

Abstract
The controlled fabrication of nanometer-scale objects is without doubt one of the central issues in current science and technology. However, existing fabrication techniques suffer from several disadvantages including size-restrictions and a general paucity of applicable materials. Because of this, the development of alternative approaches based on supramolecular self-assembly processes is anticipated as a breakthrough methodology. This review article aims to comprehensively summarize the salient aspects of self-assembly through the introduction of the recent challenges and breakthroughs in three categories: (i) types of self-assembly in bulk media; (ii) types of components for self-assembly in bulk media; and (iii) self-assembly at interfaces.
Keywords: self-assembly, nanomaterials, interfaces, supermolecules, bottom–up
Introduction
Technologies involving nanometer-scale objects continue to improve the quality of our daily lives because the down-sizing of functional units can result in a significant decrease in device energy consumption and more efficient production processes. In addition, novel phenomena have been revealed at the nanometer-scale. Shrinking component size advances nanotechnology, while the related phenomena represent nanoscience. Therefore, the regimented fabrication of nanometer-scale objects is undoubtedly one of the central issues in current science and technology. To date, several excellent top–down-type approaches including photolithography and electron-beam lithography have been used to produce nanostructures. However, in addition to the inherent size, parallelization, and 2-dimensional limitations of lithography and of top–down approaches in general, these relatively new patterning methods lack the well-developed technology that enabled the previous generations of lithography. Because of these challenges to traditional bottom–up lithography, interest has been growing in an alternate means, known as the bottom–up approach. Methods of bottom–up fabrication rely on molecular self-assembly in supramolecular processes. Supramolecular chemistry, which was originally a branch of fundamental science, has now become an important concept in nanotechnology.
Although numerous excellent articles and reviews of independent topics have been published [1–10], a comprehensive review of self-assembly is rare. Therefore, this review article aims to illustrate and summarize all the aspects of self-assembly through a description of recent challenges and breakthroughs. This review describes self-assembly from three different perspectives: (i) types of self-assembly in bulk media; (ii) types of components for self-assembly in bulk media; (iii) self-assembly at interface. In each section, we have tried to select the most appropriate examples and complemented those selections with a comprehensive range of cited research. Despite this categorization the examples contained in (i)–(iii) are actually interrelated. However, here we have attempted to make self-contained descriptions of each subject so that the reader can concentrate on sections relevant to his/her interest.
Self-assembly in bulk media 1; assembly type
Small molecule assembly
There are many types of self-assembly ranging in dimensions from molecular level (nanometric) to macroscopic size (visible to the naked eye). In this section, the smallest oligomolecular self-assemblies, consisting of just a few molecules will be described. Surprisingly, several of them exhibited molecular-level machine-like functions [11–14].
Aida and coworkers designed and developed porphyrin conjugates that induce self-assembly of external guests and possess functions related to their structures. For example, they reported that dynamic host–guest supramolecular interactions resulted in stereochemical harmonization of helical chains contained in the host molecule [15]. In the host structures depicted (figure 1, Host and Host (Phe)), two zinc porphyrin units are connected by two oligo (aminoisobutyric acid) chains, which adopt helical conformations because of the steric requirements of the C(CH3)2 groups and despite their lack of chiral centers. Therefore, thermodynamic interconversion of the chains results in a mixture of right- and left-handed helices. Binding of a pyridyl-substituted helical guest containing an (L)-leucine residue at its center generated an intense chiroptical signal through dynamic stereochemical harmonization between the helical chains. From the sign of the split Cotton effects it is apparent that the oligopeptide chains of the guest-complexed host molecules adopt the same helical conformation as that of the guest. This phenomenon demonstrates that self-assembly through binding of an appropriate guest modulates the dynamics of the helix structures in the host.
Figure 1.

Stereochemical harmonization based on guest binding to chromophoric cyclic hosts with two zinc porphyrin units and two oligo (aminoisobutyric acid) chain connectors.
In other work, Tashiro, Aida, and coworkers reported preferential assembly of hetero-guest pairs by an inter-guest electronic communication through a π-conjugated host molecule (figure 2) [16]. This host compound contained two fused zinc porphyrin dimers bridged by aliphatic chains giving a cofacial arrangement of dimers, and which displayed a strong negative cooperativity in the binding of 4,4′-bipyridine (bpy). Similarly, binding of a second fullerene C60 guest to the host was subject to negative cooperativity despite the easy accommodation of a single C60 guest by the host. The C60 dimer, C120, may also be bound by this host molecule suggesting that from a spatial point-of-view alone, the host should be able to accommodate two C60 molecules. Thus, it was concluded that the negative cooperativity in the binding of the C60 guest is due to a conflicting electronic interaction. On the other hand, the same cyclic host could selectively include one each of C60 and bpy. Equimolar binding of C60 and TMHDA (N,N,N′,N″-tetramethylhexane-1,6-diamine) was due to a similar positive heterotropic cooperativity. Peculiarities in the assembly of these host–guest complexes are probably due to electronic communication occurring between the C60 and diamine guests and mediated by the π-conjugated fused metalloporphyrin arrays. Apart from this prime example, several other new concepts including chiral fullerene detection [17], chemical friction during rotary guest motion [18], and supramolecular polychromatic thermometers [19] have been proposed by the same research group, all by using porphyrin conjugate molecules.
Figure 2.

Preferential assembly of hetero-guest pairs by electronic communication between the two guests through a p-conjugated host molecule.
Fujita and coworkers have performed pioneering research on assembly of coordination capsules using square-cornered Pd(II) complexes and designer co-ligands [20]. For example, assembly of a stable, nanosized capsule from 18 metal ions and six triangular ligands was achieved [21]. The molecular capsule, comprised of six edge-sharing triangles with two metal ions at each edge, is approximately hexahedral with a fully closed 0.9 nm3 internal space and is impervious to all but very small molecules. More recently the same authors reported the ability to control the Diels–Alder reaction of certain substrates within the molecular capsules (figure 3) [22]. The Diels–Alder reaction between anthracene and phthalimide yields two major products. However, if performed within the molecular capsule, the process preferentially yields the product due to reaction of phthalimide at the 1,4 position of anthracene. The site of reaction varies from that observed in the solution state. Use of a half-bowl shaped molecular capsule gave the expected product, as a result of reaction at 9,10 positions of anthracene, although catalyst-like turnover was confirmed. Reaction mediation by the capsule framework could be also demonstrated by mediation of alkane oxidation through the photochemical excitation of a molecular capsule [23].
Figure 3.

Control of a Diels–Alder reaction within pre-designed molecular capsules.
The main advantage of this strategy lies in the versatility of morphology design since an appropriate choice of ligand molecules can result in unique nano-objects of various shapes, such as coordination nanotubes [14, 25], nanoballs [26], nanoboxes [27], and self-assembled cages [28]. These self-assembled objects provide nanospaces where new physical chemistries might be observed. For example, molecular ice formation [29] or color sensing of guest inclusion [30] have been investigated. Coating of self-assembled capsules and aggregate formation with multivalent proteins [31] has also been demonstrated.
Capsules and cages formed through self-assembled processes have been investigated by other research groups. Crego-Camala, Reinhoudt, and coworkers reported calix[4]arene-based molecular boxes and their guest binding capabilities [32]. Atwood and coworkers prepared supramolecular capsules by using large numbers of hydrogen bonds. They reported a chiral spherical molecular assembly held together by 60 hydrogen bonds, consisting of six calix[4]resorcinarenes and eight water molecules [33], and later reported molecular capsules containing 48 intermolecular hydrogen bonds which were stable in polar media [34]. Rebek and coworkers developed molecular capsule formation through self-assembly of cavitands [35–37]. For example, they reported an unusual hetero-guest inclusion by a hydrogen-bonded molecular capsule, with preferential accommodation of a molecule each of benzene and p-xylene [38]. Their self-assembled capsule also provides a medium for stabilization of reactive intermediates [39], to shift the equilibrium between two stereoisomers [40], or to regulate the conformation of a guest species [41]. They expanded their self-assembled cavitand capsules by the addition of appropriate spacers, permitting variation of guest selectivity, while also demonstrating attachment of a rotating door unit to the cavitands [42, 43]. They synthesized an enzyme reaction cavity mimic [44]. Kobayashi and coworkers reported selective formation of self-assembling homo- or hetero-cavitand cages [45]. Stang and coworkers reported synthesis of a cuboctahedral capsule from twenty subunits in a single-step self-assembly [46]. Using similar methods a molecular capsule with a trigonal prismatic framework was prepared by means of spontaneous self-assembly of a predesigned molecular clip with tritopic pyridyl subunits [47]. Recently, Shionoya and coworkers reported molecular switching between fluorescent coordination capsule and nonfluorescent cage [48].
Various components and different interactions have been used in the formation of capsules, cages and closed squares, in which several novel phenomena have been observed. For example, Candau, Lehn, and coworkers reported gel formation through formation of self-assembled G (guanine)-quartets, which are strongly dependent on the nature of both the binding cation and the decorating aldehyde (figure 4) [49]. The interaction of the decorating aldehyde is of particular interest since it indicates a possible method to control the supramolecular self-assembly process by dynamic covalent decoration. Pluth et al reported unusual basicity changes of amines encapsulated in a self-assembled coordination cavity [50]. Mirkin and coworkers demonstrated reversible interconversion of homochiral macrocycles and helical coordination polymers [51]. Severin and coworkers reported formation of an expanded helicate from a ruthenium complex and a piperazine-bridged bis(dihydroxypyridine) ligand, which is able to bind phosphate and acetate anions in aqueous solution at neutral pH [52]. Ballester and coworkers reported solid-state self-assembly of a calix[4]pyrrole-resorcinarene hybrid into a hexameric cage [53]. On–off switching of self-assembly through dynamic covalent bond formation between boronic acids and alcohols has been demonstrated by Iwasawa and Takahagi [54]. Inclusion of fullerene into self-assembled capsules is an attractive research topic. Beer and coworkers proposed use of a polymetallic resorcinarene host for binding of fullerene [55]. Inclusion of metallofullerene into a cavitand-based self-assembled cage was realized by Pirondini et al [56]. Haino and coworkers reported fullerene encapsulation by calix[5]arene [57]. They also reported encapsulation of various guests within similar hosts [58]. A unique inclusion complexation was proposed by Inomata and Konishi who demonstrated preparation of a hexaporphyrin cage using a gold nanocluster template [59].
Figure 4.

G-quartet formation upon binding of cation and a decorating aldehyde.
Catenanes and rotaxanes make up a family of compounds that are exceptional in the world of supramolecular chemistry. Rotaxanes consist of molecular rings threaded by molecular wires that have stoppers at both ends to prevent unthreading. Of the various cyclic molecules used as the molecular ring in rotaxanes, cyclodextrin and crown ethers are the most useful because of their self-assembling nature based on molecular recognition properties. In particular, cyclodextrin can accommodate linear polymers such as polyethylene glycol in an aqueous phase. Harada and coworkers reported that polymer chains thread cyclodextrin rings upon the addition of a water-soluble polymer into an aqueous cyclodextrin solution (figure 5A) [60–62]. Subsequent stoppering of the polymer results in the formation of rotaxanes. The cyclodextrins self-align in a head-to-head (tail-to-tail) array, stabilized by hydrogen bond formation between primary hydroxyl groups and between secondary hydroxyl groups. While rotaxanes are composed of wires and rings, catenanes consist of two or more interlocked rings. The example shown in figure 5B illustrates catenane preparation through tetrahedral coordination of Cu(I) [63]. In the first process, two phenanthroline ligands coordinate to Cu(I). Each ligand is individually cyclized through hydroxyl groups attached to the ligand unit. Removal of Cu(I) completes catenane formation. Syntheses of molecular knots and doubly locked catenanes based on a similar strategy have also been reported [64]. Furthermore, molecules with five interlocked rings (similar to the symbol used for the Olympic Games) have also been synthesized [65].
Figure 5.
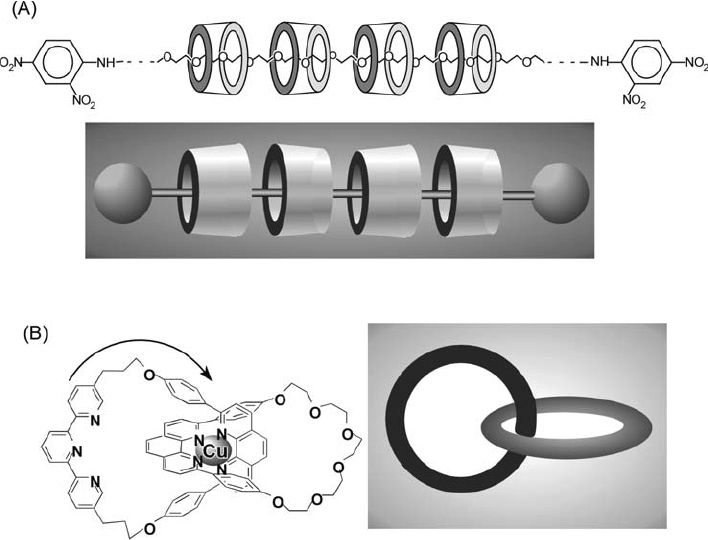
Typical examples of (A) a rotaxane and (B) a catenane.
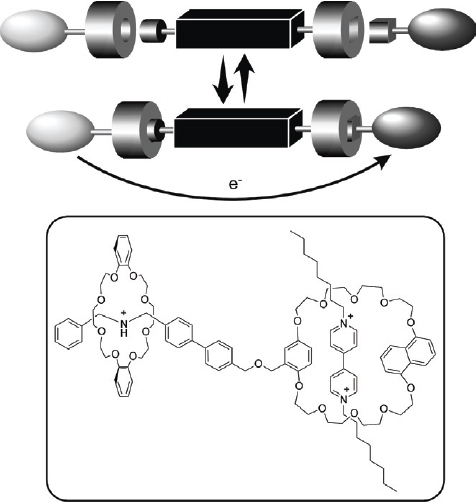
Rotaxanated or catenated objects are often considered as molecular machines. Rotaxane structures are attractive entities for the design of molecular devices because the relative positions of their component parts can be influenced by external stimuli. Controlling the relative position of the rings on the wire permits design of molecular shuttles [66]. In the example shown in figure 6, a rotaxane exhibits translational isomerism with the tetracationic cyclophane ring preferentially occupying the π-electron-rich benzidine station (upper). Electrochemical oxidation of the benzidine unit to the monocationic radical, results in translation of the cyclophane ring to the biphenol station (lower), driven by electrostatic repulsion. Shuttling of the cyclophane is completely reversible depending on the oxidation state of the benzidine unit. Stoddart, Credi, and coworkers used this process to realize preparation of a molecular elevator as illustrated in figure 7 [67, 68]. The design of the molecular elevator relies on two mechanically interlocked molecules that incorporate the features of a pH-switchable [2] rotaxane. At low pH the CH2NH2 sites bind the macrocycles preferentially while at high pH deprotonation occurs disrupting hydrogen bonding so that the macrocycles move to the bipyridinium dication sites, because of stabilization by π–π stacking interactions. These mechanically interlocked molecules were assembled from a trifurcated ‘rig-like’ component containing thread components of three [2] rotaxanes each fused at alternate positions (1,3,5) of a benzenoid core. This rig-like component is mechanically interlocked with a platform based on a tritopic receptor, containing three oligo-ether macrocycles fused to a hexaoxatriphenylene core.
Figure 6.

A molecular shuttle.
Figure 7.

A molecular elevator.
Very recently, Stoddart and coworkers presented a molecular plug–socket connector [69]. Figure 8 illustrates the three components of the molecular level plug–socket connector: (i) a secondary dialkylammonium center which plays the role of a plug for dibenzo[24]crown-8; (ii) a rigid and conducting biphenyl spacer; (iii) 1,4-benzo- 1,5-naphtho[36]crown-10 capable of behaving as a socket for a 4,4′-bipyridinium dicationic plug. Two connections of the three-component assembly were shown to be reversibly controllable by using external acid/base and red/ox inputs. These results represented a key step in the design and construction of a self-assembling supramolecular system in which a molecular electron source could be connected to the molecular electron drain through a molecular elongation cable. Similarly, photoinduced electron flow in a self-assembling supramolecular extension cable has also been reported [70]. In one unique usage of a rotaxane, the same research group reported tethering of pseudorotaxanes at the entrances of the cylindrical pores of mesostructured silica thus creating nanovalves or gates capable of trapping luminescent molecules at the mesopore interior, and able to release them on demand (figure 9) [71]. For the [2] pseudorotaxane gatekeeper structure, a tethered l,5-dioxynaphthalene-containing derivative, acted as the gatepost, and cyclobis(paraquat-p-phenylene), which recognizes dioxynaphthalene units, served as the gate controlling access in and out of the nanopores. Luminescent molecules could be released by introduction of an external reducing reagent, NaCNBH3, which opens the nanovalve. The same research group also demonstrated reversible electrochemical operation of a nanovalve [72]; i.e. molecules could be trapped and released from the maze of nanoscopic passageways in silica by controlling the state of redox-activated bistable[2]rotaxane molecules tethered at the of surface nanopore openings of the nanoscale reservoir.
Figure 8.
Molecular plug–socket connector.
Figure 9.

Pseudorotaxane gates at the entrances of cylindrical pores.
Apart from these examples, a great variety of machine-like self-assembled objects have been developed by Stoddart and [73–75]. Cyclodextrin-based rotaxanes have also seen recent developments [76–82]. Use of other components such as crown ethers [83–85] and fullerenes [86] as components of rotaxane structures have been demonstrated.
Self-assembled structures with few components have been widely reported and some so far unlisted recent examples are also available [87–90]. However, we would like to here mention one important subject of this category. Appropriate molecular design of a host structure and self-assembly with a target guest sometimes induces a specific material conversion similar to that observed for enzyme function so that these examples are often referred to as artificial enzymes [91–96]. Self-assembly operation of functional groups can be observed in naturally-occurring enzyme mechanisms. For example, RNA hydrolysis by ribonuclease, as depicted in figure 10A, is achieved through sequential interaction with self-aggregated amino acid residues. By analogy with this and other similar systems, molecular spaces provided by self-assembly have been employed as artificial enzymes. Molecular spaces for artificial enzymes are designed to recognize specific substrate molecules, stabilize reaction intermediates, and provide reactive groups in appropriate proximity to the trapped substrate molecules. The example shown in figure 10B is of a molecular-cavity-type artificial enzyme. Desper and Breslow designed a cyclodextrin host carrying two imidazole moieties that can hydrolyze a model substrate [97]. Inclusion of the hydrophobic part of the cyclic phosphodiester (the model substrate) at the core of the cyclodextrin disposed the phosphodiester between the two histidines. Activation of a water molecule through deprotonation by the neutral histidine (left), was followed by nucleophilic attack by the activated water at a phosphate group. Protonation of the phosphodiester by protonated histidine (right) assisted this nucleophilic attack. The maximum activity of the artificial ribonuclease was obtained at around the pKa of the histidines (ca. pH 7) because of the essential cooperative function of the neutral and protonated histidines. Figure 10C illustrates a molecular-cleft-type artificial enzyme, which was developed by Anslyn and coworkers, where guanidinium residues connected to a rigid backbone could immobilize an RNA phosphodiester moiety [98–100]. Imidazole in solution activated 2′-OH groups with subsequent nucleophilic attack at the phosphate group. Electrostatic hydrogen bonding with the guanidinium groups stabilized the transition state during hydrolysis. The reaction rate of RNA cleavage was increased by this stabilization.
Figure 10.

(A) Functional relay in ribonuclease; (B) molecular-cavity-type artificial enzyme; (C) molecular-cleft-type artificial enzyme.
Porous crystals by metal coordination and hydrogen bonding
Self-assembly processes based on directionally-defined interactions such as metal cation coordination or hydrogen bonding can be used for syntheses of materials with precise nanostructures. In particular, rigid porous frameworks have been constructed through a coordination-type self-assembly process. Some of these are known as metal organic frameworks (MOFs) and/or coordination polymers. As illustrated in figure 11A, appropriate selection of organic ligands can lead to regular porous structures with various sizes and geometries. For example, Yaghi and coworkers extensively developed MOF families using oxo-bridged coordination complexes and organic ligands (figure 11B) [101]. Because this research field has been recently rapidly expanding, only a brief outline with several highlighted topics will be presented here. Related research has been adequately described in several excellent reviews [102, 103].
Figure 11.
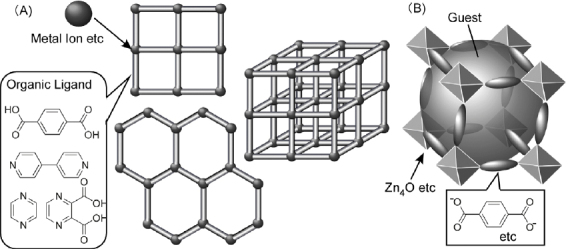
(A) general structure of metal organic frameworks (MOFs); (B) MOF with zinc oxide complex and organic ligands.
Yaghi and coworkers have extensively developed novel types of MOF structures and some of them have even reached commercialization. They recently reported preparation of three-dimensional covalent organic frameworks [104]. These materials are anticipated to have excellent capacities for material storage. Yaghi and coworkers reported use of MOFs for storage of gaseous guests such as methane [105] and hydrogen [106].
Kitagawa and coworkers have developed various coordination polymers and investigated the unusual properties of trapped guest molecules [107]. They reported the direct observation of dioxygen molecules physisorbed in the nanochannels of a microporous copper coordination polymer [108]. The one-dimensional nanochannels of the coordination polymer result in a confinement effect and restricted geometry leading to a specific molecular assembly. The one-dimensional ladder structure of O2 dimers thus obtained is unlikely to exist as a bulk fluid and/or solid. The same research group reported that a combination of framework-builder (Cu(II) ion and 4,4′-bipyridine ligand) and framework-regulator (AF6 type anions; A=Si, Ge, and P) provided a series of novel porous coordination polymers [109]. Immersion of some of the coordination polymer compounds in water stimulated a conversion of three-dimensional networks to interpenetrated networks (two-dimensional interpenetration). The latter network exhibited unique dynamic anion exchange properties, concurrent with drastic structural conversions.
Kitagawa and coworkers also reported high levels of selective sorption of acetylene molecules, relative to the similar molecule carbon dioxide, onto the functionalized surface of some coordination polymers [110]. Hydrogen bonding between two non-coordinated oxygen atoms of the framework material and the two hydrogen atoms of the acetylene molecule resulted in adsorption of acetylene molecules with a regular periodicity in the structure. Using this phenomenon at room temperature, acetylene could be stored at a density 200 times greater than the safe compression limit of free acetylene. Figure 12 illustrates the controlled and selective polymerization of substituted acetylenes in one-dimensional specific nanochannels containing basic carboxylate oxygen atoms as catalytic interaction sites on the pore walls [111].
Figure 12.

Controlled and selective polymerizations of substituted acetylenes in one-dimensional specific nanochannels of a coordination polymer.
Specific interactions and physical phenomena have also been investigated in these structures. Kitagawa and coworkers found an efficient photo-induced charge transfer between confined electron donor guests and anthracene moieties of a porous coordination framework [112]. Hanton and coworkers reported anion encapsulation through anion-π interactions within cordination polymers prepared from Ag(I) salts and a pyrimidine ligand [113]. Molnár, Bousseksou, and coworkers proposed a method for patterning of spin-crossover coordination polymers [114]. Selective material adsorption such as kinetic separation of hexane isomers by Rodrigues, Chen, and coworkers is also an interesting research target for this kind of material [115]. Fundamental studies using the BET method for surface area determination [116] and gas sorption behaviors [117] have also been researched.
Hydrogen bonding can result in formation of self-assembled porous solids. Aoyama and coworkers investigated formation and guest inclusion phenomena of porous hydrogen-bonded networks composed of bisresorcinol derivatives [118]. As shown in figure 13, hydroxyl groups in the derivatives can form hydrogen bonds of specific geometry, resulting in inclusion of appropriate guests. Since design of components for the network structures is not limited an acridinylresorcinol could also be used as a self-complementary building block in formation of a robust hydrogen-bonded two-dimensional network [119]. Guest inclusion properties were statically and kinetically monitored, and host-only cast films of the bisresorcinol on a quartz crystal microbalance were used for analyses [120, 121]. Very recently, Dalrymple and Shimizu reported optimization of both metal complex and organosulfonate pillars enabling an idealized hexagonal hydrogen bonding motif, which in turn provided a permanently porous solid constructed exclusively by charge-assisted hydrogen bonds [122].
Figure 13.

Porous hydrogen-bonded networks composed of bisresorcinol derivatives.
Hydrogen-bonded porous crystals offer a medium for reaction control as can be seen in molecular capsules and coordination polymers. Aoyama and coworkers demonstrated that an anthracene-bisresorcinol derivative, when used as an organic network material, showed a novel catalysis of the acrolein-cyclohexadiene Diels–Alder reaction in the solid state [123–125]. In that reaction, the two reagents have differing polarities and so could be assembled within the same cavity. The subsequent intracavity reactions exhibited high stereoselectivities as well as remarkable rate enhancements. Depending on their structures, the products were either retained or exited the cavities, resulting in either deactivation of the catalyst or turnover, respectively.
Generally speaking, these porous crystals provide well-defined nanospaces, which can be adjusted by appropriate selection of organic ligands and interactive modules (hydrogen bond unit and coordination metals). Therefore, unknown sciences for molecules and supermolecules entrapped within the formed nanospaces will be thoroughly investigated. Supermolecular chemistry in dimension-controlled spaces would become a new challenges.
Apart from the porous crystalline materials, well designed hydrogen bonding arrays provide other types of self-assembled objects. One representative example is nanotube formation by designer cyclic peptides prepared by Ghadiri and coworkers [126, 127]. These self-assembled materials can operate as artificial membrane channels. They are one example amongst a variety of bio-related materials such as peptides, saccharides, proteins, and nucleic acids that can be used as molecular modules for hydrogen-bond-based self-assembled structures. These topics are described later in this review.
Lipid assembly: liposome, vesicle, fiber, tube
Distinct from the direction-defined interactions such as metal coordination and hydrogen bonding are the solvophilic and solvophobic effects available in appropriate media. Therefore, the self-assembled structures based on these effects should have dynamic nature, which is reflected in both dynamical changes of assembled structures and mechanical flexibility of macroscopic shapes. Typical examples are liposomes and vesicles formed by self-assembly of lipids or related amphiphilic compounds. The amphiphilic natures of the naturally-occurring lipids, including phospholipids and glycolipids (figure 14A), and certain types of synthetic lipids (figure 14B) often result in formation of lipid bilayer structures in aqueous phases [128]. The lipid bilayer structure extends two-dimensionally and forms the ‘skin’ of a closed sphere that contains an aqueous pool (figure 14, upper). This capsule-like structure can be thought of as a simplified model of a cell. The term liposome is coined when the constituent components are naturally-occurring lipids. Kunitake and Okahata found that artificial compounds possessing an appropriate structural balance between hydrophilic and hydrophobic moieties can form similar spherical objects [129], which are usually referred to as vesicles. Examples of artificially designed amphiphiles are shown in figure 14B. A dialkyl structure is often used for the tail, but trialkyl structures, tetraalkyl structures and azobenzene-type rigid structures are also available [130–132].
Figure 14.

Liposome or vesicle structures prepared from: (A) naturally-occurring lipids; (B) synthetic lipids.
Liposomes and vesicles are often proposed for use in biological applications such as drug delivery [133–136] because of their structural similarity to biomembranes. They also provide a medium for studying basic biological phenomena such as the flip-flop motion within lipid bilayers [137] and membrane fusion [138, 139]. Various mimics of biologically important functions such as ion-channeling [140], photosynthetic processes [141], and proton gradient formation [142] have been constructed using these artificial cell membranes. Other artificial systems such as vesicle-based rechargeable batteries have also been proposed [143].
In spite of substantial developments in the science and technology of liposomes and vesicles, several features of the systems still rely on traditional concepts of amphiphile design and useful medium (still usually an aqueous solution). Therefore, the conceptual expansion in molecular design of amphiphiles that are capable of forming vesicilar structures under unusual conditions should be a breakthrough development. For example, Katagiri et al developed a novel concept for fusion of an inorganic framework with lipidic vesicles. They investigated the covalent linkage of a siloxane framework to a lipid bilayer vesicle [144–146]. The resulting vesicles had a siloxane network covalently attached to the bilayer membrane surface and are referred to as cerasomes (ceramics + soma) (figure 15A). Initial dispersion of the alkoxysilane-bearing amphiphiles in an acidic aqueous solution was achieved using a vortex mixer. This translucent sample was morphologically stable over long periods. The formation of the siloxane bonds was confirmed by infrared spectroscopy, which indicated the presence of Si–O–Si and Si–OH groups. Formation of vesicular structures was confirmed by using transmission electron microscopy (TEM) where images of multi-lamellar cerasomes with a bilayer thickness of about 4 nm and vesicular diameter of 150 nm were obtained (figure 15A). Interestingly, TEM images of vesicular aggregates were observed in the same specimen (figure 15B). Suppression of the collapse and fusion of the cerasomes is implied from the fact that some of the aggregates maintained their original spherical structure, probably through formation of an intra- and intermembrane siloxane network. This indicates that a multi-cellular model could be obtained by association of these stable cerasomes.
Figure 15.

(A) Cerasome structure and (B) cerasome assembly.
Kimizuka and coworkers developed the formation of aqueous bilayer structures from separate components linked through hydrogen bonding (figure 16) [147]. Thus, alkyl tails were connected to a melamine unit and a polar ammonium head was linked to a cyanuric acid moiety. The melamine and cyanuric acid parts formed infinitely linked ribbon structures by complementary hydrogen bonding, thus leading to a bilayer structure. By using this concept, individually synthesized hydrophobic and hydrophilic components can be combined with a great degree of freedom.
Figure 16.

Aqueous bilayer structures from separated components through hydrogen bonding.
For expansion of vesicle technology to non-aqueous media, Kunitake and coworkers recognized the immiscibility of fluorocarbon components in general organic solvents and subsequently synthesized compounds possessing both hydrocarbon and fluorocarbon segments. These were then studied for their self-assembly behaviors in nonaqueous media (figure 17) [148]. Using appropriate solvents, some compounds containing both fluorocarbon and hydrocarbon parts formed bilayer-like assemblies. The low affinity that the fluorocarbon part has for the organic solvent leads to the label solvophobic, and is in contrast to the solvophilic characteristics exhibited by the hydrocarbon parts. These conceptually novel amphiphilic molecules assembled so that the solvophobic component is hidden inside the assembly while the solvophilic component is exposed to the solvent. Bilayer structures are obtained if there is a good structural balance between the solvophilic and solvophobic parts. Although studies of lipid bilayers and vesicle formation were originally initiated in order to mimic cell membranes in aqueous media, appropriate molecular design has extended this concept to a wide range of molecules. In further advanced developments, Nakashima and Kimizuka demonstrated formation of a vesicle structure using dialkyldimethylammonium salts in ionic liquid media, known as the ‘vesicle in salt’ [149].
Figure 17.

Self-assembling behavior of compounds possessing both hydrocarbon and fluorocarbon segments in nonaqueous media.
Bilayer membrane structures can provide a fluidic environment where immobilized components can function as relays. For example, Kikuchi and coworkers demonstrated logic-gate-like activity control of an enzyme self-assembled on lipid bilayer structures [150–153]. Figure 18 displays a functional relay at an artificial thin film between an amphiphilic amine receptor and an effector lactate dehydrogenase (LDH). LDH activity is inhibited by the Cu ion so it is in the OFF state. Subsequent addition of an appropriate signal molecule (1-hydroxy-2-naphthaldehyde) removes the Cu ion from LDH and forms a signal-receptor complex (Schiff's base). This removal of the Cu ion reactivates the LDH, restoring the ON state. Photo-regulation of the LDH activity could also be achieved through receptor photo-isomerization. The association behavior of the receptor molecules and binding ability toward the Cu ion changes upon photo-isomerization of the azobenzene moiety. LDH activity was completely suppressed for the cis-isomer system resulting from visible light irradiation (OFF state); afterwards, the high-level of activity for the system (ON state) could be restored by UV irradiation, which gives the trans-isomer. This system can be regarded as a switching device where a primary chemical switching signal, is followed by reversible switching of enzyme activity by using a photosignal. It can also operate as a logic device since activation of the enzymatic reaction (AND-type logic gate) requires appropriate application of both chemical and photo-signals. This is a primitive example but it might provide inspiration for construction of functional relays contained within artificial thin films.
Figure 18.

Logic-gate-like activity control of an enzyme self-assembled at a lipid bilayer membrane.
Lipid design allows us to introduce various functional groups into fundamental hydrophilic/hydrophobic amphiphile structures. In particular, the introduction of a hydrogen-bond-forming unit into an amphiphilic structure often leads to formation of higher-order structures with unique shapes through self-assembly processes [154–156], as illustrated in figure 19. These structures include disc-like micelles and rod-like supermolecules. Further development of micelle and vesicle structures sometimes results in sheet structures consisting of a bilayer unit. When the sheet has curvature, helical ribbons and tubules are formed by twisting and rolling of the sheets. This flexibility in structural formation is highly advantageous for the bottom–up fabrication of nano- and micro-objects.
Figure 19.
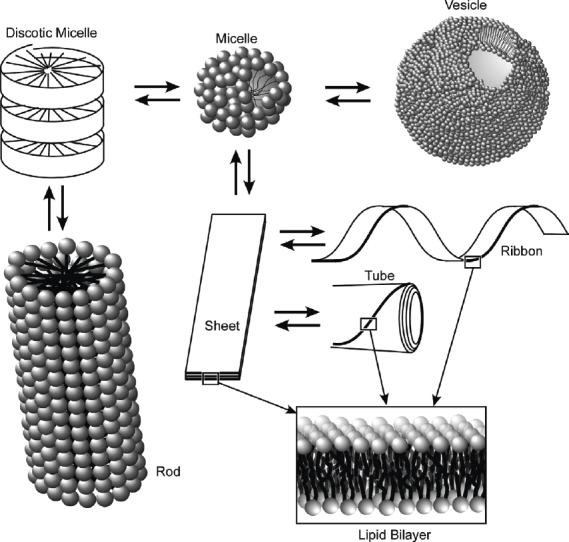
Formation of higher order structures of amphiphile assemblies with unique shapes.
Typical examples are described in the following few figures. Yamada, Ariga, and coworkers exercised control over the morphologies of self-assembled structures using lipids with a tripeptide moiety [157–163]. These peptide lipids can form aggregates not only in water but also in nonpolar organic solvents forming β-sheet structures. Infrared spectra of tripeptide derivatives used in their research in CCl4 solutions contain the amide A, amide I, and amide II bands at ca. 3300, ca. 1630, and ca. 1540 cm−1, respectively, and no peak at ca. 1690 cm−1 indicating that these derivatives form parallel β-sheet structures. The TEM image shown in figure 20A indicates the presence of thin fibers, which are aligned in a parallel arrangement. Self-assembled structures observed in cast films of the peptide lipids obtained from CCl4 solutions are different from those conatined in the films prepared from aqueous solution. AFM images of the cast films from CCl4 solution are shown in figure 20B. Incubation time strongly influences the structure of the fibers. In films prepared immediately after dispersion, formation of numerous entangled thin fibers of widths 20–40 nm is observed. Conversely, thick fibers or rods with widths of 300–400 nm were found in films prepared following a 3-day incubation of the solution. Figure 20C shows the proposed self-assembly mechanism of the peptide lipids in CCl4 solution and in their cast films. Parallel β-sheet structure formation together with exposure of the hydrophobic tails to the nonpolar solvent requires formation of a fibrous structure bearing the cross section of a single or a multiple reversed micelle. Following an appropriate incubation time the fibers become aligned as is apparent from the TEM image. When the solvent is evaporated, fusion of the aligned fibers through solidification of the alkyl chains, results in the broader structure.
Figure 20.
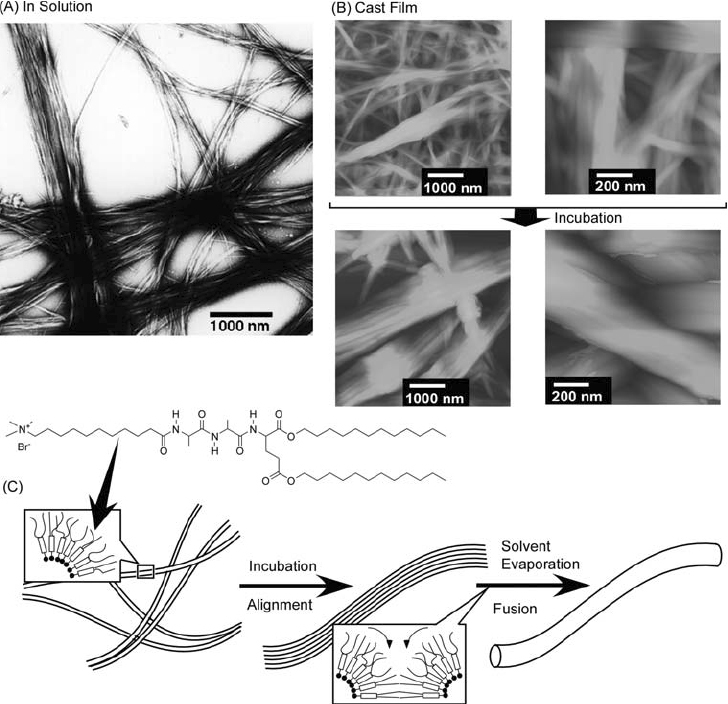
(A) Self-assembled structures of peptide lipids in CCl4; (B) self-assembled structures in cast films; (C) proposed self-assembly mechanism of peptide lipids. © 2000, American Chemical Society, Langmuir 16 (2000) 4929.
A slight increase in the solvent polarity for self-assembly results in cast films that differ substantially from those observed from CCl4. Figure 21 shows AFM images of cast films obtained from CHCl3 solutions of tripeptide-containing lipids. All films possessed a reasonably flat texture with specific patterns. Infrared data for these films indicate that the tripeptide group is in a non-interacting state in CHCl3 solution so that the morphologies observed by AFM were gradually assembled during solvent evaporation. Inspired by this flat film formation, Yamada and coworkers significantly strengthened their self-assembled supramolecular films by utilizing the interaction between oligoleucine side chains [164]. Figure 22 shows preparation of self-standing thin films of a Leu–Leu–Leu–Glu-type peptide lipid by casting and drying from CHCl3 solution. Films prepared by this method are robust and can be manipulated using tweezers or folded without fracture despite the lack of covalent linkages between peptides. This mechanical strength results from the perpendicular arrangement of the parallel β-sheet and leucine ‘fastener’ structures within the films.
Figure 21.

Self-assembled structures of peptide lipids in films cast from CHCl3 solutions. © 2000, American Chemical Society, Langmuir 16 (2000) 4929.
Figure 22.

Self-assembled supramolecular films of Lue-Leu-Ley-Glu-type peptide lipid.
Shimizu and coworkers extensively developed formation of fibrous and tubular structures through self-assembly of bola-type amphiphiles [165–168]. Tube structures require an uneven curvature of opposing sides of a precursor self-assembled membrane. Thus, bola-amphiphiles with terminal groups of differing size should be better candidates for construction of lipid tubes. By careful consideration of lipid structure design, unsymmetrical lipid nanotubes with differing interior and exterior surface structures can be prepared. As shown in figure 23, Masuda and Shimizu realized this concept by designing unsymmetrical bola-amphiphiles, which self-assembled in water to form lipid nano- and microtubes [169]. The nanotubes obtained encapsulated the staining reagent phosphotungstate revealing a hollow cylindrical morphology several hundred micrometers long with 30–43 nm and 14–29 nm outer and inner diameters, respectively. In their pioneering work Shimizu and coworkers realized formation of vesicle-containing tubular structures from a bola-amphiphile where glycylglycine segments are attached at both the end of an aliphatic chain (figure 23) [170, 171]. Infrared spectroscopy revealed that the glycylglycine functional groups in the monolayer form a networked polyglycine II hydrogen bond instead of the conventional β-sheet motif. Detailed AFM observations of the tubes revealed a hierarchical ordering within the structure of this assembly. The structures formed were robust even stable against heating at 100 °C in water or to ultrasonic irradiation. Even after evaporation to dryness, the structure could be regenerated simply by addition of water.
Figure 23.

Tubular structures of self-assembled bola-amphiphiles.
Figure 24.
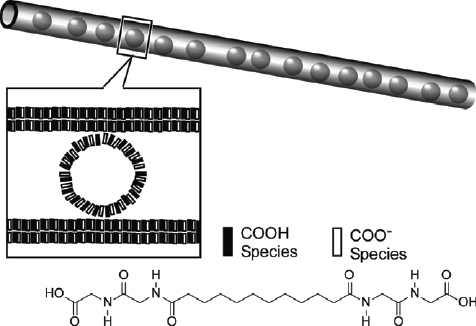
Tubular structures including vesicles from a bola-amphiphile with glycylglycine segments.
Hybridization of organic self-assembled structures with inorganic materials can also lead to development of novel properties and functions that are not available in the individual components. Figure 25 illustrates formation of an inorganic nanowire within a lipid envelope in work performed by Kimizuka and coworkers [172, 173]. One class of halogen-bridged one-dimensional mixed valence complexes is of great interest due to its unique physicochemical properties. These properties include intense intervalence charge-transfer absorption, semiconductivity, and large third-order nonlinear optical susceptibilities. Chloro-bridged linear platinum chains were dispersed as aggregates of a polyion complex with a sulfonate amphiphile whose lipophilic alkyl chains are oriented toward the organic media. Dispersion of the low-dimensional structures is due to the organized lipid molecules, which can stabilize the mixed valence chains in solution. Heating the linear platinum complex causes dissociation into its individual complexes, which are then re-assembled upon cooling to lower temperatures. A reversible structural transition between aggregated mesoscopic and individual molecular metal complexes was thus realized by the formation of a self-assembling amphiphilic supramolecular structure.
Figure 25.

Inorganic nanowire within a lipid envelope.
Because a huge variety of shapes is accessible using self-assembly of lipids and related amphiphiles through the wide choice of commercial and designer components, and because of their soft, malleable natures, such structures have attracted the attention of many researchers who have reported many excellent results. Other relevant work has been reported [174–184].
Gels and liquid crystals
Unlike many supermolecules that may still remain within basic scientific areas, gels and liquid crystals are well recognized as practical materials, as seen in water preserver/absorber and optical displays. In particular, gels and liquid crystals formed through self-assembly of low molecular-weight components have attracted significant attention [185–188]. In those materials, appropriate design of small molecular structures can lead to drastic changes in the properties of the bulk materials.
Ajayaghosh and coworkers have extensively investigated microstructure formation from conjugate oligomers and their properties of gelation. For example, they have reported the formation of micrometer-size supramolecular tapes and helices through self-assembly of oligo (p-phenylenevinylene) derivatives (figure 26A) [189]. Formation of superstructures of micrometer size in a dried assembly obtained from decane was revealed by SEM imaging. Birefringence exhibited by the gel when viewed through crossed polarizers indicated the molecular anisotropy within the aggregates. The tunable size and morphology of the assemblies resulted in preparation of nanoparticles, microspheres, and superstructured blue light-emitting organogels. These workers also reported self-assembly transformations of squaraine dyes (figure 26B), which could form vesicular structures or helical architectures [190]. Hollow spherical structures of the tripodal squaraines first self-assembled from acetonitrile were transformed into linear helical structures upon introduction of Ca2+ or Mg2+. An interesting target for functionalization of self-assemblies of aromatic oligomers is the potential control of optical properties based on supramolecular structure variation. Ajayaghosh et al also investigated fluorescence resonance energy transfer (FRET) between the tape-like structure of a few tailor-made oligo (p-phenylenevinylene) derivatives (donor, figure 26C) and entrapped rhodamine B (acceptor) [191]. The efficiency of FRET was influenced considerably by the ability to form self-assembled aggregates of the oligo (p-phenylenevinylene) compound and so control could be realized by variation of solvent polarity and/or temperature.
Figure 26.

Gel-forming conjugate oligomers.
Structural control of conductive polymers within gel structures was achieved by Fujita, Shinkai, and coworkers. They proposed molecular design of gel-forming molecules with controllability of effective conjugation length in poly(diacetylene) structure [192]. As shown in figure 27, they attached the well known gel-formation promoting substituents, 3,4,5-trialkoxybenzoic acid moieties, to the diacetylene using flexible alkyl chains. The photopolymerizable unit containing flexible linkers was expected to be strongly stabilized by intermolecular hydrogen bonding involving two amide linkages, which were surrounded by the gelation-promoting moieties. The multiplicity (odd or even) number of alkyl chains was found to be a key factor in determining whether the blue or red phases of poly(diacetylene) was obtained.
Figure 27.

Gel-forming molecules with controllability of effective conjugation length in poly(diacethylene) structure.
A variety of designs of low molecular-weight gelators have been reported. Ihara and coworkers reported chirality control of a self-assembled organogel [193] and self-assembled fibrillar network formation [194] based on molecular design using a glutamate backbone. They have also recently reported enhanced fluorescence emission and photochromism in organogel structures of salicylideneaniline derivatives [195]. Hanabusa and coworkers have worked extensively on low molecular- weight gelators based on molecular structures of ureylene [196], diaminocyclohexane [197], and cyclohexanetricarboxamide [198]. Sakurai and coworkers reported gel fibers from cyclic bisurea derivatives [199]. Shinkai and coworkers developed organogels with cholesterol-based [200] and sugar-based [201] molecular designs.
As a final example of gelatinous self-assembly one very unique topic is shown in figure 28. Fukushima, Aida, and coworkers found serendipitous gel formation on intimate mixing of an ionic liquid and carbon nanotubes [202, 203]. Grinding of a mixture of pristine single-walled carbon nanotubes and an imidazolium room-temperature ionic liquid resulted in stable gels. The heavily entangled nanotube bundles were found to untangle within the gel to form much finer bundles. Non-volatility of the ionic liquids imparted on the gels thermal stability and resistance to shriveling even under reduced pressure although they would readily undergo a gel-to-solid transition when physically placed on absorbent materials.
Figure 28.
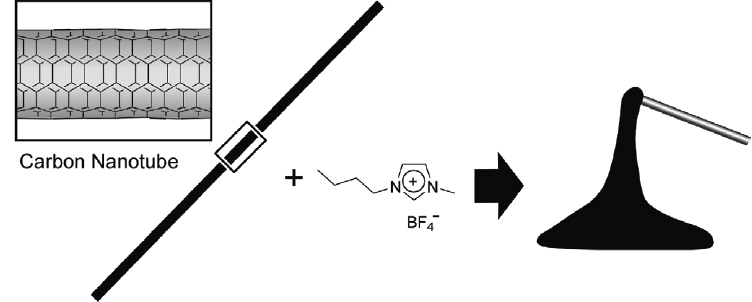
Gel formation from an ionic liquid and carbon nanotubes.
In common with gel materials, liquid crystals have many practical uses. Phase transitions and orientation control of liquid crystalline materials are attractive subjects, especially in relation to possible photonic applications, because of their molecular alignments. Kato and coworkers developed liquid crystalline materials that are stabilized by hydrogen bonding [204, 205]. They demonstrated variation in the self-assembled liquid crystalline structures of folic acid derivatives. Addition of alkali metal salts to a thermotropic folic acid derivative resulted in a transition from smectic to hexagonal columnar phase (figure 29) [206]. With this phase change the hydrogen bond pattern was altered from ribbon to disk because the columnar liquid crystalline structures were stabilized by ion-dipolar interactions between the added sodium ions and the discotic assemblies.
Figure 29.

Change from smectic to hexagonal columnar phases of folic acid derivatives by the addition of alkali metal salts
Ichimura and coworkers pioneered the novel concept of a ‘command surface’ where a single monolayer controls orientation within bulk liquid crystalline films [207]. They developed magnification of structural information by combining photoisomerization of surface monolayer and liquid crystalline materials. In the example shown in figure 30, a photoisomerizable ‘command’ monolayer was immobilized on a solid surface and liquid crystalline materials were then deposited on the monolayer. Orientation of the thick liquid crystalline layer could then be achieved by photoisomerization of the command monolayer so that a phenomenon occurring at the molecular level was amplified leading to a bulk structural change. This concept could be applied in the development or improvement of display devices.
Figure 30.

Orientational changes of a liquid crystal by photoisomerization of a surface monolayer.
Furumi et al investigated electrical control [208] and photonic control [209–112] of the photonic bandgap in chiral liquid crystals for possible use in laser devices. Shape-memory fibers consisting of self-assembled liquid crystalline polymers were proposed by Terentjev and coworkers [213]. Photo-induced polarization inversion in ferroelectric liquid crystals was realized by Lemieux et al using an ambidextrous chiral thioindigo dopant [214]. Schmidt-Mende et al developed high efficiency organic photovoltaics using self-organized discotic liquid crystals of hexa-perihexabenzocoronene derivatives [215]. Clark and coworkers reported a class of fluid polar smectic liquid crystals in which local splay prevails in the form of periodic supermolecular-scale polarization modulation stripes coupled to layer undulation waves [216]. Other recent examples on self-assembled liquid crystal materials are also available [217–221].
Structure-transcribed material
Self-assembled materials consisting of organic or biological materials are often mechanically fragile. If one could use these structures as templates for nanostructure syntheses of robust stable formations of metallic or inorganic materials, stable objects bearing morphologies reflecting the original self-assembled structures might be obtainable. In fact this strategy has been applied and is referred to as structure transcription from self-assembled objects. It is one of the best methods to produce self-assembled structures for use under harsh conditions.
Research based on this concept has recently received attention [222, 223]. Transcription of fibrous and tubular structures of organic gels into inorganic substances has been extensively reported by the groups of Shinkai, Shimizu, and Hanabusa [224–233]. For example, in appropriate media the compound shown in figure 31 forms tubular structures, which, upon sol–gel condensation of the co-reactant tetraethyl orthosilicate and subsequent calcination, are transcribed as double-walled silica nanotubes [234]. Helical ribbon aggregates composed of crown-appended cholesterol derivatives have been used to transcribe chiral silica [235]. Hollow fibers of titanium oxide have been prepared from self-assembled gels of cyclohexanediamide derivatives [236]. With similar synthetic concepts, silica nanostructures containing fullerene [237] and porphyrin [238] moieties have also been reported. Shimizu and coworkers reported metal nanowire formation, where a synthetic glycolipid N-(11-cis-octadecennoyl)-y-D- glucopyranosylamine in a tubular structure was used as a template [239]. First, capillary force was used to encapsulate gold or silver nanoparticles in the hollow cylinders of the lipid nanotubes, followed by removal of the organic components by calcination of the the nanocomposite, yielding gold nanowires of controlled width. Additionally, glycolipid nanotubes were used to prepare helical arrays of CdS nanoparticles [240]. Modulation of structure of the transcribed materials, including regulation of silica nanotube diameter, can be realized using solvent-sensitive morphological changes of the template peptidic-lipid nanotubes [241].
Figure 31.
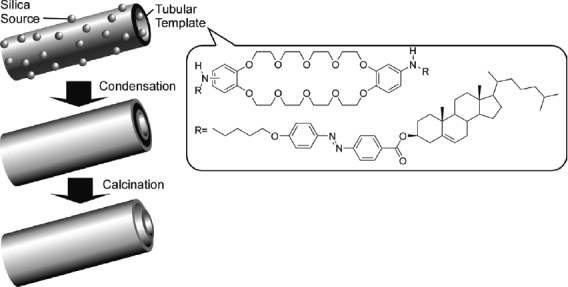
Transcription of tubular structures of organic gels into double-walled silica nanotubes.
A great deal of other research into fabrication of various types of nano- and micro-structures based on structure transcription has also been reported. For example, Price et al. reported preparation of Cu spiral/helical nanostructures through selective electroless metallization using a phospholipid microtubule template [242]. Ras et al also reported synthesis of hollow nanospheres and nanotubes of Al2O3, having tuned the wall thicknesses by atomic layer deposition on self-assembled polymeric templates [243]. Synthesis of vanadium oxide nanotubes using a self-assembled alkyl amine template was demonstrated by Li and coworkers [244]. One atypical morphology prepared by the structure transcription technique is that of the carbon nano-test tubes fabricated using a cre-sheath Te@carbon nanocomposite as a template, as reported by Yu and coworkers [245]. Corking of silica nano-test tube using chemical self-assembly processes has been reported by Stewart, Martin, and coworkers [246]. By using concepts of self-assembly processes various inorganic nanostructures have been prepared. For example, boron nitride nanotubes [247], Si-core microwires coated with aligned SiO2 nanowires [248], silica-shielded Ga–ZnS nanowires [249], and boron nitride nanomesh [250].
Of the known structure-transcribed objects, mesoporous materials the most popular and many applications are anticipated [251–263]. Mesoporous silica materials can be prepared using micellar assemblies of surfactants or block-copolymers as soft templates (figure 32A). In 1990, Kuroda and coworkers first reported the preparation of mesoporous silica with a uniform pore size distribution through intercalation of cetyltrimethylammonium cations into the layered polysilicate kanemite followed by calcination to remove the organic moiety (FSM-16) [264, 265]. Later, Mobil scientists supplied materials having large uniform pore structures, high specific surface area, with specific pore volume and hexagonal geometry MCM-41 [266, 267], cubic geometry MCM-48 [268], or lamellar geometry MCM-50 [269]. Tanev et al prepared HMS using a neutral amine as template [270], and Bagshaw et al similarly synthesized a disordered mesoporous material designated as MSU-1 using polyethylene oxide (PEO) [271]. Stucky and coworkers developed highly ordered large pore mesoporous silica SBA-15 with thicker pore walls and a two dimensional hexagonal structure using an amphiphilic triblock copolymer of poly(ethylene oxide) and poly(propylene oxide) in highly acidic media [272, 273]. The same group also prepared MCF type materials where triblock copolymers stabilizing oil in water microemulsions were used as templates [274]. Mesoporous carbon materials can be synthesized through carbonization within mesoporous silica as a hard template and subsequent selective removal of the silica (figure 32B). Ryoo et al first realized synthesis of ordered mesoporous carbon CMK-1 using MCM-48 silica as a template and sucrose as the carbon source [275]. The first well ordered mesoporous carbon CMK-3 that was a faithful replica of the template was synthesized using SBA-15 as a template [276]. Hyeon et al reported, independently and somewhat later, a similar approach for preparation of well-ordered mesoporous carbon materials designated SNU-x [277–279].
Figure 32.

Synthesis of (A) mesoporous silica and (B) mesoporous carbon.
Although preparation of mesoporous carbon and related materials has been extensively researched [280–284], breakthrough concepts have been recently proposed by Vinu and coworkers. They applied a similar strategy for the synthesis of mesoporous carbon nitride with combined carbon and nitrogen sources (figure 33A) [285–287]. More recently, they have pioneered a third method for synthesis of mesoporous materials, called the ‘elemental substitution method’ [288]. In this method, component elements are substituted by other elements with retention of the mesoporous structure. For example, they successfully realized the first synthesis of mesoporous boron nitride and mesoporous boron carbon nitride (figure 33B) [289–291].
Figure 33.
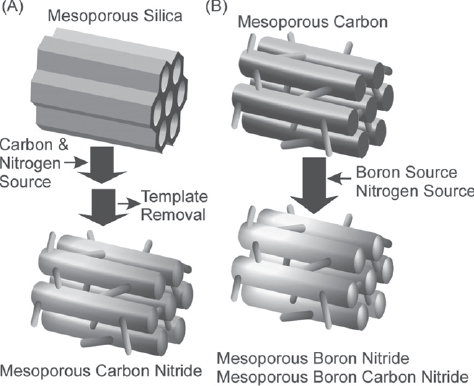
Synthesis of (A) mesoporous carbon nitride and (B) mesoporous boron nitride and mesoporous boron carbon nitride.
Vinu and coworkers recently reported synthesis of the novel nanocarbon, ‘carbon nanocage’ [292–295], through template synthesis using a large three-dimensional cage-type face centered cubic mesoporous silica material (KIT-5) as the inorganic template (figure 34A). The specific surface area and specific pore volume of carbon nanocage greatly exceeds those reported for conventional mesoporous carbon materials. Therefore, the capacity for lysozyme adsorption on the carbon nanocage is much larger than that observed with mesoporous carbon CMK-3. The carbon nanocage also exhibits excellent capabilities in the separation of small molecules [296]. In figure 34B, the superior adsorption capability of the carbon nanocage in removal of a dyestuff (Alizarin Yellow) is demonstrated. An aqueous solution was passed through a bed of the respective carbon material deposited on top of a cotton plug in a pipette and with application of a slight pressure. When compared with the control test without carbon (a), the carbon nanocage materials completely removed the dye (c), while activated carbon powder (b) and CMK-3 (d) were not effective for removal of the dye under these conditions. A highly selective separation of catechin and tannic acid by the carbon nanocage material in a one-pot process was also found. Use of carbon nanocage as adsorbent provided a highly selective adsorption of tannic acid (ca. 95%) in a simple one-pot process. This result could originate from the very unique geometry of the carbon nanocage materials.
Figure 34.

(A) Synthesis of carbon nanocage; (B) its TEM image; (C) filtration of aqueous Alizarin Yellow: (a) without carbon, (b) with activated carbon; (c) with carbon nanocage; (d) with mesoporous carbon CMK-3. © 2007, American Chemical Society, J. Am. Chem. Soc. 129 (2007) 11022.
Because chemical modifications of mesoporous materials (especially mesoporous silica) are well established [297], further fabrication and functionalization of mesoporous materials has been extensively researched. Recent breakthroughs on fabrication of mesoporous materials involved synthesis of chiral mesoporous silica materials and preparation of periodic mesoporous organosilicates (PMO). Chirality was introduced in mesoporous materials by using a chirally-defined surfactant template, which was demonstrated by Che et al [298–300] who used an N-acyl-type alanine-based surfactant to prepare mesoporous silica containing chiral pores (figure 35). SEM observation of the obtained mesoporous silica revealed that the materials possessed regularly twisted rod-like structures with diameters of 130–180 nm and lengths of 1–6 mm. TEM observation confirmed the presence of hexagonally aligned mesoscopic channels with diameters of 2.2 nm wound together in a particular direction. This finding attracted some researchers to this field who have further developed the synthesis of chiral mesoporous objects [301–303]. As one of the other significant breakthrough materials for this area, PMO materials were introduced by three groups working independently: Inagaki group [304], Ozin group [305], and Stein group [306]) all in 1999. As illustrated in figure 36, this category of materials is synthesized using organic molecules having multiple alkoxysilane groups such as bis(triethoxysilyl)ethene and bis(triethoxysilyl)benzene. Although much work has been done on PMO materials [307–309], one of the most fantastic findings in PMO technology is the formation of crystalline pore walls [310]. Inagaki et al synthesized a PMO material using benzene-bridged organosilane, 1,4-bis(triethoxysilyl)benzene. The powder XRD patterns showed a set of peaks assignable to a two-dimensional hexagonal lattice. Four sharp peaks corresponding to periodic structure with a spacing of 0.76 nm were detected in the small angle region. Many lattice fringes, stacked along the channel axes with a uniform spacing of 0.76 nm, were confirmed by TEM imaging. These results are consistent with a crystal-like pore-wall structure, with an alternate arrangement of hydrophilic silicate layers and hydrophobic benzene layers.
Figure 35.

Mesoporous silica with chiral motifs.
Figure 36.

Periodic mesoporous organosilicates (PMO) with various organic moieties.
As summarized in a recent review by Ariga et al [311], supramolecular functions of mesoporous materials make up a significant new trend in this research field. Of various reported functions, controlled release from mesopores is one of the most attractive topics (one example is described in figure 9 of the previous section). In pioneering work, Fujiwara and coworkers achieved photo-controlled regulation of drug storage and release from mesoporous silica [312, 313]. They functionalized MCM-41 with photoactive coumarin, which showed photo-responsive dimerization resulting in reversible gate operation. Lin and coworkers realized a controlled-release delivery system using colloid capping of mesoporous silica [314]. The same research group demonstrated gate-controlled molecular recognition by selective functionalization at the external and internal surfaces of mesoporous silicates with polylactate and o-phthalic hemithioacetal, respectively [315]. They synthesized a series of room temperature ionic liquid-containing mesoporous silica nanoparticulate materials with various particle morphologies, including spheres, ellipsoids, rods, and tubes [316] and investigated controlled release profiles using ionic liquids as antibacterial agents against the Gram negative microbe Escherichia coli K12. Martnez-Mez and coworkers used an MCM-41 mesoporous solid support functionalized at its external surface with polyamines for controlled entrapment and release of guest anions [317]. Xiao and coworkers also reported a pH-responsive carrier system based on carboxylic acid modified SBA-15 silica rods and poly(dimethyldiallylammonium chloride) for storage and release of drug molecules from pore voids [318]. Vallet-Reg and coworkers used two types of hexagonally ordered mesoporous materials, MCM-41 and SBA-15 as matrices for alendronate (bisphosphonate) adsorption and release [319]. The same research group also reported controlled delivery of macrolide-type antibiotics using SBA-15 [320]. Hyeon and coworkers report a synthetic procedure for the fabrication of mesoporous silica spheres, which was applied to the uptake and controlled release of drugs [321]. A unique example combining molecular assembly techniques and mesoporous materials for controlled release was reported by Wang and Caruso where enzyme immobilization and encapsulation was accomplished in bimodal mesoporous silica spheres with the aid of layer-by-layer (LbL) assembly [322, 323].
Mesoporous materials provide nanospaces of predictable structural dimensions where novel functions as well as unknown phenomena can be investigated in depth. Confinement of polymeric substances in mesopores has been researched [324–328]. Several examples of polymerization in mesoporous media using monomeric templates for preparation of mesoporous materials with conductive polymers contained within the pores have been reported (figure 37). Aida and Tajima used a few kinds of hexadecadiynyl trimethylammonium bromide as templates to synthesize mesoporous silica containing diacetylene in micro-fibrous form [329]. Lu et al used oligo (ethylene glycol)-functionalized diacetylenic surfactants as structure-directing reagents for mesoporous silica films through casting, spin-coating, or dip-coating methods [330]. Diacetylene can be then polymerized upon exposure to UV light. Aida and coworkers also developed poly(pyrrole)-containing mesoporous silica films in hexagonal and lamellar phases [331]. The films obtained were immersed in a solution containing anhydrous FeCl3 to polymerize the pyrrole monomer. The polypyrrole chains are highly constrained and insulated when incorporated within the hexagonal nanoscopic channels and the possibility of polarons recombining into bipolarons is suppressed significantly. In contrast, the two-dimensional lamellar phase afforded spatial freedom for electron recombination. Similarly thiophene polymerization in mesoporous silica channels was reported by Fuhrhop and coworkers [332].
Figure 37.

Polymerization of conjugate polymers in mesopores.
Supramolecular self-assemblies can also be confined in mesopores. Aida and coworkers reported the first example of immobilization of one-dimensional columnar charge-transfer assemblies within mesoporous silica films through the sol–gel reaction involving charge-transfer complexes of an amphiphilic triphenylene donor and various acceptors (figure 38) [333]. The films obtained were highly transparent and color-tunable by varying the intercalated acceptor. The molar ratio of donor and acceptor, related to the photoconductive properties, could be varied over a wide range from 1 : 1 to 9 : 1. In hexagonal mesoporous silica, the silica wall segregates the individual charge-transfer columns, which display neither solvatochromism nor guest-exchange activity. Also, they exhibited red-shifted absorption bands, which are possibly due to a long-range structural ordering.
Figure 38.
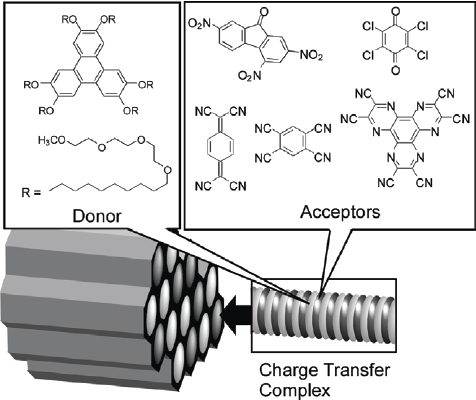
Charge-transfer complexes of amphiphilic triphenylene donor and various acceptors within mesoporous channel.
Introduction of biological components is a new challenge in mesoporous science and technology. Ariga and coworkers developed mesoporous silica with densely packed peptide segments known as proteosilica (figure 39) [334, 335]. This nano-composite material provides an asymmetric medium for photochemical reactions [336]. Photochromic dye molecules such as spiropyran can be doped into the chiral environments of the proteosilica films, where asymmetric photoreactions have been demonstrated using alanylalanine-type surfactants. Isomerization between the spiropyran form and the merocyanine form can be repeated upon alternate irradiation of visible light (420 nm) and UV light (280 nm) to the films, respectively. Only negligible CD signals originating from the guest could be observed for the film containing the merocyanine form, while the film with the spiropyran form showed clear CD activity in the region from 250 nm to 400 nm. A complete mirror image of the CD spectrum was obtained depending on whether the L-peptide or D-peptide host was used as the host surfactant. Alternate irradiation with UV and visible light induced repeated changes in the CD spectra with a small degradation in the intensity. The reported system could be applied for development of non-destructive memory devices.
Figure 39.

Isomerization between the spiropyran and merocyanine forms of a merocyanine dye in proteosilica.
A new synthetic method that addresses the issues of both dense functionalization of the pore interior and increased accessibility for external guests was developed by Ariga, Aida, and coworkers (figure 40A) [337, 338]. The template amphiphile, which had dialkoxysilane functionality as part of the head group, was covalently attached to the silica framework by sol–gel reaction with tetraethyl orthosilicate; this resulted in densely filled mesoporous silica channels. Subsequent cleavage of the alkyl tail by hydrolysis of the ester at the C-terminal left open pores with an internal surface functionalized with alanine moieties. In this case the template behaves like a ‘lizard,’ that bites the silica wall and then sheds its tail. Reactor applications were introduced for similarly prepared hybrid structures. The catalytic capability of these materials prior to hydrolysis on the acetalization of a ketone, such as cyclohexanone, in ethanol (EtOH) under mild conditions was demonstrated by Aida and coworkers (figure 40B) [339]. Simultaneous incorporation into the silica channels of cyclohexanone and EtOH, at the hydrophobic inner domain and hydrophilic (ionic) outer shell, respectively, caused their activation through hydrogen bonding with the amide NH and carbonyl groups of the peptide functionalities at the core-shell interface. Carbocationic intermediates that are probably transiently involved in the acetalization, and generation of these intermediates is favored by the highly polar environment of concentrated ammonium salt functionalities. This work appears to provide a novel and general strategy for rational molecular design of novel types of artificial enzymes as bio-inspired solid catalysts.
Figure 40.
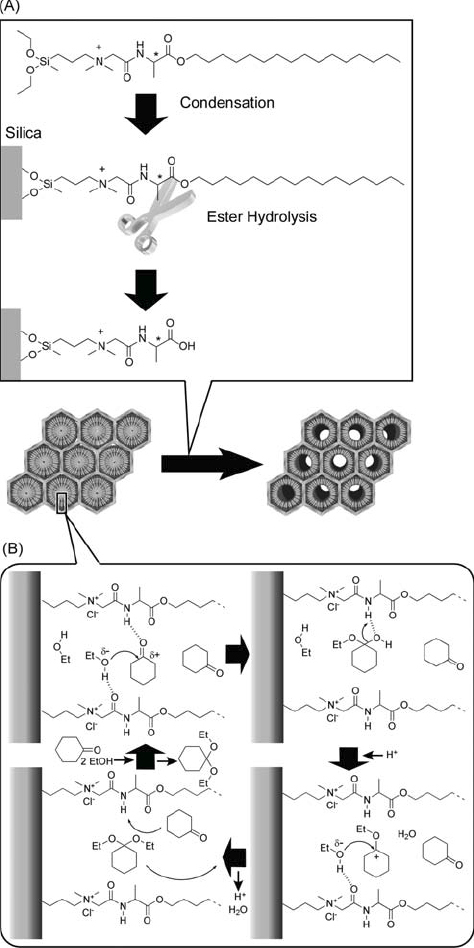
(A) Functionalization of the pore interior by lizard template method and (B) acetalization of a ketone within unhydrolyzed pores. © 2004, American Chemical Society, J. Am. Chem. Soc. 126 (2004) 988.
Immobilization of more complicated biomaterials has been recently investigated with one typical example being the inclusion of proteins in mesopore channels [340–345]. Vinu and coworkers performed systematic investigations on protein adsorption onto mesoporous materials, mesoporous silica and mesoporous carbon [346–358]. Typically, protein adsorption obeys Langmuir monolayer adsorption and much depends on pore size and geometry of the mesoporous adsorbents. Because the charged states of proteins and/or mesoporous materials vary depending on pH, adsorption behaviors of proteins could be regulated by using pH control. Adsorption near the protein isoelectric point tends to be optimum probably because of minimized electronic repulsions.
Macroscopic assembly
Since self-assembly is a very general concept, material formation through self-assembly is not limited to molecular-scale and nanometer-level objects. It can be applied even to macro-sized objects. In this section, unique examples of this will be briefly summarized.
Mirkin et al reported self-assembly processes of metal-polymer amphiphiles fabricated by sequential electrodeposition of gold and polypyrrole into a porous aluminum template (figure 41) [359]. These micron-sized assemblies had outside diameters of 400 nm for the gold and 360 nm for the polypyrrole portions. The hydrophobic attraction between polymer moieties in water caused contraction of the polypyrrole domains resulting in a designed curvature of the self-assembled microstructures of gold-polypyrrole amphiphilic rods. They mostly formed tubular structures with diameters in the range 10–100 mm, depending on the ratio between gold and polypyrrole domains. The rod units themselves can behave like well designed amphiphilic molecules. Yan et al reported the macroscopic molecular self-assembly of an amphiphilic hyperbranched copolymer in acetone, which generated multi-walled tubes millimeters in diameter and centimeters in length (figure 42) [360]. The thickness of the tube walls approached 400 nm, and the walls had an inhomogeneous lamellar structure that alternates between amorphous, partly irregular hydrophilic domains and well-ordered hydrophilic domains.
Figure 41.

Micron-sized assemblies from metal-polymer amphiphiles.
Figure 42.

Macroscopic molecular self-assembly of an amphiphilic hyperbranched copolymer in acetone.
In this particular field of self-assembly, Whitesides and coworkers have been making extremely significant contributions [361–363]. They have reported a series of results based on visual models of supramolecular self-assembly. One example of their work is shown in figure 43 and is based on millimeter-sized hexagonal sections bearing hydrophilic and hydrophobic faces in an alternating arrangement [364]. Upon positioning of these pieces at an interface of hydrophobic perfluorodecalin and water, the pieces assembled spontaneously so that hydrophobic faces grouped together thus avoiding an unfavorable exposure of these faces to water. This assembly process resulted in the formation of a regular honeycomb structure. Altering the shape of the unit pieces changed the arrangement of the hydrophobic faces and led to creation of a variety of regular macroscopic assemblies. Using four different pieces with differently shaped hydrophobic faces so that four pieces form two kinds of pair by contact between complementary shapes, matching of linked structures could also be demonstrated [365]. This macromodel can be regarded as a mimic of hybridization processes of nucleic acids in the visible regime. Such approaches are not limited to self-assembly within two-dimensional space, and construction of three-dimensional systems has also been reported [366–368]. For example, spontaneous folding of three-dimensional tape-like objects was demonstrated and can be regarded as mimicry of folding in protein and peptide segments [369, 370].
Figure 43.

Visible models of supramolecular self-assembly on millimeter-sized hexagonal pieces with hydrophilic and hydrophobic faces at the interface of perfluorodecalin and water.
Self-assembly in bulk media 2; component type
In the following sections, we have selected several of these molecular types and objects and summarized recent research of their self-assembling behaviors and properties.
Porphyrin
Porphyrins and related molecules play essential roles in many biological systems. For example, they are involved in the crucial reactions within photosynthetic systems that are composed of sophisticated supramolecular assemblies of proteins and dyes. The antenna complexes of photosynthetic bacteria consist of a core light-harvesting antenna (LH1) and a peripheral light-harvesting antenna (LH2) that contribute to the collection of light energy. The excitation energy migrates within the wheel-like arrays of chlorophylls in the LH1 and LH2 complexes and is finally funneled into the chlorophyll dimer (special pair) at the photosynthetic reaction center. These elegant processes associated with assemblies of porphyrin molecules have attracted many organic, biological, and supramolecular chemists [371–383]. In particular, the formation of structures containing multiple porphyrin molecules using self-assembly processes is an attractive research target. A wide variety of structures can be obtained through supramolecular self-assembly thanks to the molecular design flexibility of porphyrins, since they can be easily modified with relatively little synthetic effort.
Kobuke and coworkers developed a non-covalent supramolecular approach for construction of porphyrin arrays from rather simple units [384], principally using coordination between central metal and imidazole groups attached at the meso-position (figure 44A) [385, 386]. The lengths of the supramolecular arrays can be controlled simply by fixing the ratio between a porphyrin dimer with two zinc centers and a porphyrin dimer with a single zinc center. A straightforward mimic of the natural light harvesting antenna system was provided using imidazolylporphyrinatozinc(II) dimers connected with a 1,3-phenylene spacer as the linking subunit. Under appropriate conditions, the conformation of this unit resulted in formation of a closed ring (figure 44B) [387, 388]. Such a model may be important for understanding the evolutional strategy for ring structures in the natural photosynthetic system.
Figure 44.
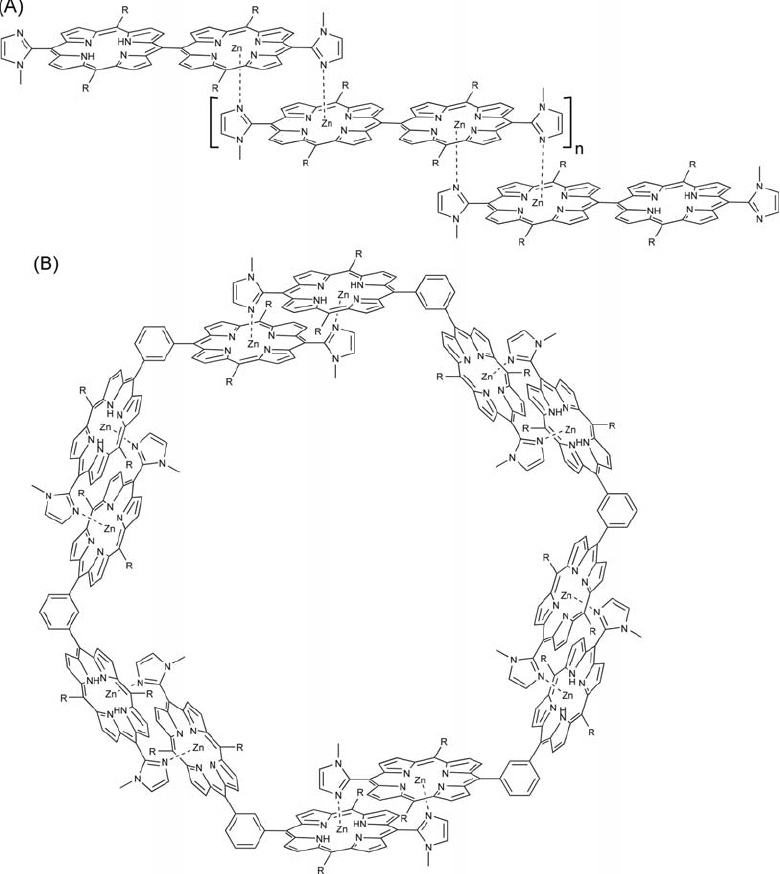
Self-assembled structures of (A) porphyrin with imidazole attached at the meso-position and (B) imidazolylporphyrinatozinc(II) dimers connected with a 1,3-phenylene spacer.
Coordination interactions have also been used for non-covalent bridging of conjugated covalent arrays of porphyrin units. A series of conjugated zinc porphyrin oligomers formed stable ladder complexes with linear bidentate ligands such as 1,4-diazabicyclo[2.2.2]octane (DABCO) and 4,4′-bipyridyl (figure 45) [389–391] as reported by Anderson and coworkers. The DABCO molecule has high basicity and this characteristic is important for stable complex formation with a rigid structure. Additionally, interesting cooperativity in their self-assembly processes was observed and formation-dissociation equilibria of the ladder complexes exhibited a large Hill constant. Formation and dissociation in this system is a representative example of ‘all-or-nothing’ behavior. A self-sorting mechanism occurs when porphyrin oligomers with different lengths are mixed and preferential formation of complexes of the same array chain-length was observed. Significant changes in the electro-optical properties of the porphyrin arrays were also found upon complex formation due to an increased planarity and overlap of π-orbitals in the ladder structures compared to the independent oligomers. This was indicated by an increased splitting in the Q-bands and from a reduction in size of their HOMO-LUMO gaps. Nonlinear optical characteristics of the arrays apparently amplify with formation of the supramolecular strands. The double-stranded ladder complex with 4,4′-bipyridyl exhibited an order of magnitude larger optical nonlinearity per macrocycle than the corresponding uncomplexed oligomers. DABCO-induced self-assembly of trisporphyrin double decker cage was also demonstraeted by Ballester, Hunter, and coworkers [392].
Figure 45.

Ladder complexes of conjugated covalent arrays of porphyrin units with linear bidentate ligands such as 1,4-diazabicyclo[2.2.2]octane (DABCO).
Because porphyrin derivatives possess excellent electronic and photonic properties, functions related to photo-electronic properties have attracted many researchers. Kuroda and coworkers reported porphyrin assemblies containing 17 porphyrin molecules [393]. In their system, the photo-energy absorbed by the entire system could be effectively converted to emission by the single central porphyrin. The central porphyrin has pyrazine arms, prepared through dimerization of carboxy groups, to coordinate the zinc porphyrin dimers. Excitation of the central porphyrin is enhanced directly by absorption by the antenna pigments even in a large scale assembly such as this. Because of this enhancement, the antenna effect for this system resulted in 77 times the fluorescence of the free central porphyrin. Prodi reported wavelength-dependent electron and energy transfer in assemblies of ruthenium porphyrin and perylene bisimide [394]. Okada and Segawa reported sustituent-controlled excitation in J-aggregated self-assembly of porphyrins [395]. Supramolecular chiral memory was demonstrated in porphyrin assemblies as reported by Purrello and coworkers [396]. Rella and coworkers demonstrated optical sensing response for vapors using porphyrin/phthalocyanine hybrid thin films [397].
Materials preparation from porphyrin components has also been researched. For example, Liu and coworkers reported gel-formation and supramolecular chirality induction [398]. Shinkai and coworkers have made extensive contributions on porphyrin gel preparation [399–401]. Takeuchi, Shinkai, and coworkers reported a series of porphyrin-based gels and one example is shown in figure 46 [402]. The porphyrin derivative used has hydrogen-bond-donating (carboxylic acid)/accepting (pyridine) substituents or electron-donating (dialkylamino)/withdrawing (pyridine) substituents at peripheral positions. The porphyrins a priori tend to assemble in a one-dimensional fashion due to their π–π stacking interactions, which can be further modulated by the combination of peripheral substituents.
Figure 46.

A packing model of porphyrin derivatives having hydrogen-bond-donating (carboxylic acid)/accepting (pyridine) substituents or electron-donating (dialkylamino)/withdrawing (pyridine) substituents at peripheral positions.
Harada and Kojima focused on developing new functional materials on the basis of distorted porphyrins, which can form unique curved surfaces rather than the normal planar structures [403]. They took advantage of the curved surface of the dication of the saddle-distorted dodecaphenylporphyrin as a building block for novel nanochannels in order to construct redox-active functional materials based on porphyrin nanostructures, as shown in figure 47. This type of cationic porphyrin channel forms through concomitant hydrogen bonding and π–π interactions and is promoted by double protonation, which in turn forms hydrogen bonding receptor sites,. Therefore, functional molecules such as quinones can be included by non-covalent interactions into the channels. This could provide a new strategy for porphyrin-based functional nanomaterials for targeting of novel photochemical devices in view of photoinduced electron and energy transfer. They also reported preparation of porphyrin nanotube and inclusion of Mo-Oxo clusters [405].
Figure 47.

Novel nanochannels from the saddle-distorted dodecaphenylporphyrin.
Ionic self-assembly of two oppositely charged porphyrins in aqueous solution for preparation of robust porphyrin nanotubes was reported by Shelnutt and coworkers (figure 48) [406]. The porphyrin nanotubes were formed by mixing aqueous solutions of the two porphyrins. The porphyrin nanotubes are micrometers in length and have diameters in the range of 50–70 nm with approximately 20nm thick walls. Images of the nanotubes serendipitously caught in vertical orientations confirmed the open-ended hollow tubular structure. Controlling the dimensions of the nanotubes was achieved by altering the structure of the porphyrin tectons. One potentially useful property of the nanotubes is their ability to respond mechanically to light illumination. Even though they are stable for months when stored in the dark, irradiation of a suspension of the tubes for just five minutes using the incandescent light from a projector lamp resulted in materials whose TEM images showed rod-like structures rather than tubes. This response to light is reversible, since the tubes reform (self-heal) when left in the dark. The same workers reported photocatalytic porphyrin nanotubes [406], photocatalytic self-metallization of porphyrin nanofiber bundles [407], and porphyrin nanosheets [408].
Figure 48.

Porphyrin nanotubes prepared by ionic self-assembly of two oppositely charged porphyrins.
Other research areas on porphyrin self-assemblies are very active. Porphyrin wheels [409] and coiled-coil aggregates of phthalocyanine derivatives [410] were demonstrated by Nolte and coworkers. They also reported surface patterning using porphyrin trimers by self-assembly and dewetting [411]. Wan presented preparation of hollow nanoprisms of self-assembled porphyrins [412]. Hybridization of porphyrin assemblies with other functional components such as fullerenes, polypeptides [413] or single-walled carbon nanotube [414] has also been reported.
Fullerene and graphene
Fullerene structures that had been proposed for the first time in 1970 by Osawa, [415] were actually detected during the now famous mass spectroscopic measurements performed by Curl, Kroto, and Smalley in 1985 [416]. Fullerenes and their derivatives exhibit new and unusual properties that have attracted much attention from scientists from various backgrounds. In particular, the extraordinary electronic and photonic properties of the fullerenes and their derivatives include unique electrochemical and optical absorption features in the excited state. By using self-assembly together with covalent chemical modifications, various functional fullerene structures and materials have been created [417–427]. In this section, various kinds of fullerene assembly and fullerene materials are surveyed.
One of the most impressive discoveries for fullerene assembly could be formation of vesicle structures. Nakamura and coworkers [428] reported formation of bilayer vesicle structures composed entirely of fullerene derivatives (figure 49). By appropriate substitution, fullerenes can be transformed into stabilized anions that convert highly hydrophobic fullerene into an amphiphile. Penta-substitution of fullerene with phenyl rings provides a stable cyclopentadienide anion, resulting in a negative charge in the 50-π electron system at the bottom of the C60 cage. The association behavior of the potassium salt of the pentaphenyl fullerene in water was studied by laser light-scattering and revealed formation of stable spherical vesicles with an average hydrodynamic radius and a radius of gyration of about 17 nm at a very low critical aggregation concentration. The fullerene-based vesicles had unique features when compared with vesicles of conventional lipids such as phospholipids. In contrast to the soft alkyl chains in the usual lipids, the rigid and well-defined structures of the hydrophobic moiety possessed a dominant intrinsic geometric constraint, which may lead to vesicular structures of well-controlled design. The same research group developed fullerene apexes and their assembly into crystalline and liquid crystalline states [429–431].
Figure 49.

Formation of bilayer vesicle structures composed entirely of fullerene derivatives.
Vesicle formation through self-assembly of fullerene derivatives has been reported by other research groups. Notably, Sano, Shinkai, and coworkers reported vesicle formation by bola-amphiphilic fullerene and discussed their fractal distribution within the self-assembled structure [432]. Shinkai and coworkers also reported encapsulation of fullerene in an organogel [433]. Ciang and coworkers reported vesicle formation with an amphiphilic diphenylaminofullerene conjugate [434]. Li et al investigated formation of self-assembled vesicles and encapsulation of pyrene [435]. Hirsch and coworkers developed fullerene-incorporated bilayer membranes and micelle assembly [436–439]. Nakashima and coworkers developed supramolecular films such as bilayer cast films and Langmuir–Blodgett films with fullerene components [440–446]. A C60 molecule can potentially serve as storage for six electrons and could play an important role in storage of electron density in molecular electronics devices. The stability of its reduction products (C60 anions) is strongly influenced by the surrounding environment including the nature of counter cations and their hydration state. The same workers synthesized C60 molecules with triple alkyl chains that could be self-assembled into cast films of lipid bilayer structures on electrodes (figure 50). Redox potential values and reduction currents can be controlled by phase transition behavior of the lipid part. Below the phase transition temperatures, the lipid bilayer is in a rigid crystalline state and electron exchange between fullerene and electrode is effectively suppressed. Smooth redox behavior was observed above the phase transition temperatures where the fluid nature of the lipid membrane allows a facile electron transfer between the fullerene and the electrode. Based on this finding, design of devices which can convert thermal stimuli to electrochemical response is anticipated.
Figure 50.

Lipid bilayer structures of synthetic C60 molecules with triple alkyl chains assembled on electrodes.
Supramolecular interactions have also been used for fullerene assembly. For example, Haino et al reported preparation of supramolecular polymeric nanonetworks through molecular-recognition-directed self-assembly between a calix[5]arene and fullerene (figure 51) [447]. The interaction between calixarene and fullerene can lead to supramolecular polymer networks. The size and morphology of the composite could be confirmed directly by SEM observation. A film was prepared by casting a solution of the 1 : 1 mixture of these two components onto a glass plate. The thicker of the entwined fibers formed had lengths of more than 100 mm and widths of 250–500 nm, suggesting that the ditopic calixarene host bound iteratively to the dumbbell fullerene to create a two-dimensional nanonetwork. AFM images of the sample cast on a mica plate also displayed the nanonetworks of fibrous assemblies with widths of 60–90 nm and heights of 1.2–1.9 nm. Judging from the calculated structure of the oligomers, the alkyl side chains have lengths of about 3.5 nm, which is longer than the observed height. This suggests that the alkyl chains adopt a structure parallel to the mica surface, and that the nanoassemblies are probably composed of bundles of 40–60 polymer chains created by entanglement of the alkyl side chains through van der Waals interactions.
Figure 51.
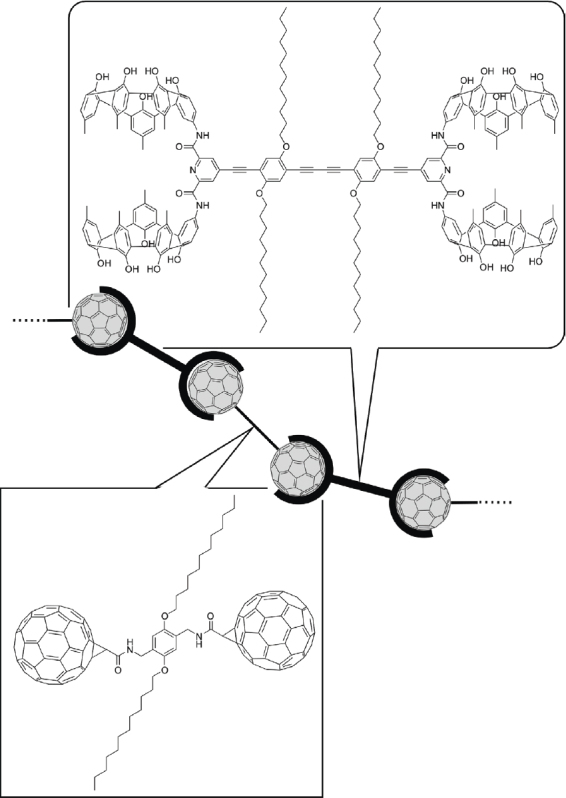
Supramolecular polymeric nanonetworks between a calix[5]arene and fullerene.
Versatile fabrication methodologies for fullerene microstructures through self-assembly processes are an important subject for the corresponding research fields. Georgakilas et al reported preparation of different nanoscale structures such as spheres, nanorods, and nanotubules in water depending on the side chain appendage of the fullerene spheroid [448]. However, one of the most promising techniques for science and technology based on self-assembled materials would be a methodology to create freely various morphologies from a single compound using a simple procedure, and this was recently realized by Nakanishi et al through research on microstructure formation of a single fullerene molecule [449–451]. As shown in figure 52, the molecule used contains two distinct portions, an sp2-carbon-rich fullerene moiety and a group containing sp3-carbon-rich aliphatic chains. These parts are both hydrophobic but show varying affinities for several solvents, resulting in different solvation behaviors for these two groups in solvents. When 1,4-dioxane was used as the self-assembly medium, the brown colored supernatant contained self-assembled single bilayer disks (figure 52(a)) in its SEM image where the diameter of the disks was observed to be 0.2–1.5 mm. The layer thickness of ca. 4.4 nm determined from tapping-mode AFM corresponds to the thickness of an alkyl-chain-interdigitated bilayer structure of this fullerene derivative. This fullerene derivative self-assembled into spherical vesicle-like aggregates with an average diameter of 250 nm in the 2-propanol/toluene solvent system (figure 52(b)) as confirmed by SEM, and the presence of an empty core and wall structure was confirmed by HRTEM observation. Use of 1-propanol resulted in one-dimensional self-assembled fibers (figure 52C) and bundles of these fibers with lengths of over 20 mm; these bundles appeared as partially twisted two-dimensional tapes in TEM images. When this fullerene derivative was dispersed in an equimolar mixture of water/THF, a turbid brown-colored dispersion was obtained. SEM and TEM micrographs revealed cone-shaped objects of suβ-micron size (figure 52D). Recently, the same research group reported preparation of flower-like objects [452] and their superhydrophobic properties [453] as well as room temperature liquid fullerene [454].
Figure 52.

Microstructure formation of a single fullerene molecule from: (a) 1,4-dioxane; (b) 2-propanol/toluene; (c) 1-propanol; (d) water/THF. © 2005, Royal Society of Chemistry, Chem. Commun. (2005) 5982.
Incorporation of fullerene molecules into useful structures is important for future applications. Liquid-liquid interfaces can act as nucleation sites for crystals and can be used to produce C60 crystals in unique morphologies. Miyazawa and coworkers used liquid-liquid interfaces in their discovery of very fine needlelike crystalline precipitates of C60 [455–476]. As shown in the optical micrograph in figure 53, long fibers of C60 with brownish, metallic color and with diameters smaller than 1 mm were obtained. These fine fibers of C60 were quite stable even against a concentrated electron beam, indicating electrical conduction. The needlelike crystal is a single crystal of C60 as determined by electron diffraction patterns of selected areas. These observations clearly show that the submicrons diameter needlelike precipitates are single-crystalline C60 fibers, i.e. whiskers of C60 called ‘C60 nanowhiskers’. Similarly, nanotube structures were obtained. Recently, they reported a similar preparation of nanoporous fullerene whiskers [475] and size-tunable fullerene hexagons [476].
Figure 53.

C60 nanowhiskers.
Other examples of fullerene assemblies [477–484] have been extensively reported, as well as preparation of polymer composites [485–491]. Fabrication of hollow carbon nanospheres from oxidized fullerenes [492] and fullerene nanoparticles by manual grinding [493] might also have interesting implications.
Graphene molecules have received much attention as partial structures of nanocarbons such as carbon nanotubes and fullerenes. Synthetic analogues of graphene have been subjected to self-assembly [494–496], which might provide properties similar to true nanocarbons. Mllen and coworkers presented a novel method to induce formation of an oriented assembly of columnar structures of hexa-peri-hexabenzocoronene (as graphene mimic) in a thin film form [497]. As depicted in figure 54, deposition of a hexa-peri-hexabenzocoronene (HBC) solution from a stationary nozzle onto a moving substrate produces temperature and concentration gradients that dictate uniaxial columnar growth of the HBC molecules. The uniaxially aligned high axial ratio columns have a single crystalline order over several square centimeters, which could be observed by AFM. The film thickness was confirmed to be approximately 15 nm and such wide area ordered films are very promising for application in high performance electronic devices where the charge transport channels are required to span the gap between two electrodes.
Figure 54.

Deposition of hexa-peri-hexabenzocoronene solution from a stationary nozzle onto a moving substrate.
Amphiphilic hexa-peri-hexabenzocoronene derivatives have been developed by Fukushima, Aida, and coworkers for self-assembly as graphene tubes. A hexa-peri -hexabenzocoronene derivative containing hydrophobic C12 chains and hydrophilic triethylene glycol chains in an appropriate substitution pattern was prepared by Hill et al in the realization of electrically conductive tubular structures through self-assembly (figure 55A) [498]. Detailed investigation by TEM revealed tubular structures that were straight and discrete with no detectable branching, having a uniform diameter of 20 nm. Although the intact tube was essentially insulating, the tubular structure showed a conducting current-voltage profile with an ohmic behavior after oxidation with NOBF4. Jin et al investigated chirality of the helical tubes assembled from similar hexa-peri-hexabenzocoronene derivatives containing chiral centers in the attached chains (figure 55B and C) [499]. The tubular structures with right- and left-handed helical senses were obtained from the (S)- and (R)-enantiomers of the amphiphile, respectively. Even though the enantiomeric excess of the chiral amphiphile was changed over a wide range from 20% to 100%, the CD spectrum of the suspension remained almost unchanged, resulting in a sigmoidal response of the CD intensity to the mixing ratio between (S)- and (R)- enatiomers. Such a nonlinear phenomenon is referred to as chirality amplification, where the major enantiomer incorporated into each tube determines the helical sense (majority rule). Yamamoto et al reported photoelectronic properties of a coaxial tubular structure formed by controlled self-assembly of a trinitrofluorenone-appended HBC amphiphile (figure 55d) [500]. Current-voltage profiles of the tubes showed that the current was enhanced markedly by a factor of >104 upon photo-irradiation. Photo-excitation of the self-assembled HBC should result in the generation of a charge-separated state involving radical cations and anions in the inner and outer layers of the tubes, respectively. This spatial separation of charge carriers prevents their rapid recombination, thereby enabling photo-conduction to occur along the tubes.
Figure 55.

Hexa-peri-hexabenzocoronene derivative for self-assembled graphene tubes.
Other small molecules
In this section, self-assemblies of small molecules, which were not described above, are briefly summarized with a classification by mode of interaction. Hydrogen bonding is a very useful interaction for design of self-assembled structures, because it possesses clear directionality and a medium strength. The latter characteristic is sometimes precious for dynamic control of assembly and disassembly processes. For example, Yagai et al reported photoresponsive self-assembly using a hydrogen-bond-directed cyclic hexamer (rosette) formed between melamine and barbituric acid derivatives (figure 56) [501, 502]. In their system, azobenzene was selected as a photo-isomeric unit. The stability of the cyclic hexamer is high when the azobenzene in the melamine unit is in the trans form. The large structural change caused by azobenzene photo-isomerization from trans to cis induced a clear decrease in complexation efficiency. The photo-regulatable complexation of the hexamer led to phototriggered formation of the rosette by irradiation of the mixture of melamine and barbituric acid derivatives with a selected wavelength of light. This strategy of photoregulation of supramolecular assemblies with a desired functionality may lead to novel types of smart nanomaterials.
Figure 56.

Photoresponsive discrete self-assembly using a hydrogen bond-directed cyclic hexamer (rosette) formed between melamine and barbituric acid derivatives.
Hydrogen bonding has been used for other self-assembled systems. Rovira and Veciana reported selfassembly of polychlorotriphenylmethyl radical derivatives substituted with six carboxylic acid groups for an investigation of its ferromagnetic properties [503]. Shimizu and corworkers synthesized porous apohost structures through self-assembly of bis-urea macrocycles and evaluated their guest inclusion properties [504]. Hydrogen-bond-based supramolecular polymers were fabricated through quadruplex-type self-assembly of ditopic guanosine macromonomers [505]. Fibrous nanostructures were found with hydrogen-bond-type self assembly of bis(2,6-diacylaminopyridine) derivatives [506], benzene-1,3,5-tricarboxamides [507], and pyromellitamide [508].
Metal-mediated coordination-type interactions have lead to important contributions in research on self-assembly of low-molecular-weight objects. For example, Aida and coworkers reported a novel photoluminescent ink for rewritable media that dichroically emits phosphorescence because of a structural bistability in the self-assembled luminophor [509]. They synthesized dendritic Cu(I) pyrazolate complexes carrying long alkyl chains (figure 57). Heat-sensitive phosphorescent paper was fabricated by coating a white polyethylene terephthalate paper with the Cu(I) complex containing a polymer support. The PET paper itself emitted blue light. The coated paper thus obtained looked white in daylight, whereas exposure to ultraviolet light resulted in luminescent pink coloration. Images were developed by using a facsimile-type thermal printer. Although the images persisted for over a year without any deterioration, they could be readily blacked out in a minute on heating to 100 °C. On the other hand, when aged at 45 °C, the papers that were printed and blacked out were completely re-initialized for writing. The same research group used self-assembled structures based on metallic interactions for control of physical properties including reversible RGB-color switching [510] and spin state switching [511].
Figure 57.

Self-assembled dendritic Cu(I) pyrazolate complexes bearing long alkyl chains.
Maeda et al demonstrated modification of microstructures of coordination polymer assemblies upon modification of the ligand structures [512]. Complexation of bisdipyrrin (dipyrromethene) ligands with Zn(II) ions providing various coordination polymers. The use of mpm-type and pmp-type ligands for coordination with Zn(II) ions in THF resulted in the formation of submicron-sized spherical structures ((a) and (b) in figure 58, respectively). Elemental mapping of the structure using high-resolution TEM energy dispersive x-ray (HRTEM-EDX) analysis(inserted image in figure 58(a)) confirmed the uniform distribution of Zn(II) ions in the former structure. Uniform distribution of the complex within the structure was illustrated using fluorescence micrographic imaging of the latter assembly (image inserted in figure 58B). The use of mixtures of THF and water (2 : 1, v/v) instead of pure THF afforded bell-shaped objects of the coordination polymer with the pmp-type ligand (figure 58C). The structures self-assembled from the mmm-type ligand and Zn(II) ions in 1 : 1 THF/water solution had an appearance similar to micron-scale golf balls (figure 58D).
Figure 58.

SEM images of coordination polymer assemblies with various ligands: (a) with pmp-ligand from THF (inset: Zn mapping HRTEM-EDX); (b) with mpm-ligand from THF (inset: fluorescence micrograph); (c) with mpm-ligand from THF/water (2/1); (d) with mmm-ligand from THF/water (1/1) (inset: TEM image). © 2006, American Chemical Society, J. Am. Chem. Soc. 128 (2006) 10024.
Many other coordination-type self-assemblies have been reported. Garca-Zarracino and Höpfl reported formation of polymeric and trinuclear macrocyclic hybrids with porous solid-state structures through self-assembly of diorganotin(IV) oxides [513]. Lehn, Gtlich, and coworkers described formation of molecular-grid-type hierarchical self-assembled structures as spintronic modules [514]. Fabrication of colloidal nanoparticles [515] and crystalline rods was accomplished by Mirkin and coworkers [516].
More ambiguous self-assembly based on solvophobic interactions involving unique molecular modules was also extensively investigated. Kim and coworkers presented vesicle formation from amphiphilic cucurbit[6]uril derivatives [517]. Similar vesicle formation using amphiphilic calix[4]arenes as cyclic host compounds was reported by Mecozzi and coworkers [518]. Fabrication of nanostructures and supramolecular polymers with cyclic host units was also demonstrated using an amino-calix[5]arene by Cohen, Parisi, and coworkers [519]. Other examples include a calix[6]arene by Rémita and coworkers [520], a bifunctional cyclodextrin by Harada and coworkers [521], and an arylene ethynylene macrocycle [522].
Self-assembly of various dye molecules has been investigated because of potential photoelectronic applications. Meijer, Schenning, and coworkers constructed thin films containing p-type oligo (p-phenylenevinylene)s and n-type perylenebisimides in a controlled morphology [523]. The films were prepared by spin-coating concentrated solutions containing stacks of the p-type component (figure 59A), mixed aggregates (figure 59B), and the hydrogen-bonded p–n dyad complexes (figure 59C). Charge-transport measurements in field-effect transistors indicated unipolar charge transport. When p- and n-type building blocks were mixed, ambipolar transport could be measured, but only in cases of aggregates with an appropriate morphology.
Figure 59.

Thin films containing p-type oligo (p-phenylenevinylene)s and n-type perylenebisimides in a controlled morphology: (A) stacks of the p-type component; (B) mixed aggregates; (C) hydrogen-bonded p–n dyad complexes.
There are many other descriptions of research based on self-assembled structures and their functions [524–532].
Polymers
Polymeric materials are valued for their physical and mechanical strengths. Therefore, fabrication of structure-defined structures based on molecularly-engineered polymers has become an important subject of research in supramolecular chemistry, especially from the viewpoint of practical applications. In particular, self-assembly of conductive polymers is an attractive target for the development of novel photoelectronic properties. In corresponding research, control of alignment and orientation is a key subject because effective conjugation length and anisotropic photoelectronic properties depend significantly on these features. Oriented polymers and/or polymer nanostructures have attracted a great deal of attention because of their potential applications as, for example, electrochemical switches, electric devices, and sensors.
Kim and Swager demonstrated the influence of structure on the charge- and energy-transport properties of conjugated polymers (figure 60) [533]. The polymer they used consisted of two building blocks. In the figure, the block shown at left contains hydrophobic dioctylamide groups and is expected to display a face-on orientation with its phenyl groups' co-facial to the air–water interface. The other block has hydrophobic and hydrophilic groups and is inclined toward an edge-on structure with the p-plane normal to the interface. Surface pressure–molecular area (π–A) isotherms and spectroscopic changes during compression and expansion cycles are completely reversible. The molecular area observed in the isotherms indicated that the monolayer of this polymer adopts a face-on conformation (figure 60A) at low pressures and a zipper conformation (figure 60B) in states of greater compression.
Figure 60.

Langmuir monolayer of synthetic conjugated polymers: (A) face-on conformation; (B) zipper conformation.
Control of polymer structure using small porphyrin oligomers has been reported recently as shown in figure 61. Takeuchi, Shinkai, and coworkers reported that ordered structures constructed using porphyrin oligomers as an aligner molecule of conjugated polymers could be efficiently converted into the corresponding poly-pseudo-rotaxane structures by the template-assisted ring-closing olefin metathesis of olefinic groups at the peripheral positions of the aligner molecule [534]. They prepared solution-cast films from the thus-formed complex on a transition electron microscopy (TEM) grid without staining. The TEM micrograph indicated the presence of immobilized assemblies aggregated as micrometer-size crystalline sheets with a multi-lamellar morphology with 2.0 and 0.2 nm periodicities. The ring-closing olefin metathesis reaction did not affect the periodicities or the resultant two-dimensional sheet morphologies. This approach could be more generally applicable to polymers bearing coordinating moieties.
Figure 61.

Control of polymer orientation using small porphyrin oligomers.
Akagi and coworkers have developed strategies for controlled alignment of conductive polymers as summarized in a recent review [535]. If the polymers used contain a substituent mesomorphic group, the polymer is not only soluble in organic solvents, but can be also easily aligned by spontaneous orientation of the liquid-crystalline group. Furthermore, it could be macroscopically aligned using external perturbation, such as shear stress or an electromagnetic field. Thus, monodomain structures of the liquid crystalline phase could be constructed at the macroscopic level. Because of this the polymer should exhibit a higher electrical conductivity, relative to those with random orientation. At the same time, the molecular orientation and hence the electrical conductivity of the polymers can be controlled using an external force. Figure 62 schematically illustrates spontaneous orientation and externally-stimulated macroscopic alignment of side chain type liquid-crystalline-conjugated polymers, where the former and the latter generate multiple and mono domains of liquid-crystalline phases, respectively [545–549].
Figure 62.
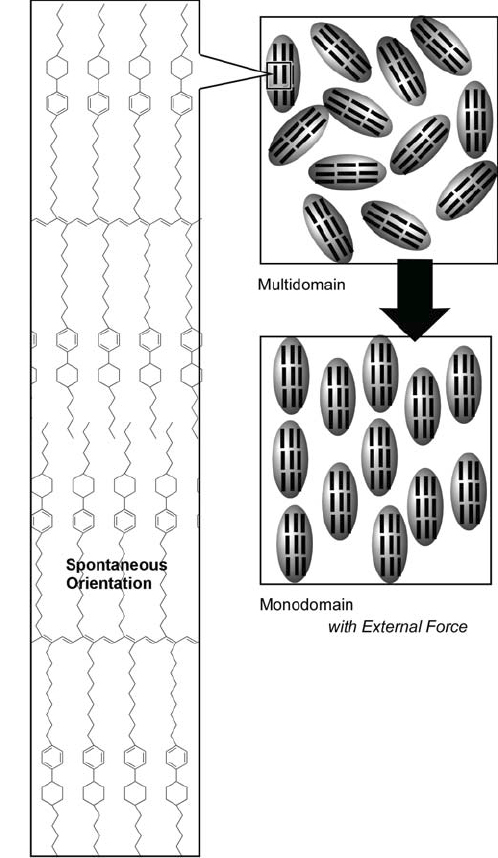
Spontaneous orientation and externally-forced macroscopic alignment of side chain type liquid-crystalline-conjugated polymers.
Apart from the above-mentioned examples, self-assembly of conjugated polymers has been widely studied- [545–549]. Additionally, other uniquely structured polymers and oligomers have also been studied with respect to their self-assembling abilities [550, 551]. Among them dendrimers make attractive structural units because the shape of the dendron units is relatively inflexible when compared with freely extending conventional polymers. Exemplary work has been presented in pioneering research by Percec and coworkers [552]. Recently, they reported self-assembly of semifluorinated dendrons attached through a flexible spacer to an electron-donor, which induced p-stacking of the donors in the center of a supramolecular helical pyramidal column. These structures self-organize into a variety of columnar liquid crystal phases; these crystal phases mediate self-processing of large monocrystalline mesophase domains with capability to self-repair their intracolumnar structural defects. Cho et al studied the self-assembly of amphiphilic dendrons extended with linear polyethylene oxide chains and their ion complexes [553]. Since a single compound possessed several mesophases, correlations could be made between charge transport, mechanical properties and structure. Self-assembly of dendrimers with pentafluorophenyl units was reported by Mllen and coworkers [554]. Self-assembly of dendron-rod-coil molecules into ribbon-like one-dimensional structures was demonstrated by Stupp and coworkers [555]. Stoddart and coworkers reported reversible control over the self-assembled structure of dendronized polyacetylenes based on acid-base interactions [556].
Formation of vesicle structures by self-assembly of polymeric materials has also been investigated. Well-designed peptides can act as amphiphilic components of polymer vesicles. Bellomo et al reported the self-assembly of diblock copolypeptides into spherical vesicles whose micron-scale diameter and structure are dictated primarily by the ordered conformations of the polymer segments [557]. As shown in figure 63, the polymer used contains oligo (ethyleneoxide)-terminated lysine sequences and leucine–lysine copolymer as hydrophilic and hydrophobic domains, respectively. In the latter, hydrophilic and electrostatic properties of the lysine residues could be tuned by varying pH. Protonation of the lysine residues in the polypeptide chain considerably enhances their hydrophilicity with a concurrent destabilization, caused by electrostatic repulsion, of the a-helical structure of the leucine-rich domain. A helix-to-coil conformational transition induced by protonation of the lysine groups also destabilizes the vesicular assembly, leading to porous membranes. Leakage of trapped materials within the vesicle core was minimized under basic conditions. However, at lower pH, a rapid disruption of the vesicle membranes occurred with release of the trapped molecules.
Figure 63.

Self-assembly of diblock copolypeptides into spherical vesicles.
Fabrication of vesicle and capsule structures [558–562], tubes [563], fibers and rods [564, 565] and porous structures [566–568] using polymeric structural units has been extensively reported. Amongst these a unique example is the fabrication of honeycomb structures developed by Shimomura and coworkers [569–572]. They proposed a method for creation of ordered patterns of polymeric materials in honeycomb-like structures by a simple solution-casting of the organic substances onto solid substrates. Figure 64 illustrates schematically the mechanism of formation of the honeycomb structure made from a polyion complex. Evaporation of the solvent from a droplet of a chloroform solution of the polyion complex induces a cooling of the solution, which allows water droplets to condense onto the chloroform solution. The surface active polyion complex reduces the surface tension between water and chloroform, and fusion of the stabilized water droplets is prevented. The droplets are either dragged into solution by convection or keep floating on the solution surface. Further evaporation of the chloroform solution facilitates condensation of water, resulting in hexagonal packing of the water droplets at the solution-air-substrate three-phase line by capillary force. The three-phase line moves over the array of water droplets, leaving behind ordered water droplets with polymer between them. The water evaporates and finally leaves a honeycomb-like structure.
Figure 64.

Mechanism for the formation of the honeycomb structure made from a polyion complex.
Biomolecules
Many of the sophisticated structures present in biological systems are constructed through self-assembly of their constituent molecules including lipids, saccharides, peptides, proteins, and nucleic acids. Therefore, biomolecules are useful for formation of self-assembled structures. Structures self-assembled from such biomimicking molecules, or from biomolecules themselves, have been also investigated [573–576].
Among the biomolecules, peptides are probably the best understood with respect to their synthesis, structure, and properties. Therefore, research on self-assemblies using peptide components is well developed. Stupp and coworkers developed supramolecular nanostructures from peptide-amphiphiles with function-oriented designs (figure 65) [577]. A hydrophobic hexadecanoyl tail was introduced at the N-terminus of a tetracysteine that can form reversibly cross-linked structures upon redox reaction. Triglycine and phosphorylated serine were connected sequentially at the C-terminal position of the tetracysteine residue. These residues play the roles of spacer and calcium ion host, respectively. Finally, a cell adhesive sequence (RGD, Arg-Gly-Asp) was introduced at the end of the oligopeptide. At lower pHs (pH<4), an aqueous solution of this amphiphilic peptide attains a gel-like structure containing fibrils with a diameter of 6–7 nm and lengths of several micrometers. Increasing the pH to approximate neutrality induced dissolution of the fibers, although dissolution could be suppressed by oxidative cross-linking of the cysteine residues. In addition, hydroxyapatite crystals could be grown in the presence of these peptide fibers and the composites obtained can be regarded as analogues of the complex structures in collagen fibrils containing hydroxyapatite in naturally occurring systems.
Figure 65.

Peptide design for supramolecular self-assembled nanostructures.
Deming and coworkers synthesized diblock copolypeptide amphiphiles containing charged and hydrophobic segments [578]. Their gelation behavior depended not only on the amphiphilic nature of the polypeptides but also on chain conformations, i.e. α-helix, β-strand or random coil. The same research group prepared vesicles composed of polyarginine and polyleucine segments that were stable in solution, could entrap water soluble species, or could be processed to different sizes and prepared in large quantities [579]. Morikawa and Kimizuka reported spatially controlled self-assembly of gold nanoparticles encased in a-helical polypeptide nanospheres [580]. Pochan, Schneider, and coworkers fabricated β-sheet fibrils exhibiting a non-twisted, stacked morphology [581]. Hentschel and Börner reported self-assembly of a polymer-peptide conjugate comprising a sequence-defined polypeptide and a poly(n-butyl acrylate) in organic media and found transformation of helical structures into superhelices [582]. Higashi and coworkers reported a novel, programmable, molecular self-assembling system for fabrication of shape-specific, three-dimensional nanoarchitectures, using three types of simple 16-mer peptides consisting of hydrophobic Leu and hydrophilic Lys [583]. Oda and coworkers have been investigating self-assembly behavior of gemini-amphiphiles and recently reported formation of twisted and helical ribbon structures based on the interpeptidic -sheet structure of gemini-oligoalanine [584]. Nilsson et al researched interactions between polythiophene derivatives with synthetic peptides, where conformation and optical properties of the polythiophene conjugated polymer can be regulated [585, 586]. Pochan, Schneider, and coworkers developed a simple light-activated hydrogelation system that employs a designed peptide whose ability to self-assemble into a hydrogel material depends on its intramolecular folded conformational state [587]. Matsuzawa et al prepared a molecular system capable of organizing and dispersing upon exposure to different wavelengths of light by using an azobenzene derivative containing three valyl units [588]. Because of their biological importance, self-assembly of amyloid fibrils and their analogues has been investigated by several groups [589–592]. Formation of gels [593] and nanotubes [594–598] has been reported as well as formation of peptide-containing inorganic structures by biomineralization [599, 600]. Larger protein structural units have also been subjected to self-assembly forming nanotubes [601], nanowires and fibers [602–604], and nanorings [605]. Mao et al proposed the use of virus assembly for the preparation of one-dimensional inorganic structures [606].
Nucleic acids and their bases are also good candidates for formation of self-assembled structures because they can be involved in very specific hydrogen bonding interactions. As structures self-assembled from simple molecular units, Kimizuka and coworkers have recently found dynamic nanowire formation through self-assembly of adenosine triphosphate (ATP) and dichloro-substituted thiacarbocyanine dyes (figure 66) [607]. An immediate color change from pink to orange was observed upon addition of ATP to an aqueous solution of the thiacarbocyanine dye. Circular dichroism (CD) spectra revealed intense peaks at 423 nm (positive) and 475 nm (negative) that were assigned to the absorption band of achiral chromospheres, and indicating the appearance of induced circular dichroism (ICD). This ICD pattern is indicative of excitonic interactions among multiple chromophores, where the formation of parallel-oriented chromophores (H-aggregates) can be inferred from the spectral blue shift to 463 nm. Observation by TEM revealed nanowires with a minimum width of ∼10 nm and lengths of several micrometers. Upon heating, the peak intensity of the H-aggregates gradually decreased until the peak due to monomeric absorption at 546 nm was obtained. At 55 °C, nanowires could not be observed in TEM images. Upon cooling the dispersion below 44 °C, a high yield of nanowires with regular width of ∼40 nm was obtained. Therefore, nanowire formation can be dynamically controlled with thermal stimuli; this suggests that molecular ordering in supramolecular assemblies may be enhanced by thermal control.
Figure 66.

Dynamic nanowire formations through self-assembly of adenosine triphosphate (ATP) and dichloro-substituted thiacarbocyanine dyes.
The highly specific complementary base-pairing of nucleic acids, variously DNA or RNA, permits them to store and transmit biological information. This unique property can be used for programmed formation of nanostructures and microstructures and has been summarized in a recent review by Seeman and Lukeman [608]. Not only structural formation but also the operation of molecular mechanical devices, i.e. nanorobots, based on programmed DNA assemblies was proposed by Ding and Seeman [609]. In their system, a robotic arm composed of DNA can rotate on a two-dimensional crystal of a DNA array. Self-assembly based on similar concepts has become an attractive goal for many researchers since custom synthesis of the desired DNA sequences can be now performed with ease [610–612]. One example was recently reported by Matsuura and coworkers who successfully prepared a nanocage using DNA three-way junctions formed from three 30-mer oligonucleotides that contain single-chained self-complementary sticky ends (figure 67) [613]. Absence of defects such as the single and double strand end structures from the spherical nanoassemblies was indicated by exo- and endo-nuclease digestion experiments, and provided evidence for the closed nanocage structure. Very recently, they developed a similar concept using artificial C3-symmetric peptide conjugates [614].
Figure 67.
Nanocage formation from DNA three-way junctions formed from three 30-mer oligonucleotides that contain single-chained self-complementary ‘sticky’ ends.
Inorganic substances
Because self-assembly is usually driven by intermolecular interactions such as hydrogen bonding and coordination, one might think that the scope of self-assembling processes is limited to organic compounds or biological molecules. However, inorganic substances such as colloidal particles or nanocarbons can be self-assembled through the mediation of organic interactions and physical interactions such as capillary force. In this section, self-assembly of inorganic substances is briefly outlined.
Self-assembly of colloidal nanoparticles has been well researched and there are several excellent reviews available [615, 616]. Therefore, only the unusual aspects of this research topic are described here. Arrays of inorganic particles can be created from self-assembled structures of biomaterials. For example, Yamashita and coworkers demonstrated use of a biomolecular array for structure transcription (figure 68) [617, 618]. Ferritin, which is composed of 24 self-assembled peptide subunits and capable of including iron oxide, was assembled as a Langmuir monolayer then transferred onto a hydrophobized silicon substrate forming a two-dimensional hexagonal array of ferritin. Organic components were then destroyed through UV/ozone treatment, which was followed by heat treatment under hydrogen leaving iron particles with diameters less than 6 nm arranged in a hexagonal arrangement. Supramolecular arrays prepared from inorganic substances can be used for device preparation, for instance, as the gate of a floating quantum dot gate transistor for multilevel logic capable of room temperature operation.
Figure 68.

Iron nanodot array synthesized from Langmuir monolayer of ferritin.
Self-assembly of inorganic particles within the pores of mesoporous materials is similar to the previous example in that inorganic particles are assembled through confinement in nanosized spaces. Petridis and coworkers entrapped magnetic particles in pores of MCM-41 mesoporous silica [619]. Propionic acid vapor was chemisorbed into an iron-impregnated MCM-41 so that iron-carboxylate species were formed through interaction of the propionic acid vapors with iron atoms entrapped in the MCM-41. Subsequent pyrolysis gave rise to γ-Fe2O3 particles. Huang, Corrigan, and coworkers demonstrated synthesis of copper-tellurate (Cu2Te) nanoparticles within a mesoporous system [620]. Fukuoka et al synthesized bimetallic nanowires and nanonecklace with high aspect ratios in one-dimensional channels of ordered mesoporous organosilica [621].
Surface modification of colloidal particles with appropriate supramolecular elements can induce controlled assembly of colloidal particles in bulk solution. Bhatia and coworkers reported logic operations in the self-aggregation of superparamagnetic Fe3O4 nanoparticles for two inputs associated with cancer invasion (MMP2 and MMP7) [622]. As illustrated in figure 69, the Fe3O4 nanoparticles were designed to coalesce in response to logical AND or OR functions. Polymers were linked to each particle using unique protease substrates so that assembly could occur only in the presence of both enzymes (AND logic gate). Furthermore, actuation of assembly in the presence of either or both of the enzyme inputs (OR logic gate) was achieved by anchoring polymers to only the ligand nanoparticles with a tandem peptide substrate (containing both enzyme cleavage motifs in series). Web and coworkers created vesicle aggregates that were susceptible to external magnetic fields by cross-linking the vesicles with magnetite nanoparticles through Cu-mediated interaction (figure 70) [623]. Patterned assemblies of vesicles with potential bionanotechnological applications could be obtained through magnetospatial manipulation of different populations of vesicle aggregates. Reversibility of cross-linking of the vesicles by magnetic nanoparticles was designed into the system by using multiple weak interactions to link the vesicle and nanoparticle surfaces.
Figure 69.

Logic operations of self-aggregation of superparamagnetic Fe3O4 nanoparticles two inputs associated with cancer invasion (MMP2 and MMP7).
Figure 70.

Vesicle aggregates cross-linked by magnetite nanoparticles through Cu-mediated interaction.
Colloid assemblies often provide regular crystal-like arrays, which can lead to potential optical applications. For example, Furumi et al presented a new potential use of high-quality colloidal crystal films for low-threshold laser applications by optical excitation [624]. Figure 71 shows a schematic illustration of the colloidal crystal laser device structure. Highly efficient laser-feedback occurs when a light-emitting polymer layer is introduced between a pair of colloidal crystal films as a planar defect because of the photonic band-gap effect in colloidal crystal films. They demonstrated operation of a flexible colloidal crystal laser device constructed of all-polymer materials.
Figure 71.

A colloidal crystal laser device.
Self-assembled structures of colloidal particles provide various micro-objects with specific shapes. Dinsmore et al proposed an approach for fabrication of solid capsules from colloidal paricles with precise control of size, permeability, mechanical strength, and compatibility [625]. As illustrated in figure 72, capsules were fabricated by the self-assembly of colloidal particles onto the interface of emulsion droplets. Following ‘locking’ together of the particles to form elastic shells, the emulsion droplets were transferred to a fresh continuous-phase fluid identical with that contained inside the droplets. The resultant structures, called ‘colloidosomes,’ are hollow, elastic shells whose permeability and elasticity can be precisely controlled. Apart from colloidsomes [626–628], self-assembly and related processes provide variously shaped inorganic objects such as tubular structures [629, 630], nanosheets [631–633], nanodiscs [634], microribbons [635], nanowires [636, 637], porous structures [638], and others [639, 640].
Figure 72.

Formation of colloidosome.
Similarly to the fullerene self-assemblies that were described in a previous section, assembled structures of nanocarbons have become an important subject in materials research. In particular, the volume of research performed based on self-assembly of carbon nanotubes for material preparation has increased rapidly. Although many approaches have been reported [641–646], only one example based on a well-known supramolecular concept is here described. Harada and coworkers demonstrated a novel chemically responsive supramolecular single-walled carbon nanotube (SWNT) hydrogel by using soluble cyclodextrin-functionalized SWNTs [647]. Since cyclodextrin has a high solubility in water, water-soluble SWNTs carrying cyclodextrins were obtained by using π–π interaction between pyrene modified β-cyclodextrins and SWNTs. Cyclodextrin forms host–guest complexes with a variety of guest compounds, so that the vacant cyclodextrin cavities surrounding the SWNTswere capable of capturing guest molecules at the SWNT surface. As illustrated in figure 73, following host–guest interactions with polymers carrying guest moieties, supramolecular SWNT hydrogels could be fabricated.
Figure 73.

Supramolecular single-walled carbon nanotube hydrogels based on guest inclusion of attached cyclodextrin.
Self-assembly at interfaces
Objects self-assembled in solution are often invisible making solid state device preparation inconvenient. Therefore, self-assembly at an interface, which immobilizes supramolecular objects on defined surfaces, should be important with respect to practical applications. Additionally, self-assembling processes at interfaces have different characteristics from those observed in bulk phases. Research on self-assembly at interfaces has now become a separate research field. In the following section, we have summarized research on self-assemblies at interfaces.
Langmuir-blodgett films
The air–water interface provides a unique hetero-dielectric environment where two phases having totally opposite dielectric constants are in contact. On the other hand, this interfacial space possesses a molecularly flat plane albeit in a dynamic state. Therefore, the nature of self-assembly processes at the air–water interface should be atypical. Resulting self-assembled structures can be immobilized by using the Langmuir–Blodgett (LB) technique. As schematically illustrated in figure 74, LB films are prepared through layer-by-layer transfer of monolayer structures spread on liquid surface such as the air–water interface.
Figure 74.

Langmuir–Blodgett (LB) film.
Assembly between monolayer components and guest materials from the aqueous subphase, i.e. molecular recognition at the air–water interface [648–653], is one of the most important research topics in this field. Efficiency of molecular recognition occurring at the air–water interface is generally greatly enhanced compared to that observed in bulk aqueous solution, was and this has been demonstrated in the following examples. As depicted in figure 75, phosphate functionality was recognized by guanidinium groups embedded at the water surface through both electrostatic interaction and hydrogen bonding [654]. Kunitake and coworkers investigated the strength of binding for this recognition pair. Binding constants for aqueous phosphates such as adenosine monophosphate (AMP) and adenosine triphosphate (ATP) are in the range of 106–107 M-1 (figure 75C), much greater than that reported for monomeric species in water (1.4 M-1, figure 75A). Binding constants of AMP to the guanidinium functionality in aqueous aggregates such as micelles or bilayer vesicles are found in the range 102–104 M-1. These values are significantly larger than that between molecularly-dispersed guanidinium and phosphate in water, but are also much smaller than those observed at the air–water interface. The magnitude of binding constants should be related to the type of interface. Sakurai and coworkers proposed application of a quantum chemical calculation to molecular recognition at the air–water interface [655–657]. Calculations were performed based on a multi-dielectric model for the guanidinium-phosphate system situated at an interface. This series of calculations revealed that, even when the binding sites are located near a non-polar phase and are exposed to the aqueous phase, the ion complex can be stabilized. This indicates that absence of water at the binding sites is not always necessary for efficient guest binding through hydrogen bonding.
Figure 75.

Binding constant between phosphate and guanidinium at (A) molecular interface, (B) mesoscopic interface, and (C) macroscopic interface.
As shown in the examples in figure 76, recognition of various biomaterials through hydrogen bonding has been demonstrated by Kunitake and coworkers. Diaminotriazine-functionalized amphiphile monolayers and thymine derivatives can form a complementary hydrogen bond pair (figure 76A) [658]. The observed binding constant (ca. 300 M-1) corresponds roughly to that for similar binding combinations in aprotic organic solvents. Similarly amphiphilic orotate recognizes adenine at the air–water interface (figure 76B) where guest binding cooperativity could be detected [659]. Conceivably, stacking of the bound adenine molecules accelerates the binding, and this can be seen as an artificial mimic of the base stacking in the double-helix formation of DNA. Recognition at the air–water interface provides median excellent medium for multi-point binding as seen in the recognition of ATP (figure 76C) [660, 661]. Mixed host components spontaneously assembled into the optimized structure for aqueous guest recognition. Multi-point molecular recognition was also demonstrated in a binary system, involving recognition of flavin mononucleotide (FMN) by mixed monolayers of dialkylmelamine and monoalkyl guanidinium where reasonably efficient with a binding constant of more than 107 M-1 [662]. A ternary recognition system was also similarly designed as shown in figure 76(d). A single molecule of flavin adenine dinucleotide (FAD) possesses the potential to bind two guanidinium molecules at phosphate groups, one orotate molecule at adenine sites, and diaminotriazine at an isoalloxiazine ring [663]. Great effort in the synthesis of complicated covalently-linked host compounds, which is often an obstacle in research on molecular recognition, is not required for formation of multi-point recognition sites in mixed monolayer.
Figure 76.

Molecular recognition of various biomaterials through hydrogen bonding at the air–water interface: (a) thymine; (b) adenine; (c) ATP; (d) FAD.
Kunitake and coworkers have made systematic studies on recognition of aqueous peptides using single-component or mixed monolayers of variously functionalized amphiphiles [664–668]. An examples is shown in figure 77 where an aqueous dipeptide (GlyLeu) was bound at a binding site dynamically formed from a GlyVal-functionalized amphiphile and a benzoic acid amphiphile. Infrared data suggested that the guest GlyLeu forms mainly a COOH dimer at its C-terminal with the benzoic acid group of the host benzoic acid amphiphile. The hydrophobic part of the monolayer accommodated the hydrophobic side chain of GlyLeu. In addition, a stable antiparallel-β-sheet-type hydrogen bonded structure is formed with the surrounding dipeptide moieties of the host GlyVal-functionalized amphiphile.
Figure 77.

Recognition of aqueous peptides by mixed monolayers.
The air–water interface has been also used for recognition of various aqueous guest molecules including nucleic acids, their bases, nucleosides, and nucleotides [669–673], barbituric acid and related derivatives [674–678], amino acids, saccharides [679–687], and others [688, 689]. However, most of these examples do not utilize one critical feature of the air–water interface, its dynamic nature. In biological systems, very sophisticated control of signal response based on molecular recognition may result from dynamic processes, and they could be mimicked and applied for the preparation of artificial devices. From these points of view, effects of dynamism of the monolayer structures at the air–water interface on molecular recognition should be more seriously considered [690]. Recent examples from the authors' group are briefly summarized below. The first example, shown in figure 78, involves monolayers of a steroid cyclophane, which contains a 1,6,20,25-tetraaza[6.1.6.1]-paracyclophane cyclic core connected to four steroid moieties (cholic acid) through a flexible L-lysine spacer [691–694]. The steroid cyclophane was reported to exhibit large binding constants for aqueous naphthalene guests such as 6-(p-toluidino)naphthalene-2-sulfonate (TNS) in a 1 : 1 stoichiometry, while the corresponding cyclophane lacking the steroidal residues only weakly binds this guest. Therefore, attainment of a cavity conformation is essential for efficient molecular recognition as illustrated in figure 78. Compression and expansion of the monolayer induces reversible formation of the cavity conformation and results in reversible binding (capture and release) of the aqueous fluorescent guest TNS.
Figure 78.

Dynamic molecular recognition of aqueous guests by monolayers of a steroid cyclophane. © 2006, Royal Society of Chemistry, Soft Matter 2 (2006) 465.
Chiral recognition is another important topic in molecular recognition. The air–water interface has been also used for chiral recognition within membrane components [695–700] and/or between amphiphile and aqueous guests [701–708]. Mechanical control of chiral recognition was recently achieved using the novel concept of molecular twisting, as shown in figure 79A, where an octacoordinate sodium complex of a polycholesteryl-substituted cyclen embedded at the air–water interface was used as a molecular host [709]. Helicity of the two possible quadruple helicate structures of this complex is influenced by chirality of the side arms especially when ordered or aggregated at the supramolecular level. Compression and expansion of the monolayer of the cholesteryl-substituted cyclen complex induced particular changes in the helix structure and there was a consequent alteration of the diastereomeric stability of complexes with guest molecules. The binding constant for L-leucine is always smaller than that of, D-leucine indicating that the cyclen monolayers have a weaker interaction with L-leucine (figure 79B). Conversely, the binding constant of L-valine is smaller than that of D-valine at low surface pressure but exceeds it at 22–23 mN m-1. Thus, upon monolayer compression chiral recognition of valine stereoisomers by the cyclen monolayer reverses from the D- to L-form. Because chiral selection is one of the major mysteries in biological processes, alteration of chiral selection based on a simple mechanical stimulus is surprising.
Figure 79.

(A) Molecular twisting of an octacoordinate sodium complex of a polycholesteryl-substituted cyclen embedded at the air–water interface. (B) Ratio of binding constant between D- and L-amino acid (Leu and Val) to the polycholesteryl-substituted cyclen monolayer. © 2006, American Chemical Society, J. Am. Chem. Soc. 128 (2006) 14478.
Monolayers and LB films are appropriate media for investigation of molecular orientation and assembly modes within size-constrained systems. For example, dye assembly in monolayers can be easily evaluated using specific spectral shifts, which can be useful for sensor applications. If aggregation of porphyrin molecules is controlled by guest binding at the air–water interface, spectral shifts upon porphyrin aggregation can result in colorimetric reporting of analytes. This idea was realized by Ariga et al for halide anion sensing using a Langmuir monolayer of an N-confused porphyrin, 5,10,15,20-tetraphenyl-2-aza-21-carbaporphyrin (N-confused tetraphenylporphyrin, NC-TPP) [710]. NC-TPP in a matrix monolayer with methyl octadecanoate on aqueous sodium halides were analyzed using UV-vis spectroscopy. On aqueous NaI, the monolayer contained a distinct peak at around 465 nm, while others gave suppressed peaks with maxima at around 420–430 nm. The red-shifted absorption band obtained from the NC-TPP monolayer on aqueous NaI indicated that NC-TPP forms J aggregates on binding of iodide ions. H aggregates were formed with the other halide ions (fluoride, chloride, and bromide) and in the absence of anions. A plausible mechanism is shown in figure 80. Iodide anions in the subphase specifically induce red shifts in the UV-vis spectra upon J-aggregate formation of the NC-TPP molecules embedded in the lipid matrix. In contrast, the NC-TPP monolayer tends to form H aggregates in the presence of the other halogen ions and it was also noted that at pH 11 free-base NC-TPP forms a J-aggregate structure. Selection of halogen ions was also permitted by transfer of these monolayers onto a solid surface. While loosely bound fluoride, chloride, and bromide ions were detached from NC-TPP so that it transformed to the stable free-base accompanied by J-aggregate formation, iodide ions were accommodated during transfer and the assembly mode of the NC-TPP molecules was not changed. Stabilization of the J-aggregate structure may be due to the size of the bound iodide ions, probably resulting in stable binding of said ions to the film.
Figure 80.

Aggregate-based anion recognition by N-confused porphyrin monolayer.
The LB technique can also provide facile contact of functional thin films onto device surfaces allowing, for example, speculation on sensor applications [711–713]. For example, Miyahara and Kurihara synthesized a novel electroconductive amphiphile functionalized with a boronic acid which could be incorporated into an LB film at an electrode (figure 81) [714]. Subsequent doping of the monolayer with iodine made the corresponding amphiphilic component conductive. Electrochemical measurements then confirmed that selective binding of a sugar derivative to the LB monolayer occurred. Conducting molecules containing a binding site can operate as molecular wires with a connecting terminal. Because of the great variety in composition and construction of self-assemblies this strategy should open up various possibilities for the design of well-defined molecular sensing systems. Immobilization of proteins as molecular thin films using the LB technique is also an attractive approach, although proteins sometimes suffer denaturation caused by the high surface tension at an air–water surface. Okahata et al prepared a lipid-coated enzyme (GOD, glucose oxidase) that could be obtained as a precipitate by mixing aqueous solutions of the enzyme and lipid, and transferred their monolayers onto a Pt electrode [715, 716]. Denaturation of the proteins even at the air–water interface was prevented by covering GOD with lipids. The electrode obtained was used in amperometric measurements as a glucose sensor (figure 82).
Figure 81.
Sensor for sugar by electroconductive amphiphile functionalized with a boronic acid.
Figure 82.

Glucose sensing by an LB film of a lipid-coated glucose oxidase.
The air–water interface promotes formation of dynamically assembled structures from component molecules. For example, Talham and coworkers reported formation of a nickel-iron cyanide grid network at the air–water interface (figure 83) [717, 718]. They synthesized a cyanide ligand-containing amphiphilic octahedral iron complex. Langmuir monolayers of this complex with aqueous nickel ions contained in the subphase resulted in the formation of coordinate covalent networks. The square grid nickel-iron cyanide network arises from the 90° bond angles about the iron cyanide complex where the amphiphilic dialkylaminopyridine confined the square grid network. Magnetic measurements indicated a ferromagnetically ordered state below 8 K; the magnetic behavior was strongly dependent on the orientation of the sample within field. In another unique example, construction of a box structure was achieved through self-assembly of amphiphilic ligands with end-capped Pd(II) components in the presence of a rod-like guest as a template as demonstrated by Aoyagi et al, who used a hydrophobic terpyridine derivative together with a biphenyl-type guest bearing a long alkyl chain (figure 84) [719]. Upon coordination with the end-capped Pd(II), a nanobox assembly structure was obtained at the air–water interface. The relationship between the molar ratio of amphiphilic components and the molecular area of the monolayers revealed that the only species present in the monolayer consists of the amphiphilic terpyridine, the biphenyl guest, and end-capped Pd(II) with 4 : 1 : 6 stoichiometry, suggesting dynamic formation of the nanobox structure. Spontaneous formation of chirally selected structures at the air–water interface was extensively researched by Liu and coworkers, who first discovered formation of spiral (chiral) structures in transferred monolayers of a non-chiral barbituric acid derivative (figure 85) [720]. Spiral nanofibers were observed by AFM observation when the film was compressed to the inflection point in the ý-A isotherm. The spirals observed were wound in both anticlockwise and clockwise directions. Circular dichroism (CD) spectral data suggested that handedness of aggregation was maintained after initial selection at the beginning of structure formation because of a cooperative interaction. Interestingly, the starting aggregate handedness was determined by chance.
Figure 83.

Formation of a nickel-iron cyanide grid network at the air–water interface.
Figure 84.

Formation of a box structure by the self-assembly of amphiphilic ligands with end-capped Pd(II) components in the presence of a rod-like guest as a template.
Figure 85.

Formation of spiral (chiral) structures in transferred monolayers of a non-chiral barbituric acid derivative.
These features of molecular recognition can be used to prepare specific structures in two-dimensional formations. The so-called ‘two-dimensional molecular patterning’ methodology [721–723] was pioneered by Kunitake and coworkers. Examples in figure 86 reported by Kunitake and coworkers reveal that aqueous dicarboxylate can bind to two dialkylguanidinium molecules, and that a spacer between the two carboxylates of the aqueous template affects the crystallinity of the guanidinium amphiphiles [724–726]. Interaction between some acidic species such as phosphate and carboxylate and guanidinium is quite strong because of the combined effect of electrostatic interactions and hydrogen bonding. Monolayers transferred from aqueous oxalate (no methylene spacer) displayed a crystalline arc (a), while only an amorphous halo was observed for the monolayer from the pure water subphase (b). The rigid oxalate template bound the two guanidinium molecules effectively weakening the electrostatic repulsion among cationic guanidinium molecules by the stoichiometric binding. When the template was changed to malonate (CH2) spacer or succinate (CH2)2 spacer, the electron diffraction pattern obtained could be assigned to the crystalline state, but the arc in the patterns broadened (c). A further increase in the spacer length glutarate with (CH2)3 spacer and adipate with (CH2)4 spacer results in an amorphous phase of the monolayer (d).
Figure 86.
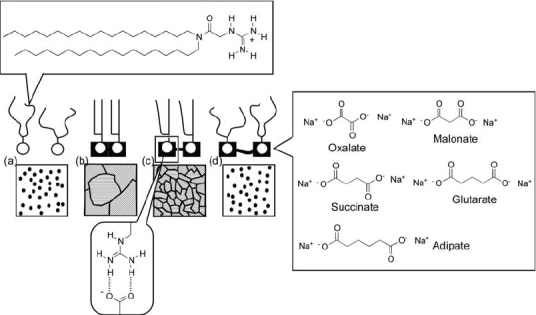
Control of crystalline structures of guanidinium monolayers by binding of aqueous dicarboxylate.
Kunitake and coworkers also demonstrated twodimensional molecular patterning upon molecular ribbon formation between amphiphilic melamine and aqueous barbituric acid derivatives (figure 87) [727]. An AFM molecular image of the didodecylmelamine monolayer bound to the barbituric acid derivative was easily observed, although the corresponding monolayer transferred from pure water was too fragile for AFM observation. The packing model derived from the AFM image indicated an oblique array of methyl terminals. With binding of thiobarbituric acid rectangular and oblique arrays of the alkyl chains of the monolayer were observed, respectively, whereas a hexagonal arrangement of the alkyl chains was observed for the monolayer transferred from pure water. Varying the concentration of thiobarbituric acid in the subphase permits some control over the mesoscopic pattern of the molecular ribbon structure. Extended structures were obtained at higher concentrations of thiobarbituric acid probably due to more efficient formation of the molecular ribbon.
Figure 87.
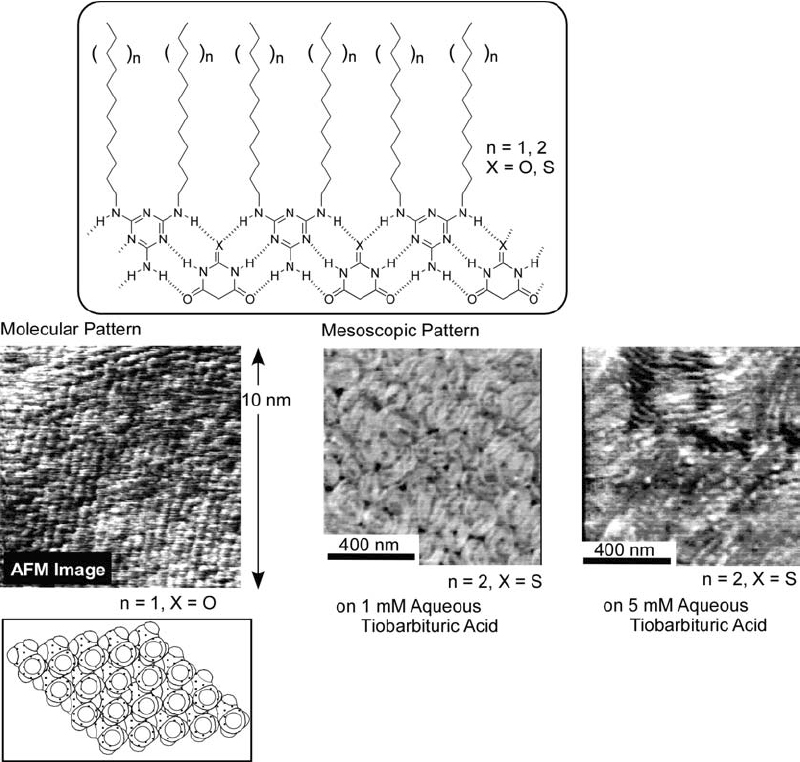
Two-dimensional molecular patterning upon molecular ribbon formation between amphiphilic melamine and aqueous barbituric acid derivatives. © 1996, Royal Society of Chemistry, Chem. Commun. (1996) 1769.
Figure 88 illustrates that binding of an aqueous guest at heterogeneous recognition sites of mixtures of amphiphiles at the air–water interface can induce a sequence-controlled array of amphiphiles [728, 729]. Molecular patterning by this method requires an aqueous template molecule with multiple binding sites, such as flavin adenine dinucleotide (FAD), to recognize the guanidinium and orotate amphiphiles. AFM observation of such patterns at a mica surface revealed that the three-dimensional profile of the transferred monolayer displays a periodic wavelike structure composed of peaks with two different amplitudes. The height difference between the two peaks is estimated to be several angstroms. Binding of FAD with the guanidinium/orotate mixed monolayer fixes the two functional units at the same level through binding to the template FAD molecule. Because of the conformational flexibility of the FAD molecule, the flavin unit in FAD may be buried under the phosphate group resulting in a height difference between the two terminal methyl groups.
Figure 88.

Molecular patterning based binding of flavin adenine dinucleotide (FAD) to the guanidinium and orotate amphiphiles. © 1997, American Chemical Society, Langmuir 13 (1997) 519.
Predictable pattern formation may not be favored by the flexible nature of the complexes formed because the complexes deform easily upon monolayer compression. To solve this problem, the novel ‘molecular tiling’ technique was proposed. Molecules first used in this method had six chains located at the apexes and one chain at the center and are referred to as ‘Heptopus’ (figure 89) [730]. Uneven positioning of the inner and outer chains of Heptopus should result in periodic holes (a) and tips (b) created by the differences in chain length. A second example of molecular tiling used cyclic phenylazomethine molecules with a rigid triangle geometry as a tiling template [731]. Subsequently, the secondary dialkylguanidinium amphiphile component was added. These amphiphiles form a stoichiometric complex on addition of an aqueous solution of 6-(p-toluidino)naphthalene-2-sulfonate (TNS). This tiling strategy provides a versatile design of patterns upon appropriate selection of component molecules.
Figure 89.

Molecular tiling with ‘Heptopus’.
A unique fabrication technique for LB films in the out-plane direction was recently developed by Regen and coworkers, and is known as the ‘glued LB film’ [732–734]. They developed glued LB films through polyanion bridging of cationic amphiphiles at an LB interlayer and investigated gas permeability through the glued LB films (figure 90). The combined amphiphile-polymer structure effectively serves as a barrier to gas transport, which would not be accomplished by a thin layer of polymer encased in the lipid bilayer. The authors also raise the significant possibility of using molecular design to create new classes of glued LB films that can discriminate among permeants on the basis of their size, shape, hydrophobicity, and polarity.
Figure 90.

Glued LB films through bridging cationic amphiphiles by polyanions at LB interlayer.
Other examples not mentioned above include monolayers and LB films of unusual amphiphiles [735–741], quantum dots [742], clay plates [743], and carbon nanotubes [744] and will be described in later sections of this review. Advanced functions such as electroactive properties [745, 746], photo-induced electron transfer [747], logic gate [748], photo-switched wettability control [749], protonation-induced rearrangement [750], and vectorial electron transfer [751] were reported as well as more fundamental science related to phase boundary [752], domain formation [753], acid-base dissociation [754, 755], elasticity [756], basic characterization [757–759]. Furthermore, other interfaces such as the air-mercury interface [760] and interfaces between water and organic solvents [761–764] have been used for self-assembly processes.
Layer-by-layer assembly
The alternate layer-by-layer (LbL) adsorption technique has been embraced as a versatile method for thin film preparation and has rapidly developed as a method for self-assembly at interfaces [765–769]. Figure 91A illustrates the method of electrostatic layer-by-layer assembly in which a cationic polyelectrolyte first adsorbs on a negatively charged surface of a solid support in such a manner that over-adsorption occurs causing surface charge reversal. The subsequent adsorption of an anionic particle again reverses the surface charge. Alternation of the surface charge permits continuous fabrication of the layered structure. Because of the simplicity of the assembly process, there is a vast choice of materials available. Various types of polymers including conventional polyelectrolytes [770–774], conductive polymers [775, 776], block-copolymers, dendrimers [777, 778]; biomaterials such as proteins [780–785], nucleic acids [786, 787], saccharides [788–792], virus particles [793], and other biomaterials [795]; inorganic materials such as colloidal particles [795–799], quantum dots [800], clay plates [801, 802], nanosheet [803–809], nanorods and nanowires [810, 811], various nanotubes [812, 813], and other inorganic substances [814–817]; molecular assemblies including dye aggregates [818–821], micelles [822, 823], vesicles [824, 825], LB films [826], and lipid membranes [827–830] are applicable in the LbL technique. Therefore, the LbL technique has become a powerful methodology for preparation of nanostructured supermolecules. Additionally, this concept can be extended to preparation of three-dimensional nano-objects, using a colloidal core as a supporting material (figure 91B) [831–833]. LbL assembly at the surface of the colloidal core can be followed by destruction of the central core resulting in hollow capsules.
Figure 91.

(A) Alternate layer-by-layer (LbL) adsorption technique for thin film preparation on flat solid support. (B) LbL assembly on colloidal particle and hollow capsule formation.
Driving forces for film assembly are not limited to electrostatic interactions alone. In pioneering work, Mallouk and coworkers initiated LbL assembly based on metal-phosphate interaction (figure 92A) [834]. A self-assembled monolayer of a phosphonate-functionalized thiol compound was anchored on a gold surface, and was then subjected to LbL assembly of zirconium ions and 1,2-ethanediylbis(phosphonic acid). Success in the assembly process was proven using analyses based on ellipsometry, XPS, and electrochemical measurements. The film obtained was of a higher order than the usual electrostatic LbL assembly. Marks and coworkers prepared out-of-plane non-centrosymmetric microstructures by layer-by-layer deposition from an air phase (figure 92B) [835]. In their design, intermolecular longitudinal triple hydrogen bonding interactions were used to prepare the non-centrosymmetric layered structure in which chromophore molecules align in a head-to-tail manner preferentially perpendicular to the substrate. They used the chromophore 5-{4-[2-(4,6-diamino-[1,3,5]triazin-2-yl)-vinyl]-benzylidene} pyrimidine-2,4,6-trione (DTPT), which contains a hydrogen-bond/electron donor and hydrogen-bond/electron acceptor module at both terminals. Ito and coworkers pioneered use of a charge transfer interaction during LbL assembly [836]. Thinly layered assemblies of a charge transfer complex yielded oriented dye arrays of specific electronic characteristics that could find use in novel types of functional photo-electronic systems. They demonstrated assembly between an electron-accepting polymer and an electron-donating polymer. As shown in figure 92C, the layers of their charge transfer complex are sandwiched between the donor and the acceptor layers. Other molecular interactions such as supramolecular inclusion [837] and bio-specific recognition [838, 839] can be exploited as well as covalent bond formation [840–842].
Figure 92.

LbL assembly based on (A) metal-phosphate interaction; (B) hydrogen bonding; (C) charge transfer complex formation.
A wide variety of film depositon techniques are also available. Char and coworkers reported an alternative method for spin-coating of an LbL assembly for making well-organized multilayer films using a very short process time (figure 93A) [843]. In the spin-coating process, the adsorption and rearrangement of adsorbed chains on the surface and the elimination of weakly bound polymer chains from the substrate are achieved almost simultaneously at high spin rates in a short time. Air shear force caused by the spinning process in the spin-coating LbL method has a significant effect on the surface roughness of prepared multilayer films. Films prepared using this spin-coating LbL process possess a highly ordered internal structure far superior to that obtained by the conventional dipping method. Decher and coworkers proposed use of a spraying process for facile LbL assembly (figure 93B) [844]. They demonstrated preparation of a polyelectrolyte film by successive spraying of polycation and polyanion solutions and compared the films obtained with those prepared by the classic dipping method. Film deposition by spraying was easily controlled and very reliable. Spray deposition permits regular multilayer growth even under conditions where dipping fails to produce homogeneous films, such as in the case of extremely short contact times. Automatic machines for film preparation have been developed by Shiratori and coworkers [845] and by Hammond and coworkers [846].
Figure 93.

(A) Spin-coating LbL assembly and (B) spraying process for facile LbL assembly.
By using a combination of the LbL assembly and template synthesis free-standing objects can be prepared. Li et al applied the LbL assembly technique to fabrication of microtubes from biocomponents such as proteins and phospholipids using porous template (figure 94) [847]. Human serum albumin is stable structurally under acidic or basic conditions (even at pH departing significantly from its isoelectric point) because its subdomains possess strong binding sites. Thus, the surface charge of human serum albumin can be made either more positive or more negative through variation of pH. This makes it possible to form an LbL assembly of human serum albumin itself. Figure 94 depicts synthetic microtubes of human serum albumin with walls that have smooth and clean surfaces and a thickness of around 30 nm. These tubes are about 60 mm in length and they are tolerant to free bending. LbL films prepared on solid supports can be peeled from the support yielding free-standing films that are useful in a wide range of applications. As illustrated in figure 95, free-standing film preparation can be achieved through LbL film assembly on a sacrificial support layer followed by dissolution of the sacrificial layer. Tsukruk and coworkers used this strategy for fabrication of freely suspended, multi-layered nanocomposite membranes containing gold nanoparticles [848]. The gold nanoparticles were initially deposited alternately with oppositely charged polyelectrolytes then covering with additional assemblies of conventional polyelectrolytes. Dissolving the sacrificial cellulose acetate layer in acetone resulted in release of the multi-layered polymer films with gold nanoparticles from the supporting silicon wafer. The free-standing film was transferred to a water surface and could be lifted using a highly polished copper holder. These unique nanofilms possess very stable micromechanical characteristics as well as a sensitivity far surpassing any existing pressure sensors based on hard inorganic materials of similar dimensions.
Figure 94.

Fabrication of microtubes from biocomponents through LbL assembly using porous template. © 2005, American Chemical Society, Langmuir 21 (2005) 1679.
Figure 95.

Preparation of free-standing LbL film.
Assembly procedures for the LbL technique are generally mild and so are suitable for incorporation of rather fragile biomaterials into a film structure so that applications based on functions of biomaterials are promising targets. Glucose oxidase (GOD) exhibits improved stability when embedded in polyelectrolyte LbL films (figure 96) [849, 850]. The storage stability of GOD-immobilized LbL films was examined under three sets of conditions (figure 96A): (a) stored in water at 25 °C, (b) stored in 0.1 M PIPES buffer (pH 7) at 4 °C, and (c) allowed to stand in air at 4 °C. The film samples stored in water at 25 °C showed drastic reductions in activity with approximately 70% of the activity lost after 4 weeks probably because of bacterial growth. In contrast, the films stored in the buffer at 4 °C showed no significant decrease in enzymatic activity after 14 weeks. A 10% decrease in activity was observed in the first week for the films kept in air at 4 °C, but activity remained constant during the following 13 weeks. The initial loss in activity of the films was probably due to drying. GOD contained in the alternating assembly was immobilized through electrostatic interaction, and activity losses caused by structural strain and deformation were absent. Thermostabilities of the enzymatic activity for LbL assembled films were also examined (figure 96B). An initial measurement of the enzymatic activity was performed at 25 °C immediately after the incubation period, while the second measurement was carried out after storing the film in air at room temperature for 30 min. Activity measurements were similarly performed for GOD dissolved in water. GOD dissolved in aqueous solution lost its activity during incubation even at 30–40 °C (a), and it became inactive at 50 °C. Activity was partially recovered by returning the solution to room temperature (b). The recovery of activity decreased with increasing incubation temperature and totally disappeared after heating at 70 °C. Assembly with PEI improved remarkably the thermostability of GOD (c). Even after incubation at 50 °C a significant decrease in activity was not detected. Interestingly, recovery of any lost activity was not observed in the film sample at any incubation temperature (d). The improved enzymatic activity and the absence of the activity recovery of GOD in the LbL film was attributed to suppression of the conformational mobility of GOD upon complex formation with the surrounding polymer chains.
Figure 96.
(A) Storage stability of glucose oxidase immobilized LbL films: (a) stored in water at 25 °C, (b) stored in 0.1 M PIPES buffer (pH 7) at 4 °C, and (c) allowed to stand in air at 4 °C. (B) Thermostability of the enzymatic activity for LbL assembled films: (a) free glucose oxidase measured immediately; (b) free glucose oxidase after 30 min; (c) glucose oxidase in LbL film measured immediately; (b) glucose oxidase in LbL film after 30 min
Flexibility in film construction allows for various sequences of layering structures. For example, a multienzyme reactor could be demonstrated, where two enzymes, glucoamylase (GA) and GOD, were assembled in the film (figure 97) [851]. The substrate starch is hydrolyzed into glucose by hydrolysis of the glycoside bond performed by GA. The glucose produced is then converted to gluconolactone by GOD with H2O2 as a by-product. Various film structures can be easily prepared by changing the dipping number and sequence, and thus the reactor structures can be rather easily optimized.
Figure 97.

Multienzyme reactor with glucoamylase and glucose oxidase on ultrafilter.
A unique example of a non-enzyme reactor was prepared by Akashi, Serizawa, and coworkers who reported alternate layer-by-layer adsorption based on stereo-complexation of hydrophobic poly(methyl methacrylate) (PMMA) [852]. Isotactic and syndiotactic PMMAs are well known to form a double-strand helical structure, in which isotactic PMMA is surrounded by double the molar amount of syndiotactic PMMA. Alternate assembly between these two kinds of PMMA polymer in acetonitrile was demonstrated. Isotactic PMMA is poorly soluble in the acetonitrile and physically adsorbs on a solid support. Syndiotactic PMMA has a higher solubility and does not adsorb on the bare substrate, but rather it can be adsorbed onto the isotactic-PMMA-exposed surface during stereo-complex formation. Alternating physical adsorption and stereo-complexation resulted in multilayer formation. They first prepared LbL films of a stereo-complex between isotactic-PMMA and syndiotactic-poly(methacrylic acid) (PMAA) with a 1 : 1 unit-molar stoichiometry. Because these polymers have differing solubilities in particular solvents, one can be extracted selectively from the assemblies, resulting in porous syndiotactic-PMAA or isotactic-PMMA films. The resulting macromolecular nanospaces in the films could be used for stereoregular template polymerization of isotactic-PMMA and syndiotactic-PMAA, respectively. Isotactic polymers of methacrylates are usually more difficult to obtain by free-radical polymerization because of steric hindrance at lateral groups; the stereoregularity of polymers synthesized by this method was absolutely dependent on that of the template polymers. These workers extended the concept of shape-recognizing LbL assembly to the field of artificial enzymes (figure 98) [853]. Basically, enzymes catalyze specific reactions in a specific pore structure where substrate molecules can be accommodated through non-covalent interactions, such as hydrogen bonds, then converted into a thermodynamically stable form. If appropriate void structures can be provided, specific reactions may proceed with this void as a template. Therefore, shape imprinting in LbL films can lead to enzyme-mimicry.
Figure 98.
LbL reactor based on stereo-complex formation.
Inorganic-organic hybrid vesicles, cerasomes (see figure 15), have been subjected to LbL assembly with counterionic polylectrolytes [854]. For more advanced assemblies, appropriate design of the amphiphilic silanes led to the preparation of both cationic and anionic cerasomes, enabling direct assembly of cerasome particles and avoiding use of interlayer polyelectrolytes (figure 99) [855]. Formation of a smaller (20–100 nm) cationic cerasomes and larger (70–300 nm) anionic cerasomes was confirmed by transmission electron microscopic (TEM) observations. QCM observation revealed both small and large steps in frequency change corresponding respectively to adsorption of the cationic and anionic cerasomes. Stone pavement-like close packing of the cerasome particles was indicated by AFM observations of the surfaces of the assembled structures, where the difference in the particle size for each layer confirmed preparation of layered structures of the cationic and anionic cerasomes. The structures presented can be regarded as novel types of artificial multi-cellular systems for use as bioreactors or biosensors. Immobilization of various biomolecules, such as enzymes and antibodies, following further functionalization of the cerasome surface could be used to create novel bio-organic-inorganic nano-hybrids.
Figure 99.

LbL assembly between cationic and anionic cerasomes.
Polyelectrolyte-based LbL films have soft and flexible constitutions, and their structures can be regulated by external stimuli. These attributes are useful for controlled release and drug delivery [856, 857]. For example, Rubner, Cohen, and coworkers introduced soft LbL films within the pores of a polymer support to prepare a pH-sensitive permeation valve (figure 100) [858]. Track-etched polycarbonate membranes (TEPC) were filled with the LbL assembled polyelectrolyte multi-layers comprising poly(allylamine hydrochloride) (PAH) and poly(styrenesulphonic acid sodium salt). Gating properties of the multilayer-modified TEPC membranes upon their swelling/deswelling were studied by measuring the flux of pH-adjusted deionized water. The multilayers within the cylindrical pores of TEPC membranes exhibit discontinuous swelling/deswelling behavior indicated by large discontinuous changes in the transmembrane flux. Reversible gating properties were exhibited by this hybrid membrane as the pH condition of the feed solution was alternated between pH 2.5 and 10.5. A ‘closed’ or ‘open’ state can be realized at the same pH condition depending on the pretreatment history because this LbL valve performs hysteretic gating. High molecular weight polymers could be selected for retention or transmission by varying the membrane pretreatment condition. Similar gating properties aimed at control of water flow would be useful in microfluidic channel devices.
Figure 100.

LbL assembly as a pH-sensitive permeation valve on track-etched polycarbonate membranes.
Hollow capsules prepared by LbL techniques are appropriate objects for drug delivery [859, 860]. Encapsulation and controlled delivery of DNA using LbL capsules have also been reported. For example, Lvov and coworkers reported an approach for DNA encapsulation using a biocompatible polyelectrolyte capsule (figure 101A) [861]. In their method, a water-insoluble DNA/spermidine complex was first precipitated onto MnCO3 particles. LbL assembly of biocompatible polyarginine and chondroitin sulfate was then performed on the mixed component cores. Dissolution of the MnCO3 template particles in deaerated 0.01 M HCl resulted in biocompatible capsules containing the DNA/spermidine complex. Further treatment with 0.1 M HCl led to decomposition of the DNA/spermidine complex, and only low molecular weight spermidine was released from the capsule interior, giving DNA-entrapped biocompatible capsules. Lvov and coworkers reported encapsulation of peroxidase inside polyelectrolyte LbL capsules as a catalytic system for the synthesis of phenolic polymers confined within microcapsules (figure 101B) [862]. Cationic and anionic polyelectrolytes were first assembled by LbL on a weakly polymerized melamine formaldehyde particle. Subsequent dissolution of the template core in an acidic medium was followed by soaking of the hollow capsules, thus obtained, in an aqueous peroxidase solution. When the pH of the bulk solution reached a value of 4.0, the shell wall allowed permeation of the peroxidase into the capsules. When the pH was subsequently adjusted to 8.5, permeability of the capsule walls was reduced leaving the peroxidase trapped mostly within the capsules, but also some contained in the walls. The closed capsule allowed permeation of the monomer, tyramine, while the resultant polymerized fluorescent polymers were confined inside the capsule.
Figure 101.

(A) Encapsulation strategy of DNA into hollow LbL capsule. (B) Encapsulation of peroxidase and polymerization in hollow capsule.
Multilayer structures on colloidal particle cores have an ideal structure for research of basic phenomena. To understand energy transfer processes, Decher and coworkers exploited the superiority of LbL assembly for structural design and synthesis (figure 102) [863]. They prepared LbL films of 13 nm gold colloids for fabrication of metal core-polymer shell capsules where the fluorescent organic dyes fluorescein isothiocyanate (FITC) and lissamine rhodamine B (LISS) were placed at various distances from the gold core. Fluorescence of the fluorescein and lissamine dyes situated in the outer polymer layers of the core-shell nanoparticles is quenched by the gold nanocore. A systematic investigation involving varying the distance between dye and metal core provided a direct method to assess the effect of the metal core on radiative rates, thus revealing strongly distance-dependent fluorescence quenching or reduction in the transition probability for radiative transitions by gold nanoparticles.
Figure 102.

LbL assembly for structural design to understand energy transfer processes.
Because the LbL assembly method is so versatile, it can be combined with other fabrication techniques. Hammond and coworkers proposed the use of chemically patterned surfaces produced by stamping techniques as templates for ionic multilayer assembly based on stamping techniques (figure 103A(a)) [864, 865]. The LbL multilayers, as a polyelectrolyte platform, were deposited on a solid support and terminated by using a layer containing an amine group. A poly(dimethylsiloxane) (PDMS) stamp was treated with an oxygen plasma, which provides a hydrophilic surface, and then inked with a solution of copolymer comprised of oligo (ethylene glycol) and maleic anhydride. The maleic anhydride group from the ink reacts with the amine group from the LbL surface, leaving only the oligo (ethylene glycol) chains exposed, which are inert to adsorption of ionic species. In this manner, stamping on the first LbL film produces a pattern resistant to further adsorption and selectively allows further LbL assembly on the unstamped areas. Combination of the LbL technique with ink-jet printing and also with photolithography was demonstrated by Yang and Rubner (figure 103A(b) and (c)) [866]. Poly(acrylic acid) (PAA) and polyacrylamide (PAAm) can be assembled using the LbL method, but they interact through hydrogen bonding. Exposure of the film to water with an appropriate pH through the ink-jet printing method led to micro-patterning with a desirable design (figure 103A(b)). Cross-linking between these two polymers by heat treatment stabilized the etched patterns. Use of photosensitive polymers in the LbL technique permits fabrication of micropatterns on LbL films. In figure 103A(c), the photoindicator part generated free radicals upon UV light irradiation, promoting cross-linking of polymers under those parts exposed to light. The latter polymer portion became insoluble, while the unexposed parts remained soluble. As a result, micropatterns could be drawn according to photomasks. An excellent example of microfabrication of LbL films into a free-standing micro-object was performed by Lvov and coworkers who demonstrated the synthesis of an LbL self-assembled ultrathin micro-cantilever consisting of clay/polymer nanocomposites [867]. They used sequenced procedures including patterning, photo-treatment, etching, and LbL assembly (figure 103B).
Figure 103.

(A) Microfabrication techniques with LbL method: (a) stamp method; (b) ink-jet method; (c) photolithography. (B) Cantilever preparation based on LbL technique.
There are countless research accounts of examples of the application of the LbL technique, and this simple review cannot cover them all. Recently, research of this method has become application-oriented as can be seen in several reports including surface modification for superhydrophobicity [868, 869], sensor preparation [870–874], electrochromic devices [875], photochromic devices [876], thin film transistors [877], fuel cells [878], solid-state photovoltaic devices [879], rectifying junctions [880], and chiral switching [881].
Self-assembled monolayers
One of the most frequently used techniques for modification of solid surfaces is that of self-assembled monolayer (SAM) formation, which is a useful method for construction of interfacial self-assembled structures on solid surfaces [882, 883–888]. As illustrated in figure 104, three main types of method for covalent bonding have been proposed. Science and technology based on surface coating has been widely developed using the SAM method [882, 883–888].
Figure 104.

Self-assembled monolayers (SAM).
SAM structures ensure strong covalent bonding between organic monolayers and solid substrates, and therefore, can be used for various applications. Permeation control through self-assembled monolayers (SAM) of dialkylsilane amphiphiles covalently immobilized within glass pores was reported by Okahata and coworkers (figure 105) [889]. Phase transitions between solid and fluid liquid crystalline states of the monolayers immobilized in the pores were used to regulate the permeation rate of NaCl across the porous glass plate. They further developed this concept by combining with the LB technique finally to demonstrate permeation control using single monolayers [890, 891].
Figure 105.

Permeation control through SAM of dialkylsilane amphiphiles on a porous glass plate.
Molecular modules with photo-electronic properties such as porphyrin and fullerene can be easily immobilized on solid surface. Imahori and coworkers have been extensively researching photoelectronic properties using SAM structures containing both porphyrin and fullerene units [892–895]. They proposed a mixed SAM system where boron dipyrrin, as the light-harvesting molecule, is combined with a ferrocene-porphyrin-fullerene triad as a charge-separation molecule, in order to mimic the light-harvesting function in antenna complexes of green plants (figure 106) [896]. A two-step electron transfer was achieved in the triad molecule (i.e. photo-induced electron transfer from the porphyrin to C60 followed by efficient charge transfer from ferrocene to the resulting porphyrin radical cation) yielding a charge separated state. The gold electrode donates one electron to the Fc+ moiety and the charge-separated complex donates one electron to a carrier such as methylviologen or O2 resulting in cathodic photocurrent generation. Emission from the boron-dipyrrin overlaps well with the absorption band of the porphyrin in the triad so that, in the mixed SAM structure, an efficient energy transfer can take place from the boron-dipyrrin moiety to the porphyrin moiety. This efficient energy transfer enables efficient photocurrent generation using this mixed SAM system.
Figure 106.

SAM with boron dipyrrin, as the light-harvesting molecule, and charge-separation molecule, a ferrocene-porphyrin-fullerene triad.
Porphyrin-based SAMs have also been used as molecular memories [897], for sensing of acid and base [898], sensing of NO2 [899], sensing of O2 [900], and as an optical pH meter [901]. SAM structures incorporating fullerene have been applied to catalysis [902] and their electronic properties were investigated [903, 904]. Also, Tour and coworkers investigated the structures of SAMs and pointed out the possibilities of multilayer formation, based on experiments using thiol-terminated fullerene derivatives (figure 107) [905]. Various polymeric units such as oligo (phenyleneethynylene) [906, 907], oligothiophene [908], polyelectrolyte brushes [909, 910], polyaniline [911], poly(ethylene glycol) [912], peptide [913], protein [914], DNA [915], nucleotide [916], carbon nanotubes [917, 918], coordination cages [919] have been immobilized in SAM structures. SAMs with cyclodextrin [920, 921] and calixarene [922] can be used for material sensing through inclusion complex formation. Inorganic structures such as nanoparticles [923], nanowires [924, 925], and nanocrystals [926, 927] have also been immobilized indirectly at appropriate SAM surfaces.
Figure 107.

Fullerene derivatives for SAM structures on gold.
Stoddart and coworkers immobilized molecular machines at a solid surface using the SAM technique. Bistable [2]rotaxanes exhibit controllable switching properties at surfaces, which can be useful in device designing. The electrochemically and electrically driven mechanical shuttling motion of the ring-shaped component, cyclobis(paraquat-p-phenylene), is the basis for these phenomena (figure 108). Goddard III and coworkers reported the structure and properties as a function of surface coverage for of a SAM of a bistable[2]rotaxane on Au(111) surfaces as a function of surface coverage. This work was based on atomistic molecular dynamics studies with a force field optimized from density functional theory calculations, but also includes several experiments that validate the computational predictions [928]. In a more advanced approach, Ho, Stoddart, and coworkers proposed a ‘molecular muscle’ [929]. For this purpose, switchable, palindromically constituted bistable[3]rotaxanes were designed and synthesized with a pair of mechanically mobile rings encircling a single dumbbell (figure 109). The site of occupation of the two cyclobis(paraquat-p-phenylene) rings can be controlled to be on either tetrathiafulvalene or naphthalene stations. For the purpose of its self-assembly at a gold surface, the active form of the bistable[3]rotaxane bears disulfide tethers attached covalently to both of the cyclobis(paraquat-p-phenylene) ring components. An array of flexible microcantilever beams, each coated on one side with a monolayer of 6 billion of the active bistable[3]rotaxane molecules, undergoes controllable and reversible bending when exposed to the synchronous addition of aqueous chemical oxidants and reductants.
Figure 108.

SAM of a bistable[2]rotaxane on Au (111) surfaces.
Figure 109.
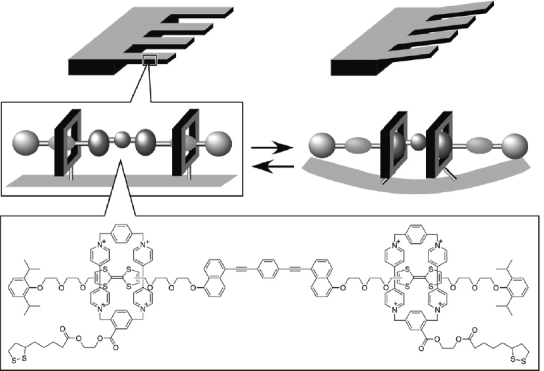
Molecular muscle based on SAM structure of bistable[3]rotaxanes on cantilever surface.
Because various functional groups can be introduced at a solid surface using the SAM method, wettability of surface by a liquid can be regulated by appropriate design. As demonstrated by Whitesides et al, hydrophilicity gradients can be generated by surface modification with belts composed of monolayers of various hydrophilicities. Because of favorable interactions with the monolayer surface, liquid droplets can move across such surfaces against gravity (figure 110) [930]. Ichimura and coworkers reported more advanced control of the motion of a liquid droplet on a SAM surface functionalized photoresponsive unit [931]. After an amine-carrying monolayer had been first covalently immobilized on a silica plate, a calix[4]resorcinarene derivative having a photochromic azobenzene unit (figure 111) was immobilized using electrostatic interactions. The macrocyclic structure of the amphiphile was designed to ensure sufficient free volume required for the photoisomerization of the azobenzene moieties. Photo-irradiation of the monolayer with UV light (365 nm) resulted in the formation of ca 90% cis isomer with a higher dipole moment. Therefore, upon UV irradiation, the surface energy increased. Photoirradiation of the cis-rich surface with blue light (365 nm) transformed the cis isomer into the trans isomer, so that the surface reverted to being of low energy. When the SAM surface was irradiated with UV light, the surface became wetted to a greater degree with olive oil. In contrast, irradiation with blue light resulted in a larger contact angle of the oil. The directional motion of a droplet on a cis-rich surface upon asymmetrical irradiation with blue light was realized. By moving the light beam, a controlled gradient of the surface energy between the advancing and receding edges of the droplet could be maintained. Motion of the droplet could be sustained by irradiation with blue light of graded intensity, but irradiation of the photoresponsive surface with a homogeneous blue light effectively prevented motion of the droplet.
Figure 110.

Control of liquid droplet motion by adjustment of surface wettability.
Figure 111.

Control of the motion of a liquid droplet on a SAM by photoirradiation.
One recent breakthrough technique in SAM technology is the invention of dip-pen nanolithography [932]. A new AFM-based soft lithography technique was created by combining self-assembled monolayer chemistry with AFM technology (figure 112). Nanoscale can be written on metal and semiconductor surfaces using a solution of monolayer-forming materials as ink. Apart from this technique, surface patterning with SAM methods has been extensively studied [933–939].
Figure 112.

Dip-pen nanolithography.
Molecular arrays and material arrays on surfaces
Fabrication techniques for thin film structures such as the LB technique and LbL method provide control of layering sequences but are not always useful for molecular arrangement within a single monolayer plane. Therefore, techniques to control molecular arrangements within a monomolecular plain with high precision are necessary in order to further develop controlled self-assembly as a breakthrough methodology. Because of advances in probe microscopies, we can now observe directly images of individual molecules and their arrangements. Excellent images of various species such as aliphatic molecules [940–942], aromatic molecules [943–951], porphyrins and their families [952–960], fullerenes [961–967] and polymers [968] are now available. In addition, manipulation of molecular level processes has been demonstrated. For example, in situ polymerization of diacetylene (10,12-nonacosadiynoic acid) molecules by using an STM tip was realized by Okawa, Aono, and coworkers [969, 970], and recently, Sakaguchi et al reported electrochemical epitaxial polymerization of single-molecular wires [971] and visualization of single-molecule conjugate copolymers [972]. Now that imaging of individual molecules and control of molecular level processes has been demonstrated, dynamic control of self-assembly should be possible. Several examples of control of molecular arrangements through self-assembled process are described in the first part of this section.
Recently, Hill et al reported dynamic behaviour in the two-dimensional molecular arrangements of a porphyrin derivative, tetrakis(3,5-di-t-butyl-4-hydroxy phenyl)porphyrin (TDtBHPP) (figure 113A) [973, 974]. Dynamic changes of the molecular arrangement from a hexagonal phase to a square phase could be observed by sequential real-time STM observation of arrays of TDtBHPP molecules at a Cu(111) surface at room temperature. Changes in the molecular arrangements appear to occur through an apparent domino effect where conversion from hexagonal to square domain structures took place with a well defined phase boundary. A molecular conformational transition from coplanarity to non-planarity of the porphyrin meso-phenyl substituents accompanies the phase transition from hexagonal to square domain structure (figure 113B). The molecules contained in the square domain have a four-fold symmetry, which is similar to that of the free molecule, i.e. non-planar conformation with a large dihedral angle. In the latter conformation, the square grid structure was induced by van der Waals contacts between t-butyl groups of the neighbouring molecules. Square-packing would be preferable from the viewpoint of the conformational energy of an individual molecule. This is because the molecules in this packing had a stress-free conformation with a large dihedral angle. However, van der Waals contacts among neighbouring molecules and those between molecule and surface were reduced when compared with those in the hexagonal packing phase. In contrast, the stability of the hexagonal packing arose from increased intermolecular and surface-molecule contacts of the co-planar conformation, although this came at the expense of an increased conformational energy of the distorted molecular conformation. Coexistence of the hexagonal and square domains indicated that both packings are in fact metastable and the energy barrier between these domains was relatively small allowing the molecules to occupy either structure.
Figure 113.

Two-dimensional molecular arrangements of tetrakis(3,5-di-t-butyl-4-hydroxyphenyl)porphyrin (TDtBHPP): (A) AFM observation; (B) molecular model. © 2006, Royal Society of Chemistry, Chem. Commun. (2006) 2320.
In other work, TDtBHPP molecules were found to adopt several morphologies through variation of dihedral angles between substituted groups and central porphyrin core. Hill et al found that molecules adapted their conformations to avoid mismatches between two different two-dimensional crystalline phases [975]. The planar conformation of the TDtBHPP molecule favors the hexagonal packing, as shown in figure 114A. Rotation of the phenyl substituents to low dihedral angles, in response to surface adsorption, causes saddle-like distortion of the tetrapyrrole core, which results in a different packing motif (figure 114B). Surprisingly, molecules with a ‘mixed’ conformation could be found at the periphery of the latter assembly and at the boundary between two different two-dimensional crystal domains. In this conformation, phenyl substituents of molecules at the domain exterior more closely approached coplanarity with the molecular mean plane, while those involved within the assembly were constrained against this. A model of the packing of the domain is shown in figure 114C together with space-filling representations of the three differing molecular conformations. The transformation between two kinds of domains was smooth because of mediation by the mixed conformation molecules at the phase boundary.
Figure 114.

(A) and (B) two-dimensional crystalline phases of TDtBHPP molecules and (C) molecular models for lattice. © 2006, Royal Society of Chemistry, Phys. Chem. Chem. Phys. 8 (2006) 5034.
Minor variation of molecular structure altered the assembly mechanism of the porphyrin molecules. For example, change of substituent group from bulky tert-butyl groups to small methyl groups in the TDtBHPP structure makes hydrogen bonding possible. The new molecule can form a hydrogen-bonded network, a Kagomé lattice (figure 115) [976]. Importance of hydrogen bonding in molecular arrangement was also demonstrated in pioneering research by Yokoyama et al [977]. They reported preparation of two-dimensional artificial molecular patterns of porphyrin derivatives through hydrogen bonding, at a Au(111) surface (figure 116). Two kinds of tetraphenylporphyrin that contain two cyano groups in different relative positions (cis-isomer and trans-isomer) were used in their research. Cyano groups introduced at the porphyrin phenyl substituents can dimerize through hydrogen bond formation. Linear molecular wires were found in STM observations of the trans-isomer while tetrameric cyclic domains were formed preferentially (figure 116A) in the case of the cis-isomer (figure 116B). This example demonstrated that only a small difference in molecular structures can be reflected in the patterns of the porphyrin array. The pattern of the porphyrin arrays could be said to be programmed by the structures of the subunit molecules.
Figure 115.
Hydrogen-bonded network, Kagome lattice structure. © 2007, American Chemical Society, J. Phys. Chem. C 111 (2007) 16174.
Figure 116.
Two-dimensional artificial molecular patterns of porphyrin derivatives via hydrogen bonding: (A) cis-isomer and trans-isomer; (B) trans-isomer.
Barth, Kern, and coworkers demonstrate that novel supramolecular nanostructures can be obtained with the aid of cooperative self-assembly between adsorbed molecules. They fabricated a one-dimensional supramolecular nanograting at a Ag(111) surface by vapor deposition of 4-[trans-2-(pyrid-4-yl-vinyl)]benzoic acid under ultrahigh vacuum [978]. Figure 117 depicts model structures of the resulting molecular pattern, on the basis of STM observations. Formation of a highly regular, one-dimensional supramolecular arrangement in a domain that extends over two neighboring terraces separated by an atomic step is revealed by an overview image. A close-up view of some molecular stripes indicated that the one-dimensional superstructure actually consists of two chains of 4-[trans-2-(pyrid-4-yl-vinyl)]benzoic acid. The model proposed in figure 117 was used to rationalize the features observed within the supramolecular structure. Orientation of the molecular chains reflected a good match between the 4-[trans-2-(pyrid-4-yl-vinyl)]benzoic acid subunits and the high-symmetry lattice positions. The proximity of the coupled groups was associated with formation of weak hydrogen bonds. This finding suggested that the self-assembly of properly designed molecules by hydrogen bonding could open up new avenues of research for the positioning of functional units in supramolecular architectures in two-dimensions.
Figure 117.

One-dimensional supramolecular nanograting of 4-[trans-2-(pyrid-4-yl-vinyl)]benzoic acid at a Ag(111) surface.
Epitaxial adsorption of alkyl chain moieties on a substrate surface can also induce regular arrangements of functional units. Nakanishi et al wisely applied this concept to form a two-dimensional fullerene array (figure 118) [979, 980]. A fullerene derivative containing three hexadecyl chains was spin-coated from chloroform solution onto freshly cleaved HOPG under conditions suitable for obtaining submonolayer coverage revealing structural details of the interfacial layers. Tapping mode AFM images revealed that this fullerene derivative formed a one-dimensional lamellar structure on HOPG. High-resolution STM imageing under the optimized conditions showed lamellae composed of C60 arranged in zigzag fashion. The periodicity of the lamellae was 6.3 nm. CV measurements performed on the fullerene derivative adsorbed on HOPG revealed a single redox event corresponding to generation of the fullerene monoanion at a potential of E1/2=−1.29 V. From the current passed during the reduction process, the electroactive quantity was calculated to be 25% of full monolayer coverage. This result is consistent with the surface coverage determined by AFM. The results of CV measurement confirmed that the C60 moieties are electroactive, opening the way to address the individual molecules and to investigate the conductivity of individual nanowires.
Figure 118.

Two-dimensional fullerene array on HOPG substrate. © 2006, American Chemical Society, J. Am. Chem. Soc. 128 (2006) 6328.
Self-assemblies of coordination complexes have also been visualized using STM. Figure 119 shows a monolayer array of an organoplatinum complex as reported by Stang and coworkers [981]. Metals such as platinum and palladium have been used for preparation of shape-defined complexes with appropriately designed organic ligands. The example shown in figure 119 used bipyridine-type ligands to give an array of square units through coordiation with platinum on a Au(111) surface. Formation of patterns with rectangle or cage units were also realized using an appropriate organic ligand and by using the same strategy. Two-dimensional supramolecular cavities should become size-tunable given selection of appropriate building blocks and coordination conditions, and could be used for selective inclusion of guest molecules. This idea was realized in pioneering work by Barth and coworkers [982]. They used 1,4-dicarboxylic benzoic (terephthalic) acid as a building block for cavity formation through coordination with Fe atoms. Formation of coordination complexes on Cu(100) surfaces produced several types of cavities depending on the coordination mode (figure 120). If Fe concentration was kept low, and in the case of terephthalic acid, ladder-type structures with elongated cavities evolved, where not all carboxylate groups could participate in coordination to Fe atoms. The remaining carboxylate groups presumably formed hydrogen-bonds with adjacent phenyl rings in order to stabilize the network structures further. Upon completion of two-dimensional Fe-carboxylate coordination, a larger square-shaped cavity is formed. C60 fullerene guests could be accommodated within these cavities with differing efficiencies.
Figure 119.

Array of square units through coordination with platinum on a Au(111) surface.
Figure 120.

Two-dimensional supramolecular cavities of 1,4-dicarboxylic benzoic acid upon coordination complexes on Cu(100) and inclusion of fullerene.
Controlled alignment of nano-sized inorganic objects is also an attractive research subject, and could make important contributions in device preparation and basic quantum science. However, objects of such high mass per unit structure cannot be subjected to vacuum deposition so that other self-assembly technique, such as the LB method, can be applied for this purpose. Typical two-dimensional surface pressures prevalent within a floating Langmuir monolayer are of the order of a few dozen megapascals. Therefore, it might be expected that some coalescence reactions could be directly and strongly affected by this surface pressure. This is along with the well-known pressure effect on the orientational order and the proximity of the molecules within the film. This concept can also be applied in the large scale alignment of 1D nanomaterials below the operating limits of conventional lithographic techniques (figure 121). The nanomaterials remains in raft-like islands at low surface pressure with large separations between the islands. Upon compression, the rods/wires organize parallel to the barrier through their reorienting in a head-to-tail alignment and resulting in a closely packed monolayer.
Figure 121.

LB methor for nanomaterial alignment on water surface.
The example shown in figure 122 was reported by Golan and coworkers [983]. A composite map comprised of large area TEM images revealed the parallel aligned PbS nanowires after transfer onto a TEM grid by using the Langmuir Blodgett technique. A monolayer of the aligned nanowires was lifted at π=23 mN/m and T=22 °C. Images a–f represent a continuous area of the TEM grid with location marks indicated by the circled numbers. The area represented here is over 3 m2, which is only part of the total aligned area of ca 15 m2. Such long range areas could be observed frequently at many different sites of the TEM grid. The nanowires retain the width of the initial PbS nanowires after alignment with an average diameterý of 1.8±0.08 nm and pitch of 2.7±0.08 nm. Similarly LB methods were used for assembly of nanoparticles [984–992], nanowires and nanorods [993–999], and quantum dots [1000].
Figure 122.

Composite map comprised of large area TEM images with the aligned parallel PbS nanowires. © 2007, American Chemical Society, Nano Lett. 7 (2007) 1459.
Assembly of colloidal particles can be accomplished by using a solution process. Teranishi and coworkers developed fabrication technique for two-dimensional superlattices [1001]. As depicted in figure 123, amino-functionalized Au nanoparticle building blocks were self-assembled with organic acids forming of two-dimensional superlattices of different symmetries. The two-dimensional superlattices of quasi-honeycomb and square structures were obtained by neutralizing amino-functionalized Au nanoparticles with 1,3,5-tribenzenecarboxylic acid and acetic acid, respectively. The results strongly suggest that different types of two-dimensional or three-dimensional superlattices can be constructed by addition of the appropriate acid to nanoparticles functionalized with amino groups. Self-assembly of various nanomaterials such as nanoparticles [1002–1006], nanorods [1007–1012], and nanocrystals [1013–1015], from solution has also been achieved. Designed surfaces prepared by appropriate method such as SAM can also assist nanomaterial assembly [1016, 1017]. Structure transcription from colloidal particle assemblies has also been reported. Xu and Goedel proposed a method using colloidal assembly at the air–water interface (figure 124) [1018]. First, a mixed monolayer of colloidal silica (diameter, 140 nm) coated with hydrophobic polymer and polyisoprene-type amphiphilic polymer was spread on water, and followed by cross-linking of the polymer using UV irradiation. Following transfer of the monolayer onto a mesh substrate and selective removal of the silica components, porous films with pore diameter 55 nm and of thickness 40 nm were obtained. The other attempts on assemblies of inorganic materials were reported [1019–1023].
Figure 123.

Two-dimensional superlattices formation of Au nanoparticles with 1,3,5-tribenzenecarboxylic acid.
Figure 124.

Porous pattern formation by fabricaton of colloidal particle assembly.
As mentioned previously, DNA is a powerful tool for construction of defined nanostructures. The DNA self-assembly technique has also been used for pattern formation in the two-dimensional plane [1024–1028]. As schematized in figure 125, Winfree et al reported formation of regular two-dimensional patterns through specific recognition between DNA fragments [1029]. One unit is composed of four custom designed DNA chains and spontaneous base-pairing between them results in a complex with four unbound DNA fragments as appended tags. The A unit has tags with TCACT, TAGAG, CATAC, TCTTG sequences, while AGAAC, GTATG, ATCTC, and AGTGA tags are attached to the B unit. These tags undergo mutual recognition, resulting in alternating patterns of A and B units. Since design and synthesis of such DNA chains is a simple matter using existing techniques, this concept could be a versatile method for fabrication of two-dimensional patterns. Patterned surface structures created using DNA self-assembly provide scaffolds for regular arrays of nanoparticles. Yan and coworkers reported the use of a self-assembled two-dimensional DNA nanogrid as a template for organization of 5 nm gold nanoparticles into periodic square lattices (figure 126) [1030]. Each particle sits on a single DNA tile. The center-to-center interparticle spacing between neighboring particles is controlled to be ∼38 nm. These evenly distributed Au nanoparticle arrangements including accurate control of interparticle spacing may find applications in nanoelectronic and nanophotonic devices.
Figure 125.

Formation of regular two-dimensional patterns through specific recognition between DNA fragments.
Figure 126.

Periodic square lattices of gold nanoparticles on a self-assembled two-dimensional DNA nanogrid.
Summary and future perspectives
In this review, various aspects of self-assembly have been presented, concentrating on the different types of self-assembly, the components involved, and the role of interfaces in directing their ordering. The examples presented are diverse in their scales, structures, and properties. Because of the broad scope of this challenging science, it is difficult to summarize the field in a single scheme. To attempt this, we have further categorized the presented topics (assembly type, component type, interface assembly) according to the scales at which they operate (figure 127). At the smallest scale, structural control over molecular arrays and materials arrays provides nanoscale structures, where molecular attributes such as morphology and functional group positioning require thoughtful design. For micro-sized objects made using self-assembly, careful design of hierarchical processes should lead to creation of more intricate structures. For fabrication of macro-sized materials, transcription of self-assembly onto desirable products should improve opportunities for creation of a variety of bulk materials with precise nanostructures at their interiors. Some techniques, such as dynamic manipulation of self-assembled molecules at the air–water interface, can bridge between the molecular world and the macro-world.
Figure 127.

Outline of ‘self-assembly world’ from nano to bulk.
Since self-assembled processes are basically molecular processes, preparation of practical materials through self-assembly needs considerable extension of interactive connections. Some of the examples, such as coordination polymers, porous crystals, liquid crystals, and gel, fulfilled this requirement, but in these objects, structural motifs are rather simple. Unlike these artificial molecular constructions, biology accomplished much sophisticated construction from molecular bottom to visible organism. Their construction is highly hierarchic. We have to learn ways to construct hierarchic structures from biological examples, in order to create multi-functional materials through self-assembled process. However, construction of hierarchic structures only from bottom–up self-assembling processes could be very tough. With the current state of self-assembly science, a successful outcome probably cannot yet be obtained by sole reliance on the bottom–up concept. Fusion with well-developed top–down-type nanofabrication is important for development of self-assembly science and technology. For example, the union of top–down nanofabrication and layer-by-layer assembly has enabled rapid development of advanced applications, as illustrated in figure 103. Overall, by addressing challenges and exploiting breakthroughs, the science of self-assembly at any scale is reaching its fruition.
Acknowledgments
We thank Dr Kun'ichi Miyazawa (NIMS), Dr Takashi Nakanishi (NIMS), Dr Hiromitsu Maeda (Ritsumeikan Univ.), and Dr Junbai Li (Chinese Academy of Sciences) for kindly providing original photographs.
The research described in this review was partially supported by Grant-in-Aid for Scientific Research on Priority Areas ‘Chemistry of Coordination Space’ and a Grant-in-Aid for Science Research in a Priority Area ‘Super-Hierarchical Structures’ from Ministry of Education, Science, Sports, and Culture, Japan, World Premier International Research Center Initiative (WPI Initiative) on Materials Nanoarchitronics, from Ministry of Education, Science, Sports, and Culture, Japan, and Grants-in-Aid for Scientific Research (B) from Japan Society for the Promotion of Science. All the authors much appreciate Dr M A for devoted supports on manuscript preparation.
Footnotes
Invited paper.
References
- Alivisatos A P. et al Adv. Mater. 1998;10:1297. doi: 10.1002/(SICI)1521-4095(199811)10:16<1297::AID-ADMA1297>3.0.CO;2-7. [DOI] [Google Scholar]
- Elemans J A A W, Rowan A E. and Nolte R J M. J. Mater. Chem. 2003;13:2661. doi: 10.1039/b304972h. [DOI] [Google Scholar]
- Martínez-Máñez R. and Sancenón F. Chem. Rev. 2003;103:4419. doi: 10.1021/cr010421e. [DOI] [PubMed] [Google Scholar]
- Vriezema D M, Aragonès M C, Elemans J A A W, Cornelissen J J L M, Rowan A E. and Nolte R J M. Chem. Rev. 2005;105:1445. doi: 10.1021/cr0300688. [DOI] [PubMed] [Google Scholar]
- Chandler D. Nature. 2005;437:640. doi: 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- Yin Y. and Alivisatos A P. Nature. 2004;437:664. doi: 10.1038/nature04165. [DOI] [PubMed] [Google Scholar]
- Lazzari M, Rodriguez-Abreu C, Rivas J. and Lopez-Quintela M A. J. Nanosci. Nanotechnol. 2006;6:892. doi: 10.1166/jnn.2006.172. [DOI] [PubMed] [Google Scholar]
- Ariga K, Nakanishi T. and Hill J P. Curr. Opin. Colloid Interface Sci. 2007;12:106. doi: 10.1016/j.cocis.2007.05.008. [DOI] [Google Scholar]
- Nandi N. and Vollhardt D. Acc. Chem. Res. 2007;40:351. doi: 10.1021/ar600057b. [DOI] [PubMed] [Google Scholar]
- De Santis P, Morosetti S. and Scipioni A. J. Nanosci. Nanotechnol. 2007;7:2230. doi: 10.1166/jnn.2007.644. [DOI] [PubMed] [Google Scholar]
- Kottas G S, Clarke L I, Horinek D. and Michl J. Chem. Rev. 2005;105:1281. doi: 10.1021/cr0300993. [DOI] [PubMed] [Google Scholar]
- Kinbara K. and Aida T. Chem. Rev. 2005;105:1377. doi: 10.1021/cr030071r. [DOI] [PubMed] [Google Scholar]
- Brown W R. and Feringa B L. Nat. Nanotechnol. 2005;1:24. [Google Scholar]
- Kay E R, Leigh D A. and Zerbetto F. J. Am. Chem. Soc. 2007;46:72. doi: 10.1002/anie.200504313. [DOI] [PubMed] [Google Scholar]
- Guo Y-M, Oike H. and Aida T. J. Am. Chem. Soc. 2004;126:716. doi: 10.1021/ja039369p. [DOI] [PubMed] [Google Scholar]
- Sato H, Tashiro K, Shinmori H, Osuka A, Murata Y, Komatsu K. and Aida T. J. Am. Chem. Soc. 2005;127:13086. doi: 10.1021/ja052993c. [DOI] [PubMed] [Google Scholar]
- Shoji Y, Tashiro K. and Aida T. J. Am. Chem. Soc. 2006;128:10690. doi: 10.1021/ja063828f. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M, Tashiro K, Yamasaki M. and Aida T. J. Am. Chem. Soc. 2007;129:11912. doi: 10.1021/ja0747526. [DOI] [PubMed] [Google Scholar]
- Tsuda A, Sakamoto S, Yamaguchi K. and Aida T. J. Am. Chem. Soc. 2003;125:15722. doi: 10.1021/ja038349k. [DOI] [PubMed] [Google Scholar]
- Fujita M, Tominaga M, Hori A. and Therrien B. Acc. Chem. Res. 2005;38:369. doi: 10.1021/ar040153h. [DOI] [PubMed] [Google Scholar]
- Takeda N, Umemoto K, Yamaguchi K. and Fujita M. Nature. 1999;398:794. doi: 10.1038/19734. [DOI] [Google Scholar]
- Yoshizawa M, Tamura M. and Fujita M. Science. 2006;312:251. doi: 10.1126/science.1124985. [DOI] [PubMed] [Google Scholar]
- Yoshizawa M, Miyagi S, Kawano M, Ishiguro K. and Fujita M. J. Am. Chem. Soc. 2004;126:9172. doi: 10.1021/ja047612u. [DOI] [PubMed] [Google Scholar]
- Aoyagi M, Tashiro S, Tominaga M, Biradha K. and Fujita M. Chem. Commun. 2002:2036. doi: 10.1039/b205194j. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Tashiro S, Aoyagi M. and Fujita M. Chem. Commun. 2002:2038. doi: 10.1039/b205196f. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Suzuki K, Murase T. and Fujita M. J. Am. Chem. Soc. 2005;127:11950. doi: 10.1021/ja054069o. [DOI] [PubMed] [Google Scholar]
- Yamanoi Y, Sakamoto Y, Kusukawa T, Fujita M, Sakamoto S. and Yamaguchi K. J. Am. Chem. Soc. 2001;123:980. doi: 10.1021/ja003043o. [DOI] [PubMed] [Google Scholar]
- Kawano M, Kobayashi Y, Ozeki T. and Fujita M. J. Am. Chem. Soc. 2006;128:6558. doi: 10.1021/ja0609250. [DOI] [PubMed] [Google Scholar]
- Yoshizawa M, Kusukawa T, Kawano M, Ohhara T, Tanaka I, Kurihara K, Niimura N. and Fujita M. J. Am. Chem. Soc. 2005;127:2798. doi: 10.1021/ja043953w. [DOI] [PubMed] [Google Scholar]
- Takaoka K, Kawano M, Ozeki T. and Fujita M. Chem. Commun. 2006:1625. doi: 10.1039/b600812g. [DOI] [PubMed] [Google Scholar]
- Kamiya N, Tominaga M, Sato S. and Fujita M. J. Am. Chem. Soc. 2007;129:3816. doi: 10.1021/ja0693082. [DOI] [PubMed] [Google Scholar]
- Corbellini F, Knegtel R M A, Grootenhuis P D J, Crego-Calama M. and Reinhoudt D N. Chem. Eur. J. 2005;11:298. doi: 10.1002/chem.200400849. [DOI] [PubMed] [Google Scholar]
- MacGillivray L R. and Atwood J L. Nature. 1997;389:469. doi: 10.1038/38985. [DOI] [Google Scholar]
- Atwood J L, Barbour L J. and Jerga A. Chem. Commun. 2001:2376. doi: 10.1039/b106250f. [DOI] [PubMed] [Google Scholar]
- Kang J, Santamaría J, Hilmersson G, Rebek J., Jr J. Am. Chem. Soc. 1998;120:7389. doi: 10.1021/ja980927n. [DOI] [Google Scholar]
- Rebek J., Jr Chem. Commun. 2000:637. doi: 10.1039/a910339m. [DOI] [Google Scholar]
- Rebek J., Jr Chem. Commun. 2007:2777. doi: 10.1039/b617548a. [DOI] [PubMed] [Google Scholar]
- Heinz T, Rudkevich D M, Rebek J., Jr Nature. 1998;394:764. doi: 10.1038/29501. [DOI] [Google Scholar]
- Iwasawa T, Hooley R J, Rebek J., Jr Science. 2007;317:493. doi: 10.1126/science.1143272. [DOI] [PubMed] [Google Scholar]
- Iwasawa T, Mann E, Rebek J., Jr J. Am. Chem. Soc. 2006;128:9308. doi: 10.1021/ja062768a. [DOI] [PubMed] [Google Scholar]
- Purse B W, Rebek J., Jr Proc. Natl. Acad. Sci. USA. 2006;103:2530. doi: 10.1073/pnas.0511149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajami D, Rebek J., Jr J. Am. Chem. Soc. 2006;128:5314. doi: 10.1021/ja060095q. [DOI] [PubMed] [Google Scholar]
- Hooley R J, Van Anda H J, Rebek J., Jr J. Am. Chem. Soc. 2006;128:3894. doi: 10.1021/ja058727g. [DOI] [PubMed] [Google Scholar]
- Butterfield S M, Rebek J., Jr J. Am. Chem. Soc. 2006;128:15366. doi: 10.1021/ja0663374. [DOI] [PubMed] [Google Scholar]
- Yamanaka M, Yamada Y, Sei Y, Yamaguchi K. and Kobayashi K. J. Am. Chem. Soc. 2006;128:1531. doi: 10.1021/ja0555365. [DOI] [PubMed] [Google Scholar]
- Olenyuk B, Whiteford J A, Fechtenkötter A. and Stang P J. Nature. 1999;398:796. doi: 10.1038/19740. [DOI] [PubMed] [Google Scholar]
- Kuehl C J, Kryschenko Y K, Radhakrishnan U, Seidel S R, Huang S D. and Stang P J. Proc. Nat. Acad. Sci. USA. 2002;99:4932. doi: 10.1073/pnas.012540799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harano K, Hiraoka S. and Shionoya M. J. Am. Chem. Soc. 2007;129:5300. doi: 10.1021/ja0659727. [DOI] [PubMed] [Google Scholar]
- Buhler E, Sreenivasachary N, Candau S-J. and Lehn J-M. J. Am. Chem. Soc. 2007;129:10058. doi: 10.1021/ja072109d. [DOI] [PubMed] [Google Scholar]
- Pluth Mi D, Bergman R G. and Raymond K N. J. Am. Chem. Soc. 2007;129:11459. doi: 10.1021/ja072654e. [DOI] [PubMed] [Google Scholar]
- Heo J, Jeon Y-M. and Mirkin C A. J. Am. Chem. Soc. 2007;129:7712. doi: 10.1021/ja0716812. [DOI] [PubMed] [Google Scholar]
- Olivier C, Grote Z, Solari E, Scopelliti R. and Severin K. Chem. Commun. 2007:4000. doi: 10.1039/b711425g. [DOI] [PubMed] [Google Scholar]
- Gil-Ramírez G, Benet-Buchholz J, Escudero-Adán E C. and Ballester P. J. Am. Chem. Soc. 2007;129:3820. doi: 10.1021/ja070037k. [DOI] [PubMed] [Google Scholar]
- Iwasawa N. and Takahagi H. J. Am. Chem. Soc. 2007;129:7754. doi: 10.1021/ja072319q. [DOI] [PubMed] [Google Scholar]
- Fox O D, Cookson J, Wilkinson E J S, Drew M G B, MacLean E J, Teat S J. and Beer P D. J. Am. Chem. Soc. 2006;128:6990. doi: 10.1021/ja060982t. [DOI] [PubMed] [Google Scholar]
- Pirondini L, Bonifazi D, Cantadori B, Braiuca P, Campagnolo M, De Zorzi R, Geremia S, Diederichd F. and Dalcanalea E. Tetrahedron. 2006;62:2008. doi: 10.1016/j.tet.2005.06.122. [DOI] [Google Scholar]
- Haino T, Yanase M, Fukunaga C. and Fukazawa Y. Tetrahedron. 2006;62:2025. doi: 10.1016/j.tet.2005.07.121. [DOI] [Google Scholar]
- Haino T, Kobayashi M. and Fukazawa Y. Chem. Eur. J. 2006;12:3310. doi: 10.1002/chem.200500976. [DOI] [PubMed] [Google Scholar]
- Inomata T. and Konishi K. Chem. Commun. 2003:1282. doi: 10.1039/b302609d. [DOI] [PubMed] [Google Scholar]
- Harada A, Li J. and Kamachi M. Nature. 1992;356:325. doi: 10.1038/356325a0. [DOI] [Google Scholar]
- Harada A, Li J. and Kamachi M. Nature. 1993;364:516. doi: 10.1038/364516a0. [DOI] [Google Scholar]
- Harada A. Acc. Chem. Res. 2001;34:456. doi: 10.1021/ar000174l. [DOI] [PubMed] [Google Scholar]
- Livoreil A, Dietrich-Buchecker C O. and Sauvage J-P. J. Am. Chem. Soc. 1994;116:9399. doi: 10.1021/ja00099a095. [DOI] [PubMed] [Google Scholar]
- Fyfe M C T. and Stoddart J F. Acc. Chem. Res. 1997;30:393. doi: 10.1021/ar950199y. [DOI] [Google Scholar]
- Amabilino D B, Ashton P R, Reder A S, Spencer N. and Stoddart J F. Angew. Chem. Int. Ed. 1994;33:1286. doi: 10.1002/anie.199412861. [DOI] [Google Scholar]
- Bissell R A, Cordova E, Kaifer A E. and Stoddart J F. Nature. 1994;369:133. doi: 10.1038/369133a0. [DOI] [Google Scholar]
- Badjic J D, Balzani V, Credi A, Silvi S. and Stoddart J F. Science. 2004;303:1845. doi: 10.1126/science.1094791. [DOI] [PubMed] [Google Scholar]
- Badjic J D, Ronconi C M, Stoddart J F, Balzani V, Silvi S. and Credi A. J. Am. Chem. Soc. 2006;128:1489. doi: 10.1021/ja0543954. [DOI] [PubMed] [Google Scholar]
- Rogez G, Ribera B F, Credi A, Ballardini R, Gandolfi M T, Balzani V, Liu Y, Northrop B H. and Stoddart J F. J. Am. Chem. Soc. 2007;129:4633. doi: 10.1021/ja067739e. [DOI] [PubMed] [Google Scholar]
- Ferrer B, Rogez G, Credi A, Ballardini R, Gandolfi M T, Balzani V, Liu Y, Tseng H-R. and Stoddart J F. Proc. Nat. Acad. Sci. USA. 2006;103:18411. doi: 10.1073/pnas.0606459103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T D, Liu Y, Saha S, Leung K C-F, Stoddart J F. and Zink J I. J. Am. Chem. Soc. 2007;9:627. doi: 10.1021/ja065485r. [DOI] [PubMed] [Google Scholar]
- Nguyen T D, Tseng H-R, Celestre P C, Flood A H, Liu Y, Stoddart J. and Zink J I. Proc. Nat. Acad. Sci. USA. 2005;102:10029. doi: 10.1073/pnas.0504109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Vignon S A, Tseng H-R. and Stoddart J F. Chem. Eur. J. 2004;10:2555. doi: 10.1002/chem.200305725. [DOI] [PubMed] [Google Scholar]
- Bunker B C. et al Langmuir. 2007;23:31. doi: 10.1021/la0615793. [DOI] [PubMed] [Google Scholar]
- Saha S, Flood A H, Stoddart J F, Impellizzeri S, Silvi S, Venturi M. and Credi A. J. Am. Chem. Soc. 2007;129:12159. doi: 10.1021/ja0724590. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Watabe N, Ooya T. and Yui N. Macromol. Chem. Phys. 2001;202:1338. doi: 10.1002/1521-3935(20010501)202:8<1338::AID-MACP1338>3.0.CO;2-O. [DOI] [Google Scholar]
- Ooya T, Eguchi M. and Yui N. J. Am. Chem. Soc. 2003;125:13016. doi: 10.1021/ja034583z. [DOI] [PubMed] [Google Scholar]
- Murakami H, Kawabuchi A, Matsumoto R, Ido T. and Nakashima N. J. Am. Chem. Soc. 2005;127:15891. doi: 10.1021/ja053690l. [DOI] [PubMed] [Google Scholar]
- Kataoka T, Kidowaki M, Zhao C, Minamikawa H, Shimizu T and Ito K. 2006. J. Phys. Chem. B 110 24377 10.1021/jp0649246 [DOI] [PubMed] [Google Scholar]
- Choi H S, Hirasawa A, Ooya T, Kajihara D, Hohsaka T. and Yui N. Chem. Phys. Chem. 2006;7:1671. doi: 10.1002/cphc.200600242. [DOI] [PubMed] [Google Scholar]
- Inoue Y, Miyauchi M, Nakajima H, Takashima Y, Yamaguchi H. and Harada A. Macromolecules. 2007;40:3256. doi: 10.1021/ma070095q. [DOI] [Google Scholar]
- Oshikiri T, Takashima Y, Yamaguchi H. and Harada A. Chem. Eur. J. 2007;13:7091. doi: 10.1002/chem.200601657. [DOI] [PubMed] [Google Scholar]
- Huang F. and Gibson H W. Chem. Commun. 2005:1696. doi: 10.1039/b417966h. [DOI] [PubMed] [Google Scholar]
- Solladié N, Walther M E, Herschbach H, Leize E, Van Dorsselaer A, Duartec T M F. and Nierengartenc J-F. Tetrahedron. 2006;62:1979. doi: 10.1016/j.tet.2005.07.120. [DOI] [Google Scholar]
- Huang F, Nagvekar D S, Zhou X. and Gibson H W. Macromolecules. 2007;40:3561. doi: 10.1021/ma062080i. [DOI] [Google Scholar]
- Sasabe H, Ikeshita K, Rajkumar G A, Watanabe N, Kihara N, Furusho Y, Mizuno K, Ogawaa A. and Takatac T. Tetrahedron. 2006;62:1988. doi: 10.1016/j.tet.2005.09.149. [DOI] [Google Scholar]
- Sessler J L. and Jayawickramarajah J. Chem. Commun. 2005:1939. doi: 10.1039/b418526a. [DOI] [PubMed] [Google Scholar]
- Telfer S G. and Kuroda R. Chem. Eur. J. 2005;11:57. doi: 10.1002/chem.200400485. [DOI] [PubMed] [Google Scholar]
- Hutin M, Schalley C A, Bernardinelli G. and Nitschke J R. Chem. Eur. J. 2006;12:4069. doi: 10.1002/chem.200501591. [DOI] [PubMed] [Google Scholar]
- Kolomiets E, Berl V. and Lehn J-M. Chem. Eur. J. 2007;13:5466. doi: 10.1002/chem.200601826. [DOI] [PubMed] [Google Scholar]
- Breslow R. Acc. Chem. Res. 1991;24:159. doi: 10.1021/ar00006a001. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Kikuchi J, Hisaeda Y. and Hayashida O. Chem. Rev. 1996;96:721. doi: 10.1021/cr9403704. [DOI] [PubMed] [Google Scholar]
- Motherwell W B, Bingham M J. and Six Y. Tetrahedron. 2001;57:4663. doi: 10.1016/S0040-4020(01)00288-5. [DOI] [Google Scholar]
- Worm K, Chu F, Matsumoto K, Best M D, Lynch V. and Anslyn E V. Chem. Eur. J. 2003;9:741. doi: 10.1002/chem.200390082. [DOI] [PubMed] [Google Scholar]
- Suh J. Acc. Chem. Res. 2003;36:562. doi: 10.1021/ar020037j. [DOI] [PubMed] [Google Scholar]
- Miller S J. Acc. Chem. Res. 2004;37:601. doi: 10.1021/ar030061c. [DOI] [PubMed] [Google Scholar]
- Desper J M. and Breslow R. J. Am. Chem. Soc. 1994;116:12081. doi: 10.1021/ja00105a070. [DOI] [Google Scholar]
- Ariga K. and Anslyn E V. J. Org. Chem. 1992;57:417. doi: 10.1021/jo00028a001. [DOI] [Google Scholar]
- Smith J, Ariga K. and Anslyn E V. J. Am. Chem. Soc. 1993;15:362. doi: 10.1021/ja00054a062. [DOI] [Google Scholar]
- Kneeland D M, Ariga K, Lynch V M, Huang C-Y. and Anslyn E V. J. Am. Chem. Soc. 1993;115:10042. doi: 10.1021/ja00075a021. [DOI] [Google Scholar]
- Yaghi O M, O'Keeffe M, Ockwig N W, Chae H K, Eddaoudi M. and Kim J. Nature. 2003;423:705. doi: 10.1038/nature01650. [DOI] [PubMed] [Google Scholar]
- Eddaoudi M, Moler D B, Li H, Chen B, Reineke T M, O'Keeffe M. and Yaghi O M. Acc. Chem. Res. 2001;34:319. doi: 10.1021/ar000034b. [DOI] [PubMed] [Google Scholar]
- Suslick K S, Bhyrappa P, Chou J H, Kosal M E, Nakagaki S, Smithenry D W. and Wilson S R. Acc. Chem. Res. 2005;38:283. doi: 10.1021/ar040173j. [DOI] [PubMed] [Google Scholar]
- El-Kaderi H M, Hunt J R, Mendoza-Cortés J L, Côté A P, Taylor R E, O'Keeffe M. and Yaghi O M. Science. 2007;316:268. doi: 10.1126/science.1139915. [DOI] [PubMed] [Google Scholar]
- Eddaoudi M, Kim J, Rosi N, Vodak D, Wachter J, O'Keeffe M. and Yaghi O M. Science. 2002;295:469. doi: 10.1126/science.1067208. [DOI] [PubMed] [Google Scholar]
- Rosi N L, Eckert J, Eddaoudi M, Vodak D T, Kim J, O'Keeffe M. and Yaghi O M. Science. 2003;300:1127. doi: 10.1126/science.1083440. [DOI] [PubMed] [Google Scholar]
- Kitagawa S. Nature. 2006;441:584. doi: 10.1038/441584a. [DOI] [PubMed] [Google Scholar]
- Kitaura R. et al Science. 2002;298:2358. doi: 10.1126/science.1078481. [DOI] [PubMed] [Google Scholar]
- Noro S, Kitaura R, Kondo M, Kitagawa S, Ishii T, Matsuzaka H. and Yamashita M. J. Am. Chem. Soc. 2002;124:9. doi: 10.1021/ja0113192. [DOI] [PubMed] [Google Scholar]
- Matsuda R. et al Nature. 2005;436:238. doi: 10.1038/nature03852. [DOI] [PubMed] [Google Scholar]
- Uemura T, Kitaura R, Ohta Y, Nagaoka M. and Kitagawa S. Angew. Chem. Int. Ed. 2006;45:4112. doi: 10.1002/anie.200600333. [DOI] [PubMed] [Google Scholar]
- Tanaka D, Horike S, Kitagawa S, Ohba M, Hasegawa M, Ozawa Y. and Toriumi K. Chem. Commun. 2007:3142. doi: 10.1039/b707947h. [DOI] [PubMed] [Google Scholar]
- Black C A, Hanton L R. and Spicer M D. Chem. Commun. 2007:3171. doi: 10.1039/b703522e. [DOI] [PubMed] [Google Scholar]
- Molnár G, Cobo S, Real J A, Carcenac F, Daran E, Vieu C. and Bousseksou A. Adv. Mater. 2007;19:2163. doi: 10.1002/adma.200700448. [DOI] [Google Scholar]
- Bárcia P S, Zapata F, Silva J A C, Rodrigues A E and Chen B. 2007. J. Phys. Chem. B 111 6101 10.1021/jp0721898 [DOI] [PubMed] [Google Scholar]
- Walton K S. and Snurr R Q. J. Am. Chem. Soc. 2007;129:8552. doi: 10.1021/ja071174k. [DOI] [PubMed] [Google Scholar]
- Kondo A, Noguchi H, Carlucci L, Proserpio D M, Ciani G, Kajiro H, Ohba T, Kanoh H. and Kaneko K. J. Am. Chem. Soc. 2007;129:12362. doi: 10.1021/ja073568h. [DOI] [PubMed] [Google Scholar]
- Endo K, Koike T, Sawaki T, Hayashida O, Masuda H. and Aoyama Y. J. Am. Chem. Soc. 1997;119:4117. doi: 10.1021/ja964198s. [DOI] [Google Scholar]
- Tanaka T, Tasaki T. and Aoyama Y. J. Am. Chem. Soc. 2002;124:12453. doi: 10.1021/ja026704l. [DOI] [PubMed] [Google Scholar]
- Matsuura K, Ariga K, Endo K, Aoyama Y. and Okahata Y. Chem. Eur. J. 2000;6:1750. doi: 10.1002/(SICI)1521-3765(20000515)6:10<1750::AID-CHEM1750>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Ariga K, Endo K, Aoyama Y. and Okahata Y. Colloid Surf. A Physicochem. Eng. Asp. 2000;169:177. doi: 10.1016/S0927-7757(00)00434-9. [DOI] [Google Scholar]
- Dalrymple S A. and Shimizu G K H. J. Am. Chem. Soc. 2007;129:12114. doi: 10.1021/ja076094v. [DOI] [PubMed] [Google Scholar]
- Endo K, Koike T, Sawaki T, Hayashida O, Masuda H. and Aoyama Y. J. Am. Chem. Soc. 1997;119:4117. doi: 10.1021/ja964198s. [DOI] [Google Scholar]
- Aoyama Y. Top. Curr. Chem. 1998;198:131. [Google Scholar]
- Sawaki T. and Aoyama Y. J. Am. Chem. Soc. 1999;121:4793. doi: 10.1021/ja9900407. [DOI] [Google Scholar]
- Ghadiri M R, Granja J R, Milligan R A, Mcree D E. and Khazanovich N. Nature. 1993;366:324. doi: 10.1038/366324a0. [DOI] [PubMed] [Google Scholar]
- Horne W S, Ashkenasy N. and Ghadiri M R. Chem. Eur. J. 2005;11:1137. doi: 10.1002/chem.200400923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunitake T. Angew. Chem. Int. Ed. Engl. 1992;31:709. doi: 10.1002/anie.199207091. [DOI] [Google Scholar]
- Kunitake T. and Okahata Y. J. Am. Chem. Soc. 1977;99:3860. doi: 10.1021/ja00453a066. [DOI] [Google Scholar]
- Kunitake T, Okahata Y, Shimomura M, Yasunami S. and Takarabe K. J. Am. Chem. Soc. 1981;103:5401. doi: 10.1021/ja00408a021. [DOI] [Google Scholar]
- Shimomura M, Ando R, Kunitake T. and Ber Busenges. Phys. Chem. Chem. Phys. 1983;87:1134. [Google Scholar]
- Kunitake T, Kimizuka N, Higashi N. and Nakashima N. J. Am. Chem. Soc. 1984;106:1978. doi: 10.1021/ja00319a014. [DOI] [Google Scholar]
- Allen T M. Drugs. 1997;54:8. doi: 10.2165/00003495-199700544-00004. [DOI] [PubMed] [Google Scholar]
- Gregoriadis G. Trend. Biotechnol. 1995;13:527. doi: 10.1016/S0167-7799(00)89017-4. [DOI] [PubMed] [Google Scholar]
- Lian T. and Ho R J Y. J. Pharm. Sci. 2001;90:667. doi: 10.1002/jps.1023. [DOI] [PubMed] [Google Scholar]
- LaVan D A, McGuire T. and Langer R. Nat. Biotechnol. 2003;21:1184. doi: 10.1038/nbt876. [DOI] [PubMed] [Google Scholar]
- Yaroslavov A A, Melik-Nubarov N S. and Menger F M. Acc. Chem. Res. 2006;39:702. doi: 10.1021/ar050078q. [DOI] [PubMed] [Google Scholar]
- Gong Y, Luo Y. and Bong D. J. Am. Chem. Soc. 2006;128:14430. doi: 10.1021/ja0644576. [DOI] [PubMed] [Google Scholar]
- Stengel G, Zahn R. and Höök F. J. Am. Chem. Soc. 2007;129:9584. doi: 10.1021/ja073200k. [DOI] [PubMed] [Google Scholar]
- Cazacu A, Tong C, van der Lee A, Fyles T M. and Barboiu M. J. Am. Chem. Soc. 2006;128:9541. doi: 10.1021/ja061861w. [DOI] [PubMed] [Google Scholar]
- Gust D, Moore T A. and Moore A L. Acc. Chem. Res. 2001;34:40. doi: 10.1021/ar9801301. [DOI] [PubMed] [Google Scholar]
- Bhosale S. et al Science. 2006;313:84. doi: 10.1126/science.1126524. [DOI] [PubMed] [Google Scholar]
- Stanish I, Lowy D A, Hung C-W. and Singh A. Adv. Mater. 2005;17:1194. doi: 10.1002/adma.200401132. [DOI] [Google Scholar]
- Katagiri K, Ariga K. and Kikuchi J. Chem. Lett. 1999:661. doi: 10.1246/cl.1999.661. [DOI] [Google Scholar]
- Katagiri K, Hamasaki R, Ariga K. and Kikuchi J. J. Sol-Gel Sci. Technol. 2003;26:393. doi: 10.1023/A:1020781400998. [DOI] [Google Scholar]
- Katagiri K, Hashizume M, Ariga K, Terashima T. and Kikuchi J. Chem. Eur. J. 2007;13:5272. doi: 10.1002/chem.200700175. [DOI] [PubMed] [Google Scholar]
- Kimizuka N, Kawasaki T, Hirata K. and Kunitake T. J. Am. Chem. Soc. 1998;120:4094. doi: 10.1021/ja974379+. [DOI] [Google Scholar]
- Ishikawa Y, Kuwahara H. and Kunitake T. J. Am. Chem. Soc. 1994;116:5579. doi: 10.1021/ja00092a008. [DOI] [Google Scholar]
- Nakashima T. and Kimizuka N. Chem. Lett. 2002:1018. doi: 10.1246/cl.2002.1018. [DOI] [Google Scholar]
- Kikuchi J, Ariga K, Miyazaki T. and Ikeda K. Chem. Lett. 1999:253. doi: 10.1246/cl.1999.253. [DOI] [Google Scholar]
- Kikuchi J, Ariga K. and Ikeda K. Chem. Commun. 1999:547. doi: 10.1039/a900083f. [DOI] [Google Scholar]
- Fukuda K, Sasaki Y, Ariga K. and Kikuchi J. J. Mol. Catal. B-Enzym. 2001;11:971. doi: 10.1016/S1381-1177(00)00012-6. [DOI] [Google Scholar]
- Kikuchi J, Ariga K, Sasaki Y. and Ikeda K. J. Mol. Catal. B-Enzym. 2001;11:977. doi: 10.1016/S1381-1177(00)00019-9. [DOI] [Google Scholar]
- Nakashima N, Asakuma S. and Kunitake T. J. Am. Chem. Soc. 1985;107:509. doi: 10.1021/ja00288a043. [DOI] [Google Scholar]
- Fuhrhop J H. and Helfrich W. Chem. Rev. 1993;93:1565. doi: 10.1021/cr00020a008. [DOI] [Google Scholar]
- Schnur J M. Science. 1993;262:1669. doi: 10.1126/science.262.5140.1669. [DOI] [PubMed] [Google Scholar]
- Ariga K, Yamada N, Naito M, Koyama E. and Okahata Y. Chem. Lett. 1998:493. doi: 10.1246/cl.1998.493. [DOI] [Google Scholar]
- Yamada N, Ariga K, Naito M, Matsubara K. and Koyama E. J. Am. Chem. Soc. 1998;120:12192. doi: 10.1021/ja981363q. [DOI] [Google Scholar]
- Ariga K, Kikuchi J, Narumi K, Koyama E. and Yamada N. Chem. Lett. 1999:787. doi: 10.1246/cl.1999.787. [DOI] [Google Scholar]
- Ariga K, Kikuchi J, Naito M, Koyama E. and Yamada N. Langmuir. 2000;16:4929. doi: 10.1021/la000249u. [DOI] [Google Scholar]
- Ariga K, Kikuchi J, Naito M. and Yamada N. Polym. Adv. Technol. 2000;11:856. doi: 10.1002/1099-1581(200008/12)11:8/12<856::AID-PAT33>3.0.CO;2-3. [DOI] [Google Scholar]
- Yamada N. and Ariga K. Synlett. 2000:575. [Google Scholar]
- Yamada N, Matsubara K, Narumi K, Sato Y, Koyama E and Ariga K. 2000. Colloid Surf. A 169 271 10.1016/S0927-7757(00)00442-8 [DOI] [Google Scholar]
- Yamada N, Komatsu T, Yoshinaga H, Yoshizawa K, Edo S. and Kunitake M. Angew. Chem. Int. Ed. 2003;42:5496. doi: 10.1002/anie.200352185. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Masuda M. and Minamikawa H. Chem. Rev. 2005;105:1401. doi: 10.1021/cr030072j. [DOI] [PubMed] [Google Scholar]
- Iwaura R. and Shimizu T. Angew. Chem. Int. Ed. 2006;45:4601. doi: 10.1002/anie.200601173. [DOI] [PubMed] [Google Scholar]
- Kameta N, Mizuno G, Masuda M, Minamikawa H, Kogiso M. and Shimizu T. Chem. Lett. 2007;36:896. doi: 10.1246/cl.2007.896. [DOI] [Google Scholar]
- Kameta N, Masuda M, Minamikawa H, Mishima Y, Yamashita I. and Shimizu T. Chem. Mater. 2007;19:3553. doi: 10.1021/cm070626p. [DOI] [Google Scholar]
- Masuda M. and Shimizu T. Langmuir. 2004;20:5969. doi: 10.1021/la049085y. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Kogiso M. and Masuda M. Nature. 1996;383:487. doi: 10.1038/383487b0. [DOI] [Google Scholar]
- Kogiso M, Ohnishi S, Yase K, Masuda M. and Shimizu T. Langmuir. 1998;14:4978. doi: 10.1021/la9802419. [DOI] [Google Scholar]
- Kimizuka N, Oda N. and Kunitake T. Inorg. Chem. 2000;39:2684. doi: 10.1021/ic000189f. [DOI] [PubMed] [Google Scholar]
- Kimizuka N. Adv. Mater. 2000;12:1461. doi: 10.1002/1521-4095(200010)12:19<1461::AID-ADMA1461>3.0.CO;2-X. [DOI] [Google Scholar]
- Oda R, Huc I. and Candau S J. J. Am. Chem. Soc. 1998;37:2689. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2689::AID-ANIE2689>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Kawasaki T, Tokuhiro M, Kimizuka N. and Kunitake T. Angew. Chem. Int. Ed. 2001;123:6792. doi: 10.1021/ja010035e. [DOI] [PubMed] [Google Scholar]
- Boettcher C, Schade B. and Fuhrhop J-H. Langmuir. 2001;17:873. doi: 10.1021/la001054p. [DOI] [Google Scholar]
- Spector M S, Singh A, Messersmith P B. and Schnur J M. Nano Lett. 2001;1:375. doi: 10.1021/nl015554u. [DOI] [Google Scholar]
- Singh A, Wong E M. and Schnur J M. Langmuir. 2003;19:1888. doi: 10.1021/la026337r. [DOI] [Google Scholar]
- Takafuji M, Ishiodori A, Yamada T, Sakurai T. and Ihara H. Chem. Commun. 2004:1122. doi: 10.1039/b316673b. [DOI] [PubMed] [Google Scholar]
- Zhan C, Gao P. and Liu M. Chem. Commun. 2005:462. doi: 10.1039/b413259a. [DOI] [PubMed] [Google Scholar]
- Moreau L, Ziarelli F, Grinstaff M W. and Barthélémy P. Chem. Commun. 2006:1661. doi: 10.1039/b601038e. [DOI] [PubMed] [Google Scholar]
- Brizard A, Aimé C, Labrot T, Huc I, Berthier D, Artzner F, Desbat B. and Oda R. J. Am. Chem. Soc. 2007;129:3754. doi: 10.1021/ja0682172. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Minamikawa H, Kamiya S, Shimizu T. and Isoda S. J. Nanosci. Nanotechnol. 2007;7:960. doi: 10.1166/jnn.2007.406. [DOI] [PubMed] [Google Scholar]
- Fang J. J. Mater. Chem. 2007;17:3479. doi: 10.1039/b705350a. [DOI] [Google Scholar]
- Terech P. and Weiss R G. Chem. Rev. 1997;97:3133. doi: 10.1021/cr9700282. [DOI] [PubMed] [Google Scholar]
- Abdallah D J. and Weiss R G. Adv. Mater. 2000;12:1237. doi: 10.1002/1521-4095(200009)12:17<1237::AID-ADMA1237>3.0.CO;2-B. [DOI] [Google Scholar]
- Ajayaghosh A. and Praveen V K. Acc. Chem. Res. 2007;40:644. doi: 10.1021/ar7000364. [DOI] [PubMed] [Google Scholar]
- Laschat S. et al Angew. Chem. Int. Ed. 2007;46:4832. doi: 10.1002/anie.200604203. [DOI] [PubMed] [Google Scholar]
- Ajayaghosh A, Varghese R, Praveen V K. and Mahesh S. Angew. Chem. Int. Ed. 2006;45:3261. doi: 10.1002/anie.200600256. [DOI] [PubMed] [Google Scholar]
- Ajayaghosh A, Chithra P. and Varghese R. Angew. Chem. Int. Ed. 2007;46:230. doi: 10.1002/anie.200603611. [DOI] [PubMed] [Google Scholar]
- Praveen V K, George S J, Varghese R, Vijayakumar C. and Ajayaghosh A. J. Am. Chem. Soc. 2006;128:7542. doi: 10.1021/ja0587594. [DOI] [PubMed] [Google Scholar]
- Fujita N, Sakamoto Y, Shirakawa M, Ojima M, Fujii A, Ozaki M. and Shinkai S. J. Am. Chem. Soc. 2007;129:4134. doi: 10.1021/ja069307+. [DOI] [PubMed] [Google Scholar]
- Ihara H, Sakurai T, Yamada T, Hashimoto T, Takafuji M, Sagawa T. and Hachisako H. Langmuir. 2002;18:7120. doi: 10.1021/la025535f. [DOI] [Google Scholar]
- Sagawa T, Fukugawa S, Yamada T. and Ihara H. Langmuir. 2002;18:7223. doi: 10.1021/la0255267. [DOI] [Google Scholar]
- Xue P, Lu R, Chen G, Zhang Y, Nomoto H, Takafuji M. and Ihara H. Chem. Eur. J. 2007;13:8231. doi: 10.1002/chem.200700321. [DOI] [PubMed] [Google Scholar]
- Hanabusa K, Shimura K, Hirose K, Kimura M. and Shirai H. Chem. Lett. 1996:885. doi: 10.1246/cl.1996.885. [DOI] [Google Scholar]
- Hanabusa K, Yamada M, Kimura M. and Shirai H. J. Am. Chem. Soc. 1996;35:1949. [Google Scholar]
- Hanabusa K, Kawakami A, Kimura M. and Shirai H. Chem. Lett. 1997:191. doi: 10.1246/cl.1997.191. [DOI] [Google Scholar]
- Jeong Y, Hanabusa K, Masunaga H, Akiba I, Miyoshi K, Sakurai S. and Sakurai K. Langmuir. 2005;21:586. doi: 10.1021/la047538t. [DOI] [PubMed] [Google Scholar]
- Shinkai S. and Murata K. J. Mater. Chem. 1998;8:485. doi: 10.1039/a704820c. [DOI] [Google Scholar]
- Yoza K, Amanokura N, Ono Y, Akao T, Shinmori H, Takeuchi M, Shinkai S. and Reinhoudt D N. Chem. Eur. J. 1999;5:2722. doi: 10.1002/(SICI)1521-3765(19990903)5:9<2722::AID-CHEM2722>3.0.CO;2-N. [DOI] [Google Scholar]
- Fukushima T, Kosaka A, Ishimura Y, Yamamoto T, Takigawa T, Ishii N. and Aida T. Science. 2003;300:2072. doi: 10.1126/science.1082289. [DOI] [PubMed] [Google Scholar]
- Fukushima T. and Aida T. Chem. Eur. J. 2007;13:5048. doi: 10.1002/chem.200700554. [DOI] [PubMed] [Google Scholar]
- Kato T. Science. 2002;295:2414. doi: 10.1126/science.1070967. [DOI] [PubMed] [Google Scholar]
- Kamikawa Y, Nishii M. and Kato T. Chem. Eur. J. 2004;10:5942. doi: 10.1002/chem.200400424. [DOI] [PubMed] [Google Scholar]
- Kato T, Kihara H, Kumar U, Uryu T. and Fréchet J M J. J. Am. Chem. Soc. 1994;33:1644. [Google Scholar]
- Ichimura K. Chem. Rev. 2000;100:1847. doi: 10.1021/cr980079e. [DOI] [PubMed] [Google Scholar]
- Furumi S, Yokoyama S, Otomo A. and Mashiko S. Appl. Phys. Lett. 2003;82:16. doi: 10.1063/1.1534613. [DOI] [Google Scholar]
- Ichimura K, Furumi S, Morino S, Kidowaki M, Nakagawa M, Ogawa M. and Nishiura Y. Adv. Mater. 2000;12:950. doi: 10.1002/1521-4095(200006)12:13<950::AID-ADMA950>3.0.CO;2-V. [DOI] [Google Scholar]
- Furumi S, Ichimura K, Sata H. and Nishiura Y. Appl. Phys. Lett. 2000;77:2689. doi: 10.1063/1.1320017. [DOI] [Google Scholar]
- Furumi S, Yokoyama S, Otomo A. and Mashiko S. Appl. Phys. Lett. 2004;84:2491. doi: 10.1063/1.1699445. [DOI] [Google Scholar]
- Furumi S. and Sakka Y. J. Nanosci. Nanotechnol. 2006;6:1819. doi: 10.1166/jnn.2006.202. [DOI] [PubMed] [Google Scholar]
- Ahir S V, Tajbakhsh A R. and Terentjev E M. Adv. Funct. Mater. 2006;16:556. doi: 10.1002/adfm.200500692. [DOI] [Google Scholar]
- Vlahakis J Z, Wand M D. and Lemieux R P. J. Am. Chem. Soc. 2003;125:6862. doi: 10.1021/ja0353309. [DOI] [PubMed] [Google Scholar]
- Schmidt-Mende L, Fechtenkötter A, Müllen K, Moons E, Friend R H. and MacKenzie J D. Science. 2001;293:1119. doi: 10.1126/science.293.5532.1119. [DOI] [PubMed] [Google Scholar]
- Coleman D A. et al Science. 2003;301:1204. doi: 10.1126/science.1084956. [DOI] [PubMed] [Google Scholar]
- Walba D M, Körblova E, Shao R, Maclennan J E, Link D R, Glaser M A. and Clark N A. Science. 2000;288:2131. doi: 10.1126/science.288.5474.2181. [DOI] [PubMed] [Google Scholar]
- Liu C-Y, Fechtenkötter A, Watson M D, Müllen K. and Bard A J. Chem. Mater. 2003;15:124. doi: 10.1021/cm020701q. [DOI] [Google Scholar]
- Shimizu Y, Oikawa K, Nakayama K. and Guillond D. J. Mater. Chem. 2007;17:4223. doi: 10.1039/b705534j. [DOI] [Google Scholar]
- Chen Z, Baumeister U, Tschierske C. and Würthner F. Chem. Eur. J. 2007;13:450. doi: 10.1002/chem.200600891. [DOI] [PubMed] [Google Scholar]
- Amano S, Ishida Y. and Saigo K. Chem. Eur. J. 2007;13:5186. doi: 10.1002/chem.200601656. [DOI] [PubMed] [Google Scholar]
- Cheng J Y, Ross C A, Smith H I. and Thomas E L. Adv. Mater. 2006;18:2505. doi: 10.1002/adma.200502651. [DOI] [Google Scholar]
- Hill J P, Alam S, Ariga K, Anson C E. and Powell A K. Chem. Commun. 2008:383. doi: 10.1039/b713201h. [DOI] [PubMed] [Google Scholar]
- Ono Y, Nakashima K, Sano M, Kanekiyo Y, Inoue K, Hojo J. and Shinkai S. Chem. Commun. 1998:1477. doi: 10.1039/a802829j. [DOI] [Google Scholar]
- Jung J H, Ono Y, Hanabusa K. and Shinkai S. J. Am. Chem. Soc. 2000;122:5008. doi: 10.1021/ja000449s. [DOI] [Google Scholar]
- Jung J H, Ono Y. and Shinkai S. Langmuir. 2000;16:1643. doi: 10.1021/la990901p. [DOI] [Google Scholar]
- Zou Y, Kamiya S, Minamikawa H. and Shimizu T. Adv. Mater. 2007;19:4194. doi: 10.1002/adma.200701189. [DOI] [Google Scholar]
- Sugiyasu K, Tamaru S, Takeuchi M, Berthier D, Huc I, Oda R. and Shinkai S. Chem. Commun. 2000:1212. doi: 10.1039/b202799m. [DOI] [PubMed] [Google Scholar]
- Jung J H, Kobayashi H, van Bommel K J C, Shinkai S. and Shimizu T. Chem. Mater. 2002;14:1445. doi: 10.1021/cm011625e. [DOI] [Google Scholar]
- Gronwald O, Snip E. and Shinkai S. Curr. Opin. Colloid Interface Sci. 2002;7:148. doi: 10.1016/S1359-0294(02)00016-X. [DOI] [Google Scholar]
- van Bommel K J C, Friggeri A. and Shinkai S. Angew. Chem. Int. Ed. 2003;42:980. doi: 10.1002/anie.200390284. [DOI] [PubMed] [Google Scholar]
- Jung J H. and Shinkai S. Top. Curr. Chem. 2004;248:223. [Google Scholar]
- Jung J H, Shimizu T. and Shinkai S. J. Mater. Chem. 2005;15:3979. doi: 10.1039/b503441h. [DOI] [Google Scholar]
- Jung J H, Lee S-H, Yoo J S, Yoshida K, Shimizu T. and Shinkai S. Chem. Eur. J. 2003;9:5307. doi: 10.1002/chem.200305008. [DOI] [PubMed] [Google Scholar]
- Jung J H, Kobayashi H, Masuda M, Shimizu T. and Shinkai S. J. Am. Chem. Soc. 2001;23:8785. doi: 10.1021/ja010508h. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Hanabusa K, Hamasaki N, Kimura M, Shirai H. and Shinkai S. Chem. Mater. 2000;12:1523. doi: 10.1021/cM000907. [DOI] [Google Scholar]
- Asai M, Fujita N. and Shinkai S. Chem. Lett. 2003;32:186. doi: 10.1246/cl.2003.186. [DOI] [Google Scholar]
- Kawano S, Tamaru S, Fujita N. and Shinkai S. Chem. Eur. J. 2004;10:343. doi: 10.1002/chem.200305042. [DOI] [PubMed] [Google Scholar]
- Yang B, Kamiya S, Shimizu Y, Koshizaki N. and Shimizu T. Chem. Mater. 2004;16:2826. doi: 10.1021/cm049695j. [DOI] [Google Scholar]
- Zhou Y, Ji Q, Masuda M, Kamiya S. and Shimizu T. Chem. Mater. 2006;18:403. doi: 10.1021/cm051928z. [DOI] [Google Scholar]
- Ji Q, Iwaura R. and Shimizu T. Chem. Mater. 2007;19:1329. doi: 10.1021/cm0625124. [DOI] [Google Scholar]
- Price R R, Dressick W J. and Singh A. J. Am. Chem. Soc. 2003;125:11259. doi: 10.1021/ja036173g. [DOI] [PubMed] [Google Scholar]
- Ras R H A, Kemell M, de Wit J, Ritala M, ten Brinke G, Leskelä M. and Ikkala O. Adv. Mater. 2007;19:102. doi: 10.1002/adma.200600728. [DOI] [Google Scholar]
- Chen X, Sun X. and Li Y. Inorg. Chem. 2002;41:4524. doi: 10.1021/ic020092o. [DOI] [PubMed] [Google Scholar]
- Hu X, Yu J C and Gong J. 2007. J. Phys. Chem. C 111 5830 10.1021/jp0716435 [DOI] [Google Scholar]
- Hillebrenner H, Buyukserin F, Kang M, Mota M O, Stewart J D. and Martin C R. J. Am. Chem. Soc. 2006;128:4236. doi: 10.1021/ja058455h. [DOI] [PubMed] [Google Scholar]
- Golberg D, Bando Y, Tang C C. and Zhi C T. Adv. Mater. 2007;19:2413. doi: 10.1002/adma.200700179. [DOI] [Google Scholar]
- Hu J, Bando Y, Zhan J, Yuan X, Sekiguchi T. and Golberg D. Adv. Mater. 2005;17:971. doi: 10.1002/adma.200401789. [DOI] [Google Scholar]
- Hu J, Bando Y, Zhan J. and Golberg D. Adv. Mater. 2005;17:1964. doi: 10.1002/adma.200500317. [DOI] [PubMed] [Google Scholar]
- Corso M, Auwärter W, Muntwiler M, Tamai A, Greber T. and Osterwalder J. Science. 2004;303:217. doi: 10.1126/science.1091979. [DOI] [PubMed] [Google Scholar]
- Raman N K, Anderson M T. and Brinker C J. Chem. Mater. 1996;8:1682. doi: 10.1021/cm960138+. [DOI] [Google Scholar]
- Sayari A. Chem. Mater. 1996;8:1840. doi: 10.1021/cm950585+. [DOI] [Google Scholar]
- Corma A. Chem. Rev. 1997;97:2373. doi: 10.1021/cr960406n. [DOI] [PubMed] [Google Scholar]
- Moller K. and Bein T. Chem. Mater. 1998;10:2950. doi: 10.1021/cm980243e. [DOI] [Google Scholar]
- Ciesla U. and Schüth F. Microporous Mesoporous Mater. 1999;27:131. doi: 10.1016/S1387-1811(98)00249-2. [DOI] [Google Scholar]
- Ying J Y, Mehnert C P. and Wong M S. Angew. Chem. Int. Ed. 1999;38:56. doi: 10.1002/(SICI)1521-3773(19990115)38:1/2<56::AID-ANIE56>3.0.CO;2-E. [DOI] [Google Scholar]
- Stein A, Melde B J. and Schroden R C. Adv. Mater. 2000;12:1403. doi: 10.1002/1521-4095(200010)12:19<1403::AID-ADMA1403>3.0.CO;2-X. [DOI] [Google Scholar]
- Sanchez C, de G J, Soler-Illia A A, Ribot F, Lalot T, Mayer C R. and Cabuil V. Chem. Mater. 2001;13:3061. doi: 10.1021/cm011061e. [DOI] [Google Scholar]
- Polarz S. and Smarsly B. J. Nanosci. Nanotechnol. 2002;2:581. doi: 10.1166/jnn.2002.151. [DOI] [PubMed] [Google Scholar]
- Davis M E. Nature. 2002;417:813. doi: 10.1038/nature00785. [DOI] [PubMed] [Google Scholar]
- Vinu A, Murugesan V. and Hartmann M. Chem. Mater. 2003;15:1385. doi: 10.1021/cm0213523. [DOI] [Google Scholar]
- Okabe A, Fukushima T, Ariga K, Niki M. and Aida T. J. Am. Chem. Soc. 2004;126:9013. doi: 10.1021/ja0478532. [DOI] [PubMed] [Google Scholar]
- Taguchi A. and Schüth F. Microporous Mesoporous Mater. 2005;77:1. doi: 10.1016/j.micromeso.2004.06.030. [DOI] [Google Scholar]
- Yanagisawa T, Shimizu T, Kuroda K. and Kato C. Bull. Chem. Soc. Jpn. 1990;63:988. doi: 10.1246/bcsj.63.988. [DOI] [Google Scholar]
- Inagaki S, Fukushima Y. and Kuroda K. J. Chem. Soc., Chem. Commun. 1993:680. doi: 10.1039/c39930000680. [DOI] [Google Scholar]
- Kresge C T, Leonowicz M E, Roth W J, Vartuli J C. and Beck J S. Nature. 1992;359:710. doi: 10.1038/359710a0. [DOI] [Google Scholar]
- Beck J S. et al J. Am. Chem. Soc. 1992;114:10834. doi: 10.1021/ja00053a020. [DOI] [Google Scholar]
- Vartuli J C. et al Chem. Mater. 1994;6:2317. doi: 10.1021/cM00048a018. [DOI] [Google Scholar]
- Dubois M, Gulik-Krzywicki T. and Cabane B. Langmuir. 1993:673. doi: 10.1021/la00027a011. [DOI] [Google Scholar]
- Tanev P T. and Pinnavaia T J. Science. 1995;267:865. doi: 10.1126/science.267.5199.865. [DOI] [PubMed] [Google Scholar]
- Bagshaw S A, Prouset E. and Pinnavaia T J. Science. 1995;269:1242. doi: 10.1126/science.269.5228.1242. [DOI] [PubMed] [Google Scholar]
- Zhao D, Feng J, Huo Q, Melosh N, Fredickson G H, Chmelka B F. and Stucky G D. Science. 1998;279:548. doi: 10.1126/science.279.5350.548. [DOI] [PubMed] [Google Scholar]
- Zhao D, Huo Q, Feng J, Chmelka B F. and Stucky G D. J. Am. Chem. Soc. 1998;120:6024. doi: 10.1021/ja974025i. [DOI] [Google Scholar]
- Schmidt-Winkel P, Lukens W W, Jr, Zhao D, Yang P, Chmelka B F. and Stucky G D. J. Am. Chem. Soc. 1999;121:254. doi: 10.1021/ja983218i. [DOI] [Google Scholar]
- Ryoo R, Joo S H and Jun S. 1999. J. Phys. Chem. B 103 7743 10.1021/jp991673a [DOI] [Google Scholar]
- Jun S, Joo S H, Ryoo R, Kruk M, Jaroniec M, Liu Z, Ohsuna T. and Terasaki O. J. Am. Chem. Soc. 2000;122:10712. doi: 10.1021/ja002261e. [DOI] [Google Scholar]
- Lee J, Yoon S, Hyeon T, Oh S M. and Kim K B. Chem. Commun. 1999:2177. doi: 10.1039/a906872d. [DOI] [Google Scholar]
- Lee J, Yoon S, Oh S M, Shin C-H. and Hyeon T. Adv. Mater. 2000;12:359. doi: 10.1002/(SICI)1521-4095(200003)12:5<359::AID-ADMA359>3.0.CO;2-1. [DOI] [Google Scholar]
- Han S, Kim S, Lim H, Choi W, Park H, Yoon J. and Hyeon T. Microporous Mesoporous Mater. 2003;58:131. doi: 10.1016/S1387-1811(02)00611-X. [DOI] [Google Scholar]
- Vinu A, Srinivasu P, Takahashi M, Mori T, Balasubramanian V V. and Ariga K. Microporous Mesoporous Mater. 2007;100:20. doi: 10.1016/j.micromeso.2006.10.008. [DOI] [Google Scholar]
- Vinu A, Hossian K Z, Srinivasu P, Miyahara M, Anandan S, Gokulakrishnan N, Mori T, Ariga K. and Balasubramanian V V. J. Mater. Chem. 2007;17:1819. doi: 10.1039/b613899c. [DOI] [Google Scholar]
- Vinu A, Srinivasu P, Mori T, Sasaki T, Asthana A, Ariga K. and Hishita S. Chem. Lett. 2007;36:770. doi: 10.1246/cl.2007.770. [DOI] [Google Scholar]
- Srinivasu P, Vinu A, Gokulakrishnan N, Anandan S, Asthana A, Mori T. and Ariga K. J. Nanosci. Nanotechnol. 2007;7:2913. doi: 10.1166/jnn.2007.631. [DOI] [PubMed] [Google Scholar]
- Vinu A, Anandan S, Anand C, Srinivasu P, Ariga K. and Mori T. Microporous Mesoporous Mater. 2008;109:398. doi: 10.1016/j.micromeso.2007.05.037. [DOI] [Google Scholar]
- Vinu A, Ariga K, Mori T, Nakanishi T, Hishita S, Golberg D. and Bando Y. Adv. Mater. 2005;17:1648. doi: 10.1002/adma.200401643. [DOI] [Google Scholar]
- Vinu A, Srinivasu P, Sawant D P, Mori T, Ariga K, Chang J-S, Jhung S-H, Balasubramanian V V. and Hwang Y K. Chem. Mater. 2007;19:4367. doi: 10.1021/cm070657k. [DOI] [Google Scholar]
- Srinivasu P, Vinu A, Hishita S, Sasaki T, Ariga K. and Mori T. Microporous Mesoporous Mater. 2008;108:340. doi: 10.1016/j.micromeso.2007.03.048. [DOI] [Google Scholar]
- Vinua A, Moria T. and Ariga K. Sci. Technol. Adv. Mater. 2006;7:753. doi: 10.1016/j.stam.2006.10.007. [DOI] [Google Scholar]
- Vinu A, Terrones M, Golberg D, Hishita S, Ariga K. and Mori T. Chem. Mater. 2005;17:5887. doi: 10.1021/cm051780j. [DOI] [Google Scholar]
- Murakami M, Shimizu T, Tansho M, Vinu A, Ariga K. and Takegoshi K. Chem. Lett. 2006;35:986. doi: 10.1246/cl.2006.986. [DOI] [Google Scholar]
- Murakami M, Shimizu T, Tansho M, Vinu A, Ariga K, Mori T. and Takegoshi K. Solid State Nucl. Magn. Reson. 2007;31:193. doi: 10.1016/j.ssnmr.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Vinu A, Miyahara M. and Ariga K. Stud. Surf. Sci. Catal. 2005;158:971. [Google Scholar]
- Vinu A, Miyahara M, Sivamurugan V, Mori T. and Ariga K. J. Mater. Chem. 2005;15:5122. doi: 10.1039/b507456h. [DOI] [Google Scholar]
- Vinu A, Miyahara M, Mori T. and Ariga K. J. Porous. Mater. 2006;13:379. doi: 10.1007/s10934-006-8034-1. [DOI] [Google Scholar]
- Srinivasu P, Balasubramanian V V, Kumaresan L, Sawant D P, Jin X, Alam S, Ariga K, Mori T. and Vinu A. J. Nanosci. Nanotechnol. 2007;7:3250. doi: 10.1166/jnn.2007.919. [DOI] [PubMed] [Google Scholar]
- Ariga K, Vinu A, Miyahara M, Hill J P. and Mori T. J. Am. Chem. Soc. 2007;129:11022. doi: 10.1021/ja074870t. [DOI] [PubMed] [Google Scholar]
- Vinu A, Hossain K Z. and Ariga K. J. Nanosci. Nanotechnol. 2005;5:347. doi: 10.1166/jnn.2005.089. [DOI] [PubMed] [Google Scholar]
- Che S, Liu Z, Ohsuna T, Sakamoto K, Terasaki O. and Tatsumi T. Nature. 2004;429:281. doi: 10.1038/nature02529. [DOI] [PubMed] [Google Scholar]
- Jin H, Liu Z, Ohsuna T, Terasaki O, Inoue Y, Sakamoto K, Nakanishi T, Ariga K. and Che S. Adv. Mater. 2006;18:593. doi: 10.1002/adma.200502038. [DOI] [Google Scholar]
- Che S. J. Nanosci. Nanotechnol. 2006;6:1557. doi: 10.1166/jnn.2006.252. [DOI] [PubMed] [Google Scholar]
- Wang B, Chi C, Shan W, Zhang Y H, Ren N, Yang W L. and Tang Y. J. Am. Chem. Soc. 2006;45:2088. doi: 10.1002/anie.200504191. [DOI] [PubMed] [Google Scholar]
- Yang Y, Suzuki M, Fukui H, Shirai H. and Hanabusa K. Chem. Mater. 2006;18:1324. doi: 10.1021/cm0519030. [DOI] [Google Scholar]
- Yang S, Zhao L, Yu C, Zhou X, Tang J, Yuan P, Chen D. and Zhao D. J. Am. Chem. Soc. 2006;128:10460. doi: 10.1021/ja0619049. [DOI] [PubMed] [Google Scholar]
- Inagaki S, Guan S, Fukushima Y, Ohsuna T. and Terasaki O. J. Am. Chem. Soc. 1999;121:9611. doi: 10.1021/ja9916658. [DOI] [Google Scholar]
- Asefa T, MacLachlan M J, Coombs N. and Ozin G A. Nature. 1999;402:867. [Google Scholar]
- Melde B J, Holland B T, Blanford C F. and Stein A. Chem. Mater. 1999;11:3302. doi: 10.1021/cm9903935. [DOI] [Google Scholar]
- Dagö Yoshina-Ishii C, Asefa T, MacLachlan M J, Grondey H, Coombs N. and Ozin G A. Adv. Funct. Mater. 2001;11:213. doi: 10.1002/1616-3028(200106)11:3<213::AID-ADFM213>3.0.CO;2-C. [DOI] [Google Scholar]
- Kuroki M, Asefa T, Whitnal W, Kruk M, Yoshina-Ishii C, Jaroniec M. and Ozin G A. J. Am. Chem. Soc. 2002;124:13886. doi: 10.1021/ja027877d. [DOI] [PubMed] [Google Scholar]
- Kapoor M P. and Inagaki S. Bull. Chem. Soc. Jpn. 2006;79:1463. doi: 10.1246/bcsj.79.1463. [DOI] [Google Scholar]
- Inagaki S, Guan S, Ohsuna T. and Terasaki O. Nature. 2002;416:304. doi: 10.1038/416304a. [DOI] [PubMed] [Google Scholar]
- Ariga K, Vinu A, Hill J P. and Mori T. Coord. Chem. Rev. 2007;251:2562. doi: 10.1016/j.ccr.2007.02.024. [DOI] [Google Scholar]
- Mal N K, Fujiwara M. and Tanaka Y. Nature. 2003;421:350. doi: 10.1038/nature01362. [DOI] [PubMed] [Google Scholar]
- Mal N K, Fujiwara M, Tanaka Y, Taguchi T. and Matsukata M. Chem. Mater. 2003;15:3385. doi: 10.1021/cm0343296. [DOI] [Google Scholar]
- Lai C-Y, Trewryn B G, Jeftinija D M, Jeftinija K, Xu S, Jeftinija S. and Lin V S-Y. J. Am. Chem. Soc. 2003;125:4451. doi: 10.1021/ja028650l. [DOI] [PubMed] [Google Scholar]
- Radu D R, Lai C-Y, Wiench J W, Pruski M. and Lin V S-Y. J. Am. Chem. Soc. 2004;126:1640. doi: 10.1021/ja038222v. [DOI] [PubMed] [Google Scholar]
- Trewyn B G, Whitman C M. and Lin V S-Y. Nano Lett. 2004;4:2139. doi: 10.1021/nl048774r. [DOI] [Google Scholar]
- Casasús R, Aznar E, Marcos M D, Martínez-Máñez R, Sancenón F. and Amorós P. J. Am. Chem. Soc. 2006;45:6661. doi: 10.1002/anie.200602045. [DOI] [PubMed] [Google Scholar]
- Yang Q, Wang S, Fan P, Wang L, Di Y, Lin K. and Xiao F-S. Chem. Mater. 2005;17:5999. doi: 10.1021/cm051198v. [DOI] [Google Scholar]
- Balas F, Manzano M, Horcajada P. and Vallet-Regí M. J. Am. Chem. Soc. 2006;128:8116. doi: 10.1021/ja062286z. [DOI] [PubMed] [Google Scholar]
- Doadrio J C, Sousa E M B, Izquierdo-Barba I, Doadrio A L, Perez-Pariente J. and Vallet-Regí M. J. Mater. Chem. 2006;16:462. doi: 10.1039/b510101h. [DOI] [Google Scholar]
- Kim J, Lee J E, Lee J, Yu J H, Kim B C, An K, Hwang Y, Shin C-H, Park J-G, Kim J. and Hyeon T. J. Am. Chem. Soc. 2006;128:688. doi: 10.1021/ja0565875. [DOI] [PubMed] [Google Scholar]
- Wang Y. and Caruso F. Chem. Mater. 2005;17:953. doi: 10.1021/cm0483137. [DOI] [Google Scholar]
- Wang Y. and Caruso F. Chem. Mater. 2006;18:4089. doi: 10.1021/cm060866p. [DOI] [Google Scholar]
- Kageyama K, Tamazawa J. and Aida T. Science. 1999;285:2113. doi: 10.1126/science.285.5436.2113. [DOI] [PubMed] [Google Scholar]
- Nguyen T-Q, Wu J, Doan V, Schwartz B J. and Tolbert S H. Science. 2000;288:652. doi: 10.1126/science.288.5466.652. [DOI] [PubMed] [Google Scholar]
- MacLachlan M J, Ginzburg M, Coombs N, Raju N P, Greedan J E, Ozin G A. and Manners I. J. Am. Chem. Soc. 2000;122:3878. doi: 10.1021/ja992006y. [DOI] [PubMed] [Google Scholar]
- Cho M S, Choi H J. and Ahn W-S. Langmuir. 2004;20:202. doi: 10.1021/la035051z. [DOI] [PubMed] [Google Scholar]
- Jang J, Lim B, Lee J. and Hyeon T. Chem. Commun. 2001:83. doi: 10.1039/b006197m. [DOI] [Google Scholar]
- Aida T. and Tajima K. J. Am. Chem. Soc. 2001;40:3803. [PubMed] [Google Scholar]
- Lu Y. et al Nature. 2001;410:913. doi: 10.1038/35073544. [DOI] [PubMed] [Google Scholar]
- Ikegame M, Tajima K. and Aida T. J. Am. Chem. Soc. 2003;42:2154. doi: 10.1002/anie.200250800. [DOI] [PubMed] [Google Scholar]
- Li G, Bhosale S, Wang T, Zhang Y, Zhu H. and Fuhrhop J-H. J. Am. Chem. Soc. 2003;42:3818. doi: 10.1002/anie.200351158. [DOI] [PubMed] [Google Scholar]
- Okabe A, Fukushima T, Ariga K. and Aida T. J. Am. Chem. Soc. 2002;41:3414. doi: 10.1002/1521-3773(20020916)41:18<3414::AID-ANIE3414>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Ariga K, Zhang Q, Niki M, Okabe A. and Aida T. Stud. Surf. Sci. Catal. 2003;146:427. [Google Scholar]
- Ariga K. Chem. Rec. 2004;3:297. doi: 10.1002/tcr.10071. [DOI] [PubMed] [Google Scholar]
- Ariga K, Aimiya T, Zhang Q, Okabe A, Niki M. and Aida T. Int. J. Nanosci. 2002;1:521. doi: 10.1142/S0219581X02000607. [DOI] [Google Scholar]
- Zhang Q, Ariga K, Okabe A. and Aida T. Stud. Surf. Sci. Catal. 2003;146:465. [Google Scholar]
- Zhang Q, Ariga K, Okabe A. and Aida T. J. Am. Chem. Soc. 2004;126:988. doi: 10.1021/ja039515r. [DOI] [PubMed] [Google Scholar]
- Otani W, Kinbara K, Zhang Q, Ariga K. and Aida T. Chem. Eur. J. 2007;13:1731. doi: 10.1002/chem.200601099. [DOI] [PubMed] [Google Scholar]
- Washmon-Kriel L, Jimenez V L, Balkus K J., Jr J. Mol. Catal. B-Enzym. 2000;10:453. doi: 10.1016/S1381-1177(99)00123-X. [DOI] [Google Scholar]
- Takahashi H, Li B, Sasaki T, Miyazaki C, Kajino T. and Inagaki S. Chem. Mater. 2000;12:3301. doi: 10.1021/cM0487a. [DOI] [Google Scholar]
- Wei Y, Xu J, Feng Q, Lin M, Dong H, Zhang W-J. and Wang C. J. Nanosci. Nanotechnol. 2001;1:83. doi: 10.1166/jnn.2001.014. [DOI] [PubMed] [Google Scholar]
- Deere J, Magner E, Wall J G and Hodnett B K. 2002. J. Phys. Chem. B 106 7340 10.1021/jp0139484 [DOI] [Google Scholar]
- Chong A S M. and Zhao X S. Appl. Surf. Sci. 2004;237:398. [Google Scholar]
- Hartmann M. Chem. Mater. 2005;17:4577. doi: 10.1021/cm0485658. [DOI] [Google Scholar]
- Vinu A, Streb C, Murugesan V and Hartmann M. 2003. J. Phys. Chem. B 107 8297 10.1021/jp035246f [DOI] [Google Scholar]
- Vinu A, Murugesan V and Hartmann M. 2004. J. Phys. Chem. B 108 7323 10.1021/jp037303a [DOI] [Google Scholar]
- Vinu A, Murugesan V, Tangermann O. and Hartmann M. Chem. Mater. 2004;16:3056. doi: 10.1021/cm049718u. [DOI] [Google Scholar]
- Vinu A, Murugesan V, Böhlmann W and Hartmann M. 2004. J. Phys. Chem. B 108 11496 10.1021/jp048411f [DOI] [Google Scholar]
- Miyahara M, Vinu A, Nakanshi T. and Ariga K. Kobunshi Ronbunshu. 2004;61:623. [Google Scholar]
- Vinu A, Miyahara M and Ariga K. 2005. J. Phys. Chem. B 109 6436 10.1021/jp050454o [DOI] [PubMed] [Google Scholar]
- Vinu A, Miyahara M, Hossain K Z, Nakanishi T. and Ariga K. Stud. Surf. Sci. Catal. 2005;156:637. [Google Scholar]
- Miyahara M, Vinu A, Hossain K Z, Nakanishi T. and Ariga K. Thin Solid Films. 2006;499:13. doi: 10.1016/j.tsf.2005.07.046. [DOI] [Google Scholar]
- Vinu A, Miyahara M. and Ariga K. J. Nanosci. Nanotechnol. 2006;6:1510. doi: 10.1166/jnn.2006.253. [DOI] [PubMed] [Google Scholar]
- Miyahara M, Vinu A. and Ariga K. J. Nanosci. Nanotechnol. 2006;6:1765. doi: 10.1166/jnn.2006.221. [DOI] [PubMed] [Google Scholar]
- Ariga K, Vinu A. and Miyahara M. Curr. Nanosci. 2006;2:197. [Google Scholar]
- Vinu A, Miyahara M, Hossain K Z, Takahashi M, Balasubramanian V V, Mori T. and Ariga K. J. Nanosci. Nanotechnol. 2007;7:828. doi: 10.1166/jnn.2007.511. [DOI] [PubMed] [Google Scholar]
- Miyahara M, Vinu A and Ariga K. 2007. Mater. Sci. Eng. C 27 232 10.1016/j.msec.2006.05.012 [DOI] [Google Scholar]
- Park S, Lim J-H, Chung S-W. and Mirkin C A. Science. 2004;303:348. doi: 10.1126/science.1093276. [DOI] [PubMed] [Google Scholar]
- Yan D, Zhou Y. and Hou J. Science. 2004;303:65. doi: 10.1126/science.1090763. [DOI] [PubMed] [Google Scholar]
- Bowden N, Terfort A, Carbeck J. and Whitesides G M. Science. 1997;276:233. doi: 10.1126/science.276.5310.233. [DOI] [PubMed] [Google Scholar]
- Bowden N B, Weck M, Choi I S. and Whitesides G M. Acc. Chem. Res. 2001;34:231. doi: 10.1021/ar0000760. [DOI] [PubMed] [Google Scholar]
- Whitesides M G. and Boncheva M. Proc. Nat. Acad. Sci. USA. 2002;99:4769. doi: 10.1073/pnas.082065899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden N, Choi I S, Grzybowski B A. and Whitesides G M. J. Am. Chem. Soc. 1999;121:5373. doi: 10.1021/ja983882z. [DOI] [Google Scholar]
- Weck M, Choi I S, Jeon N L. and Whitesides G M. J. Am. Chem. Soc. 2000;122:3546. doi: 10.1021/ja994099p. [DOI] [Google Scholar]
- Breen T L, Tien J, Oliver S R J, Hadzic T. and Whitesides G M. Science. 1999;284:948. doi: 10.1126/science.284.5416.948. [DOI] [PubMed] [Google Scholar]
- Gracias D H, Tien J, Breen T L, Hsu C. and Whitesides G M. Science. 2000;289:1170. doi: 10.1126/science.289.5482.1170. [DOI] [PubMed] [Google Scholar]
- Clark T D, Tien J, Duffy D C, Paul K E. and Whitesides G M. J. Am. Chem. Soc. 2001;123:7677. doi: 10.1021/ja010634l. [DOI] [PubMed] [Google Scholar]
- Boncheva M. and Whitesides G M. Adv. Mater. 2005;17:553. doi: 10.1002/adma.200400940. [DOI] [Google Scholar]
- Bruzewicz D A, Boncheva M, Winkleman A, St Clair J M, Engel G S. and Whitesides G M. J. Am. Chem. Soc. 2006;128:9314. doi: 10.1021/ja062973q. [DOI] [PubMed] [Google Scholar]
- Burrell A K, Officer D L, Plieger P G. and Reid D C W. Chem. Rev. 2001;101:2751. doi: 10.1021/cr0000426. [DOI] [PubMed] [Google Scholar]
- Hill J P, Schmitt W, McCarty A L, Ariga K. and D'Souza F. Eur. J. Org. Chem. 2005:2893. doi: 10.1002/ejoc.200500257. [DOI] [Google Scholar]
- Balaban T S. Acc. Chem. Res. 2005;38:612. doi: 10.1021/ar040211z. [DOI] [PubMed] [Google Scholar]
- Hill J P, Schumacher A L, D'Souza F, Labuta J, Redshaw C, Elsegood M R, Aoyagi M, Nakanishi T. and Ariga K. Inorg. Chem. 2006;45:8288. doi: 10.1021/ic0611591. [DOI] [PubMed] [Google Scholar]
- Iengo E, Zangrando E. and Alessio E. Acc. Chem. Res. 2006;39:841. doi: 10.1021/ar040240+. [DOI] [PubMed] [Google Scholar]
- Hill J P, Sandanayaka A S D, McCarty A L, Karr P A, Zandler M E, Charvet R, Ariga K, Araki Y, Ito O. and D'Souza F. Eur. J. Org. Chem. 2006:595. doi: 10.1002/ejoc.200500626. [DOI] [Google Scholar]
- Elemans J A A W, van Hameren R, Nolte R J M. and Rowan A E. Adv. Mater. 2006;18:1251. doi: 10.1002/adma.200502498. [DOI] [Google Scholar]
- Schumacher A L, Sandanayaka A S D, Hill J P, Ariga K, Karr P A, Araki Y, Ito O. and D'Souza F. Chem. Eur. J. 2007;13:4628. doi: 10.1002/chem.200601854. [DOI] [PubMed] [Google Scholar]
- Scandola F, Chiorboli C, Prodi A, Iengo E. and Alessio E. Coord. Chem. Rev. 2006;250:1471. doi: 10.1016/j.ccr.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Hill J P, Ariga K, Schumacher A L, Karr P A, D'Souza F. and Porphyr J. Phthalocyanines. 2007;11:390. [Google Scholar]
- Xie Y, Hill J P, Charvet R. and Ariga K. J. Nanosci. Nanotechnol. 2007;7:2969. doi: 10.1166/jnn.2007.910. [DOI] [PubMed] [Google Scholar]
- Schumacher A L, Hill J P, Ariga K. and D'Souza F. Electro. Chem. Commun. 2007;9:2751. [Google Scholar]
- Xie Y, Hill J P, Schumacher A L, Karr P A, D'Souza F, Anson C E, Powell A K. and Ariga K. Chem. Eur. J. 2007;13:9824. doi: 10.1002/chem.200701428. [DOI] [PubMed] [Google Scholar]
- Kobuke Y. Eur. J. Inorg. Chem. 2006:2333. doi: 10.1002/ejic.200600161. [DOI] [Google Scholar]
- Ogawa K, Zhang T, Yoshihara K. and Kobuke Y. J. Am. Chem. Soc. 2002;124:22. doi: 10.1021/ja0169015. [DOI] [PubMed] [Google Scholar]
- Ogawa K, Ohashi A, Kobuke Y, Kamada K. and Ohta K. J. Am. Chem. Soc. 2003;125:13356. doi: 10.1021/ja035056i. [DOI] [PubMed] [Google Scholar]
- Takahashi R. and Kobuke Y. J. Am. Chem. Soc. 2003;125:2372. doi: 10.1021/ja028325y. [DOI] [PubMed] [Google Scholar]
- Ikeda C, Satake A. and Kobuke Y. Org. Lett. 2003;26:4935. doi: 10.1021/ol0357191. [DOI] [PubMed] [Google Scholar]
- Taylor P N. and Anderson H L. J. Am. Chem. Soc. 1999;121:11538. doi: 10.1021/ja992821d. [DOI] [Google Scholar]
- Anderson H L. Chem. Commun. 1999:2323. doi: 10.1039/a904209a. [DOI] [Google Scholar]
- Screen T E O, Thorne J R G, Denning R G, Bucknall D G. and Anderson H L. J. Mater. Chem. 2003;13:2796. doi: 10.1039/b305053j. [DOI] [Google Scholar]
- Ballester P, Oliva A I, Costa A, Deyà P M, Frontera A, Gomila R M. and Hunter C A. J. Am. Chem. Soc. 2006;128:5560. doi: 10.1021/ja060608t. [DOI] [PubMed] [Google Scholar]
- Sugou K, Sasaki K, Kitajima K, Iwaki T. and Kuroda Y. J. Am. Chem. Soc. 2002;124:1182. doi: 10.1021/ja017470t. [DOI] [PubMed] [Google Scholar]
- Prodi A, Chiorboli C, Scandola F, Iengo E, Alessio E, Dobrawa R. and Würthner F. J. Am. Chem. Soc. 2005;127:1454. doi: 10.1021/ja045379u. [DOI] [PubMed] [Google Scholar]
- Okada S. and Segawa H. J. Am. Chem. Soc. 2003;125:2792. doi: 10.1021/ja017768j. [DOI] [PubMed] [Google Scholar]
- Mammana A, D'Urso A, Lauceri R. and Purrello R. J. Am. Chem. Soc. 2007;129:8062. doi: 10.1021/ja071447b. [DOI] [PubMed] [Google Scholar]
- Spadavecchia J, Ciccarella G, Stomeo T, Rella R, Capone S. and Siciliano P. Chem. Mater. 2004;16:2083. doi: 10.1021/cm034871u. [DOI] [Google Scholar]
- Li Y, Wang T. and Liu M. Soft Matter. 2007;3:1312. doi: 10.1039/b710165a. [DOI] [PubMed] [Google Scholar]
- Ishi-i T, Jung J H. and Shinkai S. J. Mater. Chem. 2000;10:2238. doi: 10.1039/b004539j. [DOI] [Google Scholar]
- Tamaru S, Uchino S, Takeuchi M, Ikeda M, Hatano T. and Shinkai S. Tetrahedron Lett. 2002;43:3751. doi: 10.1016/S0040-4039(02)00541-5. [DOI] [Google Scholar]
- Kishida T, Fujita N, Sada K. and Shinkai S. Chem. Lett. 2004;33:1002. doi: 10.1246/cl.2004.1002. [DOI] [Google Scholar]
- Tanaka S, Shirakawa M, Kaneko K, Takeuchi M. and Shinkai S. Langmuir. 2005;21:2163. doi: 10.1021/la047070u. [DOI] [PubMed] [Google Scholar]
- Harada R. and Kojima T. Chem. Commun. 2005:716. doi: 10.1039/b414483j. [DOI] [PubMed] [Google Scholar]
- Kojima T, Harada R, Nakanishi T, Kaneko K. and Fukuzumi S. Chem. Mater. 2007;19:51. doi: 10.1021/cm062031k. [DOI] [Google Scholar]
- Wang Z, Medforth C J. and Shelnutt J A. J. Am. Chem. Soc. 2004;126:15954. doi: 10.1021/ja045068j. [DOI] [PubMed] [Google Scholar]
- Wang Z, Medforth C J. and Shelnutt J A. J. Am. Chem. Soc. 2004;126:16720. doi: 10.1021/ja044148k. [DOI] [PubMed] [Google Scholar]
- Wang Z, Ho K J, Medforth C J. and Shelnutt J A. Adv. Mater. 2006;18:2557. doi: 10.1002/adma.200600539. [DOI] [Google Scholar]
- Wang Z, Li Z, Medforth C J. and Shelnutt J A. J. Am. Chem. Soc. 2007;129:2440. doi: 10.1021/ja068250o. [DOI] [PubMed] [Google Scholar]
- Schenning A P H J, Benneker F B G, Gearts H P M, Liu X Y. and Nolte R J M. J. Am. Chem. Soc. 1996;118:8549. doi: 10.1021/ja961234e. [DOI] [Google Scholar]
- Engelkamp H, Middelbeek S. and Nolte R J M. Science. 1999;284:785. doi: 10.1126/science.284.5415.785. [DOI] [PubMed] [Google Scholar]
- van Hameren R. et al Science. 2006;314:1433. doi: 10.1126/science.1133004. [DOI] [PubMed] [Google Scholar]
- Hu J-S, Guo Y-G, Liang H-P, Wan L-J. and Jiang L. J. Am. Chem. Soc. 2005;127:17090. doi: 10.1021/ja0553912. [DOI] [PubMed] [Google Scholar]
- Kovaric B C, Kokona B, Schwab A D, Twomey M A, de Paula J C. and Fairman R. J. Am. Chem. Soc. 2006;128:4166. doi: 10.1021/ja056357q. [DOI] [PubMed] [Google Scholar]
- D'Souza F, Chitta R, Sandanayaka A S D, Subbaiyan N K, D'Souza L, Araki Y. and Ito O. Chem. Eur. J. 2007;13:8277. doi: 10.1002/chem.200700583. [DOI] [PubMed] [Google Scholar]
- Osawa E. Kagaku. 1970;25:854. [Google Scholar]
- Kroto H W, Heath J R, O'Brien S C, Curl R F. and Smalley R E. Nature. 1985;318:162. doi: 10.1038/318162a0. [DOI] [Google Scholar]
- Diederich F. and Gómez-Lopéz M. Chem. Soc. Rev. 1999;28:263. doi: 10.1039/a804248i. [DOI] [Google Scholar]
- Guldi D M. and Martín N. J. Mater. Chem. 2002;12:1978. doi: 10.1039/b203007a. [DOI] [Google Scholar]
- Armaroli N. Phoitochem. Photobiol. Sci. 2003;2:73. doi: 10.1039/b210569a. [DOI] [PubMed] [Google Scholar]
- Nakamura E. and Isobe H. Acc. Chem. Res. 2003;36:807. doi: 10.1021/ar030027y. [DOI] [PubMed] [Google Scholar]
- Guldi D M, Zerbetto F, Georgakilas V. and Prato M. Acc. Chem. Res. 2005;38:38. doi: 10.1021/ar040222s. [DOI] [PubMed] [Google Scholar]
- Boyd P D W. and Reed C A. Acc. Chem. Res. 2005;38:235. doi: 10.1021/ar040168f. [DOI] [PubMed] [Google Scholar]
- Segura J L, Martín N. and Guldi D M. Chem. Soc. Rev. 2005;34:31. doi: 10.1039/b402417f. [DOI] [PubMed] [Google Scholar]
- Martín N. Chem. Commun. 2006:2093. doi: 10.1039/b601582b. [DOI] [PubMed] [Google Scholar]
- Thilgen C. and Diederich F. Chem. Rev. 2006;106:5049. doi: 10.1021/cr0505371. [DOI] [PubMed] [Google Scholar]
- Patnaik A. J. Nanosci. Nanotechnol. 2007;7:1111. doi: 10.1166/jnn.2007.303. [DOI] [PubMed] [Google Scholar]
- Ravi P, Dai S, Wang C. and Tam K C. J. Nanosci. Nanotechnol. 2007;7:1176. doi: 10.1166/jnn.2007.456. [DOI] [PubMed] [Google Scholar]
- Zhou S, Burger C, Chu B, Sawamura M, Nagahama N, Toganoh M, Hackler U E, Isobe H. and Nakamura E. Science. 2001;291:1944. doi: 10.1126/science.291.5510.1944. [DOI] [PubMed] [Google Scholar]
- Sawamura M, Kawai K, Matsuo Y, Kanie K, Kato T. and Nakamura E. Nature. 2002;419:702. doi: 10.1038/nature01110. [DOI] [PubMed] [Google Scholar]
- Matsuo Y, Muramatsu A, Hamasaki R, Mizoshita N, Kato T. and Nakamura E. J. Am. Chem. Soc. 2004;126:432. doi: 10.1021/ja038816y. [DOI] [PubMed] [Google Scholar]
- Zhong Y-W, Matsuo Y. and Nakamura E. J. Am. Chem. Soc. 2007;129:3052. doi: 10.1021/ja068780k. [DOI] [PubMed] [Google Scholar]
- Sano M, Oishi K, Ishi-i T. and Shinkai S. Langmuir. 2000;16:3773. doi: 10.1021/la991550h. [DOI] [Google Scholar]
- Shirakawa M, Fujita N, Shimakoshi H, Hisaeda Y. and Shinkai S. Tetrahedron. 2006;62:2016. doi: 10.1016/j.tet.2005.07.125. [DOI] [Google Scholar]
- Verma S, Hauck T, El-Khouly M E, Padmawar P A, Canteenwala T, Pritzker K, Ito O. and Chiang L Y. Langmuir. 2005;21:3267. doi: 10.1021/la047082f. [DOI] [PubMed] [Google Scholar]
- Li J, Sun N, Zhang P, Guo Z-X. and Zhu D. Chem. Phys. Lett. 2005;406:425. doi: 10.1016/j.cplett.2005.03.021. [DOI] [Google Scholar]
- Braun M. and Hirsch A. Carbon. 2000;38:1565. doi: 10.1016/S0008-6223(99)00288-2. [DOI] [Google Scholar]
- Hao J, Li H, Liu W. and Hirsch A. Chem. Commun. 2004:602. doi: 10.1039/b312549a. [DOI] [PubMed] [Google Scholar]
- Burghardt S, Hirsch A, Schade B, Ludwig K. and Böttcher C. Angew. Chem. Int. Ed. 2005;44:2976. doi: 10.1002/anie.200462465. [DOI] [PubMed] [Google Scholar]
- Schade B, Ludwig K, Böttcher C, Hartnagel U. and Hirsch A. J. Am. Chem. Soc. 2007;46:4393. doi: 10.1002/anie.200604784. [DOI] [PubMed] [Google Scholar]
- Nakanishi T, Murakami H. and Nakashima N. Chem. Lett. 1988:1219. [Google Scholar]
- Mouri E, Nakanishi T, Nakashima N. and Matsuoka H. Langmuir. 2002;18:10042. doi: 10.1021/la0204895. [DOI] [Google Scholar]
- Murakami H, Watanabe Y. and Nakashima N. J. Am. Chem. Soc. 1996;118:4484. doi: 10.1021/ja960037c. [DOI] [Google Scholar]
- Nakanishi T, Murakami H, Sagara T and Nakashima N. 1999. J. Phys. Chem. B 103 304 10.1021/jp9833101 [DOI] [Google Scholar]
- Nakanishi T, Morita M, Murakami H, Sagara T. and Nakashima N. Chem. Eur. J. 2002;8:1641. doi: 10.1002/1521-3765(20020402)8:7<1641::AID-CHEM1641>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Nakanishi T, Kouzai H, Morita M, Murakami H, Sagara T, Ariga K. and Nakashima T. J. Nanosci. Nanotechnol. 2006;6:1779. doi: 10.1166/jnn.2006.214. [DOI] [PubMed] [Google Scholar]
- Nakanishi T, Ariga K, Morita M, Kozai H, Taniguchi N, Murakami H, Sagara T. and Nakashima N. Colloid Surf. A-Physicochem. Eng. Asp. 2006;284/285:607. doi: 10.1016/j.colsurfa.2005.10.063. [DOI] [Google Scholar]
- Haino T, Matsumoto Y. and Fukazawa Y. J. Am. Chem. Soc. 2005;127:8936. doi: 10.1021/ja0524088. [DOI] [PubMed] [Google Scholar]
- Georgakilas V, Pellarini F, Prato M, Guldi D M, Melle-Franco M. and Zerbetto F. Proc. Nat. Acad. Sci. USA. 2002;99:5075. doi: 10.1073/pnas.072006599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi T, Schmitt W, Michinobu T, Kurth D G. and Ariga K. Chem. Commun. 2005:5982. doi: 10.1039/b512320h. [DOI] [PubMed] [Google Scholar]
- Nakanishi T, Takahashi H, Michinobu T, Hill J, Jonathan Teranishi T and Ariga K. Thin Solid Films, in press [Google Scholar]
- Nakanishi T, Wang J, Möhwald H, Kurth D G, Michinobu T, Takeuchi M and Ariga M K. J. Nanosci. Nanotechnol. in press [DOI] [PubMed] [Google Scholar]
- Nakanishi T, Ariga K, Michinobu T, Yoshida K, Takahashi H, Teranishi T, Möhwald H. and Kurth D G. Small. 2007;3:2019. doi: 10.1002/smll.200700647. [DOI] [PubMed] [Google Scholar]
- Nakanishi T, Michinobu T, Yoshida K, Shirahata N, Ariga K, Möhwald H and Kurth D G. Adv. Mater. in press. DOI: 10.1002/adma.200701537 [DOI] [Google Scholar]
- Michinobu T, Nakanishi T, Hill J P, Funahashi M. and Ariga K. J. Am. Chem. Soc. 2006;128:10384. doi: 10.1021/ja063866z. [DOI] [PubMed] [Google Scholar]
- Miyazawa K, Obayashi A. and Kuwabara M. J. Am. Ceram. Soc. 2001;84:3037. [Google Scholar]
- Miyazawa K, Kuwasaki Y, Obayashi A. and Kuwabara M. J. Mater. Res. 2002;17:83. doi: 10.1557/JMR.2002.0014. [DOI] [Google Scholar]
- Miyazawa K. and Hamamoto K. J. Mater. Res. 2002;17:2205. doi: 10.1557/JMR.2002.0325. [DOI] [Google Scholar]
- Miyazawa K. J. Am. Ceram. Soc. 2002;85:1297. [Google Scholar]
- Miyazawa K, Akaishi M, Kuwasaki Y. and Suga T. J. Mater. Res. 2003;18:166. doi: 10.1557/JMR.2003.0023. [DOI] [Google Scholar]
- Miyazawa K, Hamamoto K, Nagata S. and Suga T. J. Mater. Res. 2003;18:1096. doi: 10.1557/JMR.2003.0151. [DOI] [Google Scholar]
- Miyazawa K, Mashino T. and Suga T. J. Mater. Res. 2003;18:2730. doi: 10.1557/JMR.2003.0380. [DOI] [Google Scholar]
- Miyazawa K, Kuwasaki Y, Hamamoto K, Nagata S, Obayashi A. and Kuwabara M. Surf. Interface Anal. 2003;35:117. doi: 10.1002/sia.1506. [DOI] [Google Scholar]
- Tachibana M, Kobayadhi K, Uchida T, Kojima K, Tanimura M. and Miyazaea K. Chem. Phys. Lett. 2003;374:279. doi: 10.1016/S0009-2614(03)00723-1. [DOI] [Google Scholar]
- Miyazawa K. and Suga T. J. Mater. Res. 2004;19:2410. doi: 10.1557/JMR.2004.0304. [DOI] [Google Scholar]
- Miyazawa K. and Suga T. J. Mater. Res. 2004;19:3145. doi: 10.1557/JMR.2004.0409. [DOI] [Google Scholar]
- Miyazawa K, Minato J, Fujino M. and Suga T. Diam. Relat. Mater. 2006;15:1143. doi: 10.1016/j.diamond.2005.10.027. [DOI] [Google Scholar]
- Miyazawa K, Minato J, Yohii T, Fujino M. and Suga T. J. Mater. Res. 2005;20:688. doi: 10.1557/JMR.2005.0091. [DOI] [Google Scholar]
- Minato J, Miyazawa K, Suga T, Kanda H, Akashi M, Yamamura K, Muromachi E. and Kakisawa H. J. Mater. Res. 2005;20:742. doi: 10.1557/JMR.2005.0095. [DOI] [Google Scholar]
- Lee S-H, Miyazawa K. and Maeda R. Carbon. 2005;43:887. doi: 10.1016/j.carbon.2004.11.020. [DOI] [Google Scholar]
- Minato J. and Miyazawa K. Carbon. 2005;43:2837. doi: 10.1016/j.carbon.2005.06.013. [DOI] [Google Scholar]
- Minato J, Miyazawa K. and Suga T. Sci. Technol. Adv. Mater. 2005;6:272. [Google Scholar]
- Miyazawa K, Minato J, Yoshii T. and Suga T. Sci. Technol. Adv. Mater. 2005;6:388. doi: 10.1016/j.stam.2005.05.019. [DOI] [Google Scholar]
- Miyazawa K, Minato J, Zhou H, Taniguchi T, Honma I. and Suga T. J. Eur. Ceram. Soc. 2006;26:429. doi: 10.1016/j.jeurceramsoc.2005.07.010. [DOI] [Google Scholar]
- Asaka K, Kato R, Yoshizaki R, Miyazawa K. and Kizuka T. Diam. Relat. Mater. 2007;16:1936. doi: 10.1016/j.diamond.2007.08.005. [DOI] [Google Scholar]
- Sathish M, Miyazawa K. and Sasaki T. Chem. Mater. 2007;19:2398. doi: 10.1021/cm070114a. [DOI] [Google Scholar]
- Sathish M. and Miyazawa K. J. Am. Chem. Soc. 2007;129:13816. doi: 10.1021/ja076251q. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Ishii N, Tashiro K. and Aida T. J. Am. Chem. Soc. 2003;125:13934. doi: 10.1021/ja038178j. [DOI] [PubMed] [Google Scholar]
- Charvet R, Jiang D-L. and Aida T. Chem. Commun. 2004;26:2664. doi: 10.1039/b409228g. [DOI] [PubMed] [Google Scholar]
- Angelini G, Cusan C, De Maria P, Fontana A, Maggini M, Pierini M, Prato M, Schergna S. and Villani C. Eur. J. Org. Chem. 2005:1884. doi: 10.1002/ejoc.200400699. [DOI] [Google Scholar]
- Brough P, Bonifazi D. and Prato M. Tetrahedron. 2006;62:2110. doi: 10.1016/j.tet.2005.08.125. [DOI] [Google Scholar]
- Liu Y, Chen G-S, Chen Y, Zhang N, Chen J. and Zhao Y-L. Nano Lett. 2006;6:2196. doi: 10.1021/nl061393o. [DOI] [PubMed] [Google Scholar]
- Hubble L J. and Raston C L. Chem. Eur. J. 2007;13:6755. doi: 10.1002/chem.200700339. [DOI] [PubMed] [Google Scholar]
- Hahn U, González J J, Huerta E, Segura M, Eckert J-F, Cardinali F, de Mendoza J. and Nierengarten J-F. Chem. Eur. J. 2005;11:6666. doi: 10.1002/chem.200500565. [DOI] [PubMed] [Google Scholar]
- Sygula A, Fronczek F R, Sygula R, Rabideau P W. and Olmstead M M. J. Am. Chem. Soc. 2007;129:3842. doi: 10.1021/ja070616p. [DOI] [PubMed] [Google Scholar]
- Ramos A M, Rispens M T, van Duren J K J, Hummelen J C. and Janssen R A J. J. Am. Chem. Soc. 2001;123:6714. doi: 10.1021/ja015614y. [DOI] [PubMed] [Google Scholar]
- Wang C, Ravi P. and Tam K C. Langmuir. 2007;23:8798. doi: 10.1021/la700600r. [DOI] [PubMed] [Google Scholar]
- Kawauchi T, Kumaki J. and Yashima E. J. Am. Chem. Soc. 2006;128:10560. doi: 10.1021/ja063252u. [DOI] [PubMed] [Google Scholar]
- Laiho A, Ras R H A, Valkama S, Ruokolainen J, Österbacka R. and Ikkala O. Macromolecules. 2006;39:7648. doi: 10.1021/ma061165g. [DOI] [Google Scholar]
- Kim Y. et al Nat. Mater. 2006;5:197. doi: 10.1038/nmat1574. [DOI] [Google Scholar]
- Campidelli S, Pérez L, Rodríguez-López J, Barberá J, Langa F. and Deschenaux R. Tetrahedron. 2006;62:2115. doi: 10.1016/j.tet.2005.10.065. [DOI] [Google Scholar]
- Bushby R J, Hamley I W, Liu Q, Lozman O R. and Lydon J E. J. Mater. Chem. 2005;15:4429. doi: 10.1039/b509492e. [DOI] [Google Scholar]
- Wang Y, Nepal D. and Geckeler K E. J. Mater. Chem. 2005;15:1049. doi: 10.1039/b413988g. [DOI] [Google Scholar]
- Deguchi S, Mukai S, Tsudome M. and Horikoshi K. Adv. Mater. 2006;18:729. doi: 10.1002/adma.200502487. [DOI] [Google Scholar]
- van de Craats A M, Warman J M, Müllen K, Geerts Y. and Brand J D. Adv. Mater. 1998;10:36. doi: 10.1002/(SICI)1521-4095(199801)10:1<36::AID-ADMA36>3.0.CO;2-A. [DOI] [Google Scholar]
- Thünemann A F, Kubowicz S, Burger C, Watson M D, Tchebotareva N. and Müllen K. J. Am. Chem. Soc. 2003;125:352. doi: 10.1021/ja0277632. [DOI] [PubMed] [Google Scholar]
- Pisula W, Kastler M, Wasserfallen D, Davies R J, García-Gutiérrez M-C. and Müllen K. J. Am. Chem. Soc. 2006;128:14424. doi: 10.1021/ja0641960. [DOI] [PubMed] [Google Scholar]
- Simpson C D, Wu J, Watson M D. and Müllen K. J. Mater. Chem. 2004;14:494. doi: 10.1039/b312789c. [DOI] [Google Scholar]
- Hill J P, Jin W, Kosaka A, Fukushima T, Ichihara H, Shimomura T, Ito K, Hashizume T, Ishii N. and Aida T. Science. 304:1481. doi: 10.1126/science.1097789. [DOI] [PubMed] [Google Scholar]
- Jin W, Fukushima T, Niki T, Kosaka A, Ishii N. and Aida T. Proc. Nat. Acad. Sci. USA. 2005;102:10801. doi: 10.1073/pnas.0500852102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Fukushima T, Suna Y, Ishii N, Saeki A, Seki S, Tagawa S, Taniguchi M, Kawai T. and Aida T. Science. 2006;314:1761. doi: 10.1126/science.1134441. [DOI] [PubMed] [Google Scholar]
- Yagai S, Nakajima T, Karatsu T, Saitow K. and Kitamura A. J. Am. Chem. Soc. 2004;126:11500. doi: 10.1021/ja047783z. [DOI] [PubMed] [Google Scholar]
- Yagai S, Iwashima T, Kishikawa K, Nakahara S, Karatsu T. and Kitamura A. Chem. Eur. J. 2006;12:3984. doi: 10.1002/chem.200501468. [DOI] [PubMed] [Google Scholar]
- Roques N, Maspoch D, Domingo N, Ruiz-Molina D, Wurst K, Tejada J, Rovira C. and Veciana J. Chem. Commun. 2005:4801. doi: 10.1039/b508952b. [DOI] [PubMed] [Google Scholar]
- Dewal M B, Lufaso M W, Hughes A D, Samuel S A, Pellechia P. and Shimizu L S. Chem. Mater. 2006;18:4855. doi: 10.1021/cm0614057. [DOI] [Google Scholar]
- Pintér G. et al Langmuir. 2007;23:5283. doi: 10.1021/la070019g. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhuang J, Liu H, Li Y, Lu F, Gan H, Jiu T, Wang N, He X. and Zhu D. Chem. Phys. Chem. 2004;5:1210. doi: 10.1002/cphc.200400165. [DOI] [PubMed] [Google Scholar]
- Bose P P, Drew M G B, Das A K. and Banerjee A. Chem. Commun. 2006:3196. doi: 10.1039/b606371c. [DOI] [PubMed] [Google Scholar]
- Webb J E A, Crossley M J, Turner P. and Thordarson P. J. Am. Chem. Soc. 2007;129:7155. doi: 10.1021/ja0713781. [DOI] [PubMed] [Google Scholar]
- Kishimura A, Yamashita T, Yamaguchi K. and Aida T. Nat. Mater. 2005;4:546. doi: 10.1038/nmat1401. [DOI] [PubMed] [Google Scholar]
- Kishimura A, Yamashita T. and Aida T. J. Am. Chem. Soc. 2005;127:179. doi: 10.1021/ja0441007. [DOI] [PubMed] [Google Scholar]
- Fujigaya T, Jiang D-L. and Aida T. J. Am. Chem. Soc. 2003;125:14690. doi: 10.1021/ja038088e. [DOI] [PubMed] [Google Scholar]
- Maeda H, Hasegawa M, Hashimoto T, Kakimoto T, Nishio S. and Nakanishi T. J. Am. Chem. Soc. 2006;128:10024. doi: 10.1021/ja0637301. [DOI] [PubMed] [Google Scholar]
- García-Zarracino R. and Höpfl H. J. Am. Chem. Soc. 2005;127:3120. doi: 10.1021/ja0437095. [DOI] [PubMed] [Google Scholar]
- Ruben M, Ziener U, Lehn J-M, Ksenofontov V, Gütlich P. and Vaughan G B M. Chem. Eur. J. 2005;11:94. doi: 10.1002/chem.200400584. [DOI] [PubMed] [Google Scholar]
- Oh M. and Mirkin C A. Nature. 2005;438:651. doi: 10.1038/nature04191. [DOI] [PubMed] [Google Scholar]
- Jeon Y-M, Heo J. and Mirkin C A. J. Am. Chem. Soc. 2007;129:7480. doi: 10.1021/ja071046w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-K, Park K M, Jeon Y J, Kim D, Oh D H, Kim H S, Park C K. and Kim K. J. Am. Chem. Soc. 2005;127:5006. doi: 10.1021/ja042172s. [DOI] [PubMed] [Google Scholar]
- Martin O M, Yu L. and Mecozzi S. Chem. Commun. 2005:4964. doi: 10.1039/b506781b. [DOI] [PubMed] [Google Scholar]
- Pappalardo S, Villari V, Slovak S, Cohen Y, Gattuso G, Notti A, Pappalardo A, Pisagatti I. and Parisi M F. Chem. Eur. J. 2007;13:8164. doi: 10.1002/chem.200601785. [DOI] [PubMed] [Google Scholar]
- Houmadi S, Coquière D, Legrand L, Fauré M C, Goldmann M, Reinaud O. and Rémita S. Langmuir. 2007;23:4849. doi: 10.1021/la700271a. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Takashima Y, Yamaguchi H. and Harada A. Chem. Lett. 2007;36:828. doi: 10.1246/cl.2007.828. [DOI] [Google Scholar]
- Balakrishnan K, Datar A, Zhang W, Yang X, Naddo T, Huang J, Zuo J, Yen M, Moore J S. and Zang L. J. Am. Chem. Soc. 2006;128:6576. doi: 10.1021/ja0618550. [DOI] [PubMed] [Google Scholar]
- Hirschberg J H K K, Brunsveld L, Ramzi A, Vekemans J A J M, Sijbesma R P. and Meijer E W. Nature. 2000;407:167. doi: 10.1038/35025027. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Nobori T, Schmutz M. and Lehn J-M. Chem. Eur. J. 2005;11:662. doi: 10.1002/chem.200400826. [DOI] [PubMed] [Google Scholar]
- Kim J-K, Lee E, Jeong Y-H, Lee J-K, Zin W-C. and Lee M. J. Am. Chem. Soc. 2007;129:6082. doi: 10.1021/ja071394y. [DOI] [PubMed] [Google Scholar]
- Sagara Y, Mutai T, Yoshikawa I. and Araki K. J. Am. Chem. Soc. 2007;129:1520. doi: 10.1021/ja0677362. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chen Z. and Würthner F. J. Am. Chem. Soc. 2007;129:4886. doi: 10.1021/ja070994u. [DOI] [PubMed] [Google Scholar]
- Jyothish K, Hariharan M. and Ramaiah D. Chem. Eur. J. 2007;13:5944. doi: 10.1002/chem.200700130. [DOI] [PubMed] [Google Scholar]
- Valentini L, Mengoni F, Kenny J M, Marrocchi A. and Taticchi A. Small. 2007;3:1200. doi: 10.1002/smll.200600623. [DOI] [PubMed] [Google Scholar]
- Metrangolo P, Meyer F, Pilati T, Proserpio D M. and Resnati G. Chem. Eur. J. 2007;13:5765. doi: 10.1002/chem.200601653. [DOI] [PubMed] [Google Scholar]
- Miyata M, Tohnai N. and Hisaki I. Acc. Chem. Res. 2007;40:694. doi: 10.1021/ar700017a. [DOI] [PubMed] [Google Scholar]
- Hu Z-Q. and Chen C-F. Chem. Commun. 2005:2445. doi: 10.1039/b501941a. [DOI] [PubMed] [Google Scholar]
- Kim J, Levitsky I A, McQuade D T. and Swager T M. J. Am. Chem. Soc. 2002;124:7710. doi: 10.1021/ja0200600. [DOI] [PubMed] [Google Scholar]
- Wakabayashi R, Kubo Y, Kaneko K, Takeuchi M. and Shinkai S. J. Am. Chem. Soc. 2006;128:8744. doi: 10.1021/ja063040x. [DOI] [PubMed] [Google Scholar]
- Akagi K. Bull. Chem. Soc. Jpn. 2007;80:649. doi: 10.1246/bcsj.80.649. [DOI] [Google Scholar]
- Akagi K, Piao G, Kaneko S, Sakamaki K, Shirakawa H. and Kyotani M. Science. 1998;282:1683. doi: 10.1126/science.282.5394.1683. [DOI] [PubMed] [Google Scholar]
- Hulvat J F, Sofos M, Tajima K. and Stupp S I. J. Am. Chem. Soc. 2005;127:366. doi: 10.1021/ja047210m. [DOI] [PubMed] [Google Scholar]
- Li C, Numata M, Bae A-H, Sakurai K. and Shinkai S. J. Am. Chem. Soc. 2005;127:4548. doi: 10.1021/ja050168q. [DOI] [PubMed] [Google Scholar]
- Gan H. et al J. Am. Chem. Soc. 2005;127:12452. doi: 10.1021/ja053352k. [DOI] [PubMed] [Google Scholar]
- Akagi K, Guo S, Mori T, Goh M, Piao G. and Kyotani M. J. Am. Chem. Soc. 2005;127:14647. doi: 10.1021/ja051548e. [DOI] [PubMed] [Google Scholar]
- McNeill C R, Watts B, Thomsen L, Belcher W J, Greenham N C. and Dastoor P C. Nano Lett. 2006;6:1202. doi: 10.1021/nl060583w. [DOI] [PubMed] [Google Scholar]
- Tajima K, Li L. and Stupp S I. J. Am. Chem. Soc. 2006;128:5488. doi: 10.1021/ja058610s. [DOI] [PubMed] [Google Scholar]
- Thomas S W III, Joly G D. and Swager T M. Chem. Rev. 2007;107:1339. doi: 10.1021/cr0501339. [DOI] [PubMed] [Google Scholar]
- Goh M, Kyotani M. and Akagi K. J. Am. Chem. Soc. 2007;129:8519. doi: 10.1021/ja070701x. [DOI] [PubMed] [Google Scholar]
- Klok H-A. and Lecommandoux S. Adv. Mater. 2001;13:1217. doi: 10.1002/1521-4095(200108)13:16<1217::AID-ADMA1217>3.0.CO;2-D. [DOI] [Google Scholar]
- Cornelissen J J L M, Rowan A E, Nolte R J M. and Sommerdijk N A J M. Chem. Rev. 2001;101:4039. doi: 10.1021/cr990126i. [DOI] [PubMed] [Google Scholar]
- Ikkala O. and Brinke G. Science. 2002;295:2407. doi: 10.1126/science.1067794. [DOI] [PubMed] [Google Scholar]
- He Y, Li Z, Simone P. and Lodge T P. J. Am. Chem. Soc. 2006;128:2745. doi: 10.1021/ja058091t. [DOI] [PubMed] [Google Scholar]
- Ishihara Y, Bazzi H S, Toader V, Godin F. and Sleiman H F. Chem. Eur. J. 2007;13:4560. doi: 10.1002/chem.200601423. [DOI] [PubMed] [Google Scholar]
- Hudson S D, Jung H-T, Percec V, Cho W-D, Johansson G, Ungar G. and Balagurusamy V S K. Science. 1997;278:449. doi: 10.1126/science.278.5337.449. [DOI] [Google Scholar]
- Percec V, Ahn C-H, Ungar G, Yeardley D J P, Müller M. and Sheiko S S. Nature. 1998;391:161. doi: 10.1038/34384. [DOI] [Google Scholar]
- Percec V. et al Chem. Eur. J. 2006;12:6298. doi: 10.1002/chem.200501195. [DOI] [PubMed] [Google Scholar]
- Cho B-K, Jain A, Gruner S M. and Wiesner U. Science. 2004;305:1598. doi: 10.1126/science.1100872. [DOI] [PubMed] [Google Scholar]
- Bauer R, Liu D, Heyen A V, De Schryver F, De Feyter S. and Müllen K. Macromolecules. 2007;40:4753. doi: 10.1021/ma0625511. [DOI] [Google Scholar]
- Zubarev E R, Sone E D. and Stupp S I. Chem. Eur. J. 2006;12:7313. doi: 10.1002/chem.200600619. [DOI] [PubMed] [Google Scholar]
- Leung K C-F, Mendes P M, Magonov S N, Northrop B H, Kim S, Patel K, Flood A H, Tseng H-R. and Stoddart J F. J. Am. Chem. Soc. 2006;128:10707. doi: 10.1021/ja058151v. [DOI] [PubMed] [Google Scholar]
- Bellomo E G, Wyrsta M D, Pakstis L, Pochan D J. and Deming T J. Nat. Mater. 2004;3:244. doi: 10.1038/nmat1093. [DOI] [PubMed] [Google Scholar]
- Napoli A, Valentini M, Tirelli N, Müller M. and Hubbell J A. Nat. Mater. 2004;3:183. doi: 10.1038/nmat1081. [DOI] [PubMed] [Google Scholar]
- Tian L, Nguyen P. and Hammond P T. Chem. Commun. 2006:3489. doi: 10.1039/b608363c. [DOI] [PubMed] [Google Scholar]
- Jones M-C, Tewari P, Blei C, Hales K, Pochan D J. and Leroux J-C. J. Am. Chem. Soc. 2006;128:14599. doi: 10.1021/ja065462c. [DOI] [PubMed] [Google Scholar]
- Shi L. and Berkland C. Macromolecules. 2007;40:4635. doi: 10.1021/ma070443o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanson K D, Huemmerich D, Scheibel T. and Bausch A R. Adv. Mater. 2007;19:1810. doi: 10.1002/adma.200602709. [DOI] [Google Scholar]
- Raez J, Manners I. and Winnik M A. J. Am. Chem. Soc. 2002;124:10381. doi: 10.1021/ja020349h. [DOI] [PubMed] [Google Scholar]
- Lee H J, Jin Z X, Aleshin A N, Lee J Y, Goh M J, Akagi K, Kim Y S, Kim D W. and Park Y W. J. Am. Chem. Soc. 2004;126:16722. doi: 10.1021/ja044615y. [DOI] [PubMed] [Google Scholar]
- Chiou N-R, Lee L J. and Epstein A J. Chem. Mater. 2007;19:3589. doi: 10.1021/cm070847v. [DOI] [Google Scholar]
- Germain J, Hradil J, Fréchet J M J. and Svec F. Chem. Mater. 2006;18:4430. doi: 10.1021/cm061186p. [DOI] [Google Scholar]
- Li X, Zhao S, Zhang S, Kim D H. and Knoll W. Langmuir. 2007;23:6883. doi: 10.1021/la7002589. [DOI] [PubMed] [Google Scholar]
- Zhang M, Yang L, Yurt S, Misner M J, Chen J-T, Coughlin E B, Venkataraman D. and Russell T P. Adv. Mater. 2007;19:1571. doi: 10.1002/adma.200602461. [DOI] [Google Scholar]
- Karthaus O, Gråsjö L, Maruyama N. and Shimomura M. Thin Solid Films. 1998;327–9:829. doi: 10.1016/S0040-6090(98)00771-8. [DOI] [Google Scholar]
- Maruyama N, Koito T, Nishida J, Sawadaishi T, Cieren X, Ijiro K, Karthaus O. and Shimomura M. Thin Solid Films. 1998;327–29:854. doi: 10.1016/S0040-6090(98)00777-9. [DOI] [Google Scholar]
- Yabu H, Takebayashi M, Tanaka M. and Shimomura M. Langmuir. 2005;21:3235. doi: 10.1021/la050013w. [DOI] [PubMed] [Google Scholar]
- Yabu H, Hirai Y. and Shimomura M. Langmuir. 2006;22:9760. doi: 10.1021/la062228r. [DOI] [PubMed] [Google Scholar]
- Stupp S I. and Braun P V. Science. 1997;277:1242. doi: 10.1126/science.277.5330.1242. [DOI] [PubMed] [Google Scholar]
- Zhang S. Nat. Biotechnol. 2003;21:1171. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- Baca H K, Carnes E, Singh S, Ashley C, Lopez D. and Brinker C J. Acc. Chem. Res. 2007;40:836. doi: 10.1021/ar600027u. [DOI] [PubMed] [Google Scholar]
- Guler M O. and Stupp S I. J. Am. Chem. Soc. 2007;129:12082. doi: 10.1021/ja075044n. [DOI] [PubMed] [Google Scholar]
- Hartgerink J D, Beniash E. and Stupp S I. Science. 2001;294:1684. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- Nowak A P, Breedveld V, Pakstis L, Ozbas B, Pine D J, Pochan D. and Deming T J. Nature. 2002;417:424. doi: 10.1038/417424a. [DOI] [PubMed] [Google Scholar]
- Holowka E P, Sun V Z, Kamei D T. and Deming T J. Nat. Mater. 2007;6:52. doi: 10.1038/nmat1794. [DOI] [PubMed] [Google Scholar]
- Morikawa M. and Kimizuka N. Chem. Commun. 2005:4866. doi: 10.1039/b507818k. [DOI] [PubMed] [Google Scholar]
- Lamm M S, Rajagopal K, Schneider J P. and Pochan D J. J. Am. Chem. Soc. 2005;127:16692. doi: 10.1021/ja054721f. [DOI] [PubMed] [Google Scholar]
- Hentschel J. and Börner H G. J. Am. Chem. Soc. 2006;128:14142. doi: 10.1021/ja0649872. [DOI] [PubMed] [Google Scholar]
- Koga T, Higuchi M, Kinoshita T. and Higashi N. Chem. Eur. J. 2006;12:1360. doi: 10.1002/chem.200500611. [DOI] [PubMed] [Google Scholar]
- Brizard A, Ahmad R K. and Oda R. Chem. Commun. 2007:2275. doi: 10.1039/b700959c. [DOI] [PubMed] [Google Scholar]
- Nilsson K P R, Rydberg J, Baltze L. and Inganäs O. Proc. Nat. Acad. Sci. USA. 2003;100:10170. doi: 10.1073/pnas.1834422100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson K P R, Rydberg J, Baltzer L. and Inganäs O. Proc. Nat. Acad. Sci. USA. 2004;101:11197. doi: 10.1073/pnas.0401853101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines L A, Rajagopal K, Ozbas B, Salick D A, Pochan D J. and Schneider J P. J. Am. Chem. Soc. 2005;127:17025. doi: 10.1021/ja054719o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa Y, Ueki K, Yoshida M, Tamaoki N, Nakamura T, Sakai H. and Abe M. Adv. Funct. Mater. 2007;17:1507. doi: 10.1002/adfm.200700052. [DOI] [Google Scholar]
- Lu K, Jacob J, Thiyagarajan P, Conticello V P. and Lynn D G. J. Am. Chem. Soc. 2003;125:6391. doi: 10.1021/ja0341642. [DOI] [PubMed] [Google Scholar]
- Dong J, Shokes J E, Scott R A. and Lynn D G. J. Am. Chem. Soc. 2006;128:3540. doi: 10.1021/ja055973j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S, Das A K, Drew M G B. and Banerjee A. Chem. Commun. 2006:4230. doi: 10.1039/b607657b. [DOI] [PubMed] [Google Scholar]
- Lepère M, Chevallard C, Hernandez J-F, Mitraki A. and Guenoun P. Langmuir. 2007;23:8150. doi: 10.1021/la701042t. [DOI] [PubMed] [Google Scholar]
- Toledano S, Williams R J, Jayawarna V. and Ulijn R V. J. Am. Chem. Soc. 2006;128:1070. doi: 10.1021/ja056549l. [DOI] [PubMed] [Google Scholar]
- Reches M. and Gazit E. Science. 2003;300:625. doi: 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- Yemini M, Reches M, Rishpon J. and Gazit E. Nano Lett. 2005;5:183. doi: 10.1021/nl0484189. [DOI] [PubMed] [Google Scholar]
- Kol N, Adler-Abramovich L, Barlam D, Shneck R Z, Gazit E. and Rousso I. Nano Lett. 2005;5:1343. doi: 10.1021/nl0505896. [DOI] [PubMed] [Google Scholar]
- Carny O, Shalev D E. and Gazit E. Nano Lett. 2006;6:1594. doi: 10.1021/nl060468l. [DOI] [PubMed] [Google Scholar]
- Hendler N, Sidelman N, Reches M, Gazit E, Rosenberg Y. and Richter S. Adv. Mater. 2007;19:1485. doi: 10.1002/adma.200602265. [DOI] [Google Scholar]
- Pouget E, Dujardin E, Cavalier A, Moreac A, Valéry C, Marchi-Artzner V, Weiss T, Renault A, Paternostre M. and Artzner F. Nat. Mater. 2007;6:434. doi: 10.1038/nmat1912. [DOI] [PubMed] [Google Scholar]
- Kröger N, Lorenz S, Brunner E. and Sumper M. Science. 2002;298:584. doi: 10.1126/science.1076221. [DOI] [PubMed] [Google Scholar]
- Hou S, Wang J. and Martin C R. Nano Lett. 2005;5:231. doi: 10.1021/nl048305p. [DOI] [PubMed] [Google Scholar]
- Hess H, Clemmens J, Brunner C, Doot R, Luna S, Ernst K-H. and Vogel V. Nano Lett. 2005;5:629. doi: 10.1021/nl0478427. [DOI] [PubMed] [Google Scholar]
- Smith A M, Banwell E F, Edwards W R, Pandya M J. and Woolfson D N. Adv. Funct. Mater. 2006;16:1022. doi: 10.1002/adfm.200500568. [DOI] [Google Scholar]
- Kitagishi H, Oohora K, Yamaguchi H, Sato H, Matsuo T, Harada A. and Hayashi T. J. Am. Chem. Soc. 2007;129:10326. doi: 10.1021/ja073295q. [DOI] [PubMed] [Google Scholar]
- Carlson J C T, Jena S S, Flenniken M, Chou T, Siegel R A. and Wagner C R. J. Am. Chem. Soc. 2006;128:7630. doi: 10.1021/ja060631e. [DOI] [PubMed] [Google Scholar]
- Mao C, Solis D J, Reiss B D, Kottmann S T, Sweeney R Y, Hayhurst A, Georgiou G, Iverson B. and Belcher A M. Science. 2004;303:213. doi: 10.1126/science.1092740. [DOI] [PubMed] [Google Scholar]
- Morikawa M, Yoshihara M, Endo T. and Kimizuka N. J. Am. Chem. Soc. 2005;127:1358. doi: 10.1021/ja043844h. [DOI] [PubMed] [Google Scholar]
- Seeman N C. and Lukeman P S. Rep. Prog. Phys. 2005;68:237. doi: 10.1088/0034-4885/68/1/R05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B. and Seeman N C. Science. 2006;314:1583. doi: 10.1126/science.1131372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desigaux L, Belkacem M B, Richard P, Cellier J, Loéne P, Cario L, Leroux F, Taviot-Guého C. and Pitard B. Nano Lett. 2006;6:199. doi: 10.1021/nl052020a. [DOI] [PubMed] [Google Scholar]
- Kuzuya A, Wang R, Sha R. and Seeman N C. Nano Lett. 2007;7:1757. doi: 10.1021/nl070828k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen P G A, Vandenbergh J, van Dongen J L J, Meijer E W. and Schenning A P H J. J. Am. Chem. Soc. 2007;129:6078. doi: 10.1021/ja0711967. [DOI] [PubMed] [Google Scholar]
- Matsuura K, Yamashita T, Igami Y. and Kimizuka N. Chem. Commun. 2003:376. doi: 10.1039/b210139d. [DOI] [PubMed] [Google Scholar]
- Matsuura K, Murasato K. and Kimizuka N. J. Am. Chem. Soc. 2005;127:10148. doi: 10.1021/ja052644i. [DOI] [PubMed] [Google Scholar]
- Sonin A S. J. Mater. Chem. 1998;8:2557. doi: 10.1039/a802666a. [DOI] [Google Scholar]
- Tang Z. and Kotov N A. Adv. Mater. 2005;17:951. doi: 10.1002/adma.200401593. [DOI] [Google Scholar]
- Yamashita I. Thin Solid Films. 2001;393:12. doi: 10.1016/S0040-6090(01)01083-5. [DOI] [Google Scholar]
- Okuda M, Iwahori Y, Yamashita I. and Yoshimura H. Biotechnol. Bioeng. 2003;84:187. doi: 10.1002/bit.10748. [DOI] [PubMed] [Google Scholar]
- Bourlinos A B, Simopoulos A, Boukos N and Petridis D. 2001. J. Phys. Chem. B 105 7432 10.1021/jp010286+ [DOI] [Google Scholar]
- Kowalchuk C M, Schmid G, Meyer-Zaika W, Huang Y. and Corrigan J F. Inorg. Chem. 2004;43:173. doi: 10.1021/ic0300868. [DOI] [PubMed] [Google Scholar]
- Fukuoka A, Sakamoto Y, Guan S, Inagaki S, Sugimoto N, Fukushima Y, Hirahara K, Iijima S. and Ichikawa M. J. Am. Chem. Soc. 2001;123:3373. doi: 10.1021/ja004067y. [DOI] [PubMed] [Google Scholar]
- von Maltzahn G, Harris T J, Park J-H, Min D-H, Schmidt A J, Sailor M J. and Bhatia S N. J. Am. Chem. Soc. 2007;129:6064. doi: 10.1021/ja070461l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping Liem K, Mart R J. and Webb S J. J. Am. Chem. Soc. 2007;129:12080. doi: 10.1021/ja075000e. [DOI] [PubMed] [Google Scholar]
- Furumi S, Fudouzi H, Miyazaki H T. and Sakka Y. Adv. Mater. 2007;19:2067. doi: 10.1002/adma.200602855. [DOI] [Google Scholar]
- Dinsmore A D, Hsu M F, Nikolaides M G, Marquez M, Bausch A R. and Weitz D A. Science. 2002;298:1006. doi: 10.1126/science.1074868. [DOI] [PubMed] [Google Scholar]
- Noble P F, Cayre O J, Alargova R G, Velev O D. and Paunov V N. J. Am. Chem. Soc. 2004;126:8092. doi: 10.1021/ja047808u. [DOI] [PubMed] [Google Scholar]
- Hu Y, Ge J, Sun Y, Zhang T. and Yin Y. Nano Lett. 2007;7:1832. doi: 10.1021/nl0708157. [DOI] [PubMed] [Google Scholar]
- Kim J-W, Fernández-Nieves A, Dan N, Utada A S, Marquez M. and Weitz D A. Nano Lett. 2007;7:2876. doi: 10.1021/nl0715948. [DOI] [PubMed] [Google Scholar]
- Remskar M, Mrzel A, Skraba Z, Jesih A, Ceh M, Demšar J, Stadelmann P, Lévy F. and Mihailovic D. Science. 2001;292:479. doi: 10.1126/science.1059011. [DOI] [PubMed] [Google Scholar]
- Ma R, Fukuda K, Sasaki T, Osada M and Bando Y. 2005. J. Phys. Chem. B 109 6210 10.1021/jp044282r [DOI] [PubMed] [Google Scholar]
- Sasaki T, Ebina Y, Kitami Y, Watanabe M and Oikawa T. 2001. J. Phys. Chem. B 105 6116 10.1021/jp010421i [DOI] [Google Scholar]
- Ozawa T C, Fukuda K, Akatsuka K, Ebina Y. and Sasaki T. Chem. Commun. 2007;19:6575. [Google Scholar]
- He C, Sasaki T, Zhou Y, Shimizu Y, Masuda M. and Koshizaki N. Adv. Funct. Mater. 2007;17:3554. doi: 10.1002/adfm.200700081. [DOI] [Google Scholar]
- Saunders A E, Ghezelbash A, Smilgies D-M, Sigman M B Jr. and Korgel B A. Nano Lett. 2006;6:2959. doi: 10.1021/nl062419e. [DOI] [PubMed] [Google Scholar]
- Du C, Falini G, Fermani S, Abbott C. and Moradian-Oldak J. Science. 2005;307:1450. doi: 10.1126/science.1105675. [DOI] [PubMed] [Google Scholar]
- Yang C, Zhong Z. and Lieber C M. Science. 2005;310:1304. doi: 10.1126/science.1118798. [DOI] [PubMed] [Google Scholar]
- Chen M. and Searson P C. Adv. Mater. 2005;17:2765. doi: 10.1002/adma.200501292. [DOI] [Google Scholar]
- Yoo S-H. and Park S. Adv. Mater. 2007;19:1612. doi: 10.1002/adma.200602551. [DOI] [Google Scholar]
- Hwang Y K, Lee J-M, Sathaye S D, Cho G, Hwang J-S. and Chang J-S. J. Nanosci. Nanotechnol. 2006;6:1850. doi: 10.1166/jnn.2006.206. [DOI] [PubMed] [Google Scholar]
- Shirahata N, Nakanishi T, Furumi S. and Sakka Y. J. Nanosci. Nanotechnol. 2006;6:1823. doi: 10.1166/jnn.2006.209. [DOI] [PubMed] [Google Scholar]
- Schlittler R R, Seo J W, Gimzewski J K, Durkan C, Saifullah M S M. and Welland M E. Science. 2001;292:1136. doi: 10.1126/science.1057823. [DOI] [PubMed] [Google Scholar]
- Sano M, Kamino A, Okamura J. and Shinkai S. Science. 2001;293:1299. doi: 10.1126/science.1061050. [DOI] [PubMed] [Google Scholar]
- Star A, Steuerman D W, Heath J R. and Stoddart J F. Angew. Chem. Int. Ed. 2002;41:2508. doi: 10.1002/1521-3773(20020715)41:14<2508::AID-ANIE2508>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Hasobe T, Fukuzumi S. and Kamat P V. J. Am. Chem. Soc. 2005;127:11884. doi: 10.1021/ja050687t. [DOI] [PubMed] [Google Scholar]
- Qiao R. and Ke P C. J. Am. Chem. Soc. 2006;128:13656. doi: 10.1021/ja063977y. [DOI] [PubMed] [Google Scholar]
- Nakashima N. and Fujigaya T. Chem. Lett. 2007;36:692. doi: 10.1246/cl.2007.692. [DOI] [Google Scholar]
- Ogoshi T, Takashima Y, Yamaguchi H. and Harada A. J. Am. Chem. Soc. 2007;129:4878. doi: 10.1021/ja070457+. [DOI] [PubMed] [Google Scholar]
- Paleos C M. and Tsiourvas D. Adv. Mater. 1977;9:695. doi: 10.1002/adma.19970090904. [DOI] [Google Scholar]
- Ariga K. and Kunitake T. Acc. Chem. Res. 1998;31:371. doi: 10.1021/ar970014i. [DOI] [Google Scholar]
- Kuzmenko I, Rapaport H, Kjaer K, Als-Nielsen J, Weissbuch I, Lahav M. and Leiserowitz L. Chem. Rev. 2001;101:1659. doi: 10.1021/cr990038y. [DOI] [PubMed] [Google Scholar]
- Ariga K, Nakanishi T. and Michinobu T. J. Nanosci. Nanotechnol. 2006;6:2278. doi: 10.1166/jnn.2006.503. [DOI] [PubMed] [Google Scholar]
- Leblanc R M. Curr. Opin. Chem. Biol. 2006;10:529. doi: 10.1016/j.cbpa.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Ariga K, Hill J P. and Endo H. Int. J. Mol. Sci. 2007;8:864. [Google Scholar]
- Onda M, Yoshihara K, Koyano H, Ariga K. and Kunitake T. J. Am. Chem. Soc. 1996;118:8524. doi: 10.1021/ja960991+. [DOI] [Google Scholar]
- Sakurai M, Tamagawa H, Furuki T, Inoue Y, Ariga K. and Kunitake T. Chem. Lett. 1995:1001. doi: 10.1246/cl.1995.1001. [DOI] [Google Scholar]
- Sakurai M, Tamagawa H, Inoue Y, Ariga K and Kunitake T. 1997. J. Phys. Chem. B 101 4810 10.1021/jp9700591 [DOI] [Google Scholar]
- Tamagawa H, Sakurai M, Inoue Y, Ariga K and Kunitake T. 1997. J. Phys. Chem. B 101 4817 10.1021/jp9700600 [DOI] [Google Scholar]
- Kurihara K, Ohto K, Honda Y. and Kunitake T. J. Am. Chem. Soc. 1991;113:5077. doi: 10.1021/ja00013a063. [DOI] [Google Scholar]
- Kawahara T, Kurihara K. and Kunitake T. Chem. Lett. 1992:1839. doi: 10.1246/cl.1992.1839. [DOI] [Google Scholar]
- Sasaki D Y, Kurihara K. and Kunitake T. J. Am. Chem. Soc. 1991;113:9685. doi: 10.1021/ja00025a051. [DOI] [Google Scholar]
- Sasaki D Y, Kurihara K. and Kunitake T. J. Am. Chem. Soc. 1992;114:10994. doi: 10.1021/ja00053a065. [DOI] [Google Scholar]
- Ariga K, Kamino A, Koyano H. and Kunitake T. J. Mater. Chem. 1997;7:1155. doi: 10.1039/a700081b. [DOI] [Google Scholar]
- Taguchi K, Ariga K. and Kunitake T. Chem. Lett. 1995:701. doi: 10.1246/cl.1995.701. [DOI] [Google Scholar]
- Cha X, Ariga K. and Kunitake T. Bull. Chem. Soc. Jpn. 1996;69:163. doi: 10.1246/bcsj.69.163. [DOI] [Google Scholar]
- Cha X, Ariga K, Onda M. and Kunitake T. J. Am. Chem. Soc. 1995;117:11833. doi: 10.1021/ja00153a003. [DOI] [Google Scholar]
- Cha X, Ariga K. and Kunitake T. J. Am. Chem. Soc. 1996;118:9545. doi: 10.1021/ja961526f. [DOI] [Google Scholar]
- Cha X, Ariga K. and Kunitake T. Chem. Lett. 1996:73. doi: 10.1246/cl.1996.73. [DOI] [Google Scholar]
- Ariga K, Kamino A, Cha X. and Kunitake T. Langmuir. 1999;15:3875. doi: 10.1021/la981047p. [DOI] [Google Scholar]
- Kitano H. and Ringsdorf H. Bull. Chem. Soc. Jpn. 1985;58:2826. doi: 10.1246/bcsj.58.2826. [DOI] [Google Scholar]
- Nakamura F, Ijiro K. and Shimomura M. Thin Solid Films. 1998;327–9:603. doi: 10.1016/S0040-6090(98)00722-6. [DOI] [Google Scholar]
- Marczak R, Hoang V T, Noworyta K, Zandler M E, Kutner W. and D'Souza F. J. Mater. Chem. 2002;12:2123. doi: 10.1039/b202461f. [DOI] [Google Scholar]
- Miao W, Du X and Liang Y. 2003. J. Phys. Chem. B 107 13636 10.1021/jp035880t [DOI] [Google Scholar]
- Haruta O. and Ijiro K. J. Nanosci. Nanotechnol. 2007;7:734. doi: 10.1166/jnn.2007.522. [DOI] [PubMed] [Google Scholar]
- Marchi-Artzner V, Artzner F, Karthaus O, Shimomura M, Ariga K, Kunitake T. and Lehn J-M. Langmuir. 1998;14:5164. doi: 10.1021/la971192n. [DOI] [Google Scholar]
- Koyano H, Bissel P, Yoshihara K, Ariga K. and Kunitake T. Langmuir. 1997;13:5426. doi: 10.1021/la970014r. [DOI] [Google Scholar]
- Koyano H, Bissel P, Yoshihara K, Ariga K. and Kumitake T. Chem. Eur. J. 1997;3:1077. doi: 10.1002/chem.19970030715. [DOI] [Google Scholar]
- Jiao T and Liu M. 2005. J. Phys. Chem. B 109 2532 10.1021/jp045258g [DOI] [PubMed] [Google Scholar]
- Kovalchuk N M, Vollhardt D, Fainerman V B and Aksenenko E V. 2007. J. Phys. Chem. B 111 8283 10.1021/jp071644x [DOI] [PubMed] [Google Scholar]
- Ikeura Y, Kurihara K. and Kunitake T. J. Am. Chem. Soc. 1991;113:7342. doi: 10.1021/ja00019a035. [DOI] [Google Scholar]
- Kurihara K, Ohto K, Tanaka Y, Aoyama Y. and Kunitake T. J. Am. Chem. Soc. 1991;113:444. doi: 10.1021/ja00002a010. [DOI] [Google Scholar]
- Ludwig R, Ariga K. and Shinkai S. Chem. Lett. 1993:1413. doi: 10.1246/cl.1993.1413. [DOI] [Google Scholar]
- Ariga K, Isoyama K, Hayashida O, Aoyama Y. and Okahata Y. Chem. Lett. 1998:1007. doi: 10.1246/cl.1998.1007. [DOI] [Google Scholar]
- Higuchi M, Taguchi K. and Kinoshita T. Chem. Lett. 1999;28:1117. doi: 10.1246/cl.1999.1117. [DOI] [Google Scholar]
- Higashi N, Koga T, Fujii Y. and Niwa M. Langmuir. 2001;17:4061. doi: 10.1021/la010127h. [DOI] [Google Scholar]
- Alonso C, Eliash R, Jensen T R, Kjaer K, Lahav M. and Leiserowitz L. J. Am. Chem. Soc. 2001;123:10105. doi: 10.1021/ja016732o. [DOI] [PubMed] [Google Scholar]
- Matsuura K. and Kobayashi K. Glycoconjgate J. 2004;21:139. doi: 10.1023/B:GLYC.0000044845.64354.a3. [DOI] [PubMed] [Google Scholar]
- Santacroce P V. and Basu A. Angew. Chem. Int. Ed. 2003;42:95. doi: 10.1002/anie.200390063. [DOI] [PubMed] [Google Scholar]
- Huo Q, Sui G, Zheng Y, Kele P, Leblanc R M, Hasegawa T, Nishijo J. and Umemura J. Chem. Eur. J. 2001;7:4796. doi: 10.1002/1521-3765(20011119)7:22<4796::AID-CHEM4796>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Plaut D J, Martin S M, Kjaer K, Weygand M J, Lahav M, Leiserowitz L, Weissbuch I E. and Ward M D. J. Am. Chem. Soc. 2003;125:15922. doi: 10.1021/ja0371404. [DOI] [PubMed] [Google Scholar]
- Ariga K, Nakanishi T. and Hill J P. Soft Matter. 2006;2:465. doi: 10.1039/b602732f. [DOI] [PubMed] [Google Scholar]
- Ariga K, Terasaka Y, Sakai D, Tsuji H. and Kikuchi J. J. Am. Chem. Soc. 2000;122:7835. doi: 10.1021/ja000924m. [DOI] [Google Scholar]
- Ariga K, Nakanishi T, Terasaka Y, Tsuji H, Sakai D. and Kukuchi J. Langmuir. 2005;21:976. doi: 10.1021/la0477845. [DOI] [PubMed] [Google Scholar]
- Ariga K, Nakanishi T, Hill J P, Terasaka Y, Sakai D. and Kikuchi J. Soft Matter. 2005;1:132. doi: 10.1039/b501945a. [DOI] [PubMed] [Google Scholar]
- Ariga K, Nakanishi T, Terasaka Y. and Kikuchi J. J. Porous Mater. 2006;13:427. doi: 10.1007/s10934-006-8041-2. [DOI] [Google Scholar]
- Arnett E M, Harvey N G. and Rose P L. Acc. Chem. Res. 1988;229:131. [Google Scholar]
- Arnett E M, Chao J, Kinzig B, Stewart M. and Thompson O. J. Am. Chem. Soc. 1978;100:5575. doi: 10.1021/ja00485a060. [DOI] [Google Scholar]
- Heath J G. and Arnett E M. J. Am. Chem. Soc. 1992;114:4500. doi: 10.1021/ja00038a009. [DOI] [Google Scholar]
- Neimert-Andersson K, Blomberg E. and Somfai P. J. Org. Chem. 2004;69:3746. doi: 10.1021/jo0357566. [DOI] [PubMed] [Google Scholar]
- Böhmer A. et al Eur. J. Org. Chem. 2007:45. doi: 10.1002/ejoc.200600437. [DOI] [Google Scholar]
- Weissbuch I, Rubinstein I, Weygand M J, Kjaer K, Leiserowitz L. and Lahav M. Helv. Chim. Acta. 2003;86:3867. doi: 10.1002/hlca.200390324. [DOI] [Google Scholar]
- Pathirana S, Neely W C, Myers L J. and Vodyanoy V. J. Am. Chem. Soc. 1992;114:1404. doi: 10.1021/ja00030a041. [DOI] [Google Scholar]
- Qian P, Matsuda M. and Miyashita T. J. Am. Chem. Soc. 1993;115:5624. doi: 10.1021/ja00066a031. [DOI] [Google Scholar]
- Kawabata H. and Shinkai S. Chem. Lett. 1994:375. doi: 10.1246/cl.1994.375. [DOI] [Google Scholar]
- Higashi N, Koga T, Fujii Y. and Niwa M. Langmuir. 2001;17:4061. doi: 10.1021/la010127h. [DOI] [Google Scholar]
- Tamura K, Sato H, Yamashita S, Yamagishi A and Yamada H. 2004. J. Phys. Chem. B 108 8287 10.1021/jp049349p [DOI] [Google Scholar]
- Badis M, Tomaszkiewicz I, Joly J-P. and Rogalska E. Langmuir. 2004;20:6259. doi: 10.1021/la049596k. [DOI] [PubMed] [Google Scholar]
- Shahgaldian P, Pieles U. and Hegner M. Langmuir. 2005;21:6503. doi: 10.1021/la0503101. [DOI] [PubMed] [Google Scholar]
- Ariga K, Michinobu T, Nakanishi T. and Hill J P. Curr. Opin. Colloid Interface Sci. 2008;13:23. doi: 10.1016/j.cocis.2007.08.010. [DOI] [Google Scholar]
- Michinobu T, Shinoda S, Nakanishi T, Hill J P, Fujii K, Player T N, Tsukube H. and Ariga K. J. Am. Chem. Soc. 2006;128:14478. doi: 10.1021/ja066429t. [DOI] [PubMed] [Google Scholar]
- Ariga K, Kunitake T. and Furuta H. J. Chem. Soc., Perkin Trans. 1996;2:667. [Google Scholar]
- Misawa Y, Kubo Y, Tokita S, Ohkuma H. and Nakahara H. Chem. Lett. 2004;33:1118. doi: 10.1246/cl.2004.1118. [DOI] [Google Scholar]
- Zheng Y, Orbulescu J, Ji X, Andreopoulos F M, Pham S M. and Leblanc R M. J. Am. Chem. Soc. 2003;125:2680. doi: 10.1021/ja0293610. [DOI] [PubMed] [Google Scholar]
- de Miguel G, Martín-Romero M T, Pedros J M, Muñoz E, Pérez-Morales M, Richardsonc T H. and Camacho L. J. Mater. Chem. 2007;17:2914. doi: 10.1039/b701811h. [DOI] [Google Scholar]
- Miyahara T. and Kurihara K. J. Am. Chem. Soc. 2004;126:5684. doi: 10.1021/ja0390101. [DOI] [PubMed] [Google Scholar]
- Okahata Y, Tsuruta T, Ijiro K. and Ariga K. Langmuir. 1988;4:1373. doi: 10.1021/la00084a030. [DOI] [Google Scholar]
- Okahata Y, Tsuruta T, Ijiro K. and Ariga K. Thin Solid Films. 1989;180:65. doi: 10.1016/0040-6090(89)90055-2. [DOI] [Google Scholar]
- Culp J T, Park J-H, Stratakis D, Meisel M W. and Talham D R. J. Am. Chem. Soc. 2002;124:10083. doi: 10.1021/ja026312e. [DOI] [PubMed] [Google Scholar]
- Culp J T, Park J-H, Meisel M W. and Talham D R. Inorg. Chem. 2003;42:2842. doi: 10.1021/ic026158x. [DOI] [PubMed] [Google Scholar]
- Aoyagi M, Minamikawa H. and Shimizu T. Chem. Lett. 2004;33:860. doi: 10.1246/cl.2004.860. [DOI] [Google Scholar]
- Huang X, Li C, Jiang S, Wang X, Zhang B. and Liu M. J. Am. Chem. Soc. 2004;126:1322. doi: 10.1021/ja036878i. [DOI] [PubMed] [Google Scholar]
- Ariga K. J. Nanosci. Nanotechnol. 2004;4:23. doi: 10.1166/jnn.2004.048. [DOI] [PubMed] [Google Scholar]
- Ariga K, Nakanishi T, Takagi N, Tanaka R. and Kikuchi J. Colloid Surf. A-Physicochem. Eng. Asp. 2006;284/285:499. doi: 10.1016/j.colsurfa.2005.11.008. [DOI] [Google Scholar]
- Ariga K, Hill J P and Wakayama Y. Phys. Stat. Sol. (a) in press [Google Scholar]
- Kamino A, Koyano H, Ariga K. and Kunitake T. Bull. Chem. Soc. Jpn. 1996;69:3619. doi: 10.1246/bcsj.69.3619. [DOI] [Google Scholar]
- Oishi Y, Kato T, Kuramori M, Suehiro K, Ariga K, Kamino A, Koyano H. and Kunitake T. Chem. Commun. 1997:1357. doi: 10.1039/a702880f. [DOI] [Google Scholar]
- Kato T, Kuramori M, Oishi Y, Suehiro K, Ariga K, Kamino A, Koyano H. and Kunitake T. Rep. Prog. Polym. Phys. Jpn. 1996;39:397. [Google Scholar]
- Koyano H, Yoshihara K, Ariga K, Kunitake T, Oishi Y, Kawano O, Kuramori M. and Suehiro K. Chem. Commun. 1996:1769. doi: 10.1039/cc9960001769. [DOI] [Google Scholar]
- Oishi Y, Torii Y, Kuramori M, Suehiro K, Ariga K, Taguchi K, Kamino A. and Kunitake T. Chem. Lett. 1996:411. doi: 10.1246/cl.1996.411. [DOI] [Google Scholar]
- Oishi Y, Torii Y, Kato T, Kuramori M, Suehiro K, Ariga K, Taguchi K, Kamino A, Koyano H. and Kunitake T. Langmuir. 1997;13:519. doi: 10.1021/la960112x. [DOI] [Google Scholar]
- Bissel P, Onda M, Yoshihara K, Koyano H, Ariga K, Kunitake T, Oishi Y. and Suehiro K. Langmuir. 1999;15:1791. doi: 10.1021/la981043k. [DOI] [Google Scholar]
- Ariga K, Tanaka R, Kikuchi J, Higuchi M. and Yamamoto K. J. Nanosci. Nanotechnol. 2002;2:669. doi: 10.1166/jnn.2002.147. [DOI] [PubMed] [Google Scholar]
- Yan X, Janout V, Hsu J T. and Regen S L. J. Am. Chem. Soc. 2003;125:8094. doi: 10.1021/ja035453t. [DOI] [PubMed] [Google Scholar]
- McCullough D H, III, Janout V, Li J, Hsu J T, Truong Q, Wilusz E. and Regen S L. J. Am. Chem. Soc. 2004;126:9916. doi: 10.1021/ja047365u. [DOI] [PubMed] [Google Scholar]
- McCullough D H, III, Grygorash R, Hsu J T. and Regen S L. J. Am. Chem. Soc. 2007;129:8663. doi: 10.1021/ja071936b. [DOI] [PubMed] [Google Scholar]
- Kamino A, Ariga K, Kunitake T, Birault V, Pozzi G, Nakatani Y and Ourisson G. 1995. Colloid Surf. A 103 183 10.1016/0927-7757(95)03244-8 [DOI] [Google Scholar]
- Ariga K, Urakawa T, Michiue A, Sasaki Y. and Kikuchi J. Langmuir. 2000;16:9147. doi: 10.1021/la000901l. [DOI] [PubMed] [Google Scholar]
- Ariga K, Tanaka K, Katagiri K, Kikuchi J, Ohshima E and Hisaeda Y. 2000. Colloid Surf. A 169 47 10.1016/S0927-7757(00)00416-7 [DOI] [Google Scholar]
- Ariga K, Urakawa T, Michiue A. and Kikuchi J. Langmuir. 2004;20:6762. doi: 10.1021/la0490238. [DOI] [PubMed] [Google Scholar]
- Ariga K, Yuki H, Kikuchi J, Dannemuller O, Albrecht-Gary A-M, Nakatani Y. and Ourisson G. Langmuir. 2005;21:4578. doi: 10.1021/la0467887. [DOI] [PubMed] [Google Scholar]
- Wang C, Micic M, Ensor M, Daunert S. and Leblanc R M. Langmuir. 2007;23:7602. doi: 10.1021/la700756e. [DOI] [PubMed] [Google Scholar]
- Karp E, Pecinovsky C S, McNevin M J, Gin D L. and Schwartz D K. Langmuir. 2007;23:7923. doi: 10.1021/la701345e. [DOI] [PubMed] [Google Scholar]
- Samokhvalov A, Gurney R W, Lahav M and Naaman R. 2002. J. Phys. Chem. B 106 9070 10.1021/jp0202340 [DOI] [Google Scholar]
- Yoshida J, Saruwatari K, Kameda J, Sato H, Yamagishi A, Sun L, Corriea M. and Villemurer G. Langmuir. 2006;22:9591. doi: 10.1021/la061668f. [DOI] [PubMed] [Google Scholar]
- Krstic V, Duesberg G S, Muster J, Burghard M. and Roth S. Chem. Mater. 1998;10:2338. doi: 10.1021/cm980207f. [DOI] [Google Scholar]
- Ochiai K, Tabuchi Y, Rikukawa M, Sanui K. and Ogata N. Thin Solid Films. 1998;327–9:454. doi: 10.1016/S0040-6090(98)00685-3. [DOI] [Google Scholar]
- N⊘rgaard K. and Bj⊘rnholm T. Chem. Commun. 2005:1812. doi: 10.1039/b417526n. [DOI] [Google Scholar]
- Vuorinen T, Kaunisto K, Tkachenko N V, Efimov A, Lemmetyinen H, Alekseev A S, Hosomizu K. and Imahori H. Langmuir. 2005;21:5383. doi: 10.1021/la050347l. [DOI] [PubMed] [Google Scholar]
- Collier C P, Wong E W, Belohradský M, Raymo F M, Stoddart J F, Kuekes P J, Williams R S. and Heath J R. Science. 1999;285:391. doi: 10.1126/science.285.5426.391. [DOI] [PubMed] [Google Scholar]
- Jiang W, Wang G, He Y, Wang X, An Y, Song Y. and Jiang L. Chem. Commun. 2005:3550. doi: 10.1039/b504479k. [DOI] [PubMed] [Google Scholar]
- Noworyta K, Kutner W, Deviprasad G R. and D'Souza F. Synth. Metal. 2002;130:221. doi: 10.1016/S0379-6779(02)00059-0. [DOI] [Google Scholar]
- Marczak R, Noworyta K, Nowakowski R, Kutner W, Desbat B, Araki Y, Ito O, Gadde S, Zandler M E. and D'Souza F. J. Nanosci. Nanotechnol. 2007;7:1455. doi: 10.1166/jnn.2007.328. [DOI] [PubMed] [Google Scholar]
- Muruganathan R M and Fischer Th M. 2006. J. Phys. Chem. B 110 22979 10.1021/jp065700q [DOI] [PubMed] [Google Scholar]
- McConlogue C W. and Vanderlick T K. Langmuir. 1997;13:7158. doi: 10.1021/la970898e. [DOI] [Google Scholar]
- Ariga K, Abe T. and Kikuchi J. Chem. Lett. 2000:82. doi: 10.1246/cl.2000.82. [DOI] [Google Scholar]
- Ariga K, Nakanishi T, Hill J P, Shirai M, Okuno M, Abe T. and Kikuchi J. J. Am. Chem. Soc. 2005;127:12074. doi: 10.1021/ja053226g. [DOI] [PubMed] [Google Scholar]
- Isenberg H, Kjaer K. and Rapaport H. J. Am. Chem. Soc. 2006;128:12468. doi: 10.1021/ja062363q. [DOI] [PubMed] [Google Scholar]
- Ariga K, Nakanishi T, Kawanami S, Kosaka T. and Kikuchi J. J. Nanosci. Nanotechnol. 2006;6:1718. doi: 10.1166/jnn.2006.230. [DOI] [PubMed] [Google Scholar]
- Chen X, Yang T, Kataoka S. and Cremer P S. J. Am. Chem. Soc. 2007;129:12272. doi: 10.1021/ja073869r. [DOI] [PubMed] [Google Scholar]
- Vollhardt D. 2007. J. Phys. Chem. C 111 6805 10.1021/jp0704822 [DOI] [Google Scholar]
- Song F, Zhang S, Bonifazi D, Enger O, Diederich F. and Echegoyen L. Langmuir. 2005;21:9246. doi: 10.1021/la051377r. [DOI] [PubMed] [Google Scholar]
- Ohashi A, Tsukahara S. and Watarai H. Langmuir. 2003;19:4645. doi: 10.1021/la0268451. [DOI] [Google Scholar]
- Liu B, Qian D-J, Huang H-X, Wakayama T, Hara S, Huang W, Nakamura C. and Miyake J. Langmuir. 2005;21:5079. doi: 10.1021/la050064t. [DOI] [PubMed] [Google Scholar]
- Luo K. and Dryfe R A W. Chem. Commun. 2007:3258. doi: 10.1039/b704638c. [DOI] [PubMed] [Google Scholar]
- Park Y-K, Yoo S-H. and Park S. Langmuir. 2007;23:10505. doi: 10.1021/la701445a. [DOI] [PubMed] [Google Scholar]
- Ariga K, Hill J P. and Ji Q. Phys. Chem. Chem. Phys. 2007;9:2319. doi: 10.1039/b700410a. [DOI] [PubMed] [Google Scholar]
- Iler R K. J. Colloid Interface Sci. 1966;21:569. doi: 10.1016/0095-8522(66)90018-3. [DOI] [Google Scholar]
- Decher G. Science. 1997;277:1232. doi: 10.1126/science.277.5330.1232. [DOI] [Google Scholar]
- Bertrand P, Jonas A, Laschewsky A. and Legras R. Macromol. Rapid Commun. 2000;21:319. doi: 10.1002/(SICI)1521-3927(20000401)21:7<319::AID-MARC319>3.0.CO;2-7. [DOI] [Google Scholar]
- Hammond P T. Adv. Mater. 2004;16:1271. doi: 10.1002/adma.200400760. [DOI] [Google Scholar]
- Decher G, Hong J D. and Schmitt J. Thin Solid Films. 1992;210:831. doi: 10.1016/0040-6090(92)90417-A. [DOI] [Google Scholar]
- Ramsden J J, Lvov Y M. and Decher G. Thin Solid Films. 1995;254:246. doi: 10.1016/0040-6090(94)06262-J. [DOI] [Google Scholar]
- Yoo D, Shiratori S S. and Rubner M F. Macromolecules. 1998;31:4309. doi: 10.1021/ma9800360. [DOI] [Google Scholar]
- Lvov Y, Ariga K, Onda M, Ichinose I and Kunitake T. 1999. Colloids Surf. A 146 337 10.1016/S0927-7757(98)00789-4 [DOI] [Google Scholar]
- Shiratori S S. and Rubner M F. Macromolecules. 2000;33:4213. doi: 10.1021/ma991645q. [DOI] [Google Scholar]
- Fou A C, Onitsuka O, Ferreira M, Rubner M. and Hsieh B R. J. Appl. Phys. 1996;79:7501. doi: 10.1063/1.362421. [DOI] [Google Scholar]
- Stockton W B. and Rubner M F. Macromolecules. 1997;30:2717. doi: 10.1021/ma9700486. [DOI] [Google Scholar]
- He J-A, Valluzzi R, Yang K, Dolukhanyan T, Sung C, Kumar J, Tripathy S K, Samuelson L, Balogh L. and Tomalia D A. Chem. Mater. 1999;11:3268. doi: 10.1021/cm990311c. [DOI] [Google Scholar]
- Khopade A J. and Caruso F. Langmuir. 2002;18:7669. doi: 10.1021/la020251g. [DOI] [Google Scholar]
- Lvov Y, Yamada S. and Kunitake T. Thin Solid Films. 1997;300:107. doi: 10.1016/S0040-6090(96)09494-1. [DOI] [Google Scholar]
- Lvov Y, Ariga K. and Kunitake T. Chem. Lett. 1994:2323. doi: 10.1246/cl.1994.2323. [DOI] [Google Scholar]
- Lvov Y, Ariga K, Ichinose I. and Kunitake T. J. Am. Chem. Soc. 1995;117:6117. doi: 10.1021/ja00127a026. [DOI] [Google Scholar]
- Lvov Y, Ariga K, Ichinose I. and Kunitake T. Thin Solid Films. 1996;285:797. doi: 10.1016/S0040-6090(95)08449-5. [DOI] [Google Scholar]
- Onda M, Lvov Y, Ariga K and Kunitake T. 1996. Jpn. J. Appl. Phys. part 1 36 L1608 10.1143/JJAP.36.L1608 [DOI] [Google Scholar]
- Ariga K, Onda M, Lvov Y. and Kunitake T. Chem. Lett. 1997:25. doi: 10.1246/cl.1997.25. [DOI] [Google Scholar]
- Caruso F, Furlong D N, Ariga K, Ichinose I. and Kunitake T. Langmuir. 1998;14:4559. doi: 10.1021/la971288h. [DOI] [Google Scholar]
- Lvov Y, Decher G. and Sukhorukov G. Macromolecules. 1993;26:5396. doi: 10.1021/ma00072a016. [DOI] [Google Scholar]
- Johnston A P R, Mitomo H, Read E S. and Caruso F. Langmuir. 2006;22:3251. doi: 10.1021/la052581h. [DOI] [PubMed] [Google Scholar]
- Lvov Y, Onda M, Ariga K. and Kunitake T. J. Biomater. Sci. Polym. Ed. 1998;9:345. doi: 10.1080/09205063.1998.9753060. [DOI] [PubMed] [Google Scholar]
- Serizawa T, Yamaguchi M. and Akashi M. Biomacromolecules. 2002;3:724. doi: 10.1021/bm0200027. [DOI] [PubMed] [Google Scholar]
- Constantine C A. et al J. Am. Chem. Soc. 2003;125:1805. doi: 10.1021/ja028691h. [DOI] [PubMed] [Google Scholar]
- Thierry B, Winnik F M, Merhi Y, Silver J. and Tabrizian M. Biomacromolecules. 2003;4:1564. doi: 10.1021/bm0341834. [DOI] [PubMed] [Google Scholar]
- Shutava T G. and Lvov Y M. J. Nanosci. Nanotechnol. 2006;6:1655. doi: 10.1166/jnn.2006.225. [DOI] [PubMed] [Google Scholar]
- Lvov Y, Haas H, Decher G, Möhwald H, Mikhailov A, Mtchedlishvily B, Morgunova E. and Vainstein B. Langmuir. 1994;10:4232. doi: 10.1021/la00023a052. [DOI] [Google Scholar]
- Jan E. and Kotov N A. Nano Lett. 2007;7:1123. doi: 10.1021/nl0620132. [DOI] [PubMed] [Google Scholar]
- Kotov N A, Dekany I. and Fendler J H. J. Phys. Chem. 1995;99:13065. doi: 10.1021/j100035a005. [DOI] [Google Scholar]
- Ariga K, Lvov Y, Onda M, Ichinose I. and Kunitake T. Chem. Lett. 1997:125. doi: 10.1246/cl.1997.125. [DOI] [Google Scholar]
- Lvov Y, Ariga K, Onda M, Ichinose I. and Kunitake T. Langmuir. 1997;13:6195. doi: 10.1021/la970517x. [DOI] [Google Scholar]
- Salgueirino-Maceira V, Correa-Duarte M A, Spasova M, Liz-Marzan L M. and Farle M. Adv. Funct. Mater. 2006;16:509. doi: 10.1002/adfm.200500565. [DOI] [Google Scholar]
- Liang Z, Dzienis K L, Xu J. and Wang Q. Adv. Funct. Mater. 2006;16:542. doi: 10.1002/adfm.200500334. [DOI] [Google Scholar]
- Zimnitsky D, Jiang C, Xu J, Lin Z, Zhang L. and Tsukruk V V. Langmuir. 2007;23:10176. doi: 10.1021/la7014644. [DOI] [PubMed] [Google Scholar]
- Lvov Y, Ariga K, Ichinose I. and Kunitake T. Langmuir. 1996;12:3038. doi: 10.1021/la951002d. [DOI] [Google Scholar]
- Mamedov A, Ostrander J, Aliev F. and Kotov N A. Langmuir. 2000;16:3941. doi: 10.1021/la990957j. [DOI] [Google Scholar]
- Sasaki T, Ebina Y, Tanaka T, Harada M, Watanabe M. and Decher G. Chem. Mater. 2001;13:4661. doi: 10.1021/cm010478h. [DOI] [Google Scholar]
- Wang L, Sasaki T, Ebina Y, Kurashima K. and Watanabe M. Chem. Mater. 2002;14:4827. doi: 10.1021/cm020685x. [DOI] [Google Scholar]
- Wang Z-S, Sasaki T, Muramatsu M, Ebina Y, Tanaka T, Wang L. and Watanabe M. Chem. Mater. 2003;15:807. doi: 10.1021/cm0204268. [DOI] [Google Scholar]
- Wang L, Omomo Y, Sakai N, Fukuda K, Nakai I, Ebina Y, Takada K, Watanabe M. and Sasaki T. Chem. Mater. 2003;15:2873. doi: 10.1021/cm034191r. [DOI] [Google Scholar]
- Zhou Y, Ma R, Ebina Y, Takada K. and Sasaki T. Chem. Mater. 2006;18:1235. doi: 10.1021/cm052284y. [DOI] [Google Scholar]
- Osada M, Ebina Y, Funakubo H, Yokoyama S, Kiguchi T, Takada K. and Sasaki T. Adv. Mater. 2006;18:1023. doi: 10.1002/adma.200501224. [DOI] [Google Scholar]
- Li L, Ma R, Ebina Y, Iyi N. and Sasaki T. Chem. Mater. 2005;17:4386. doi: 10.1021/cm0510460. [DOI] [Google Scholar]
- Vial S, Pastoriza-Santos I, Pérez-Juste J. and Liz-Marzán L M. Langmuir. 2007;23:4606. doi: 10.1021/la063753t. [DOI] [PubMed] [Google Scholar]
- Gunawidjaja R, Ko H, Jiang C. and Tsukruk V V. Chem. Mater. 2007;19:2007. doi: 10.1021/cm063022e. [DOI] [Google Scholar]
- Paloniemi H, Lukkarinen M, Ääritalo T, Areva S, Leiro J, Heinonen M, Haapakka K. and Lukkari J. Langmuir. 2006;22:74. doi: 10.1021/la051736i. [DOI] [PubMed] [Google Scholar]
- Ma R, Sasaki T. and Bando Y. J. Am. Chem. Soc. 2004;126:10382. doi: 10.1021/ja048855p. [DOI] [PubMed] [Google Scholar]
- Ariga K, Lvov Y, Ichinose I. and Kunitake T. Appl. Clay Sci. 1999;15:137. doi: 10.1016/S0169-1317(99)00012-5. [DOI] [Google Scholar]
- Lee G S, Lee Y-J. and Yoon K B. J. Am. Chem. Soc. 2001;123:9769. doi: 10.1021/ja010517q. [DOI] [PubMed] [Google Scholar]
- Mitsui T, Yamaguchi K, Takeoka Y, Rikukawa M. and Sanui K. Chem. Commun. 2002:1094. doi: 10.1039/b200965j. [DOI] [PubMed] [Google Scholar]
- Ji Q, Miyahara M, Hill J P, Acharya S, Vinu A, Yoon S B, Yu J-S, Sakamoto K. and Ariga K. J. Am. Chem. Soc. 2008;130:2376. doi: 10.1021/ja076139s. [DOI] [PubMed] [Google Scholar]
- Cooper T M, Campbell A L. and Crane R L. Langmuir. 1995;11:2713. doi: 10.1021/la00007a061. [DOI] [Google Scholar]
- Ariga K, Lvov Y. and Kunitake T. J. Am. Chem. Soc. 1997;119:2224. doi: 10.1021/ja963442c. [DOI] [Google Scholar]
- Araki K, Wagner M J. and Wrigton M S. Langmuir. 1996;12:5393. doi: 10.1021/la960024c. [DOI] [Google Scholar]
- Tang T, Qu J, Müllen K. and Webber S E. Langmuir. 2006;22:26. doi: 10.1021/la052766o. [DOI] [PubMed] [Google Scholar]
- Emoto K, Iijima M, Nagasaki Y. and Kataoka K. J. Am. Chem. Soc. 2000;122:2653. doi: 10.1021/ja993821g. [DOI] [Google Scholar]
- Qi B, Tong X. and Zhao Y. Macromolecules. 2006;39:5714. doi: 10.1021/ma0605393. [DOI] [Google Scholar]
- Michel M, Vautier D, Voegel J-C, Schaaf P. and Ball V. Langmuir. 2004;20:4835. doi: 10.1021/la049736q. [DOI] [PubMed] [Google Scholar]
- Michel M, Izquierdo A, Decher G, Voegel C-G, Schaaf P. and Ball V. Langmuir. 2005;21:7854. doi: 10.1021/la050497w. [DOI] [PubMed] [Google Scholar]
- Lvov Y, Essler F. and Decher G. J. Phys. Chem. 1993;97:13773. doi: 10.1021/j100153a055. [DOI] [Google Scholar]
- Decher G. and Hong J D. Makromol. Chem. Macromol. Symp. 1991;46:3217. [Google Scholar]
- Ramsden J J, Lvov Y M. and Decher G. Thin Solid Films. 1995;254:246. doi: 10.1016/0040-6090(94)06262-J. [DOI] [Google Scholar]
- Toutianoush A. and Tieke B. Macromol. Rapid Commun. 1998;19:591. doi: 10.1002/(SICI)1521-3927(19981101)19:11<591::AID-MARC591>3.0.CO;2-4. [DOI] [Google Scholar]
- Locklin J, Youk J H, Xia C, Park M-K, Fan X. and Advincula R C. Langmuir. 2002;18:877. doi: 10.1021/la011219+. [DOI] [Google Scholar]
- Caruso F, Caruso R A. and Möhwald H. Science. 1998;282:1111. doi: 10.1126/science.282.5391.1111. [DOI] [PubMed] [Google Scholar]
- Caruso F. and Möhwald H. J. Am. Chem. Soc. 1998;121:6039. doi: 10.1021/ja990441m. [DOI] [Google Scholar]
- Caruso F. Adv. Mater. 2001;13:11. doi: 10.1002/1521-4095(200101)13:1<11::AID-ADMA11>3.0.CO;2-N. [DOI] [Google Scholar]
- Lee H, Kepley L J, Hong H G. and Mallouk T E. J. Am. Chem. Soc. 1988;110:618. doi: 10.1021/ja00210a062. [DOI] [Google Scholar]
- Zhu P, Kang H, Facchetti A, Evmenenko G, Dutta P. and Marks T J. J. Am. Chem. Soc. 2003;125:11496. doi: 10.1021/ja037164a. [DOI] [PubMed] [Google Scholar]
- Shimazaki Y, Nakamura R, Ito S. and Yamamoto M. Langmuir. 2001;17:953. doi: 10.1021/la001230u. [DOI] [Google Scholar]
- Ikeda A, Hatano T, Shinkai S, Akiyama T. and Yamada S. J. Am. Chem. Soc. 2001;123:4855. doi: 10.1021/ja015596k. [DOI] [PubMed] [Google Scholar]
- Lvov Y, Ariga K, Ichinose I. and Kunitake T. J. Chem. Soc., Chem. Commun. 1995:2313. doi: 10.1039/c39950002313. [DOI] [Google Scholar]
- Sato K, Imoto Y, Sugama J, Seki S, Inoue H, Odagiri T, Hoshi T. and Anzai J. Langmuir. 2005;21:797. doi: 10.1021/la048059x. [DOI] [PubMed] [Google Scholar]
- Chen J, Huang L, Ying L, Luo G, Zhao X. and Cao W. Langmuir. 1999;15:7208. doi: 10.1021/la981668i. [DOI] [Google Scholar]
- Kohli P. and Blanchard G J. Langmuir. 2000;16:4655. doi: 10.1021/la000120k. [DOI] [Google Scholar]
- Such G K, Quinn J F, Ouinn A, Tjipto E. and Caruso F. J. Am. Chem. Soc. 2006;128:9318. doi: 10.1021/ja063043+. [DOI] [PubMed] [Google Scholar]
- Cho J, Char K, Hong J-D. and Lee K-B. Adv. Mater. 2001;13:1076. doi: 10.1002/1521-4095(200107)13:14<1076::AID-ADMA1076>3.0.CO;2-M. [DOI] [Google Scholar]
- Izquierdo A, Ono S S, Schaaf J-C. and Decher G. Langmuir. 2005;21:7558. doi: 10.1021/la047407s. [DOI] [PubMed] [Google Scholar]
- Yamada M and Shiratori S S. 2000. Sens. Actuators B 64 124 10.1016/S0925-4005(99)00494-3 [DOI] [Google Scholar]
- Clark S L. and Hammond P T. Adv. Mater. 1998;10:1515. doi: 10.1002/(SICI)1521-4095(199812)10:18<1515::AID-ADMA1515>3.0.CO;2-E. [DOI] [Google Scholar]
- Lu G, Ai S. and Li J. Langmuir. 2005;21:1679. doi: 10.1021/la047771r. [DOI] [PubMed] [Google Scholar]
- Jiang C, Markutsya S, Pikus Y. and Tsukruk V V. Nat. Mater. 2004;3:721. doi: 10.1038/nmat1212. [DOI] [PubMed] [Google Scholar]
- Onda M, Lvov Y, Ariga K. and Kunitake T. Biotechnol. Bioeng. 1996;51:163. doi: 10.1002/(SICI)1097-0290(19960720)51:2<163::AID-BIT5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Onda M, Ariga K. and Kunitake T. J. Biosci. Bioeng. 1999;87:69. doi: 10.1016/S1389-1723(99)80010-3. [DOI] [PubMed] [Google Scholar]
- Onda M, Lvov Y, Ariga K. and Kunitake T. J. Ferment. Bioeng. 1996;82:502. doi: 10.1016/S0922-338X(97)86992-9. [DOI] [PubMed] [Google Scholar]
- Serizawa T, Hamada K, Kitayama T, Fujimoto N, Hatada K. and Akashi M. J. Am. Chem. Soc. 2000;122:1891. doi: 10.1021/ja9913535. [DOI] [Google Scholar]
- Serizawa T, Hamada K. and Akashi M. Nature. 2004;429:52. doi: 10.1038/nature02525. [DOI] [PubMed] [Google Scholar]
- Katagiri K, Hamasaki R, Ariga K. and Kikuchi J. Langmuir. 2002;18:6709. doi: 10.1021/la025772i. [DOI] [Google Scholar]
- Katagiri K, Hamasaki R, Ariga K. and Kikuchi J. J. Am. Chem. Soc. 2002;124:7892. doi: 10.1021/ja0259281. [DOI] [PubMed] [Google Scholar]
- Zhang J, Chua L S. and Lynn D M. Langmuir. 2004;20:8015. doi: 10.1021/la048888i. [DOI] [PubMed] [Google Scholar]
- Park M-K, Deng S. and Advincula R C. J. Am. Chem. Soc. 2004;126:13723. doi: 10.1021/ja0484707. [DOI] [PubMed] [Google Scholar]
- Lee D, Nolte A J, Kunz A L, Rubner M F. and Cohen R E. J. Am. Chem. Soc. 2006;128:8521. doi: 10.1021/ja0608803. [DOI] [PubMed] [Google Scholar]
- Mauser T, Déjugnat C, Möhwald H. and Sukhorukov G B. Langmuir. 2006;22:5888. doi: 10.1021/la060088f. [DOI] [PubMed] [Google Scholar]
- De Geest B G, Jonas A M, Demeester J. and De Smedt S C. Langmuir. 2006;22:5070. doi: 10.1021/la053368o. [DOI] [PubMed] [Google Scholar]
- Shchukin D G, Patel A A, Sukhorukov G B. and Lvov Y M. J. Am. Chem. Soc. 2004;126:3374. doi: 10.1021/ja036952x. [DOI] [PubMed] [Google Scholar]
- Ghan R, Shutava T, Patel A, John V T. and Lvov Y. Macromolecules. 2004;37:4519. doi: 10.1021/ma035896h. [DOI] [Google Scholar]
- Schneider G, Decher G, Nerambourg N, Praho R, Werts M H V. and Blanchard-Desce M. Nano Lett. 2006;6:530. doi: 10.1021/nl052441s. [DOI] [PubMed] [Google Scholar]
- Jiang X. and Hammond P T. Langmuir. 2000;16:8501. doi: 10.1021/la0004146. [DOI] [Google Scholar]
- Tokuhisa H. and Hammond P T. Adv. Funct. Mater. 2003;13:831. doi: 10.1002/adfm.200304404. [DOI] [Google Scholar]
- Yang S Y. and Rubner M F. J. Am. Chem. Soc. 2002;124:2100. doi: 10.1021/ja017681y. [DOI] [PubMed] [Google Scholar]
- Hua F, Cui T. and Lvov Y M. Nano Lett. 2004;4:823. doi: 10.1021/nl0498031. [DOI] [Google Scholar]
- Zhai L, Berg M C, Cebeci F Ç, Kim Y, Milwid J M, Rubner M F Ç. and Cohen R E. Nano Lett. 2006;6:1213. doi: 10.1021/nl060644q. [DOI] [PubMed] [Google Scholar]
- Lee D, Rubner M F. and Cohen R E. Nano Lett. 2006;6:2305. doi: 10.1021/nl061776m. [DOI] [PubMed] [Google Scholar]
- Oliveria O N, Riul A. and Leite V B P. Braz. J. Phys. 2004;34:73. [Google Scholar]
- Kim J-H, Fujita S. and Shiratori S. Thin Solid Films. 2006;499:83. doi: 10.1016/j.tsf.2005.07.001. [DOI] [Google Scholar]
- Dennany L, Forster R J, White B, Smyth M. and Rusling J F. J. Am. Chem. Soc. 2004;126:8835. doi: 10.1021/ja048615+. [DOI] [PubMed] [Google Scholar]
- Han B-H, Manners I. and Winnik M A. Chem. Mater. 2005;17:3160. doi: 10.1021/cm047770k. [DOI] [Google Scholar]
- Xu Y, Xu C, Shvarev A, Becker T, De Marco R. and Bakker E. Anal. Chem. 2007;79:7154. doi: 10.1021/ac071201p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E. and Jung S. Chem. Mater. 2005;17:6381. doi: 10.1021/cm051492n. [DOI] [Google Scholar]
- Liu S, Möhwald H, Volkmer D. and Kurth D G. Langmuir. 2006;22:1949. doi: 10.1021/la0523863. [DOI] [PubMed] [Google Scholar]
- Stricker J T, Gudmundsdóttir A D, Smith A P, Taylor B E and Durstock M F. 2007. J. Phys. Chem. B 111 6322 10.1021/jp0688862 [DOI] [PubMed] [Google Scholar]
- Jiang S P, Liu Z. and Tian Z Q. Adv. Mater. 2006;18:1068. doi: 10.1002/adma.200502462. [DOI] [Google Scholar]
- Tokuhisa H. and Hammond P T. Adv. Funct. Mater. 2003;13:831. doi: 10.1002/adfm.200304404. [DOI] [Google Scholar]
- Kovtyukhova N I, Martin B R, Mbindyo J K N, Smith P A, Razavi B, Mayer T S and Mallouk T E. 2001. J. Phys. Chem. B 105 8762 10.1021/jp010867z [DOI] [Google Scholar]
- Jiang S. and Liu M. Chem. Mater. 2004;16:3985. doi: 10.1021/cm0491476. [DOI] [Google Scholar]
- Ulman A. Chem. Rev. 1996;96:1533. doi: 10.1021/cr9502357. [DOI] [PubMed] [Google Scholar]
- Shirahata N, Seo W-S, Kinoshita T, Yonezawa T, Hozumi A, Yokogawa Y, Kameyama T, Masuda Y. and Koumoto K. Langmuir. 2004;20:8942. doi: 10.1021/la036362c. [DOI] [PubMed] [Google Scholar]
- Yang L, Lua Y-Y, Lee M V. and Linford M R. Acc. Chem. Res. 2005;38:933. doi: 10.1021/ar040242u. [DOI] [PubMed] [Google Scholar]
- Love J C, Estroff L A, Kriebel J K, Nuzzo R G. and Whitesides G M. Chem. Rev. 2005;105:1103. doi: 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- Shirahata N, Hozumi A. and Yonezawa T. Chem. Rec. 2005;5:145. doi: 10.1002/tcr.20041. [DOI] [PubMed] [Google Scholar]
- Lua Y-Y, Fillmore W J J, Yang L, Lee M V, Savage P B, Asplund M C. and Linford M R. Langmuir. 2005;21:2093. doi: 10.1021/la047338h. [DOI] [PubMed] [Google Scholar]
- Tao F. and Bernasek S L. Chem. Rev. 2007;107:1408. doi: 10.1021/cr050258d. [DOI] [PubMed] [Google Scholar]
- Okahata Y, Ariga K. and Shimizu O. Langmuir. 1986;2:538. doi: 10.1021/la00070a028. [DOI] [Google Scholar]
- Okahata Y, Ariga K, Nakahara H. and Fukuda K. J. Chem. Soc., Chem. Commun. 1986:1069. doi: 10.1039/c39860001069. [DOI] [Google Scholar]
- Ariga K. and Okahata Y. J. Am. Chem. Soc. 1989;111:5618. doi: 10.1021/ja00197a018. [DOI] [Google Scholar]
- Hasobe T, Imahori H, Kamat P V, Ahn T K, Kim S K, Kim D, Fujimoto A, Hirakawa T. and Fukuzumi S. J. Am. Chem. Soc. 2005;127:1216. doi: 10.1021/ja047768u. [DOI] [PubMed] [Google Scholar]
- Imahori H. et al Chem. Eur. J. 2005;11:7265. doi: 10.1002/chem.200500610. [DOI] [PubMed] [Google Scholar]
- Imahori H, Mitamura K, Umeyama T, Hosomizu K, Matano Y, Yoshida K. and Isoda S. Chem. Commun. 2006:406. doi: 10.1039/b513707a. [DOI] [PubMed] [Google Scholar]
- Hasobe T, Hattori S, Kamat P V. and Fukuzumia S. Tetrahedron. 2006;62:1937. doi: 10.1016/j.tet.2005.05.113. [DOI] [Google Scholar]
- Imahori H. 2004. J. Phys. Chem. B 108 6130 10.1021/jp038036b [DOI] [PubMed] [Google Scholar]
- Liu Z, Yasseri A A, Lindsey J S. and Bocian D F. Science. 2003;302:1543. doi: 10.1126/science.1090677. [DOI] [PubMed] [Google Scholar]
- De Luca G, Pollicino G, Romeo A. and Scolaro L M. Chem. Mater. 2006;18:2005. doi: 10.1021/cm0524300. [DOI] [Google Scholar]
- Gulino A, Bazzano S, Mineo P, Scamporrino E, Vitalini D. and Fragalà I. Chem. Mater. 2005;17:521–6. doi: 10.1021/cm048130k. [DOI] [Google Scholar]
- Gulino A, Giuffrida S, Mineo P, Purrazzo M, Scamporrino E, Ventimiglia G, van der Boom M E and Fragalà I. 2006. J. Phys. Chem. B 110 16781 10.1021/jp062967g [DOI] [PubMed] [Google Scholar]
- Gulino A, Mineo P, Scamporrino E, Vitalini D. and Fragalà I. Chem. Mater. 2006;18:2404. doi: 10.1021/cm060086g. [DOI] [Google Scholar]
- Jensen A W. and Daniels C. J. Org. Chem. 2003;68:207. doi: 10.1021/jo025926z. [DOI] [PubMed] [Google Scholar]
- Otsubo T, Aso Y. and Takimiya K. J. Mater. Chem. 2002;12:2565. doi: 10.1039/b203780g. [DOI] [Google Scholar]
- Liu W, Zhang Y. and Gao X. J. Am. Chem. Soc. 2007;129:4973. doi: 10.1021/ja067056z. [DOI] [PubMed] [Google Scholar]
- Shirai Y, Cheng L, Chen B. and Tour J M. J. Am. Chem. Soc. 2006;128:13479. doi: 10.1021/ja063451d. [DOI] [PubMed] [Google Scholar]
- Fan F-R F, Yang J, Cai L, Price D W, Jr, Dirk S M, Kosynkin D V, Yao Y, Rawlett A M, Tour J M. and Bard A J. J. Am. Chem. Soc. 2002;124:5550. doi: 10.1021/ja017706t. [DOI] [PubMed] [Google Scholar]
- Moore A M, Dameron A A, Mantooth B A, Smith R K, Fuchs D J, Ciszek J W, Maya F, Yao Y, Tour J M. and Weiss P S. J. Am. Chem. Soc. 2006;128:1959. doi: 10.1021/ja055761m. [DOI] [PubMed] [Google Scholar]
- Kim D H, Han J T, Park Y D, Jang Y, Cho J H, Hwang M. and Cho K. Adv. Mater. 2006;18:719. doi: 10.1002/adma.200502442. [DOI] [Google Scholar]
- Ballauff M. and Borisov O. Curr. Opin. Colloid Interface Sci. 2006;11:316. doi: 10.1016/j.cocis.2006.12.002. [DOI] [Google Scholar]
- Azzaroni O, Brown A A, Cheng N, Wei A, Jonas A M. and Huck W T S. J. Mater. Chem. 2007;17:3433. doi: 10.1039/b704849a. [DOI] [Google Scholar]
- Zhong W, Wang Y, Yan Y, Sun Y, Deng J and Yang W. 2007. J. Phys. Chem. B 111 3918 10.1021/jp0678296 [DOI] [PubMed] [Google Scholar]
- Shirahata N. and Hozumi A. J. Nanosci. Nanotechnol. 2006;6:1695. doi: 10.1166/jnn.2006.238. [DOI] [PubMed] [Google Scholar]
- Cavalli S, Handgraaf J-W, Tellers E E, Popescu D C, Overhand M, Kjaer K, Vaiser V, Sommerdijk N A J M, Rapaport H. and Kros A. J. Am. Chem. Soc. 2006;128:13959. doi: 10.1021/ja065479v. [DOI] [PubMed] [Google Scholar]
- Shirahata N, Hozumi A, Miura Y, Kobayashi K, Sakka Y. and Yonezawa T. Thin Solid Films. 2006;499:213. doi: 10.1016/j.tsf.2005.07.048. [DOI] [Google Scholar]
- Ito Ma, Nakamura F, Baba A, Tamada K, Ushijima H, Lau K H A, Manna A and Knoll W. 2007. J. Phys. Chem. C 111 11653 10.1021/jp070524m [DOI] [Google Scholar]
- Turygin D S, Subat M, Raitman O A, Arslanov V V, König B. and Kalinina M A. Angew. Chem. Int. Ed. 2006;45:5340. doi: 10.1002/anie.200600450. [DOI] [PubMed] [Google Scholar]
- Liu Z, Shen Z, Zhu T, Hou S, Ying L, Shi Z. and Gu Z. Langmuir. 2000;16:3569. doi: 10.1021/la9914110. [DOI] [Google Scholar]
- Weitz R T, Zschieschang U, Effenberger F, Klauk H, Burghard M. and Kern K. Nano Lett. 2007;7:22. doi: 10.1021/nl061534m. [DOI] [PubMed] [Google Scholar]
- Busi M, Laurenti M, Condorelli G G, Motta A, Favazza M, Fragalà I L, Montalti M, Prodi L. and Dalcanale E. Chem. Eur. J. 2007;13:6891. doi: 10.1002/chem.200700496. [DOI] [PubMed] [Google Scholar]
- Beulen M W J. et al Chem. Eur. J. 2000;6:1176. doi: 10.1002/(SICI)1521-3765(20000403)6:7<1176::AID-CHEM1176>3.3.CO;2-T. [DOI] [Google Scholar]
- Endo H, Nakaji-Hirabayashi T, Morokoshi S, Gemmei-Ide M. and Kitano H. Langmuir. 2005;21:1314. doi: 10.1021/la048595p. [DOI] [PubMed] [Google Scholar]
- Achyerbaum K D, Weiss T, van Velzen E U T, Reinhoudt J F J. and Göpel W. Science. 1994;265:1413. doi: 10.1126/science.265.5177.1413. [DOI] [PubMed] [Google Scholar]
- Hozumi A, Inagaki M. and Shirahata N. Surf. Sci. 2006;600:4044. doi: 10.1016/j.susc.2006.01.120. [DOI] [Google Scholar]
- Kovtyukhova N I. and Mallouk T E. Chem. Eur. J. 2002;8:4354. doi: 10.1002/1521-3765(20021004)8:19<4354::AID-CHEM4354>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Yamanoi Y. and Nishihara H. Chem. Commun. 2007:3983. doi: 10.1039/b703342g. [DOI] [PubMed] [Google Scholar]
- Li H. and Qu F. J. Mater. Chem. 2007;17:3536. doi: 10.1039/b705743a. [DOI] [Google Scholar]
- De Girolamo J, Reiss P and Pron A. 2007. J. Phys. Chem. C 111 14681 10.1021/jp0741758 [DOI] [Google Scholar]
- Jang S S. et al J. Am. Chem. Soc. 2005;127:1563. doi: 10.1021/ja044530x. [DOI] [PubMed] [Google Scholar]
- Liu Y. et al J. Am. Chem. Soc. 2005;127:9745. doi: 10.1021/ja051088p. [DOI] [PubMed] [Google Scholar]
- Chaudhury M K. and Whitesides G M. Science. 1992;256:1539. doi: 10.1126/science.256.5063.1539. [DOI] [PubMed] [Google Scholar]
- Ichimura K, Oh S-K. and Nakagawa M. Science. 2000;288:1624. doi: 10.1126/science.288.5471.1624. [DOI] [PubMed] [Google Scholar]
- Hong S, Zhu J. and Mirkin C A. Science. 1999;286:523. doi: 10.1126/science.286.5439.523. [DOI] [PubMed] [Google Scholar]
- Shirahata N, Shin W, Murayama N, Hozumi A, Yokogawa Y, Kameyama T, Masuda Y. and Koumoto K. Adv. Funct. Mater. 2004;14:580. doi: 10.1002/adfm.200305060. [DOI] [Google Scholar]
- Mendes P M. and Preece J A. Curr. Opin. Colloid Interface Sci. 2004;9:236. doi: 10.1016/j.cocis.2004.06.005. [DOI] [Google Scholar]
- Chen M-S, Dulcey C S, Chrisey L A. and Dressick W J. Adv. Funct. Mater. 2006;16:774. doi: 10.1002/adfm.200500744. [DOI] [Google Scholar]
- Speets E A, te Riele P, van den Boogaart M A F, Doeswijk L M, Ravoo B J, Rijnders G, Brugger J, Reinhoudt D N. and Blank D H A. Adv. Funct. Mater. 2006;16:1337. doi: 10.1002/adfm.200500933. [DOI] [Google Scholar]
- Lim H S, Han J T, Kwak D, Jin M. and Cho K. J. Am. Chem. Soc. 2006;128:14458. doi: 10.1021/ja0655901. [DOI] [PubMed] [Google Scholar]
- Chowdhury D, Maoz R. and Sagiv J. Nano Lett. 2007;7:1770. doi: 10.1021/nl070842x. [DOI] [PubMed] [Google Scholar]
- Rhinow D. and Hampp N A. Adv. Mater. 2007;19:1967. doi: 10.1002/adma.200602387. [DOI] [Google Scholar]
- Rabe J P. and Buchholz S. Science. 1991;253:424. doi: 10.1126/science.253.5018.424. [DOI] [PubMed] [Google Scholar]
- Oishi Y, Kato T, Kuramori M, Suehiro K, Ariga K, Kamino A, Koyano H. and Kunitake T. Chem. Lett. 1996:857. doi: 10.1246/cl.1996.857. [DOI] [Google Scholar]
- Kim K B, Plass K E. and Matzger A J. Langmuir. 2005;21:647. doi: 10.1021/la048299c. [DOI] [PubMed] [Google Scholar]
- Tao F. and Bernasek S L. J. Am. Chem. Soc. 2005;127:12750. doi: 10.1021/ja050365p. [DOI] [PubMed] [Google Scholar]
- Plass K E, Engle K M, Cychosz K A. and Matzger A J. Nano Lett. 2006;6:1178. doi: 10.1021/nl0605061. [DOI] [PubMed] [Google Scholar]
- Mamdouh W, Uji-i H, Ladislaw J S, Dulcey A E, Percec V, De Schryver F C. and De Feyter S. J. Am. Chem. Soc. 2006;128:317. doi: 10.1021/ja056175w. [DOI] [PubMed] [Google Scholar]
- Puigmartí-Luis J, Minoia A, Uji-i H, Rovira C, Cornil J, De Feyter S, Lazzaroni R. and Amabilino D B. J. Am. Chem. Soc. 2006;128:12602. doi: 10.1021/ja0640288. [DOI] [PubMed] [Google Scholar]
- Gong J-R, Yan H-J, Yuan Q-H, Xu L-P, Bo Z-S. and Wan L-J. J. Am. Chem. Soc. 2006;128:12384. doi: 10.1021/ja062533z. [DOI] [PubMed] [Google Scholar]
- Otsuki J, Shimizu S. and Fumino M. Langmuir. 2006;22:6056. doi: 10.1021/la060924l. [DOI] [PubMed] [Google Scholar]
- Rädari H J, Rouhanipour A, Talarico A M, Palermo V, Samorí P. and Müllen K. Nat. Mater. 2006;5:276. doi: 10.1038/nmat1597. [DOI] [PubMed] [Google Scholar]
- Chiaravalloti F, Gross L, Rieder K-H, Stojkovic S M, Gourdon A, Joachim C. and Moresco F. Nat. Mater. 2007;6:30. doi: 10.1038/nmat1802. [DOI] [PubMed] [Google Scholar]
- Klymchenko A S, Furukawa S, Müllen K, Van der Auweraer M. and De Feyter S. Nano Lett. 2007;7:791. doi: 10.1021/nl0700752. [DOI] [PubMed] [Google Scholar]
- Ogaki K, Batina N, Kunitake M. and Itaya K. J. Phys. Chem. 1996;1100:7185. doi: 10.1021/jp953517j. [DOI] [Google Scholar]
- Sekiguchi T, Wakayama Y, Yokoyama S, Kamikado T. and Mashiko S. Thin Solid Films. 2004;464–5:393. doi: 10.1016/j.tsf.2004.06.084. [DOI] [Google Scholar]
- Otsuki J, Nagamine E, Kondo T, Iwasaki K, Asakawa M. and Miyake K. J. Am. Chem. Soc. 2005;127:10400. doi: 10.1021/ja0531778. [DOI] [PubMed] [Google Scholar]
- Otsuki J, Kawaguchi S, Yamakawa T, Asakawa M. and Miyake K. Langmuir. 2006;22:5708. doi: 10.1021/la0608617. [DOI] [PubMed] [Google Scholar]
- Kamikado T, Sekiguchi T, Yokoyama S, Wakayama Y. and Mashiko S. Thin Solid Films. 2006;499:329. doi: 10.1016/j.tsf.2005.06.074. [DOI] [Google Scholar]
- Li W-S, Kim K S, Jiang D-L, Tanaka H, Kawai T, Kwon J H, Kim D. and Aida T. J. Am. Chem. Soc. 2006;128:10527. doi: 10.1021/ja063081t. [DOI] [PubMed] [Google Scholar]
- Katsonis N, Vicario J, Kudernac T, Visser J, Pollard M M. and Feringa B L. J. Am. Chem. Soc. 2006;128:15537. doi: 10.1021/ja065823o. [DOI] [PubMed] [Google Scholar]
- Hoeben F J M, Wolffs M, Zhang J, De Feyter S, Leclère P, Schenning A P H J. and Meijer E W. J. Am. Chem. Soc. 2007;129:9819. doi: 10.1021/ja072548c. [DOI] [PubMed] [Google Scholar]
- Lensen M C, Elemans J A A W, van Dingenen S J T, Gerritsen J W, Speller S, Rowan A E. and Nolte R J M. Chem. Eur. J. 2007;13:7948. doi: 10.1002/chem.200700131. [DOI] [PubMed] [Google Scholar]
- Yoshimoto S, Narita R, Tsutsumi E, Matsumoto M, Itaya K, Ito O, Fujiwara K, Murata Y. and Komatsu K. Langmuir. 2002;18:8518. doi: 10.1021/la0205147. [DOI] [Google Scholar]
- Yoshimoto S, Honda Y, Murata Y, Murata M, Komatsu K, Ito O and Itaya K. 2005. J. Phys. Chem. B 109 8547 10.1021/jp051112l [DOI] [PubMed] [Google Scholar]
- Pan G-B, Cheng X-H, Höger S. and Freyland W. J. Am. Chem. Soc. 2006;128:4218. doi: 10.1021/ja060469f. [DOI] [PubMed] [Google Scholar]
- Shirai Y. et al J. Am. Chem. Soc. 2006;128:4854. doi: 10.1021/ja058514r. [DOI] [PubMed] [Google Scholar]
- Yoshimoto S, Tsutsumi E, Narita R, Murata Y, Murata M, Fujiwara K, Komatsu K, Ito O. and Itaya K. J. Am. Chem. Soc. 2007;129:4366. doi: 10.1021/ja0684848. [DOI] [PubMed] [Google Scholar]
- Feng M, Lee J, Zhao J, Yates J T Jr. and Petek H. J. Am. Chem. Soc. 2007;129:12394. doi: 10.1021/ja075239v. [DOI] [PubMed] [Google Scholar]
- Deak D S, Porfyrakis K. and Castell M R. Chem. Commun. 2007:2941. doi: 10.1039/b703081a. [DOI] [PubMed] [Google Scholar]
- Schenning A P H J. and Meijer E W. Chem. Commun. 2005:3245. doi: 10.1039/b501804h. [DOI] [PubMed] [Google Scholar]
- Okawa Y. and Aono M. Nature. 2001;409:683. doi: 10.1038/35055625. [DOI] [PubMed] [Google Scholar]
- Takajo D, Okawa Y, Hasegawa T. and Aono M. Langmuir. 2007;23:5247. doi: 10.1021/la700241z. [DOI] [PubMed] [Google Scholar]
- Sakaguchi H, Matsumura H. and Gong H. Nat. Mater. 2004;3:551. doi: 10.1038/nmat1176. [DOI] [PubMed] [Google Scholar]
- Sakaguchi H, Matsumura H, Gong H. and Abouelwafa A M. Science. 2005;310:1002. doi: 10.1126/science.1117990. [DOI] [PubMed] [Google Scholar]
- Hill J P, Wakayama Y, Schmitt W, Tsuruoka T, Nakanishi T, Zandler M L, McCarty A L, D'Souza F, Milgrom L R. and Ariga K. Chem. Commun. 2006:2320. doi: 10.1039/b517668a. [DOI] [PubMed] [Google Scholar]
- Wakayama Y, Hill J P. and Ariga K. Surf. Sci. 2007;601:3984. doi: 10.1016/j.susc.2007.04.068. [DOI] [Google Scholar]
- Hill J P, Wakayama Y. and Ariga K. Phys. Chem. Chem. Phys. 2006;8:5034. doi: 10.1039/b608935f. [DOI] [PubMed] [Google Scholar]
- Hill J P, Wakayama Y, Akada M and Ariga K. 2007. J. Phys. Chem. C 111 16174 10.1021/jp0745945 [DOI] [Google Scholar]
- Yokoyama T, Yokoyama S, Kamikado T, Okuno Y. and Mashiko S. Nature. 2001;413:619. doi: 10.1038/35098059. [DOI] [PubMed] [Google Scholar]
- Barth J V, Weckesser J, Cai C, Günter P, Bürgi L, Jeandupeux O. and Kern K. Angew. Chem. Int. Ed. 2000;39:1230. doi: 10.1002/(SICI)1521-3773(20000403)39:7<1230::AID-ANIE1230>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Nakanishi T, Miyashita N, Michinobu T, Wakayama Y, Tsuruoka T, Ariga K. and Kurth D G. J. Am. Chem. Soc. 2006;128:6328. doi: 10.1021/ja061450f. [DOI] [PubMed] [Google Scholar]
- Nakanishi T, Takahashi H, Michinobu T, Takeuchi M, Teranishi T and Ariga K. 2007. Colloid Surf. A-Physicochem. Eng. Asp., in press. doi: 10.1016/j.colsurfa.2007.11.029 [DOI] [Google Scholar]
- Yuan Q-H, Wan L-J, Jude H. and Stang P J. J. Am. Chem. Soc. 2005;127:16279. doi: 10.1021/ja0549300. [DOI] [PubMed] [Google Scholar]
- Stepanow S, Lingenfelder M, Dmitriev A, Spillmann H, Delvigne E, Lin N, Deng X, Cai C, Barth J V. and Kern K. Nat. Mater. 2004;3:229. doi: 10.1038/nmat1088. [DOI] [PubMed] [Google Scholar]
- Patla I, Acharya S, Zeiri L, Israelachvili J, Efrima S. and Golan Y. Nano Lett. 2007;7:1459. doi: 10.1021/nl070001q. [DOI] [PubMed] [Google Scholar]
- Santhanam V, Liu J, Agarwal R. and Andres R P. Langmuir. 2003;19:7881. doi: 10.1021/la0341761. [DOI] [PubMed] [Google Scholar]
- Lin Y, Skaff H, Emrick T, Dinsmore A D. and Russell T P. Science. 2003;299:226. doi: 10.1126/science.1078616. [DOI] [PubMed] [Google Scholar]
- Bagkar N, Ganguly R, Choudhury S, Hassan P A, Sawanta S. and Yakhmi J V. J. Mater. Chem. 2004;14:1430. doi: 10.1039/b315866g. [DOI] [Google Scholar]
- Lu Y, Liu G L. and Lee L P. Nano Lett. 2005;5:5. doi: 10.1021/nl048965u. [DOI] [PubMed] [Google Scholar]
- Bala T, Joshi B, Iyer N, Sastry M. and Prasad B L V. J. Nanosci. Nanotechnol. 2006;6:3736. doi: 10.1166/jnn.2006.615. [DOI] [PubMed] [Google Scholar]
- Lee D K, Kim Y H, Kim C W, Cha H G and Kang Y S. 2007. J. Phys. Chem. B 111 9288 10.1021/jp072612c [DOI] [PubMed] [Google Scholar]
- Wei H, Ma N, Song B, Yin S and Wang Z. 2007. J. Phys. Chem. C 111 5628 10.1021/jp068843l [DOI] [Google Scholar]
- Pradhan S, Xu L. and Chen S. Adv. Funct. Mater. 2007;17:2385. doi: 10.1002/adfm.200601034. [DOI] [Google Scholar]
- Clemente-León M, Ito T, Yashiro H. and Yamase T. Chem. Mater. 2007;19:2589. doi: 10.1021/cm070436e. [DOI] [Google Scholar]
- Yang P. and Kim F. Chem. Phys. Chem. 2002;3:503. doi: 10.1002/1439-7641(20020617)3:6<503::AID-CPHC503>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Tao A, Kim F, Hess C, Goldberger J, He R, Sun Y, Xia Y. and Yang P. Nano Lett. 2003;3:1229. doi: 10.1021/nl0344209. [DOI] [Google Scholar]
- Panda A B, Acharya S. and Efrima S. Adv. Mater. 2005;17:2471. doi: 10.1002/adma.200500551. [DOI] [Google Scholar]
- Acharya S. and Efrima S. J. Am. Chem. Soc. 2005;127:3486. doi: 10.1021/ja044934p. [DOI] [PubMed] [Google Scholar]
- Acharya S, Patla I, Kost J, Efrima S. and Golan Y. J. Am. Chem. Soc. 2006;128:9294. doi: 10.1021/ja062404i. [DOI] [PubMed] [Google Scholar]
- Acharya S, Baran Panda A, Belman N, Efrima S. and Golan Y. Adv. Mater. 2006;18:210. doi: 10.1002/adma.200501234. [DOI] [Google Scholar]
- Acharya S, Panda A B, Efrima S. and Golan Y. Adv. Mater. 2007;19:1105. doi: 10.1002/adma.200602057. [DOI] [Google Scholar]
- Collier C P, Saykally R J, Shiang J J, Henrichs S E. and Heath J R. Science. 1997;277:1978. doi: 10.1126/science.277.5334.1978. [DOI] [Google Scholar]
- Kanehara M, Oumi Y, Sano T. and Teranishi T. J. Am. Chem. Soc. 2003;125:8708. doi: 10.1021/ja035187j. [DOI] [PubMed] [Google Scholar]
- Zeng H, Li J, Liu J P, Wang Z L. and Sun S. Nature. 2002;420:395. doi: 10.1038/nature01208. [DOI] [PubMed] [Google Scholar]
- Rabani E, Reichman D R, Geissler P L. and Brus L E. Nature. 2003;426:271. doi: 10.1038/nature02087. [DOI] [PubMed] [Google Scholar]
- Bigioni T P, Lin X-M, Nguyen T T, Corwin E, Witten T A. and Jaeger H M. Nat. Mater. 2006;5:265. doi: 10.1038/nmat1611. [DOI] [PubMed] [Google Scholar]
- Shevchenko E V, Talapin D V, Murray C B. and O'Brien S. J. Am. Chem. Soc. 2006;128:3620. doi: 10.1021/ja0564261. [DOI] [PubMed] [Google Scholar]
- Kanehara M, Kodzuka E. and Teranishi T. J. Am. Chem. Soc. 2006;128:13084. doi: 10.1021/ja064510q. [DOI] [PubMed] [Google Scholar]
- Kan S, Mokari T, Rothemberg E. and Banin U. Nat. Mater. 2003;2:155. doi: 10.1038/nmat830. [DOI] [PubMed] [Google Scholar]
- Sun B. and Sirringhaus H. Nano Lett. 2005;5:2408. doi: 10.1021/nl051586w. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Gupta S, Emrick T. and Russell T P. J. Am. Chem. Soc. 2006;128:3898. doi: 10.1021/ja058615p. [DOI] [PubMed] [Google Scholar]
- Sun B. and Sirringhaus H. J. Am. Chem. Soc. 2006;128:16231. doi: 10.1021/ja065242z. [DOI] [PubMed] [Google Scholar]
- Chen M, Pica T, Jiang Y-B, Li P, Yano K, Liu J P, Datye A K. and Fan H. J. Am. Chem. Soc. 2007;129:6349. doi: 10.1021/ja069057x. [DOI] [PubMed] [Google Scholar]
- Carbone L. et al Nano Lett. 2007;7:2942. doi: 10.1021/nl0717661. [DOI] [PubMed] [Google Scholar]
- Liu B. and Zeng H C. J. Am. Chem. Soc. 2005;127:18262. doi: 10.1021/ja055734w. [DOI] [PubMed] [Google Scholar]
- Tang Z, Zhang Z, Wang Y, Glotzer S C. and Kotov N A. Science. 2006;314:274. doi: 10.1126/science.1128045. [DOI] [PubMed] [Google Scholar]
- Chen Y, Johnson E. and Peng X. J. Am. Chem. Soc. 2007;129:10937. doi: 10.1021/ja073023n. [DOI] [PubMed] [Google Scholar]
- Yamanoi Y, Shirahata N, Yonezawa T, Terasaki N, Yamamoto N, Matsui Y, Nishio K, Masuda H, Ikuhara Y. and Nishihara H. Chem. Eur. J. 2006;12:314. doi: 10.1002/chem.200500455. [DOI] [PubMed] [Google Scholar]
- Li H, Wang Y, Zhang W, Liu B. and Calzaferri G. Chem. Commun. 2007:2853. doi: 10.1039/b702175e. [DOI] [PubMed] [Google Scholar]
- Xu H. and Goedel W A. Langmuir. 2002;18:2363. doi: 10.1021/la011026m. [DOI] [Google Scholar]
- Mano T. et al Nano Lett. 2005;5:425. doi: 10.1021/nl048192+. [DOI] [PubMed] [Google Scholar]
- Owen J H G, Miki K. and Bowler D R. J. Mater. Sci. 2006;41:4568. doi: 10.1007/s10853-006-0246-x. [DOI] [Google Scholar]
- Shirahata N, Nakanishi T, Furumi S. and Sakka Y. J. Nanosci. Nanotechnol. 2006;6:1823. doi: 10.1166/jnn.2006.209. [DOI] [PubMed] [Google Scholar]
- Yoo P J, Nam K T, Qi J, Lee S-K, Park J, Belcher A M. and Hammond P T. Nat. Mater. 2006;5:234. doi: 10.1038/nmat1596. [DOI] [PubMed] [Google Scholar]
- Kim J-H, Rahman M S, Lee J-S. and Park J-W. J. Am. Chem. Soc. 2007;129:7756. doi: 10.1021/ja072412e. [DOI] [PubMed] [Google Scholar]
- Chworos A, Severcan I, Koyfman A Y, Weinkam P, Oroudjev E, Hansma H G. and Jaeger L. Science. 2004;306:2068. doi: 10.1126/science.1104686. [DOI] [PubMed] [Google Scholar]
- Le J D, Pinto Y, Seeman N C, Musier-Forsyth K, Taton T A. and Kiehl R A. Nano Lett. 2004;4:2343. doi: 10.1021/nl048635+. [DOI] [PubMed] [Google Scholar]
- Pinto Y Y, Le J D, Seeman N C, Musier-Forsyth K, Taton T A. and Kiehl R A. Nano Lett. 2005;5:2399. doi: 10.1021/nl0515495. [DOI] [PubMed] [Google Scholar]
- He Y, Chen Y, Liu H, Ribbe A E. and Mao C. J. Am. Chem. Soc. 2005;127:12202. doi: 10.1021/ja0541938. [DOI] [PubMed] [Google Scholar]
- Lund K, Liu Y, Lindsay S. and Yan H. J. Am. Chem. Soc. 2005;127:17606. doi: 10.1021/ja0568446. [DOI] [PubMed] [Google Scholar]
- Winfree E, Liu F, Wenzler L A. and Seeman N C. Nature. 1998;394:539. doi: 10.1038/28998. [DOI] [PubMed] [Google Scholar]
- Zhang J, Liu Y, Ke Y. and Yan H. Nano Lett. 2006;6:248. doi: 10.1021/nl052210l. [DOI] [PubMed] [Google Scholar]