

Abstract
Recent advances in direct-acting antivirals against Hepatitis C Virus (HCV) have led to the development of potent inhibitors, including MK-5172, that target the viral NS3/4A protease with relatively low susceptibility to resistance. MK-5172 has a P2–P4 macrocycle and a unique binding mode among current protease inhibitors where the P2 quinoxaline packs against the catalytic residues H57 and D81. However, the effect of macrocyclization on this binding mode is not clear, as is the relation between macrocyclization, thermodynamic stabilization, and susceptibility to the resistance mutation A156T. We have determined high-resolution crystal structures of linear and P1–P3 macrocyclic analogs of MK-5172 bound to WT and A156T protease and compared these structures, their molecular dynamics, and experimental binding thermodynamics to the parent compound. We find that the “unique” binding mode of MK-5172 is conserved even when the P2–P4 macrocycle is removed or replaced with a P1–P3 macrocycle. While beneficial to decreasing the entropic penalty associated with binding, the constraint exerted by the P2–P4 macrocycle prevents efficient rearrangement to accommodate the A156T mutation, a deficit alleviated in the linear and P1–P3 analogs. Design of macrocyclic inhibitors against NS3/4A needs to achieve the best balance between exerting optimal conformational constraint for enhancing potency, fitting within the substrate envelope and allowing adaptability to be robust against resistance mutations.
Graphical abstract
Hepatitis C virus (HCV) causes chronic liver infection that affects about 3% of the global population and is the main cause of hepatitis, cirrhosis, and liver cancer.1–3 HCV has highly error prone replication and therefore is a rapidly evolving, highly diverse virus with six known genotypes and multiple subtypes.4,5 Before the recent availability of direct-acting antivirals (DAAs), the standard of care consisted of pegylated-interferon α and ribavirin with moderate to low rates of cure across genotypes and low tolerability.3,6 Current efforts aim to determine the best-in-class DAAs that target several viral proteins including the viral entry protein, the NS3/4A protease, the NS5A and NS5B non structural proteins,7 and host microRNAs8 either individually or in combination. Four FDA-approved HCV inhibitors (telaprevir,9,10 boceprevir,11 simeprevir,12 and most recently, paritaprevir13) target the NS3/4A protease. The NS3/4A protein is a bifunctional enzyme containing an N-terminal serine protease domain (amino acids 1–180) with the classic catalytic triad (S139, H57, D81) of the chymotrypsin superfamily and a C-terminal DExH/D-box helicase of superfamily II with NTPase activity.14–16 The NS3/4A protease is responsible for cleaving the viral polyprotein and host factors involved in the innate immune response, including TRIF and MAVS. Thus, targeting the NS3/4A protease achieves a two-pronged attack on the virus by preventing viral maturation and restoring the immune response.17–20
As the HCV NS3/4A inhibitors are a key component of combination therapy and increasing the cure rate across HCV genotypes, many more are currently in advanced clinical trials. Among these inhibitors, MK-5172 stands out with relatively high pan-genotypic potency.21 MK-5172 shares the same peptidomimetic core P1′–P3 scaffold as several other HCV PIs (danoprevir, asunaprevir, and vaniprevir) but is distinct in its P4 capping, P2 quinoxaline moiety connected to the P2 proline via an ether linkage, and finally, its P2–P4 macrocycle (Figure 1A).22,23 While the majority of NS3/4A inhibitors are susceptible to single site mutations R155K, A156T, and D168A, MK-5172 is more robust against resistance with the exception of A156T in vitro.21,24 These single site mutations are able to confer resistance to most inhibitors, depending mostly on the inhibitor P2 moiety:21,24,25 The large P2 moiety of inhibitors protrude out from the substrate envelope and extensively contact R155 and D168 residues that mutate to confer resistance.24,26 The cocrystal structure we determined showed that MK-5172 thwarts resistance at these sites as the P2 quinoxaline moiety largely interacts with residues of the catalytic triad.24 This binding mode explains MK-5172’s relatively high potency against R155K and D168A and provides a unique barrier to resistance, as the catalytic residues cannot mutate without compromising proteolytic activity.
Figure 1.
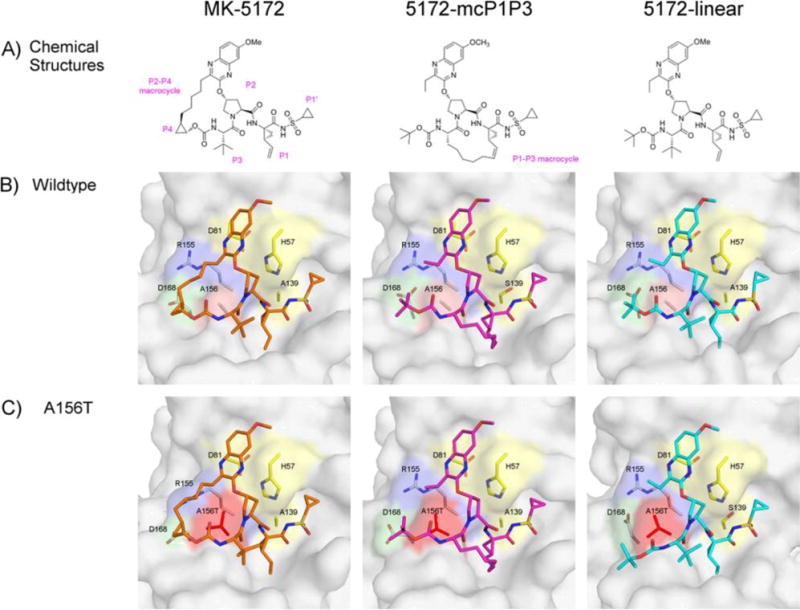
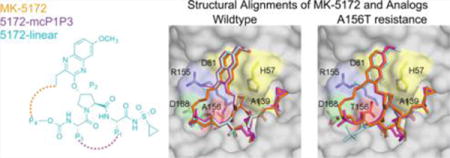
MK-5172 and analogs binding to HCV NS3/4A protease. (A) The chemical structures of MK-5172, 5172-mcP1P3, and 5172-linear. The P1′ to P4 moieties are indicated on the MK-5172 structure. Binding of the compounds at the active site in (B) WT and (C) A156T protease. The absence of the P2–P4 macrocycle does not disrupt the P2 quinoxaline’s interactions with the catalytic aspartate/histidine residues. The catalytic triad is highlighted in yellow, R155 in blue, D168 in green, and A/T156 in red.
Another distinct chemical feature of MK-5172 is the macrocycle that connects the P2 quinoxaline moiety to the P4 capping group. Macrocyclization is a general strategy employed to increase inhibitor potency in a wide array of drug targets.27,28 By introducing a conformational constraint to the inhibitor, the structure can be preorganized in a binding-competent conformation to decrease the entropic penalty associated with target association. However, the effect of macrocyclization on the energetics of binding for MK-5172 has not been quantified. In fact, there are no previously published data on the thermodynamics of binding for any active-site/competitive HCV protease inhibitor. For NS3/4A protease, we have previously found by enzymatic and antiviral assays that P1–P3 and P2–P4 macrocyclic inhibitors are more potent compared to their linear analogs and that the location of the macrocycle significantly affects resistance profiles.25 In particular, the P1–P3 macrocyclic analog of MK-5172 is more robust against the A156T resistance mutation; however, whether the unique binding mode on the catalytic triad is retained and the thermodynamic and energetic implications of alternative macrocyclization status were not known.
To elucidate the structural basis of resistance and activity for MK-5172, its linear (5172-linear) and P1–P3 macrocyclic (5172-mcP1P3) analogs (Figure 1A), and their susceptibility to A156T, we determined high-resolution inhibitor cocrystal structures with both WT and A156T mutant genotype 1a NS3/4A protease domain. In addition to analyzing the structures of the six protease–inhibitor complexes, we determined how binding energetics and conformational dynamics change with macrocyclization status using calorimetry and molecular dynamics simulations. We find that the unique binding mode of MK-5172 is independent of the macrocycle and that the rigidity imposed by the P2–P4 macrocycle prevents inhibitor rearrangement to be able to efficiently accommodate the A156T mutation. The flexibility of the linear analog allows subtle conformational rearrangements to accommodate the mutation, but the associated entropic disadvantage results in lower potency. Last, 5172-mcP1P3 benefits from the entropic advantage of macrocyclization while still allowing conformational rearrangements to reduce susceptibility to A156T. The preservation of the parent compound’s binding mode in the absence of the P2–P4 macrocycle provides further opportunities to optimize this class of inhibitors, while avoiding known sites of drug resistance.
RESULTS
We have determined high-resolution crystal structures of MK-5172 analogs in complex with the WT genotype 1a NS3/4A protease and A156T variant (WT–5172-linear, A156T–5172-linear, WT–5172-mcP1P3, and A156T–5172-mcP1P3; Table S1). Three of these structures are in the same P212121 space group while the A156T–5172-linear is in P21 with four molecules in the asymmetric unit (only one molecule was chosen for analysis; see Materials and Methods). These four new and the two previously determined crystal structures of MK-5172 (PDB IDs 3SUD and 3SUG, for WT and A156T variants, respectively)24 were analyzed comparatively, specifically for details of inhibitor–protease interactions.
The Unique Binding Mode of MK-5172 Is Independent of the Macrocycle
Unlike all other HCV PIs with known cocrystal structures, MK-5172 interacts with the catalytic triad in a unique conformation where the P2 quinoxaline moiety packs largely against the catalytic residues H57 and D81 (Figure 1). The P1–P4 peptidomimetic inhibitor scaffold spans the S1–S4 binding pockets interacting with the carbonyl oxygens of R155 and A157 as well as the Nε of A157. The P1′ acylsulfonamide is positioned in the oxyanion hole and hydrogen bonds to H57, G137, and S139. This binding mode is unchanged when the P2–P4 macrocycle is removed (5172-linear) or replaced with a P1–P3 macrocycle (5172-mcP1P3). Therefore, the binding mode of MK-5172 is a function of the P2 moiety rather than the macrocycle.
Despite conservation of the overall binding mode, the potency of MK-5172 and its analogs varies greatly against WT and A156T variants.24,25 MK-5172 inhibits WT protease with a Ki of 0.14 nM, lower than those of linear and P1–P3 analogs (9.5 and 2.0 nM respectively), but loses 74-fold potency against the A156T variant. The P1–P3 and linear analogs lose 29- and 230-fold potency against A156T relative to WT protease. To gain insights into the molecular basis of this difference, next we analyzed how the details of protease–inhibitor interactions are affected by the macrocyclization status and the A156T drug resistance mutation.
A156T Alters the Packing of the Macrocyclic Analogs at the Active Site
To analyze the details of inhibitor packing at the active site, the relative van der Waals (vdW) interaction energies between the inhibitor and protease residues in the crystal structures were calculated (Figure 2 and S1). In line with the conservation of the overall binding mode, the strongest inhibitor–protease interaction occurs with the catalytic H57 and D81 in all WT complex structures (Figure 2B), with similar total vdW interaction energies (WT–MK-5172, −43.2; WT–5172-linear, −42.3; WT–5172-mcP1P3, −45.1 kcal/mol). Nevertheless, there are subtle alterations in the details of inhibitor packing. For instance, compared to the parent compound, vdW interactions with D81 are 0.8 kcal/mol weaker in the linear analog, indicating subtle changes in the stacking of the P2 quinoxaline moiety. In addition, inhibitor contacts with R155, Y56, and Q41 are weaker in the analogs compared to the parent MK-5172, in agreement with decreased potency against the WT protease (Figure S1).
Figure 2.
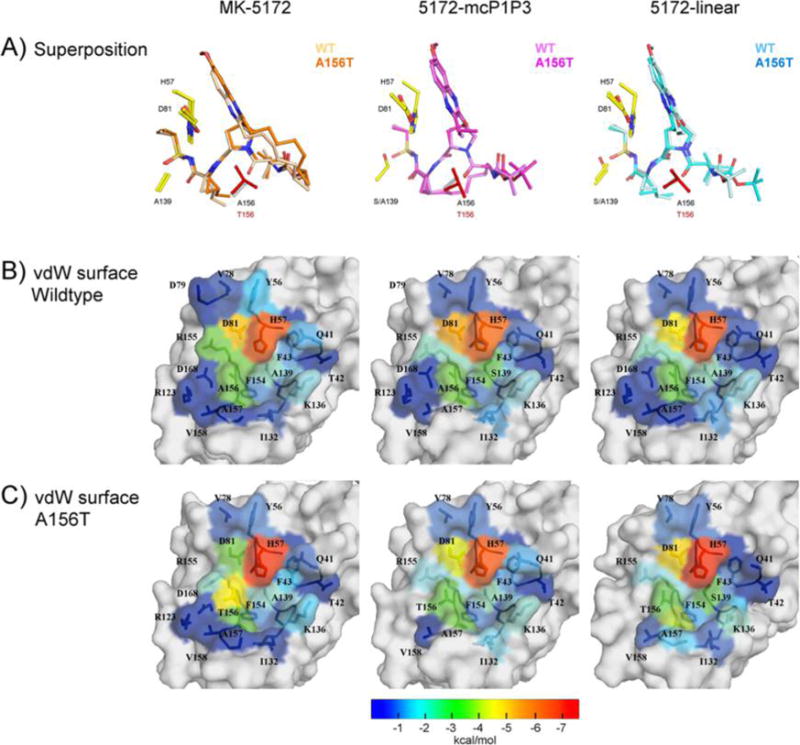
Changes in the packing of the inhibitors due to A156T at the HCV NS3/4A active site evaluated by the vdW contact energies. (A) Superposition of the inhibitor bound to WT and A156T protease. The vdW contact energy with the inhibitor mapped onto the protease surface in (B) WT and (C) A156T variants. The warmer (red) and cooler (blue) colors indicate more and less contacts with the inhibitor, respectively.
The primary drug resistance mutation A156T alters packing of MK-5172 and its analogs at the active site to varying extents (Figure 2C). In complexes with the mutant protease, the inhibitor needs to rearrange to avoid a steric clash with the larger Thr side chain. In the WT complex conformation, the P2–P4 macrocycle of MK-5172 would sterically clash with the larger Thr side chain. As a result, the entire inhibitor shifts relative to the WT complex in the A156T–MK-5172 structure, with the macrocycle moving away from residues R155 and A157 and the P2 quinoxaline away from D81 toward the catalytic histidine (Figures 2, 3). We have previously described this steric effect as the molecular mechanism behind MK-5172’s susceptibility to A156T.24 When the P2–P4 macrocycle is removed, the shift due to the A156T mutation is much more subtle with the 5172-linear’s P2 quinoxaline maintaining stacking interactions with H57 and D81. Instead, the P4 tert-butyl moiety adapts a different conformation to accommodate the larger Thr side chain and loses packing against residues 155 and 156 but establishes more contacts with residues 157–159. Similarly, in the A156T–5172-mcP1P3 complex, rearrangement of the P4 moiety is able to accommodate the larger Thr side chain to avoid a steric clash without severely compromising the stacking interactions of the P2 quinoxaline. Overall, 5172-mcP1P3 maintains WT-like contacts at the protease active site with the exception of decreased packing against D81, but to a lesser extent than that of the parent MK-5172. Hence, the 5172-mcP1P3 analog benefits from stabilization by the P1–P3 macrocyclization without paying a steric penalty due to the Thr mutation at position 156.
Figure 3.
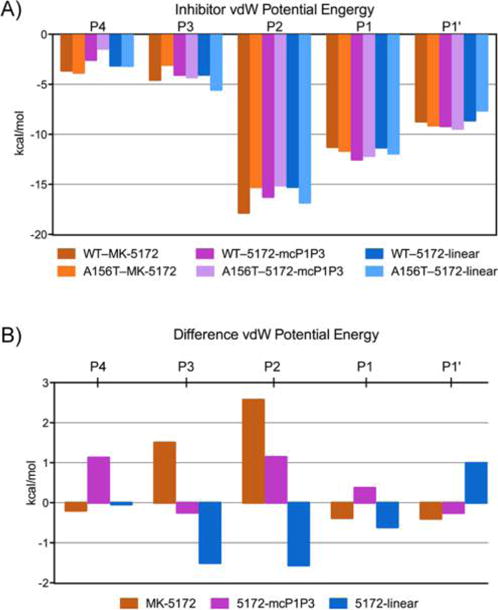
Inhibitor packing at the active site in HCV NS3/4A protease crystal structures. (A) Relative van der Waals interactions of P4−P1′ inhibitor moieties. (B) Changes in vdW interactions in A156T relative to WT protease.
Active Site Hydrogen Bonding Pattern Is Dependent on the Macrocyclization Status and Gets Disrupted with A156T Mutation
Unlike in the apo state or when binding weak ligands, high-affinity substrates and inhibitors stabilize an extensive electrostatic network at the HCV protease active site, involving side chains of protease residues D81, R155, D168, and R123.24,29 Within this network, R155–D168 interaction is particularly key for maintaining an electrostatic surface that accommodates high-affinity ligand binding.30 This interaction is present in all WT complex crystal structures with two hydrogen bonds (H-bonds) between the side chains of R155 and D168 (Figures 4, S2, and S3 and Table S3). In the A156T–MK-5172 protease complex, however, R155 loses both these H-bonds, and D168 instead interacts with R123. A similar rearrangement of side chains underlies the resistance mechanism of asunaprevir to R155K mutation.30 Mutation of A156, while not directly part of this electrostatic network, still results in a similar disruption of the active site binding surface in the MK-5172 complex, correlating with MK-5172’s susceptibility to A156T.
Figure 4.
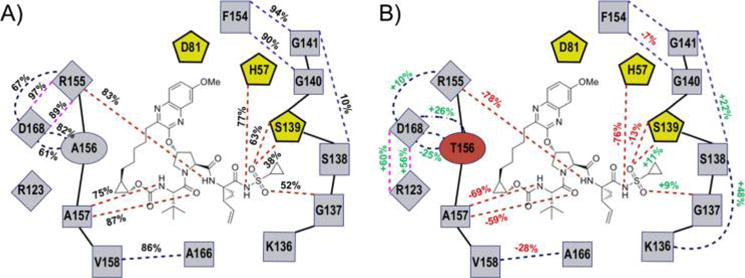
Changes in hydrogen bonding patterns across 100 ns molecular dynamic simulations. 2D schematic representation of inter- and intramolecular (black and red dashed lines, respectively) hydrogen bonding interactions during MD simulations with (A) the percentage of time the bond is present in WT–MK-5172 and (B) the percent changes with respect to WT in the A156T–MK-5172.
To assess the stability of the active site hydrogen bonding, we calculated the average time H-bonds were observed during the MD simulations. Although present in the MK-5172 crystal structure, the H-bond between D81 and R155 does not persist during the MD simulations, suggesting that this interaction may not be critical to high-affinity inhibitor binding. In contrast, the side chains of R155 and D168 form highly stable H-bonds in all WT complexes (present >89% of the time), consistent with its key role in optimal active site electrostatics. The MD simulations also revealed the persistence of protease–inhibitor H-bonds over time, elucidating the relative bond stability and overall contribution to intermolecular interactions. Most intermolecular H-bonds are at the P1–P1′ moieties (Figures 4 and S3), where the inhibitors also have strong vdW interactions with the active site (Figure 3A). Although the hydrogen-bonding patterns of the three analogs with the active site residues are very similar in the complex crystal structures (Table S2), the stability of specific H-bonds varies (Figures 4 and S3 and S4). MK-5172’s P1 amide N forms a H-bond with R155 (83% time). This interaction is lost in 5172-mcP1P3 but present in 5172-linear (68% time). At the P1′ moiety, MK-5172 forms four H-bonds with H57, S139, and G137 (77%, 63%, 38% and 52%, respectively). 5172-linear forms the same four H-bonds as MK-5172 but at a higher frequency (50%, 87%, 90%, and 86%). This improved stability of H-bonds in 5172-linear correlates with the more favorable enthalpy of binding seen in 5172-linear relative to both macrocyclic analogs (Figure 5A).
Figure 5.
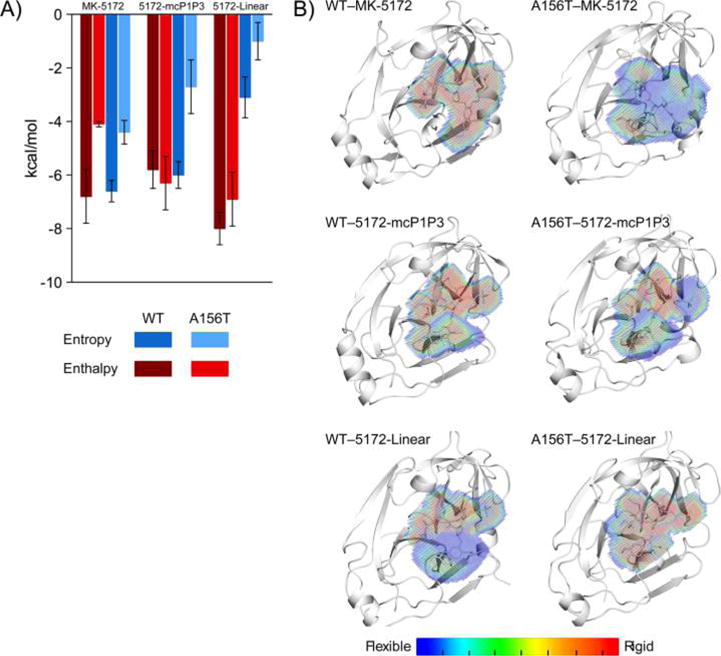
Energetic and dynamic effects of macrocyclization on inhibitor binding to HCV NS3/4A protease. (A) The entropy and enthalpy of binding of MK-5172 and analogs to WT and A156T protease variants. (B) The dynamic inhibitor envelope when bound to WT and A156T protease. Red and blue indicate less and more flexible regions of the inhibitor, respectively.
The presence of the A156T drug resistance mutation considerably alters the network of hydrogen bonding patterns at the active site. Just as in the crystal structure, during the MD simulations the A156T–MK-5172 complex loses R155–D168 interactions for the less stable D168–R123 H-bonds (97% and 89% versus 60% and 56%, respectively; Figure 4B). In contrast to the parent compound, both 5172–mcP1P3 and 5172-linear analogs maintain the key R155–D168 H-bonds (Figure S3), which partially correlates with the lower fold-loss in potency due to A156T mutation.
Compared to the protease active site changes, losses in intermolecular hydrogen bonding in the mutant complexes are more pronounced across all three inhibitors. MK-5172 loses all H-bonds at P3 and P1 and has a weaker P1′ amide N–H57 interaction. 5172-linear experiences the worst drops in H-bond stability, affecting all of its intermolecular interactions. 5172-mcP1P3, while experiencing decreases in H-bond stability throughout the inhibitor, is able to maintain WT-like interactions. The A156T drug resistance mutation therefore has a disruptive effect on both inter and intramolecular hydrogen bonding; however, the inhibitor’s macrocyclization status can differentially attenuate this effect. Overall, the P1–P3 analog is able to maintain a more WT-like hydrogen-bonding pattern.
Enthalpic Loss Due to A156T Underlies Differences in Inhibitor Susceptibility
The thermodynamics of protease binding for MK-5172 and its analogs were assessed to interrogate the underlying energetic effects of macrocyclization on inhibitor potency and susceptibility to A156T mutation (Figure 5A and Table S4) using isothermal titration calorimetry. Although a weaker binder, interestingly the linear analog had more favorable enthalpic interactions. While the macrocyclic analogs MK-5172 and 5172-mcP1P3 have a more favorable (negative) entropic contribution than 5172-linear (−TΔS of −6.6 and −6.0, respectively compared to −3.1 kcal/mol) in binding WT protease, presumably due to the lack of entropic penalty from the rigidity of the macrocyclization. This enhancement in entropy more than compensates for the decrease in the enthalpy of binding, underlying the increased potency of macrocyclic inhibitors compared to their linear counterparts.
All three inhibitors lose considerable potency in the presence of the A156T mutation compared to binding the WT protease. The entropic loss due to this mutation is similar for all inhibitors (2.2, 3.3, and 2.3 kcal/mol for MK-5172, 5172-mcP1P3, and 5172-linear, respectively, Table S4), suggesting the loss may be partially related to the greater loss in the degrees of freedom of the larger Thr side chain compared to Ala. Unlike entropy, the enthalpic changes vary greatly among the three inhibitors (2.7, −0.5, and 1.1 kcal/mol for MK-5172, 5172-mcP1P3, and 5172-linear, respectively Table S4) and largely correlate with the changes in inhibitor packing presented above and susceptibility to A156T. Although 5172-mcP1P3 loses a similar amount of entropy due to the A156T mutation, unlike the parent MK-5172, the enthalpic contribution to binding actually is better (−5.8 and −6.3 kcal/mol, respectively, for binding WT vs A156T protease). 5172-mcP1P3 can better accommodate the larger Thr side chain to enhance inhibitor packing at the active site, which results in maintaining the favorable binding enthalpy and hence potency against A156T. Thus, binding thermodynamics is consistent with the steric clash of A156T with MK-5172 causing the greatest loss of affinity.
Enzyme and Inhibitor Conformational Dynamics Correlate with Binding Thermodynamics
To further access the impact of A156T mutation on inhibitor binding and reveal any dynamic changes in the complexes underlying potency loss, we carried out fully solvated and extensive molecular dynamics (MD) simulations of all six complexes starting from each of the crystal structures we determined. The dynamics of inhibitors bound to the protease were assessed by the dynamic inhibitor envelope,31 which can be visualized as a probability distribution for inhibitor occupancy at the active site during MD trajectories (Figure 5B). When the inhibitor is rigid, the occupancy is high (red), while more flexible moieties sample different conformations resulting in low occupancy values (blue). The conformational flexibility of the bound inhibitor can affect both the entropy and enthalpy, and we find that the changes in flexibility mostly correlate with the enthalpy of binding. Comparing the two macrocyclic inhibitors binding to the WT protease, MK-5172 is more rigid and maintains more stable intermolecular interactions, which correlates with its more favorable enthalpy compared to 5172-mcP1P3. In the presence of the A156T mutation, these interactions are destabilized, resulting in a less occupied (more blue) dynamic inhibitor envelope, and worse enthalpy of binding for MK-5172 (−6.8 versus −4.1 kcal/mol). This destabilization is less pronounced for the P1–P3 analog (more rigid (or red) in Figure 5B), which maintains its enthalpy of binding. As expected, the linear analog is more flexible even in the bound state in the WT complex compared to the macrocyclic analogs. However, the linear analog pays a higher entropic penalty upon binding due to the lack of a macrocycle constraining conformational flexibility in the free state (as revealed by the smaller magnitude of favorable entropy than those of the macrocyclic compounds). However, although not favorable for entropic considerations, the lack of a macrocyclic constraint allows 5172-linear to adapt to the mutated surface for optimal packing. This adaptation likely results in the largely maintained favorable enthalpy of binding (−6.9 kcal/mol) in the presence of A156T mutation. Indeed, MD simulations revealed that the linear analog adapts to the A156T mutation by sampling a substantially different conformation than that in the WT complex or the starting crystallographic binding pose (Figure S6). By sampling this alternate conformation, 5172-linear sacrifices entropically favorable flexibility of the P2 group in the bound state, resulting in a high-occupancy (red) dynamic inhibitor envelope (Figure 5B). Hence, alterations in conformational flexibility and related changes in the entropy and enthalpy of binding due to A156T depend on the macrocyclization status of the analogs.
In addition to inhibitor flexibility, the backbone dynamics of the protein were assessed by Cα root-mean-square fluctuations (RMSF) during the MD simulations (Figures 6 and S5). Nine highly flexible structural regions were identified: (1) the N-terminal NS4A linker; (2) α0 helix spanning residues 12–15; the loops 3, 5, 7, and 8 spanning residues 37–43, 67–72, 88–93, and 157–163 respectively; (4) α1 helix spanning residues 56–61 and containing the catalytic His; (6) F1 β strand spanning residues 76–82 and containing the catalytic Asp; and (9) the C terminal α3 helix spanning residues 173–179. These flexible regions mainly correspond to turns/loops or terminal portions of secondary structural elements throughout the protein structure. The WT protease displays similar backbone dynamics with flexible N and C termini and a stable core when bound to MK-5172 and the two analogs. However, the introduction of the A156T mutation alters these dynamic signatures. In MK-5172 (Figures 6 and S5A), the A156T variant increases the backbone flexibility at the N-terminus as well as regions 2, 3, 4, 6, 7, and 8. In 5172-mcP1P3 complex (Figure S5B), the A156T mutation has the opposite effect of reducing flexibility, in particular at regions 1 and 3, relative to the WT complex. As the P1–P3 macrocycle does not sterically clash with the larger Thr side chain, the backbone fluctuations of the mutant complex are not considerably affected by this mutation. In 5172-linear (Figure S5C), the A156T mutation perturbs the backbone Cα at the N and C termini as well as at regions 2, 3, 6, and 7. Thus, the mutation causes disturbance to the protein backbone dynamics both at and away from the active site in complexes of MK-5172 and the linear analog, while the protease dynamics is more robust against the dynamic effects of the mutation when bound to the P1–P3 analog. Overall, changes in both inhibitor and protease dynamics underpin the observed impact of A156T mutation on the thermodynamics of binding and losses in inhibitor affinity.
Figure 6.
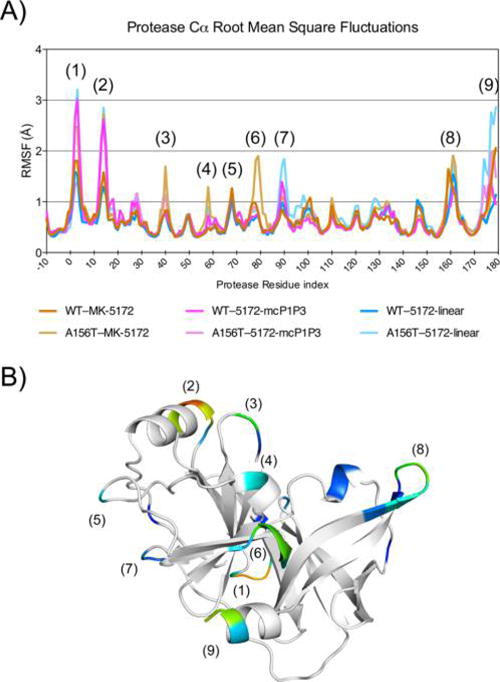
Effects of macrocyclization status on protein backbone dynamics. (A) Root mean square fluctuations (RMSF) of backbone Cα during 100 ns MD simulation are shown, with regions in the protease displaying high Cα RMSF numbered. Residues −10 through 0 represent the NS4A cofactor and linker. (B) RMSF mapped onto the NS3/4A structure, with stable core (<0.7 Å RMSF) in white and regions with peak in RMSF in color. Numbers in parentheses correspond to RMSF peaks in A.
DISCUSSION
The high potency of MK-5172 (sub-nM for WT protease) and robustness to R155K and D168A mutations depend on MK-5172’s unique binding mode where the P2 quinoxaline moiety packs against the catalytic H57 and D81. Albeit beneficial for potency, the P2–P4 macrocycle can be regarded as MK-5172’s Achilles heel as the rigidity of this region sterically clashes with the multidrug-resistant mutation A156T resulting in ~3800 fold change in Ki relative to WT. However, whether the macrocycle could be removed without affecting the binding mode was unclear. In this study, the structural and thermodynamic analyses were probed with the linear and P1–P3 analogs of MK-5172 we previously designed,25 revealing that macrocyclization does not alter the packing of the quinoxaline moiety but influences susceptibility to the A156T mutation.
The HCV NS3/4A protease structure has an extensive electrostatic network at the active site connecting the catalytic H57 and D81 with residues R155, D168, and R123.24,26,31–33 Drug resistance mutations such as R155K and D168A are detrimental because they (1) disrupt this electrostatic network reducing the protein’s binding ability and (2) weaken inhibitor binding by destabilizing the interaction of the inhibitor’s P2 heterocyclic moiety. PIs with P2 moieties stacking against the electrostatic network, such as vaniprevir, danoprevir, and asunaprevir, experience decreases in inhibitor affinity when this subsite is altered by mutations at R155 and D168. MK-5172, however, has a P2 moiety that stacks against the catalytic residues, making the inhibitor less susceptible to the canonical resistance mutations. This binding mode is beneficial for the pan-genotypic activity of MK-5172, as the triad is invariant across genotypes. Our crystal structures show that the stacking of the P2 quinoxaline in MK-5172 is maintained in linear and P1–P3 macrocyclic analogs. Thus, P2 stacking against the catalytic triad is driven primarily by the quinoxaline moiety and does not rely on the restraint exerted by the P2–P4 macrocycle on the quinoxaline.
Macrocyclization increases inhibitor potency mostly by decreasing the entropic penalty of binding by constraining the unbound inhibitors’ degrees of freedom and stabilizing the inhibitor in a binding-competent conformation. Even without any stabilizing macrocycle, the 5172 “core,” 5172-linear, has a Ki of 9.5 nM against WT protease (Table S4). This low nanomolar inhibition constant relies on the highly favorable enthalpy of binding due to the preservation of the parent compound’s binding mode and efficient packing of the quinoxaline moiety at the catalytic site. Without the macrocycle though, the linear analog is likely highly flexible in the unbound state. The presence of a macrocycle prearranges the inhibitor in binding competent conformation, favorably contributing to the entropy of binding to further increase the potency to 1.96 nM in 5172-mcP1P3 and 0.14 nM in MK-5172. In addition to contributing directly to inhibitor–protease hydrophobic interactions, as is the case for the P2–P4 macrocycle in MK-5172, the hydrophobic character of the macrocycle can increase lipophilicity and improve pharmacokinetic properties such as cellular permeability and stability.27,34,35
Protease inhibitors with a P2–P4 macrocycle, such as MK-5172 and vaniprevir, are susceptible to A156T mutation as the larger Thr side chain can engage in a steric clash with the macrocycle, displacing the inhibitor in the mutant complex structures.24 In the A156T–MK-5172 complex, the whole inhibitor including the P2–P4 macrocycle is pushed away from the active site due to A156T. Furthermore, the quinoxaline moiety loses critical stacking interactions with H57 and D81. When the P2–P4 macrocycle is not present, in the A156T–5172-linear and A156T–5172-mcP1P3 complexes, the inhibitor can conformationally adapt to accommodate the A156T mutation. Thus, the 5172-mcP1P3 analog has the advantage of macrocyclization while still maintaining critical contacts with the catalytic residues.
Second generation protease inhibitors, such as MK-5172, have broader antiviral activity and higher barriers to resistance than first generation protease inhibitors.36 However, even these inhibitors are not completely robust against drug resistance. In a recent clinical trial,37 A156T variants with an additional V36M/V or I170V mutation were detected in patients who failed therapy. Although the A156T viral variant is debilitated for replication in vitro,38 coexisting compensatory mutations, as often seen in HIV-1 protease,39 may recover viral fitness and lead to virological failure. Therefore, the development of future NS3/4A PIs needs to encompass optimal strategies to best balance inhibitor potency and barrier to resistance. The 5172-mcP1P3 analog provides opportunities for further optimization of compounds with stable stacking interactions with the catalytic residues, while staying within the substrate envelope. As the “unique” binding mode of MK-5172 is preserved even without the P2–P4 macrocycle, the removal of the macrocyclic linkage at the P2 quinoxaline enables exploration of the chemical space to modify this group for an even more potent inhibitor robust against resistance.
In rational drug design, the effect of macrocyclization on the entropy and enthalpy of binding is not straightforward to predict, especially in the presence of resistance mutations. While the macrocyclic constraint is beneficial for entropy, as we have demonstrated here for MK-5172, the location of the macrocycle should be designed for optimal enthalpy as well, to preserve the desired binding mode and ideally stay within the substrate envelope.26,29 The protrusion of the P2–P4 macrocycle in MK-5172 outside the substrate envelope effectively surrounding A156 causes vulnerability to mutations at this residue. In the presence of A156T, while the rigidity brought about by the macrocycle is still beneficial for entropy, the same rigidity is detrimental to enthalpy. Design of macrocycles should not only fit within the substrate envelope but also consider both inhibitor rigidity and adaptability to potential resistance mutations, to achieve optimal entropy–enthalpy balance required for robustness to avoid susceptibility to resistance mutations.
MATERIALS AND METHODS
Protein Constructs
The HCV genotype 1a NS3/4A protease domain gene24 was synthesized by GenScript and cloned into the pET28a expression vector (Novagen). The highly soluble single-chain construct consists of NS3/4A protease domain (residues 4–181) fused to a fragment of the cofactor NS4A (residues 12–23) via an SD linkage. A similar protease construct exhibited catalytic activity comparable to that of the authentic full-length protein.40 All protease variants were generated using the QuikChange Site-Directed Mutagenesis Kit from Stratagene.
Protein Expression and Purification
Protein expression and purification were carried out as previously described.29 Briefly, transformed BL21(DE3) E. coli cells were grown at 37 °C and induced at an optical density of 0.6 by adding 1 mM IPTG. Cells were harvested after 5 h of expression, pelleted, and frozen at −80 °C for storage. Cell pellets were thawed, resuspended in 5 mL/g of resuspension buffer (50 mM phosphate buffer, 500 mM NaCl, 10% glycerol, 2 mM β-ME, pH 7.5) and lysed with a cell disruptor. The soluble fraction was retained, applied to a nickel column (Qiagen), washed with resuspension buffer, and eluted with resuspension buffer supplemented with 200 mM imidazole. The eluent was dialyzed overnight (MWCO 10 kDa) to remove the imidazole, and the His-tag was simultaneously removed with thrombin treatment. The nickel-purified protein was then flash frozen and stored at −80 °C.
Crystallization
The above-mentioned protein solution was thawed, concentrated to ~3 mg mL−1, and loaded on a HiLoad Superdex75 16/60 column equilibrated with gel filtration buffer (25 mM MES, 500 mM NaCl, 10% glycerol, 30 mM zinc chloride, and 2 mM DTT, pH 6.5). The protease fractions were pooled and concentrated to 20–25 mg mL−1 with an Amicon Ultra-15 10 kDa device (Millipore). The concentrated samples were incubated for 1 h with 1–3 molar excess of inhibitor. Diffraction-quality crystals were obtained overnight by mixing equal volumes of concentrated protein solution with precipitant solution (20–26% PEG-3350, 0.1 M sodium MES buffer, 4% ammonium sulfate, pH 6.5) in 24-well VDX hanging drop trays.
Data Collection and Structure Solution
X-ray diffraction data were collected either at Advanced Photon Source LS-CAT 21-ID-F or at our in-house Rigaku Saturn X-ray system. Diffraction intensities were indexed, integrated, and scaled using the program HKL2000.41 All structure solutions were generated using molecular replacement with PHASER.42 The B chain model of viral substrate product 4A–4B (3M5M)26 was used as the starting model for all structure solutions. Initial refinement was carried out in the absence of modeled ligand, which was subsequently built in during later stages of refinement. Subsequent crystallographic refinement was carried out within the Phenix program suite, with iterative rounds of TLS or restrained refinement until convergence was achieved.43 The protein crystals of the A156T protease in complex with 5172-mcP1P3 grew as pseudomerohedral twins. X-Triage was used to determine the twin operator, −k,−h,−l; thus refinement was carried out using twin law = −k,−h,−l. This structure has four molecules in the asymmetric unit with the active sites of molecules A and C superposed onto molecules B and D, respectively. Molecule B was used in all of the analysis, as it possessed the lowest B factors representing the most stable and consistent conformer. The final structures were evaluated with MolProbity44 prior to deposition in the Protein Data Bank. To limit the possibility of model bias throughout the refinement process, 5% of the data were reserved for the free R-value calculation.45 Interactive model building and electron density viewing was carried out using the program COOT.46
Structural Analysis
Superpositions were performed in PyMOL47 using the Cα atoms of the active site protease residues 137–139 and 154–160. The A chain of WT–MK-5172 complex was used as the reference structure for each alignment. Two conformations are observed in the A156T–5172-linear structure, and the one with the lowest B factors within the P21 unit cell was used in the structural analysis. The van der Waals contact energies between protease residues and inhibitors were computed using a simplified Lennard–Jones potential as described previously.48
MD Simulations
Molecular dynamics simulations were carried out, in triplicate, following previously published protocols31 using Desmond49,50 with the OPLS2005 force field.51,52 After equilibration, each trajectory was run for 100 ns at 300 K and the coordinates recorded every 5 ps. The dynamic inhibitor envelope was calculated using the van der Waals volume of inhibitor conformers from the MD trajectories mapped onto the three-dimensional grid placed on the binding site of the enzyme and normalized by the total number of conformers to obtain a probability distribution, as detailed previously.31,53
Isothermal Titration Calorimetry
ITC experiments were performed as previously described.54,55 Briefly, NS3/4A protease was purified via gel filtration in 50 mM HEPES, 300 mM NaCl, 10% glycerol, and 1 mM TCEP at pH 7.4 and concentrated to 10–60 μM. Inhibitor stock was prepared in 100% DMSO and diluted to working conditions in the gel filtration buffer with final DMSO concentration not exceeding 3%. Experiments were performed with a highly sensitive Microcal ITC200 (Malvern) at 25 °C with the protein in the sample cell and inhibitor solution in the syringe. The data were analyzed using Origin 7.0, and the change in enthalpy (ΔH) and corresponding dissociation constant (Kd) were determined via nonlinear regression, with a one-binding site model. Due to the potency of MK-5172, the inhibition constant from enzyme kinetics assay (Ki) of WT–MK-5172 from our previous work25 was used to calculate the Gibbs Free Energy (ΔG), as the Kd is too low to be reliably determined by ITC without a competing ligand,56 which we were unable to successfully attain. All other ΔG calculations used ITC derived Kd values.
Supplementary Material
Acknowledgments
We thank D. Smith from the Advanced Photon Source, LS-CAT beamline for data collection; W. Royer, A. Ozen, and M. Bohn for helpful discussions. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). This work was supported by the National Institute of Allergy and Infectious Disease (R01-AI085051). DIS was also supported by National Institute of General Medical Sciences of the National Institutes of Health (F31-GM103259) as well as the HOPE scholarship sponsored by the Biomedical Science Career Program (BSCP).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.5b00647.
Tables S1–S4, Figures S1–S6 (PDF)
Notes
The authors declare the following competing financial interest(s): Overlapping with the time this research was performed, we received a sponsored research grant from Merck, which did not directly overlap with the work described in this manuscript.
References
- 1.Alter MJ. Epidemiology of hepatitis C virus infection. World J Gastroenterol. 2007;13:2436–2441. doi: 10.3748/wjg.v13.i17.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Modi AA, Liang TJ. Hepatitis C: a clinical review. Oral Dis. 2008;14:10–14. doi: 10.1111/j.1601-0825.2007.01419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization. Guidelines for the screening, care and treatment of persons with hepatitis C infection. 5th. 2014. [PubMed] [Google Scholar]
- 4.Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, Halfon P, Inchauspe G, Kuiken C, Maertens G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky JM, Penin F, Sablon E, Shin IT, Stuyver LJ, Thiel HJ, Viazov S, Weiner AJ, Widell A. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962–973. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- 5.Gower E, Estes C, Blach S, Razavi-Shearer K, Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol. 2014;61:S45–57. doi: 10.1016/j.jhep.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 6.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, Jr, Haussinger D, Diago M, Carosi G, Dhumeaux D, Craxi A, Lin A, Hoffman J, Yu J. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 7.Bartenschlager R, Lohmann V, Penin F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat Rev Microbiol. 2013;11:482–496. doi: 10.1038/nrmicro3046. [DOI] [PubMed] [Google Scholar]
- 8.Szabo G, Bala S. MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol. 2013;10:542–552. doi: 10.1038/nrgastro.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwong AD, Kauffman RS, Hurter P, Mueller P. Discovery and development of telaprevir: an NS3–4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat Biotechnol. 2011;29:993–1003. doi: 10.1038/nbt.2020. [DOI] [PubMed] [Google Scholar]
- 10.Perni RB, Almquist SJ, Byrn RA, Chandorkar G, Chaturvedi PR, Courtney LF, Decker CJ, Dinehart K, Gates CA, Harbeson SL, Heiser A, Kalkeri G, Kolaczkowski E, Lin K, Luong YP, Rao BG, Taylor WP, Thomson JA, Tung RD, Wei Y, Kwong AD, Lin C. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3–4A serine protease. Antimicrob Agents Chemother. 2006;50:899–909. doi: 10.1128/AAC.50.3.899-909.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malcolm BA, Liu R, Lahser F, Agrawal S, Belanger B, Butkiewicz N, Chase R, Gheyas F, Hart A, Hesk D, Ingravallo P, Jiang C, Kong R, Lu J, Pichardo J, Prongay A, Skelton A, Tong X, Venkatraman S, Xia E, Girijavallabhan V, Njoroge FG. SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob Agents Chemother. 2006;50:1013–1020. doi: 10.1128/AAC.50.3.1013-1020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenquist Å, Samuelsson B, Johansson PO, Cummings MD, Lenz O, Raboisson P, Simmen K, Vendeville S, de Kock H, Nilsson M, Horvath A, Kalmeijer R, de la Rosa G, Beumont-Mauviel M. Discovery and Development of Simeprevir (TMC435), a HCV NS3/4A Protease Inhibitor. J Med Chem. 2014;57:1673–1693. doi: 10.1021/jm401507s. [DOI] [PubMed] [Google Scholar]
- 13.Andreone P, Colombo MG, Enejosa JV, Koksal I, Ferenci P, Maieron A, Mullhaupt B, Horsmans Y, Weiland O, Reesink HW, Rodrigues L, Jr, Hu YB, Podsadecki T, Bernstein B. ABT-450, ritonavir, ombitasvir, and dasabuvir achieves 97% and 100% sustained virologic response with or without ribavirin in treatment-experienced patients with HCV genotype 1b infection. Gastroenterology. 2014;147:359–365.e351. doi: 10.1053/j.gastro.2014.04.045. [DOI] [PubMed] [Google Scholar]
- 14.Yao N, Reichert P, Taremi SS, Prosise WW, Weber PC. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure (Oxford, U K) 1999;7:1353–1363. doi: 10.1016/s0969-2126(00)80025-8. [DOI] [PubMed] [Google Scholar]
- 15.Love Ra, Parge HE, Wickersham Ja, Hostomsky Z, Habuka N, Moomaw EW, Adachi T, Hostomska Z. The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell. 1996;87:331–342. doi: 10.1016/s0092-8674(00)81350-1. [DOI] [PubMed] [Google Scholar]
- 16.Appleby TC, Anderson R, Fedorova O, Pyle AM, Wang R, Liu X, Brendza KM, Somoza JR. Visualizing ATP-dependent RNA translocation by the NS3 helicase from HCV. J Mol Biol. 2011;405:1139–1153. doi: 10.1016/j.jmb.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 18.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M, Jr, Lemon SM. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A. 2005;102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 20.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 21.Summa V, Ludmerer SW, McCauley JA, Fandozzi C, Burlein C, Claudio G, Coleman PJ, Dimuzio JM, Ferrara M, Di Filippo M, Gates AT, Graham DJ, Harper S, Hazuda DJ, McHale C, Monteagudo E, Pucci V, Rowley M, Rudd MT, Soriano A, Stahlhut MW, Vacca JP, Olsen DB, Liverton NJ, Carroll SS. MK-5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother. 2012;56:4161–4167. doi: 10.1128/AAC.00324-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liverton NJ, Holloway MK, McCauley JA, Rudd MT, Butcher JW, Carroll SS, DiMuzio J, Fandozzi C, Gilbert KF, Mao SS, McIntyre CJ, Nguyen KT, Romano JJ, Stahlhut M, Wan BL, Olsen DB, Vacca JP. Molecular modeling based approach to potent P2–P4 macrocyclic inhibitors of hepatitis C NS3/4A protease. J Am Chem Soc. 2008;130:4607–4609. doi: 10.1021/ja711120r. [DOI] [PubMed] [Google Scholar]
- 23.Harper S, McCauley JA, Rudd MT, Ferrara M, DiFilippo M, Crescenzi B, Koch U, Petrocchi A, Holloway MK, Butcher JW, Romano JJ, Bush KJ, Gilbert KF, McIntyre CJ, Nguyen KT, Nizi E, Carroll SS, Ludmerer SW, Burlein C, DiMuzio JM, Graham DJ, McHale CM, Stahlhut MW, Olsen DB, Monteagudo E, Cianetti S, Giuliano C, Pucci V, Trainor N, Fandozzi CM, Rowley M, Coleman PJ, Vacca JP, Summa V, Liverton NJ. Discovery of MK-5172, a Macrocyclic Hepatitis C Virus NS3/4a Protease Inhibitor. ACS Med Chem Lett. 2012;3:332–336. doi: 10.1021/ml300017p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romano KP, Ali A, Aydin C, Soumana D, Ozen A, Deveau LM, Silver C, Cao H, Newton A, Petropoulos CJ, Huang W, Schiffer CA. The molecular basis of drug resistance against hepatitis C virus NS3/4A protease inhibitors. PLoS Pathog. 2012;8:e1002832. doi: 10.1371/journal.ppat.1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ali A, Aydin C, Gildemeister R, Romano KP, Cao H, Ozen A, Soumana D, Newton A, Petropoulos CJ, Huang W, Schiffer CA. Evaluating the Role of Macrocycles in the Susceptibility of Hepatitis C Virus NS3/4A Protease Inhibitors to Drug Resistance. ACS Chem Biol. 2013;8:1469. doi: 10.1021/cb400100g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romano KP, Ali A, Royer WE, Schiffer CA. Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc Natl Acad Sci U S A. 2010;107:20986–20991. doi: 10.1073/pnas.1006370107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Driggers EM, Hale SP, Lee J, Terrett NK. The exploration of macrocycles for drug discovery–an underexploited structural class. Nat Rev Drug Discovery. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 28.Marsault E, Peterson ML. Macrocycles are great cycles: applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J Med Chem. 2011;54:1961–2004. doi: 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]
- 29.Romano KP, Laine JM, Deveau LM, Cao H, Massi F, Schiffer CA. Molecular mechanisms of viral and host cell substrate recognition by hepatitis C virus NS3/4A protease. J Virol. 2011;85:6106–6116. doi: 10.1128/JVI.00377-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soumana DI, Ali A, Schiffer CA. Structural Analysis of Asunaprevir Resistance in HCV NS3/4A Protease. ACS Chem Biol. 2014;9:2485. doi: 10.1021/cb5006118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ozen A, Sherman W, Schiffer CA. Improving the Resistance Profile of Hepatitis C NS3/4A Inhibitors: Dynamic Substrate Envelope Guided Design. J Chem Theory Comput. 2013;9:5693–5705. doi: 10.1021/ct400603p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halfon P, Locarnini S. Hepatitis C virus resistance to protease inhibitors. J Hepatol. 2011;55:192–206. doi: 10.1016/j.jhep.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 33.Pan D, Xue W, Zhang W, Liu H, Yao X. Understanding the drug resistance mechanism of hepatitis C virus NS3/4A to ITMN-191 due to R155K, A156V, D168A/E mutations: a computational study. Biochim Biophys Acta, Gen Subj. 2012;1820:1526–1534. doi: 10.1016/j.bbagen.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 34.Doak BC, Over B, Giordanetto F, Kihlberg J. Oral druggable space beyond the rule of 5: insights from drugs and clinical candidates. Chem Biol. 2014;21:1115–1142. doi: 10.1016/j.chembiol.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 35.Giordanetto F, Kihlberg J. Macrocyclic drugs and clinical candidates: what can medicinal chemists learn from their properties? J Med Chem. 2014;57:278–295. doi: 10.1021/jm400887j. [DOI] [PubMed] [Google Scholar]
- 36.Clark VC, Peter JA, Nelson DR. New therapeutic strategies in HCV: second-generation protease inhibitors. Liver Int. 2013;33:80–84. doi: 10.1111/liv.12061. [DOI] [PubMed] [Google Scholar]
- 37.Lawitz E, Sulkowski MS, Ghalib R, Rodriguez-Torres M, Younossi ZM, Corregidor A, DeJesus E, Pearlman B, Rabinovitz M, Gitlin N, Lim JK, Pockros PJ, Scott JD, Fevery B, Lambrecht T, Ouwerkerk-Mahadevan S, Callewaert K, Symonds WT, Picchio G, Lindsay KL, Beumont M, Jacobson IM. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: the COSMOS randomised study. Lancet. 2014;384:1756–1765. doi: 10.1016/S0140-6736(14)61036-9. [DOI] [PubMed] [Google Scholar]
- 38.He Y, King MS, Kempf DJ, Lu L, Lim HB, Krishnan P, Kati W, Middleton T, Molla A. Relative replication capacity and selective advantage profiles of protease inhibitor-resistant hepatitis C virus (HCV) NS3 protease mutants in the HCV genotype 1b replicon system. Antimicrob Agents Chemother. 2008;52:1101–1110. doi: 10.1128/AAC.01149-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nijhuis M, Schuurman R, de Jong D, Erickson J, Gustchina E, Albert J, Schipper P, Gulnik S, Boucher CA. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS (London, U K) 1999;13:2349–2359. doi: 10.1097/00002030-199912030-00006. [DOI] [PubMed] [Google Scholar]
- 40.Taremi SS, Beyer B, Maher M, Yao N, Prosise W, Weber PC, Malcolm BA. Construction, expression, and characterization of a novel fully activated recombinant single-chain hepatitis C virus protease. Protein Sci. 1998;7:2143–2149. doi: 10.1002/pro.5560071011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Charles W Carter., Jr, editor. Methods Enzymol. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 42.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 46.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 47.Schrodinger L. The AxPyMOL Molecular Graphics Plugin for Microsoft PowerPoint, Version 1.0 2010 [Google Scholar]
- 48.Nalam MN, Ali A, Altman MD, Reddy GS, Chellappan S, Kairys V, Ozen A, Cao H, Gilson MK, Tidor B, Rana TM, Schiffer CA. Evaluating the substrate-envelope hypothesis: structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance. J Virol. 2010;84:5368–5378. doi: 10.1128/JVI.02531-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schrödinger Release 2014–3: Desmond Molecular Dynamics System, v., D. E. Shaw Research, New York, 2014. Maestro-Desmond Interoperability Tools, version 3.9, Schrödinger, New York, 2014.
- 50.Bowers KJ, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. Proceedings of the 2006 ACM/IEEE conference on Supercomputing. ACM; Tampa, FL: 2006. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters; p. 746. [Google Scholar]
- 51.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 52.Shivakumar D, Williams J, Wu Y, Damm W, Shelley J, Sherman W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J Chem Theory Comput. 2010;6:1509–1519. doi: 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
- 53.Ozen A, Haliloglu T, Schiffer CA. Dynamics of preferential substrate recognition in HIV-1 protease: redefining the substrate envelope. J Mol Biol. 2011;410:726–744. doi: 10.1016/j.jmb.2011.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saalau-Bethell SM, Woodhead AJ, Chessari G, Carr MG, Coyle J, Graham B, Hiscock SD, Murray CW, Pathuri P, Rich SJ, Richardson CJ, Williams PA, Jhoti H. Discovery of an allosteric mechanism for the regulation of HCV NS3 protein function. Nat Chem Biol. 2012;8:920–925. doi: 10.1038/nchembio.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abian O, Vega S, Sancho J, Velazquez-Campoy A. Allosteric inhibitors of the NS3 protease from the hepatitis C virus. PLoS One. 2013;8:e69773. doi: 10.1371/journal.pone.0069773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Velazquez-Campoy A, Kiso Y, Freire E. The Binding Energetics of First- and Second-Generation HIV-1 Protease Inhibitors: Implications for Drug Design. Arch Biochem Biophys. 2001;390:169–175. doi: 10.1006/abbi.2001.2333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.