scholar]