

Abstract
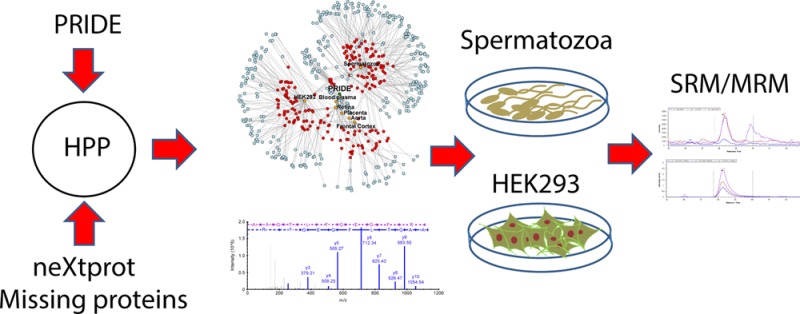
The current catalogue of the human proteome is not yet complete, as experimental proteomics evidence is still elusive for a group of proteins known as the missing proteins. The Human Proteome Project (HPP) has been successfully using technology and bioinformatic resources to improve the characterization of such challenging proteins. In this manuscript, we propose a pipeline starting with the mining of the PRIDE database to select a group of data sets potentially enriched in missing proteins that are subsequently analyzed for protein identification with a method based on the statistical analysis of proteotypic peptides. Spermatozoa and the HEK293 cell line were found to be a promising source of missing proteins and clearly merit further attention in future studies. After the analysis of the selected samples, we found 342 PSMs, suggesting the presence of 97 missing proteins in human spermatozoa or the HEK293 cell line, while only 36 missing proteins were potentially detected in the retina, frontal cortex, aorta thoracica, or placenta. The functional analysis of the missing proteins detected confirmed their tissue specificity, and the validation of a selected set of peptides using targeted proteomics (SRM/MRM assays) further supports the utility of the proposed pipeline. As illustrative examples, DNAH3 and TEPP in spermatozoa, and UNCX and ATAD3C in HEK293 cells were some of the more robust and remarkable identifications in this study. We provide evidence indicating the relevance to carefully analyze the ever-increasing MS/MS data available from PRIDE and other repositories as sources for missing proteins detection in specific biological matrices as revealed for HEK293 cells.
Keywords: C-HPP, missing proteins, MS/MS proteomics, PRIDE database
Introduction
The Human Proteome Project (HPP)1 is an international project to characterize the human proteome through two programs: a chromosome-based strategy (C-HPP) designed in 20102,3 and the biology/disease-driven strategy (B/D-HPP).4,5 Researchers from the chromosome-based strategy have used high throughput proteomics state-of-the-art technology, but major difficulties have arisen in the detection of a set of proteins, the so-called ”missing proteins”.6−8 These proteins lack experimental evidence obtained by mass spectrometry or antibody-based techniques, and their existence is based on bioinformatic predictions or transcriptomic analyses. In the C-HPP initiative, the reference database for the annotation of human proteins is neXtProt.9 This database assigns experimental evidence to each human protein using a scale with five levels, from PE1 (experimental evidence at protein level) to PE5 (uncertain protein). The missing proteins are annotated as PE2 (experimental evidence at the transcript level), PE3 (protein inferred from homology), or PE4 (protein predicted). As a reference, the database version used in this study (release 01.09.2015) contained 20061 proteins, 16791 of them annotated as PE1 (83.70% of protein entries). The number of missing proteins was 2680, corresponding to 13.36% of the total entries in the database.
Several possibilities have been proposed to explain the difficulties in the detection of these proteins, including their low abundance, their tissue expression specificity, and their stimulation dependent or development associated expression. In fact, the different methodological approaches applied to characterize missing proteins have confirmed that the selection of the tissue or cell type is critical to the success of these experiments.10−13 One of the most widely used methods for the identification of the samples in which the probability of detection of missing proteins is higher takes into account the expression level of the corresponding transcripts. Therefore, the integration of genomics, transcriptomics, and proteomics is widely used among HPP groups in order to design the experiments needed to improve the annotation of the human proteome.8 In particular, the Spanish Consortium of the HPP (spHPP), responsible for the study of chromosome 16, made a considerable effort to incorporate transcriptomic experiments as a tool for the analysis of the proteome. Public data sets from different resources such as the Gene Expression Omnibus (GEO) database14 and the ENCODE project15 were analyzed in depth to define the set of expressed genes in thousands of samples, including different biological sources (cell lines, normal tissues, and cancer samples) and technologies (microarrays and RNA-Seq).16 In addition, a bayesian classifier was developed to score the probability of expression of the missing proteins in more than 3400 microarray experiments.17 According to this study, testis, brain, and skeletal muscle were the best tissue candidates to detect the higher number of missing proteins using shotgun proteomics.
However, even when the analyzed sample is enriched in missing proteins, their identification is still challenging, especially when the bioinformatics methods and the statistical thresholds required impose stringent criteria to ensure the reliability of the observations resulting from the automatic MS data analysis and sequence assignments. Basically, the MS evidence for a protein is considered valid when the following conditions are fulfilled: 1% FDR at PSM, peptide and protein level, more than 1 peptide detected (9 or more amino acids in length) and at least two of which are not shared among the other proteins of the reference database (proteotypic peptides). The recent analysis of the human spermatozoa proteome13 is a good example. In this study those proteins with only one peptide identification were filtered using the set of unique peptides of the missing proteins obtained from the in silico digestion of the neXtProt database. The remaining PSMs were manually evaluated by three independent experts, allowing the assignment of 94 new missing proteins. Finally, the expression of C2orf57 and TEX37 was validated by immunohistochemistry. This excellent result allowed us to reach two important conclusions: the high accuracy of the available methods to predict the sample of interest based on public transcriptomics and proteomics experiments and the need to develop new bioinformatic workflows and new methods of experimental validation able to circumvent the constraints inherent in the identification of the missing proteins.
In the field of proteomics, a huge amount of shotgun experiments are publicly available in different data repositories.18 The most commonly used resources are the Global Proteome Machine Database (GPMDB, gpmdb.thegpm.org),19 PeptideAtlas (www.peptideatlas.org),20 the ProteomeXchange consortium (http://www.proteomexchange.org/),21 and the PRIDE database.22 More specifically, the members of the ProteomeXchange Consortium are working to standardize data submission and dissemination practises in the field. All proteomic experimental data sets in the HPP must be submitted to any of the ProteomeXchange resources. The stored data types include raw mass spectra data, peak lists, sample metadata, and the results of the original analyses (identification and quantification of peptides and proteins). Only the PRIDE Archive database contains at present more than 5000 data sets, including more than 60000 assays.
In this manuscript, we used public MS experiments to obtain guidance in the search for missing proteins. Initially, we assessed the possibility of obtaining information about the samples in which the number of missing proteins is enriched using the PRIDE database. This approach confirmed the results obtained using transcriptome profiles and provided new biological sources to be explored. The experiments selected were downloaded from the database and studied using two data analysis workflows. The number of missing proteins identified by our bioinformatics workflow, based on the analysis of the intersection of the PSM FDR filtering of the experimental results with the proteotypic peptides obtained from the in silico analysis of the reference database (without FDR filtering at protein level), was higher than the number of missing proteins detected applying the HPP guidelines. Upon manual inspection and curation, the best spectral assignments corresponding to chromosome 16 or detected in the HEK293 cell line were validated using SRM. Data are provided supporting the detection of DNAH3 in the spermatozoa sample. Moreover, ATAD3C and UNCX proteins, previously related to embryonic development, were also detected in the shotgun experiments, and more interestingly, ATAD3C was confirmed by the LC-SRM experiments.
Materials and Methods
Analysis Workflow
We applied an analysis approach based on the detection of proteotypic peptides in shotgun experiments using FDR filtering at the PSM level13 (Figure 1), and the results obtained in terms of the number of missing proteins were compared with those resulting from the analysis recommended in the HPP Data Interpretation Guidelines version 2.0.1 (approved 2015-12-01). However, a major issue to be previously addressed was the selection of the samples to be analyzed in order to increase the chance of successful missing protein identifications. Different approaches had been previously described to select the biological source in which this probability is higher based on gene transcription profiles.8,17 We propose a new prediction which is based on publicly available MS/MS experiments. The PRIDE database was examined22 to obtain the set of experiments in which the number of peptide candidates from the missing proteins is higher (Figure 1).
Figure 1.

(A) Overall scheme of the analysis pipeline developed to identify missing proteins using the PRIDE database. (B) Summary of the numbers of proteins and peptides in each step of the analysis pipeline developed.
Data Processing of PRIDE and neXtProt Databases
This study was based on the data mining of public human data sets in the PRIDE Archive database (April 2015), which contained at the time 47409216 PSMs, distributed in 242 projects and 7295 assays. The database included 6001962 unique human peptides and 559405 different protein accession codes obtained using several search engines, including Mascot, Sequest, X!Tandem, OMSSA, and Phenyx. Although we performed a complete proteome analysis of the samples selected for the study of the missing proteins, the selection of the proper experiments was carried out using only the human PSMs from the missing proteins of chromosome 16. We expected there to be a certain proportionality between the number of peptides from the missing proteins detected in a shotgun experiment and the number of missing proteins present in the sample, although the information about the search engine and the statistical reliability of the identifications was not considered.
Proteogest software23 was used to perform the in silico digestion of all the proteins contained in the reference database (neXtProt release 20150901). We applied the standard rules of trypsin digestion and allowed oxidation of methionine and two missed cleavages. The processing of the set of tryptic peptides obtained allowed us to find all the proteotypic peptides. In this manuscript, we use the theoretical definition of proteotypic peptide: a peptide generated after the digestion of a protein using a certain enzyme (commonly trypsin) that can only be detected in one protein, without taking into account experimental data or a bioinformatics prediction of MS detectability of the peptide.
Shotgun Data Analysis Using HPP Guidelines
The selected data sets were analyzed for protein identification following the HPP guidelines. We searched all the mgf files downloaded from PRIDE against the neXtProt database (release 20150901) using the target-decoy strategy with an in-house Mascot Server v. 2.3 (Matrix Science, London, U.K.) search engine. A decoy database was created using the peptide pseudoreversed method, and separate searches were performed for target and decoy databases.
For each sample, searching parameters were fixed on the basis of the information provided in the metadata associated with the project in PRIDE or by the methods described in the referenced article. False Discovery Rates at the PSM level and protein level using Mayu24 were calculated, and protein identifications were obtained applying the criteria of PSM FDR < 1% and protein FDR < 1%. Protein inference was performed using the PAnalyzer algorithm.25 Only those missing proteins labeled as conclusive by this algorithm and with at least 2 proteotypic peptides were considered as observed missing proteins in the sample.
Detection of Proteotypic Peptides in Shotgun Experiments
We propose an alternative analysis of the proteomics experiments to increase the number of missing proteins detected without a significant loss of the quality of the results (Figure 1). This pipeline used the PSMs with PSM FDR < 1%, and the peptides identified using this criteria were intersected with the set of proteotypic peptides obtained after the in silico digestion of all the amino acid sequences of the neXtProt database. This approach ensured that the proteins obtained had at least one peptide capable of discriminating them from the rest of the proteins in the reference database. Finally, the spectra assignments of the peptides potentially corresponding to missing proteins were manually curated to select the best candidates. Further verification by SRM was conducted in the indicated matrices. Nevertheless, an estimation of the protein FDR value was obtained by processing the results against the decoy database in a similar way. We performed the in silico digestion of the decoy database and extracted the proteotypic peptides. We used the minimum Mascot ion score of the target proteotypic peptides with PSM FDR < 1% to estimate the number of false protein identifications using the decoy proteotypic peptides with a higher score. The FDR at the protein level was calculated as the ratio between the number of decoy proteins and the number of target proteins detected.
Sample Collection and Preparation
Sperm samples (more than 30 million cells) and HEK293 cells were centrifuged at 800g for 10 min. The supernatant of sperm samples (seminal plasma) was removed and saved in a cryotube. The cellular pellet was washed twice with 1.5 mL of PBS, frozen in liquid nitrogen, and stored at −20 °C until use. The pelleted cells were thawed and disrupted by addition of lysis buffer (8 M urea, 2 M thiourea, and 4% CHAPS) and vigorous agitation in a vortex for 30 min at room temperature. Cell debris was removed by centrifugation at 24100g for 10 min. The supernatants were stored at −20 °C until use. The protein concentration of the supernatant was determined using the Bio-Rad RC DC Protein Assay Kit (#500-0122).
Targeted Proteomic Analyses (SRM/MRM)
Total cell extracts were loaded into 1D SDS-PAGE gel and run until the sample just entered the resolving gel. Gels were fixed (50% methanol/10% acetic acid), stained with Coomassie (Simply Blue Safe Stain, Invitrogen), washed to reveal the unique band containing the whole proteome, and subjected to in gel trypsin digestion. Briefly, the gel section was destained twice with AcN for 5 min at 40 °C, removing the liquid to complete dryness of the gel. Proteins were reduced and alkylated with 10 mM DTT/100 mM ammonium bicarbonate and 28 mM iodoacetamide/100 mM ammonium bicarbonate, respectively, for 10 min at 40 °C. Subsequently, gel pieces were dried with AcN for 5 min at 40 °C, removing the supernatant to complete dryness. Proteins were digested with trypsin (Promega) using a 1:20 trypsin/protein ratio overnight at 37 °C. Peptide extraction was performed with consecutive incubations (30 min, room temperature) with 1% formic acid/2% AcN; 05% formic acid/50% AcN; 100% AcN. All supernatants were combined and evaporated to dryness in a speed-vac. Peptides were solubilized in 1% trifluoroacetic acid and further extracted using a C18 reverse phase sorvent (Pierce C18 Spint Tips) following the manufacturer’s protocol. Extracted peptides were dried in a speed-vac before nLC ESI-MS/MS analysis.
A total of 17 proteotypic peptides were selected, and isotopically labeled standards were synthesized. Peptide standards were prepared at 500, 125, 25, and 5 fmol/μL in 2% acetonitrile, 0.1% FA. Two microliters of the solutions were analyzed in a Qtrap5500 (ABSciex) coupled to a nanoflow high performance HPLC (Eksigent) equipped with a nanoelectrospray ion source. Mobile phases were A (100% H2O and 0.1% formic acid) and B (100% AcN and 0.1% formic acid). Peptides were separated by C18 reverse phase chromatography at a flow rate of 0.3 μL/min in an Acclaim Peptide Map RSLC 75 μm (column ID) × 150 mm (column length) × 2 μm (particle size) analytical column, using the gradient: 0 min, 3%B; 3 min, 3%B; 90 min, 40%B; 100 min, 50%B; 102 min, 90%B; 108 min, 90%B; 110 min, 3%B; 125 min, 3%B. Electrospray parameters used were: CUR = 20; CAD = high; IS = 2800; GS1 = 20; GS2 = 0; and IHT = 150. The collision energy and declustering potential applied to each peptide was calculated with the skyline software. The dwell time for each transition was 20 ms for the synthetic heavy peptides and 100 ms for the endogenous peptides.
The raw MS proteomics data have been deposited in PeptideAtlas20 PASSEL with accession code PASS00925.
Results and Discussion
Sample Selection Based on the PRIDE Database Content
We found 601 proteotypic peptide candidates in the neXtProt (release 20150101) in 65 PRIDE projects, which suggests the presence of 102 missing proteins of chromosome 16 with 2630 PSMs. The number of detected peptides in each project is shown in Figure 2. This bar plot was used to select the project accession codes in which the expected number of missing proteins of chromosome 16 was higher (at least 50 peptides associated with missing proteins).
Figure 2.

Number of proteotypic peptides of chromosome 16 missing proteins in the neXtProt database that were detected in the shotgun MS/MS experiments stored in the PRIDE database. The experiments selected for further analyses are highlighted in red.
However, the PRIDE database is constantly changing, incorporating experiments as new proteomic data sets are submitted. We tried to consider this dynamic behavior as far as possible and included new samples in the study during the development of the project. Consequently, we included 4 samples from rare biological sources, since it had been proved that these samples can be used to detect missing proteins:13 spermatozoid,13 seminal plasma,26 retina,27 and placenta.28 In addition to that, we included a most recent proteome characterization of the HEK293 cell line29 in replacement of the experiment with PRIDE accession number PXD001383. The list of projects selected from the PRIDE database for analysis is shown in Table 1.
Table 1. Project Accessions of the PRIDE Database Selected for the Identification of Missing Proteinsa.
| Project Accession | Tissue | Instrument | ⧧ samples | ⧧ fractions |
|---|---|---|---|---|
| PXD001468 | HEK293 | Q Exactive | 1 | 24 |
| PXD002367 | Spermatozoid | LTQ Orbitrap | 1 | 21 |
| PXD001242 | Retina | LTQ Orbitrap Elite | 5 | 60 |
| PXD000754 | Placenta | LTQ Orbitrap | 2 | 47 |
| PXD000605 | Blood plasma | LTQ Orbitrap | 3 | 146 |
| PXD000004 | Frontal cortex | Q Exactive | 5 | 14 |
| PRD000269 | Aorta thoracica | LTQ Orbitrap | 1 | 108 |
| PXD002145 | Seminal plasma | LTQ Orbitrap Elite | 2 | 96 |
The number of samples and fractions analyzed in this study are shown.
In Silico Analysis of the neXtProt Database
The total number of peptides obtained was 7031853 (2958508 unique peptides), and 8.81% of the unique peptides corresponded to missing proteins. The mean number of peptides per protein for the missing proteins was 116, whereas the mean number of peptides for the nonmissing proteins was 180. This was in accordance with a previous analysis of the features of the missing proteins,17 in which it is shown that these proteins are shorter. The set of proteotypic peptides (tryptic peptides not shared among proteins of the neXtProt database) was generated using in-house scripts. The number of proteotypic peptides ranging from 9 to 30 amino acids in length was 826137, 10.59% of which were assigned to missing proteins (87545 peptides).
The number of tryptic and proteotypic peptides discovered using the amino acid sequences of the neXtProt database for each chromosome is shown in Figure 3A and Figure 3B, respectively. The mean number of proteotypic peptides per chromosome was 3498 for the missing proteins and 29552 for the nonmissing proteins. The number of proteins that contained at least one tryptic peptide with a length between 9 and 30 amino acids was 20028. Interestingly, 19410 proteins, almost all of the proteins detectable with tryptic peptides, had also at least one proteotypic peptide. The number of missing proteins that could be detected by at least one proteotypic peptide was 2533, 94.94% of the missing proteins in the neXtProt database. There were 2496 with two or more proteotypic peptides, 37 with only one and 135 without any predictable tryptic and proteotipic peptide, which will not be detectable according to the HPP guidelines using trypsin (Supporting Information Table 1). For these 95 proteins, other experimental approaches must be developed, for example the use of other enzymes for protein digestion.
Figure 3.

(A) Distribution of tryptic peptides deduced from in silico digestion of the neXtProt database (release 20150901) along chromosomes. (B) Distribution of proteotypic peptides deduced from the in silico digestion of the neXtProt database (release 20150901) along chromosomes. (C) Distribution of proteins with at least one tryptic peptide after the in silico digestion of the neXtProt database (release 20150901) along chromosomes. (D) Distribution of proteins with at least one proteotypic peptide after the in silico digestion of the neXtProt database (release 20150901) along chromosomes.
In Figure 3C and Figure 3D we represent the distribution of these proteins across chromosomes. The mean number of proteins with at least one tryptic peptide per chromosome was 801, and that with at least one proteotypic peptide was 777. In the case of the missing proteins, the average number of proteins per chromosome with at least one proteotypic peptide was reduced to 101 proteins.
With regard to chromosome 16, there are 836 proteins with at least one tryptic peptide and 813 with at least one proteotypic peptide with a length between 9 and 30 amino acids. 11.12% of tryptic proteins and 11.19% of proteotypic proteins are still considered missing proteins (93 tryptic and 91 proteotypic proteins).
Identification of Conclusive Missing Proteins
In order to perform the analysis of the missing proteins for all the chromosomes, we used more than 5 million spectra that were available in the selected projects from the PRIDE database. After the independent analysis of each of the experiments downloaded from the PRIDE database following the HPP guidelines, we assigned 503054 of these spectra (9.77%) and we identified 5284 proteins with 1 or more proteotypic peptides and 3950 proteins with 2 or more proteotypic peptides. We detected 58 missing proteins with 1 or more proteotypic peptides and 32 proteins with 2 or more proteotypic peptides (Supporting Information Table 3).
The results from each sample analysis are summarized in Table 3. Spermatozoid (PXD002367) and the HEK293 cell line (PXD001468) were the samples with the higher number of missing proteins detected. This result was consistent with previous analyses of the spermatozoid proteome,13 and it revealed the HEK293 cell line as a new biological source of missing proteins. However, we did not find any evidence of the presence of missing proteins in placenta (PXD000754), blood plasma (PXD000605), frontal cortex (PXD000004), aorta thoracica (PRD000269), and seminal plasma (PXD002145) samples.
Table 3. Number of PSMs, Peptides, and Proteins Identified Using the HPP Guidelines (PSM FDR < 1%, protein FDR < 1%) in the Samples Selected from PRIDE for the Analysis of the Missing Proteinsa.
| PXD001468 | PXD002367 | PXD001242 | PXD000754 | PXD000605 | PXD000004 | PRD000269 | PXD002145 | Total | |
|---|---|---|---|---|---|---|---|---|---|
| Spectra | 836145 | 114970 | 452880 | 519326 | 1299378 | 357899 | 370218 | 1198042 | 5148858 |
| Total PSMs | 328554 | 48609 | 110624 | 80213 | 19086 | 136506 | 21969 | 6676 | 752237 |
| FP PSMs | 161 | 34 | 136 | 201 | 5 | 154 | 11 | 116 | 818 |
| Total Peptides | 68377 | 9848 | 14413 | 10122 | 1228 | 16679 | 2001 | 199 | 93012 |
| Total Peptides (proteotypic) | 24510 | 3990 | 5393 | 4226 | 788 | 5737 | 746 | 56 | 33756 |
| Total Peptides (nonproteotypic) | 43867 | 5858 | 9020 | 5896 | 440 | 10942 | 1255 | 143 | 59256 |
| FP Peptides | 70 | 12 | 20 | 46 | 2 | 41 | 3 | 8 | 202 |
| Total Proteins | 7206 | 1437 | 2681 | 2127 | 363 | 2340 | 351 | 54 | 8712 |
| Total Conclusive Prot | 4539 | 909 | 1501 | 1140 | 146 | 1069 | 193 | 29 | 5626 |
| FP Proteins | 33 | 8 | 15 | 11 | 2 | 33 | 1 | 8 | 111 |
| Total Assigned Spectra | 191095 | 24736 | 66707 | 51392 | 18091 | 133602 | 14936 | 2495 | 503054 |
| Missing PSMs | 798 | 473 | 117 | 0 | 0 | 0 | 0 | 0 | 1388 |
| Missing Peptides | 83 | 258 | 25 | 0 | 0 | 0 | 0 | 0 | 357 |
| Missing Proteins | 10 | 47 | 5 | 0 | 0 | 0 | 0 | 0 | 60 |
| Missing Assigned Spectra | 479 | 367 | 68 | 0 | 0 | 0 | 0 | 0 | 914 |
| Total Proteins HPP (≥1 peptide) | 4276 | 888 | 1450 | 1115 | 146 | 1053 | 188 | 28 | 5284 |
| Total Proteins HPP (≥2 peptides) | 3326 | 750 | 1260 | 1000 | 120 | 924 | 169 | 22 | 3950 |
| Missing Proteins HPP (≥1 peptide) | 10 | 45 | 5 | 0 | 0 | 0 | 0 | 0 | 58 |
| Missing Proteins HPP (≥2 peptides) | 5 | 27 | 1 | 0 | 0 | 0 | 0 | 0 | 32 |
FP = false positives.
Detection of Missing Proteins Using Proteotypic Peptides
Our objective is to increase the number of missing protein detections in the human proteome using the selected PRIDE data sets with an alternative bioinformatics pipeline based on the identification of proteotypic peptides deduced from the proteins of interest. In this strategy, we retained the protein identifications that failed to pass the FDR criteria at a protein level of 1%. The PSMs obtained with the Mascot search engine (search parameters were previously shown in Table 2) with PSM FDR < 1% were used to identify all potential tryptic peptides from the proteins present in the samples (Supporting Information Table 2). Finally, this set of peptides were intersected with the proteotypic peptides found after the in silico digestion of the neXtProt database.
Table 2. Parameters Used in the Mascot Search Engine for the Analysis of Each Downloaded Project from the PRIDE Database.
| Project Accession | Precursor mass tolerance (ppm) | Fragment mass tolerance (Da) | Missed cleavages | Fixed modifications | Variable modifications |
|---|---|---|---|---|---|
| PXD001468 | 20 | 0.05 | 2 | Carbamidomethyl (C) | Oxidation (M) |
| PXD002367 | 10 | 0.5 | 2 | Carbamidomethyl (C) | Oxidation (M) |
| Acetyl (Protein N-term) | |||||
| PXD001242 | 20 | 0.05 | 2 | Carbamidomethyl (C) | Oxidation (M) |
| PXD000754 | 20 | 1 | 2 | Carbamidomethyl (C) | Oxidation (M) |
| PXD000605 | 20 | 0.05 | 2 | iTRAQ4plex114 (K) | iTRAQ4plex114 (Y) |
| Methylthio (C) | Oxidation (M) | ||||
| PXD000004 | 20 | 0.05 | 2 | Carbamidomethyl (C) | Oxidation (M) |
| Label: 13C(6) (K) | |||||
| PRD000269 | 20 | 0.05 | 2 | Carbamidomethyl (C) | Oxidation (M) |
| PXD002145 | 10 | 0.5 | 2 | Carbamidomethyl (C) | Oxidation (M) |
| Acetyl (Protein N-term) |
This approach allowed us to detect a total of 6333 proteins, 1049 more than the proteins identified with the HPP guideline analysis. With regard to the number of peptides identified, we obtained 35922 proteotypic peptides with PSM FDR < 1%, representing an increase of 6.42% over the peptides detected with the previous method. In order to achieve these results, 515506 spectra were assigned, a slight increase (0.24%) in the percentage of spectra used from the total number of spectra available in the data sets. This led to the inclusion of 12452 new spectra in the analysis (Table 4).
Table 4. Number of PSMs, Peptides, and Proteins Observed Using the Identifications of Proteotypic Peptides from the neXtProt Database (PSM FDR < 1%) in the Samples Selected from PRIDE for the Analysis of the Missing Proteins.
| PXD001468 | PXD002367 | PXD001242 | PXD000754 | PXD000605 | PXD000004 | PRD000269 | PXD002145 | Total | |
|---|---|---|---|---|---|---|---|---|---|
| Spectra | 836145 | 114970 | 452880 | 519326 | 1299378 | 357899 | 370218 | 1198042 | 5148858 |
| Total PSMs | 332417 | 49100 | 115861 | 82856 | 19182 | 138704 | 23199 | 6676 | 767995 |
| Total Peptides | 71277 | 10311 | 16329 | 11739 | 1271 | 17570 | 2521 | 199 | 98319 |
| Total Peptides (proteotypic) | 25734 | 4187 | 6259 | 4848 | 804 | 6092 | 988 | 56 | 35922 |
| Total Peptides (nonproteotypic) | 45543 | 6124 | 10070 | 6891 | 467 | 11478 | 1533 | 143 | 62397 |
| Total Proteins (≥1 peptide) | 5341 | 1293 | 2420 | 2208 | 245 | 1929 | 569 | 41 | 6333 |
| Total Proteins (≥2 peptides) | 3326 | 750 | 1260 | 1000 | 120 | 924 | 169 | 22 | 3950 |
| Total Assigned Spectra | 193971 | 25083 | 71118 | 53398 | 18187 | 135285 | 15969 | 2495 | 515506 |
| Missing PSMs | 96 | 246 | 33 | 22 | 0 | 14 | 4 | 0 | 415 |
| Missing Peptides | 48 | 163 | 14 | 10 | 0 | 8 | 4 | 0 | 242 |
| Missing Proteins (≥1 peptide) | 30 | 67 | 14 | 10 | 0 | 8 | 4 | 0 | 122 |
| Missing Proteins (≥2 peptides) | 8 | 30 | 3 | 2 | 0 | 2 | 2 | 0 | 39 |
| Missing Assigned Spectra | 62 | 195 | 29 | 16 | 0 | 14 | 4 | 0 | 320 |
The mean value of the FDR estimation at protein level was 8%. This value is higher than the threshold recommended by the HPP guidelines, but it provided high quality results after a manual curation of the assigned spectra.
Focusing on missing proteins, 122 were potentially identified (Supporting Information Table 4), 62 proteins more than those detected as conclusive proteins by PAnalyzer, and only 242 peptides were needed compared with the 357 peptides obtained after the protein inference process. Seminal plasma (PXD002145) and blood plasma (PXD000605) were the only samples where we did not find any evidence of the presence of missing proteins. We also observed differences in the number of spectra assigned, 320 in this analysis and 914 in the previously described. This result is consistent with the basis of our method, since it only allows for proteotypic peptide detection.
Peptide distribution along chromosomes showed that the number of proteotypic peptides was a small fraction of the total number of peptides observed (Figure 4A). Moreover, we obtained a statistically significant lower Mascot ion score for the peptides from the missing proteins compared with the ion score of the peptides from the nonmissing proteins (t test statistic with a p-value <1 × 10–16, see Figure 4B). Unsurprisingly, the proportion of missing proteins detected per chromosome was very small, although assignments were made in all chromosomes, with the only exception that of mitochondria (Figure 4C). As might be expected, the comparison of the missing proteins detected by the two bioinformatic pipelines (Figure 4D) showed that the majority of proteins detected using HPP guidelines were included in the set of missing proteins with proteotypic peptides.
Figure 4.

(A) Distribution of tryptic and proteotypic peptide candidates detected in the analyzed samples along the different chromosomes. (B) Boxplot with the distribution of Mascot ion scores obtained for the PSMs assigned to missing and nonmissing proteins. The difference between these distributions is statistically significant with a p-value < 1 × 10–12. (C) Distribution of missing and nonmissing proteins potentially detected in the analyzed samples using the identification of proteotypic peptides along chromosomes. (D) Venn diagram with the missing proteins observed using the HPP guidelines and the workflow proposed here and with the missing proteins in neXtProt database release 20150901.
In spite of the tissue specificity of the missing proteins, we found a few examples of shared proteins between samples (Figure 5A). The diagonal of the heat map represents the number of missing proteins identified in each sample, and the rest of the matrix is filled with the number of missing proteins common to each pair of samples. It is easy to verify that the majority of the protein identifications were sample specific. The visualization of the results was improved using a network to represent in an effective way the relations between the samples studied, the missing proteins observed, and the peptides detected (Figure 5B). This graph could be completed including the results of the analysis of more proteomic data sets in order to generate the network of the missing proteins of the human proteome.
Figure 5.

(A) Heat map with the missing proteins potentially detected in each sample and the missing proteins shared between each pair of samples analyzed. (B) Network representation of the results obtained for the study of the missing proteins using the PRIDE database. Nodes represent the database of experiments used (green), the tissue (orange), the proteins observed (red), and the identified peptides (blue). (C) Network for the missing proteins potentially observed in the HEK293 sample. Nodes represent the sample selected (green), the chromosome (blue), and the identified protein (red). (D) Network for the missing proteins potentially detected in chromosome 16. Nodes represent the sample (orange), the proteins observed (red), and the identified peptides (blue).
Missing Proteins Identified in the HEK293 Cell Line or from Chromosome 16
The network of the missing proteins was used to extract information about the proteins observed to perform functional analysis of protein sets and to select peptides for validation. In our case, we decided to continue the analysis of the missing proteins detected in the HEK293 cell line (PXD001468, shown in Figure 5C), as it is different from previous studies focused on testis and sperm or encoded by chromosome 16, genes as this is the chromosome adopted by the Spanish team in chromosome 16 (Figure 5D). In Table 5 the 34 proteins corresponding to 33 known genes found in the HEK293 cell line or chromosome 16 are shown. These proteins are a subset of the total of the proteins obtained using the detection of proteotypic peptides (Supporting Information Table 4).
Table 5. Missing Proteins Potentially Identified Using Proteotypic Peptide Candidates in the HEK293 Cell Line or in Chromosome 16.
| Protein | Name | Chr | no. PSMs | no. Peptides | Ion score | HPP guidelines (2 proteotypic peptides) | Sample |
|---|---|---|---|---|---|---|---|
| NX_A6NJT0 | UNCX | 7 | 8 | 4 | 113.22 | HEK | |
| NX_B2RXH8 | HNRNPCL2 | 1 | 276 | 15 | 102.61 | HEK,Retina | |
| NX_Q9BQ87 | TBL1Y | Y | 76 | 10 | 100.66 | HEK | |
| NX_Q2VIQ3 | KIF4B | 5 | 46 | 19 | 99.77 | √ | HEK |
| NX_Q6IS14 | EIF5AL1 | 10 | 298 | 17 | 95.03 | √ | HEK |
| NX_Q5T2N8 | ATAD3C | 1 | 56 | 8 | 85.06 | √ | HEK,Retina |
| NX_Q56UQ5 | - | X | 55 | 4 | 81.79 | √ | HEK |
| NX_Q8TD57 | DNAH3 | 16 | 27 | 25 | 80.77 | √ | Spermatozoa,Retina |
| NX_Q6URK8 | TEPP | 16 | 17 | 10 | 79.62 | Spermatozoa | |
| NX_Q9NRJ5 | PAPOLB | 7 | 10 | 3 | 77.63 | √ | HEK |
| NX_Q6ZR08 | DNAH12 | 3 | 34 | 23 | 75.04 | √ | Placenta,HEK,Spermatozoa |
| NX_A8K0S8 | MEIS3P2 | 17 | 6 | 1 | 58.75 | √ | HEK |
| NX_Q6ZMV8 | ZNF730 | 19 | 3 | 3 | 58.21 | √ | HEK |
| NX_Q14585 | ZNF345 | 19 | 1 | 1 | 57.3 | √ | HEK |
| NX_Q52M93 | ZNF585B | 19 | 1 | 1 | 54.08 | √ | HEK |
| NX_Q9UJN7 | ZNF391 | 6 | 4 | 3 | 53.79 | √ | HEK |
| NX_P58180 | OR4D2 | 17 | 3 | 1 | 52.28 | √ | HEK,Spermatozoa |
| NX_Q8NGL6 | OR4A15 | 11 | 3 | 1 | 52.28 | Spermatozoa,HEK | |
| NX_P59817 | ZNF280A | 22 | 1 | 1 | 48.17 | HEK | |
| NX_A6NHN6 | NPIPB15 | 16 | 7 | 5 | 47.7 | √ | Spermatozoa |
| NX_Q9Y2H8 | ZNF510 | 9 | 1 | 1 | 45.02 | HEK | |
| NX_Q96KX1 | C4orf36 | 4 | 1 | 1 | 44.7 | √ | HEK |
| NX_Q96M86 | DNHD1 | 11 | 1 | 1 | 44.16 | HEK | |
| NX_Q5VTU8 | ATP5EP2 | 13 | 1 | 1 | 43.65 | √ | HEK |
| NX_Q8N0W5 | IQCK | 16 | 1 | 1 | 43.57 | √ | Spermatozoa |
| NX_Q4AC99 | ACCSL | 11 | 1 | 1 | 43.57 | √ | HEK |
| NX_A6NNF4 | ZNF726 | 19 | 2 | 1 | 43.39 | √ | HEK |
| NX_P0CW27 | CCDC166 | 8 | 1 | 1 | 40.88 | √ | HEK |
| NX_A6NCM1 | IQCA1L | 7 | 1 | 1 | 40.63 | √ | HEK |
| NX_Q8NDH2 | CCDC168 | 13 | 1 | 1 | 40.58 | √ | HEK |
| NX_Q6R2W3 | ZBED9 | 6 | 1 | 1 | 40.51 | √ | HEK |
| NX_A6NN73 | GOLGA8CP | 15 | 1 | 1 | 40.35 | √ | HEK |
| NX_Q9H2H0 | CXXC4 | 4 | 1 | 1 | 39.19 | √ | HEK |
| NX_Q9BXX2 | ANKRD30B | 18 | 3 | 2 | 39.01 | √ | Aorta,HEK |
Functional Analysis of the Missing Proteins
The functional analysis of the list of the 182 missing proteins detected was performed, and a good correlation between the results obtained and the sample types analyzed was found. We used DAVID v6.7 software30 for the analysis of GO terms, INTERPRO domains, KEGG pathways, PANTHER pathways, and UNIGENE quantile expression level gene sets using the whole human proteome as the background list of proteins. The statistical analysis was performed using default parameters, and although the p-value was corrected using the multiple hypothesis methods (including FDR), the selection of enriched categories was based on a criterion of EASE Score < 0.1, as suggested by the bioinformatics tool. Using these recommended settings, we found a list of enriched categories related with specific functions carried out by these proteins in the samples analyzed (Supporting Information Table 5). First, the tissue specific expression gene ontology analysis for these genes using the ”UNIGENE EST QUARTILE” expression profile database showed statistical enrichment in ”brain normal” with 44 genes and a p-value = 0.003, ”embryo development” with 51 genes and a p-value = 0.01, and ”testis normal” with 67 genes and a p-value = 2.73 × 10–14, confirming the sample specificity of the missing proteins detected. The results of the enrichment analysis of GO terms showed categories previously related to spermatozoa function,13 such as ”microtubule-based movement”, ”sexual reproduction”, ”integral to membrane”, or ”motor activity”. Other enriched categories were related to brain tissues or neurological processes, such as ”sensory perception”, ”neurological system process”, ”cognition”, or ”postsynaptic membrane”. Finally, others were involved in cell differentiation (”transcription” or ”DNA binding”). We also compared the categories obtained with those previously defined with a similar functional analysis of all the missing proteins,17 and many overlaps were found: ”G-protein coupled receptor protein signaling pathway”, ”integral to membrane”, ”olfactory receptor activity”, or some Interpro domains (”zinc finger, C2H2-type”, ”GPCR, rhodopsin-like superfamily”).
A complementary functional and pathway analysis of this protein set was carried out using QIAGEN Ingenuity Pathway Analysis (www.ingenuity.com). As expected, we found a lack of enrichments or networks of interest due to the curated database on which this software is based. The missing proteins are proteins without experimental evidence, and in most of the cases, this is linked to scarce bibliographic information about them or their coding genes. However, interesting relationships were found between the protein WBP2NK (a sperm-specific WW domain-binding protein that promotes meiotic resumption and pronuclear development during oocyte fertilization) and ”reproductive system development and function”; proteins CNGA2 (Cyclic Nucleotide Gated Channel Alpha 2) and PLCZ1 (Phospholipase C, Zeta 1, a protein that localizes to the acrosome in spermatozoa and elicits Ca(2+) oscillations and egg activation during fertilization) and ”sperm mobility”; and UNXC (UNC Homeobox, a transcription factor involved in somitogenesis and neurogenesis and required for the maintenance and differentiation of particular elements of the axial skeleton) and ”embryonic development”.
Manual Evaluation of PSMs and Selection of Peptides
As we have previously mentioned, the estimated protein FDR for the proteins selected was 8%. In order to minimize the influence of this value on the quality of the results, we performed additional filtering steps to select the peptides for experimental validation. First, we selected the peptides with less than 20 amino acids in length, due to the limitation in the synthesis of heavy peptides for SRM/MRM experiments. For each remaining peptide of chromosome 16 or the HEK293 cell line, we chose its best PSM using the maximum Mascot ion score. This resulted in a total of 59 peptides, 43 of which were observed in the HEK293 cell line and 16 of which were observed chromosome 16.
The last stage consisted of a manual curation of the assigned spectra by three mass spectrometry experts. The 59 spectra were visualized and evaluated using the software SeeMS 3.0.7106.0 from ProteoWizard platform for proteomics data analysis,31 according to the following features:32 (a) the quality of the y-ion and b-ion series assignments; (b) the peak intensities and observed signal-to-noise ratio; (c) the number of nonassigned peaks. Only the PSMs considered as ”high quality” by the three experts were considered for further analysis. For illustrative purposes, we show in Figure 6 four PSMs corresponding to two peptides selected from chromosome 16 (Figure 6A and Figure 6B) and one peptide from the HEK293 cell line (Figure 6C and Figure 6D). The complete list of the 17 peptides selected for validation by SRM/MRM can be found in Table 6.
Figure 6.

(A) Spectra assignment of peptide LYSSLLDEIR from protein NX_Q8TD57 (DNAH3, chromosome 16) detected with Mascot ion score 75.99 in spermatozoa. (B) Spectra assignment of peptide TQTISLGQGQGPIAAK from protein NX_Q8TD57 (DNAH3, chromosome 16) detected with Mascot ion score 80.77 in spermatozoa. (C) Spectrum assignment of peptide DAASCGPGAAVAAVER from protein NX_A6NJT0 (UNCX, chromosome 7) detected with Mascot ion score 113.22 in HEK293 cell line.
Table 6. Peptides Selected for Validation Using Targeted Proteomics (SRM/MRM).
| Protein | Name | Peptide | Chr | Sample | Ion score | Missing in neXtProt20160111 | HPP guidelines (2 proteotypic peptides) |
|---|---|---|---|---|---|---|---|
| NC_A6NJT0 | UNCX | DAASCGPGAAVAAVER | 7 | HEK | 113.22 | √ | √ |
| NC_Q9BQ87 | TBL1Y | IWTENGNLASTLGQHK | Y | HEK | 93.62 | √ | √ |
| NC_Q8TD57 | DNAH3 | TQTISLGQGQGPIAAK | 16 | Spermatozoa | 80.77 | √ | |
| NC_Q8TD57 | DNAH3 | LYSSLLDEIR | 16 | Spermatozoa | 75.99 | √ | |
| NC_Q2VIQ3 | KIF4B | EMCDMEQVLSK | 5 | HEK | 67.29 | √ | √ |
| NC_Q5T2N8 | ATAD3C | AAGTLFGEGFR | 1 | HEK | 66.45 | √ | |
| NC_Q2VIQ3 | KIF4B | NLELEVINLQK | 5 | HEK | 64.73 | √ | √ |
| NC_A8K0S8 | MEIS3P2 | MVQPMIDQSNR | 17 | HEK | 58.75 | √ | |
| NC_Q8TD57 | DNAH3 | EANVAAAIAQGIK | 16 | Spermatozoa | 49.37 | √ | |
| NC_A6NHN6 | NPIPB15 | ADEVEQSPKPK | 16 | Spermatozoa | 47.7 | √ | |
| NC_Q8N0W5 | IQCK | AGEPFTEFFSIPFVEER | 16 | Spermatozoa | 43.57 | √ | |
| NC_B2RXH8 | HNRNPCL2 | MIASQVAVINLAAEPK | 1 | HEK | 43.42 | √ | √ |
| NC_Q8TD57 | DNAH3 | VESVLFPELK | 16 | Spermatozoa | 39.34 | √ | |
| NC_Q8TD57 | DNAH3 | DFDLEEVMK | 16 | Spermatozoa | 37.96 | √ | |
| NC_Q8TD57 | DNAH3 | AVVFVDDLNMPAK | 16 | Spermatozoa | 36.67 | √ | |
| NC_Q8TD57 | DNAH3 | GNILEDETAIK | 16 | Spermatozoa | 36.09 | √ | |
| NC_Q6URK8 | TEPP | YCLSQNPSLDR | 16 | Spermatozoa | 31.36 |
Although all the analyses and the selection of the peptides for validation were carried out using the neXtProt database 20150901, the release of a new version (20160111) compelled us to compare the results at this stage with the new list of missing proteins. As shown in Table 6, all the selected proteins except DNAH3 and TEPP (with new evidence in the spermatozoa sample) were still considered missing proteins in the new release.
Validation of Missing Protein Identifications Using SRM/MRM
In order to validate the identifications of the missing proteins, two experimental strategies were designed. First, a sample with a mixture of the heavy peptides for the 17 peptides selected for validation was analyzed using MIDAS (MRM-initiated detection and sequencing), a method in which the mass spectrometer (ABSciex QTrap5500) switches from MRM to enhanced product ion scanning mode when an individual MRM is detected. Data were examined manually to verify the chromatographic peaks and the transitions detected for each peptide, and 15 peptides were used for further analysis (Supporting Information Table 6). The MS/MS spectra of the heavy precursors were searched with Mascot against the neXtProt database using the target-decoy strategy with the following parameters: precursor tolerance 0.8 Da, fragment tolerance 0.6 Da, two missed cleavages, carbamidomethyl cysteine as a fixed modification, and oxidized methionine as variable modification. The identification of proteins was performed with a criterion of PSM FDR < 1%, protein FDR < 1%, and PAnalyzer to select only conclusive proteins. We detected 13 of the synthetic peptides, corresponding to 8 missing proteins (Table 7). In Figure 7 we show the comparison between a selection of the fragmentation spectra obtained for the heavy peptides and the corresponding endogenous spectra found in the shotgun experiments for the peptides VESVCFPELK (DNAH3), EANVAAAIAQGIK (DNAH3), and AAGTLFGEGFR (ATAD3C) using the SDPScore.33
Table 7. Results of the Mascot Search of the Heavy Peptide Sample Using the neXtProt Databasea.
| Peptide | Protein | Chr | Name | Max ion score | Missing in (neXtProt20160111) |
|---|---|---|---|---|---|
| DAASCGPGAAVAAVER | NX_A6NJT0 | 7 | UNCX | 78.49 | √ |
| VESVLFPELK | NX_Q8TD57 | 16 | DNAH3 | 75.29 | |
| AAGTLFGEGFR | NX_Q5T2N8 | 1 | ATAD3C | 56.93 | √ |
| MIASQVAVINLAAEPK | NX_B2RXH8 | 1 | HNRNPCL2 | 55.83 | √ |
| ADEVEQSPKPK | NX_A6NHN6 | 16 | NPIPB15 | 54.39 | √ |
| EANVAAAIAQGIK | NX_Q8TD57 | 16 | DNAH3 | 52.72 | |
| EMCDMEQVLSK | NX_Q2VIQ3 | 5 | KIF4B | 48.67 | √ |
| YCLSQNPSLDR | NX_Q6URK8 | 16 | TEPP | 46.26 | |
| MVQPMIDQSNR | NX_A8K0S8 | 17 | MEIS3P2 | 45.03 | √ |
| LYSSLLDEIR | NX_Q8TD57 | 16 | DNAH3 | 44.68 | |
| TQTISLGQGQGPIAAK | NX_Q8TD57 | 16 | DNAH3 | 44.15 | |
| AVVFVDDLNMPAK | NX_Q8TD57 | 16 | DNAH3 | 43.37 | |
| DFDLEEVMK | NX_Q8TD57 | 16 | DNAH3 | 39.71 | |
| GNILEDETAIK | NX_Q8TD57 | 16 | DNAH3 | 39.6 |
Conclusive proteins according to PAnalyzer were selected (PSM FDR < 1%, Protein FDR < 1%).
Figure 7.

(A) Comparison of the MS/MS spectrum of peptide EANVAAAIAQGIK from DNAH3 protein obtained in the shotgun experiment (lower) and the MS/MS spectrum for its synthetic heavy peptide (upper) obtained in the LC–SRM experiment (SDPScore = 0.88). (B) Comparison of the MS/MS spectrum of peptide VESVCFPELK from DNAH3 protein obtained in the shotgun experiment (lower) and the MS/MS spectrum for its synthetic heavy peptide (upper) obtained in the LC–SRM experiment (SDPScore = 0.90). (C) Comparison of the MS/MS spectrum of peptide AAGTLFGEGFR from ATAD3C protein obtained in the shotgun experiment (lower) and the MS/MS spectrum for its synthetic heavy peptide (upper) obtained in the LC–SRM experiment (SDPScore =0.89).
The final step of the validation process was the targeting of the selected peptides by SRM to detect them in the biological samples of interest (spermatozoa and the HEK293 cell line). This approach allowed us to confirm the presence of four peptides in spermatozoa (DNAH3) and an additional peptide for the protein ATAD3C in the HEK293 cell line, as can be seen in Figure 8A. As we have mentioned before, the protein DNAH3 changed its evidence from missing protein to PE1 during the development of our study (neXtProt release 20160111). The evidence at the protein level was obtained from PeptideAtlas using a reanalysis of the spermatozoa proteome.13 We validated this evidence in a set of independent samples with the detection of four proteotypic peptides. In Figure 8C–E we show the SRM/MRM signal for three of these peptides, and in Figure 8B we show the SRM/MRM signal for the peptide detected from ATAD3C in the HEK293 cell line.
Figure 8.

(A) Venn diagram with the peptides selected for detection and the results of the different stages of the validation analysis. (B) Endogenous (upper) and synthetic heavy peptide (lower) LC-SRM signals measured for the peptide AAGTLFGEGFR from ATAD3C in the HEK293 cell line. (C) Endogenous (upper) and synthetic heavy peptide (lower) LC-SRM signals measured for the peptide EANVAAAIAQGIK from DNAH3 in the spermatozoa sample. (D) Endogenous (upper) and synthetic heavy peptide (lower) LC-SRM signals measured for the peptide GNILEDETAIK from DNAH3 in the spermatozoa sample. (E) Endogenous (upper) and synthetic heavy peptide (lower) LC-SRM signals measured for the peptide VESVLFPELK from DNAH3 in the spermatozoa sample.
Although we found only one peptide using LC-SRM in the HEK293 cell line, we suggest increasing the number of experiments in this sample in order to validate the presence of a large number of missing proteins using other experimental protocols or other proteomic techniques, for example antibody-based technologies. We consider this finding as an opportunity to characterize proteins with potential interest in molecular and biology research. For example, the proteins ATAD3C (ATPase Family, AAA Domain Containing 3C), validated using the LC–SRM approach, and UNCX (UNC Homeobox), detected in the shotgun data analysis following the HPP guidelines, have been previously related to embryonic development34,35 and tumorigenesis.36 More specifically, mutations of the ATAD3C gene have been associated with colorectal cancer (COSMIC accession codes 2230025 and 2230026).
Collectively, the validation experiments carried out proved the success of the strategy described in this manuscript to detect missing proteins using the analysis of public high throughput proteomic data sets. The analysis of the shotgun experiments of the samples enriched in missing proteins from chromosome 16 was able to detect high quality spectra assigned to a set of proteins defined as missing proteins in neXtProt release 20150901, although a small fraction of them are now considered as PE1 proteins in the current release (20160111). The analysis of synthetic heavy peptides for 17 selected peptides using a MIDAS approach and the LC-SRM analysis performed in the spermatozoa sample and the HEK293 cell line provided support to the proteomics evidence for missing protein candidates. Interestingly, we were able to confirm the presence of the protein DNAH3 in spermatozoa and ATAD3C in the HEK293 cell line using SRM/MRM. The protein DNAH3 was a missing protein in the neXtProt release used in this study, but is considered as PE1 in the current release. This observation confirms the value of the developed strategy for the annotation of missing proteins. We also provided robust evidence supporting HEK293 cells as a promising source of missing proteins.
Conclusions
The complete characterization of the human proteome is an ambitious task which is being carried out jointly by proteomics laboratories worldwide in the framework of the HPP project.37 Despite the efforts made and the resources devoted to this issue since its start in 2001, no experimental evidence for 14.70% of human proteins (neXtProt release 20160201) has yet been detected in any biological matrix. The detection of this set of proteins, known as the ”missing proteins”, is a huge challenge from the proteomics, bioinformatics, and statistical points of view.8 In recent years, the analysis of the expression level of the protein coding genes and their tissue specificity has revealed a map with the most probable location of each missing protein in a wide variety of samples.17 However, the biochemical characteristics of these proteins make their detection extremely challenging, especially if stringent statistical thresholds are applied to established the likelihood of the observations.13,32 The contents of a variety of databases of proteomic experiments have been gradually incorporated into the project to define the reference human proteome, for example the PRIDE database. Using the information about all the human PSMs stored in the this database, we selected a set of target samples (we found human spermatozoa and the HEK293 cell line samples specially enriched in missing proteins), and we compared two different methods of analysis of shotgun data sets for the identification of missing proteins at the proteome level.
In an attempt to provide new horizons and guidance on how and where missing proteins should be hunted for, we propose here a nonconventional bioinformatic pipeline that relies on the use of PRIDE data sets relaxing the statistical constraints to allow the selection of PSMs that suggest the presence of peptides from missing proteins, followed by a robust validation process. We used the in silico digestion of the protein reference database and the selection of unique peptides (proteotypic peptides) for all the proteome to filter those spectra assignments with PSM FDR < 1%. With this method, without the need for protein inference and protein FDR filtering, we found 182 missing protein candidates. However, in this case, the results had to be carefully analyzed by mass spectrometry experts to remove low quality assignments, and hence, the remaining PSM entered the experimental validation process based on SRM. From our findings, 17 peptides were selected for validation, and heavy peptides were synthesized to validate the identification of 13 missing proteins with SRM/MRM experiments. We identified four proteotypic peptides from the protein DNAH3 in the spermatozoa sample and one proteotypic peptide from the protein ATAD3C in the HEK293 cell line using LC-SRM assays. Therefore, we have demonstrated the feasibility of the study of missing proteins using an alternative method that combines the proper selection of the target sample based on MS experiments from public databases and a statistical analysis based on the detection of certain peptides that uniquely defined the missing proteins.
Acknowledgments
CIMA, UV, and CNIO laboratories are members of the PRBB-ISCIII platform. This study was supported by PRBB and the Carlos III National Health Institute Agreement, PRBB-ISCIII; grants SAF2014-5478-R from Ministerio de Ciencia e Innovación and ISCIII-RETIC RD06/0020 to F.J.C., grants 33/2015 from Dpto. de Salud of Gobierno de Navarra and DPI2015-68982-R from Ministerio de Ciencia e Innovación to V.S., and grant BFU2012-39482 from Ministerio de Economía y Competitividad to M.S.P. J.A.V. and N.d.T. are supported by the Wellcome Trust [grant number WT101477MA].
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.6b00437.
Supporting Information Table 1: List of missing proteins with the number of proteotypic peptides using the in silico digestion of neXtProt (release 20150901) with Proteogest software (XLSX)
Supporting Information Table 2: Mascot search engine results obtained for the different projects from the PRIDE database analyzed (PSM FDR < 1%) (XLSX)
Supporting Information Table 3: Summary of the HPP guideline results (XLSX)
Supporting Information Table 4: List of the peptides from the missing proteins obtained in the analysis of the PRIDE samples after PSM FDR < 1% filtering, intersected with the proteotypic peptides of neXtProt (XLSX)
Supporting Information Table 5: Functional characterization of the missing proteins detected using the analysis of the proteotypic peptides of the neXtProt database (183 proteins) with DAVID software (XLSX)
Supporting Information Table 6: List of SRM/MRM transitions designed for the detection of the peptides observed from the missing proteins in the shotgun experiments with the ABSciex Qtrap5500. (XLSX)
Supporting Information File 1: Supplemental methods (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Legrain P.; et al. The human proteome project: Current state and future direction. Mol. Cell. Proteomics 2011, 10.1074/mcp.O111.009993. [DOI] [PubMed] [Google Scholar]
- Paik Y.-K.; et al. Standard guidelines for the chromosome-centric human proteome project. J. Proteome Res. 2012, 11, 2005–2013. 10.1021/pr200824a. [DOI] [PubMed] [Google Scholar]
- Paik Y.-K.; et al. The Chromosome-Centric Human Proteome Project for cataloging proteins encoded in the genome. Nat. Biotechnol. 2012, 30, 221–223. 10.1038/nbt.2152. [DOI] [PubMed] [Google Scholar]
- Aebersold R.; Bader G. D.; Edwards A. M.; van Eyk J. E.; Kussmann M.; Qin J.; Omenn G. S. The Biology/Disease-driven Human Proteome Project (B/D-HPP): Enabling Protein Research for the Life Sciences Community. J. Proteome Res. 2013, 12, 23–27. 10.1021/pr301151m. [DOI] [PubMed] [Google Scholar]
- Aebersold R.; Bader G. D.; Edwards A. M.; van Eyk J. E.; Kussmann M.; Qin J.; Omenn G. S. Highlights of B/D-HPP and HPP Resource Pillar Workshops at 12th Annual HUPO World Congress of Proteomics. Proteomics 2014, 14, 975–988. 10.1002/pmic.201400041. [DOI] [PubMed] [Google Scholar]
- Nilsson T.; Mann M.; Aebersold R.; Yates J. R. 3rd; Bairoch A.; Bergeron J. J. M. Mass spectrometry in high-throughput proteomics: ready for the big time. Nat. Methods 2010, 7, 681–685. 10.1038/nmeth0910-681. [DOI] [PubMed] [Google Scholar]
- Segura V.; et al. Surfing transcriptomic landscapes. A step beyond the annotation of chromosome 16 proteome. J. Proteome Res. 2014, 13, 158–172. 10.1021/pr400721r. [DOI] [PubMed] [Google Scholar]
- Horvatovich P.; et al. Quest for Missing Proteins: Update 2015 on Chromosome-Centric Human Proteome Project. J. Proteome Res. 2015, 14, 3415–3431. 10.1021/pr5013009. [DOI] [PubMed] [Google Scholar]
- Gaudet P.; Michel P.-A.; Zahn-Zabal M.; Cusin I.; Duek P. D.; Evalet O.; Gateau A.; Gleizes A.; Pereira M.; Teixeira D.; Zhang Y.; Lane L.; Bairoch A. The neXtProt knowledgebase on human proteins: current status. Nucleic Acids Res. 2015, 43, D764–D770. 10.1093/nar/gku1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane L.; Bairoch A.; Beavis R. C.; Deutsch E. W.; Gaudet P.; Lundberg E.; Omenn G. S. Metrics for the Human Proteome Project 2013–2014 and strategies for finding missing proteins. J. Proteome Res. 2014, 13, 15–20. 10.1021/pr401144x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Djureinovic D.; Fagerberg L.; Hallström B.; Danielsson A.; Lindskog C.; Uhlén M.; Pontén F. The human testis-specific proteome defined by transcriptomics and antibody-based profiling. Mol. Hum. Reprod. 2014, 20, 476–488. 10.1093/molehr/gau018. [DOI] [PubMed] [Google Scholar]
- Jumeau F.; Com E.; Lane L.; Duek P.; Lagarrigue M.; Lavigne R.; Guillot L.; Rondel K.; Gateau A.; Melaine N.; Guével B.; Sergeant N.; Mitchell V.; Pineau C. Human Spermatozoa as a Model for Detecting Missing Proteins in the Context of the Chromosome-Centric Human Proteome Project. J. Proteome Res. 2015, 14, 3606–3620. 10.1021/acs.jproteome.5b00170. [DOI] [PubMed] [Google Scholar]
- Clough E.; Barrett T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. 10.1007/978-1-4939-3578-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium:Dunham I.; Birney E.; Bernstein B. E.; Green E. D.; Gunter C.; Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas-Madrid D.; Alves-Cruzeiro J.; Segura V.; Guruceaga E.; Vialas V.; Prieto G.; García C.; Corrales F. J.; Albar J. P.; Pascual-Montano A. Proteogenomics Dashboard for the Human Proteome Project. J. Proteome Res. 2015, 14, 3738–3749. 10.1021/acs.jproteome.5b00466. [DOI] [PubMed] [Google Scholar]
- Guruceaga E.; Sanchez del Pino M. M.; Corrales F. J.; Segura V. Prediction of a missing protein expression map in the context of the human proteome project. J. Proteome Res. 2015, 14, 1350–1360. 10.1021/pr500850u. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Alpi E.; Wang R.; Hermjakob H.; Vizcaíno J. A. Making proteomics data accessible and reusable: current state of proteomics databases and repositories. Proteomics 2015, 15, 930–949. 10.1002/pmic.201400302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R.; Cortens J. P.; Beavis R. C. Open source system for analyzing, validating, and storing protein identification data. J. Proteome Res. 2004, 3, 1234–1242. 10.1021/pr049882h. [DOI] [PubMed] [Google Scholar]
- Farrah T.; Deutsch E. W.; Omenn G. S.; Sun Z.; Watts J. D.; Yamamoto T.; Shteynberg D.; Harris M. M.; Moritz R. L. State of the human proteome in 2013 as viewed through PeptideAtlas: comparing the kidney, urine, and plasma proteomes for the biology- and disease-driven Human Proteome Project. J. Proteome Res. 2014, 13, 60–75. 10.1021/pr4010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ternent T.; Csordas A.; Qi D.; Gómez-Baena G.; Beynon R. J.; Jones A. R.; Hermjakob H.; Vizcaíno J. A. How to submit MS proteomics data to ProteomeXchange via the PRIDE database. Proteomics 2014, 14, 2233–2241. 10.1002/pmic.201400120. [DOI] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Csordas A.; Del-Toro N.; Dianes J. A.; Griss J.; Lavidas I.; Mayer G.; Perez-Riverol Y.; Reisinger F.; Ternent T.; Xu Q.-W.; Wang R.; Hermjakob H. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagney G.; Amiri S.; Premawaradena T.; Lindo M.; Emili A. In silico proteome analysis to facilitate proteomics experiments using mass spectrometry. Proteome Sci. 2003, 1, 5. 10.1186/1477-5956-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter L.; Claassen M.; Schrimpf S. P.; Jovanovic M.; Schmidt A.; Buhmann J. M.; Hengartner M. O.; Aebersold R. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol. Cell. Proteomics 2009, 8, 2405–2417. 10.1074/mcp.M900317-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto G.; Aloria K.; Osinalde N.; Fullaondo A.; Arizmendi J. M.; Matthiesen R. PAnalyzer: a software tool for protein inference in shotgun proteomics. BMC Bioinf. 2012, 13, 288. 10.1186/1471-2105-13-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva B. F.; Meng C.; Helm D.; Pachl F.; Schiller J.; Ibrahim E.; Lynne C. M.; Brackett N. L.; Bertolla R. P.; Kuster B. Towards Understanding Male Infertility After Spinal Cord Injury Using Quantitative Proteomics. Mol. Cell. Proteomics 2016, 15, 1424–1434. 10.1074/mcp.M115.052175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Dufresne C.; Turner R.; Ferri S.; Venkatraman V.; Karani R.; Lutty G. A.; Van Eyk J. E.; Semba R. D. The proteome of human retina. Proteomics 2015, 15, 836–840. 10.1002/pmic.201400397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.-J.; et al. Comprehensive genome-wide proteomic analysis of human placental tissue for the Chromosome-Centric Human Proteome Project. J. Proteome Res. 2013, 12, 2458–2466. 10.1021/pr301040g. [DOI] [PubMed] [Google Scholar]
- Chick J. M.; Kolippakkam D.; Nusinow D. P.; Zhai B.; Rad R.; Huttlin E. L.; Gygi S. P. A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat. Biotechnol. 2015, 33, 743–749. 10.1038/nbt.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D. W.; Sherman B. T.; Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008, 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Chambers M. C.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carapito C.; et al. Computational and Mass-Spectrometry-Based Workflow for the Discovery and Validation of Missing Human Proteins: Application to Chromosomes 2 and 14. J. Proteome Res. 2015, 14, 3621–3634. 10.1021/pr5010345. [DOI] [PubMed] [Google Scholar]
- Ye D.; Fu Y.; Sun R.-X.; Wang H.-P.; Yuan Z.-F.; Chi H.; He S.-M. Open MS/MS spectral library search to identify unanticipated post-translational modifications and increase spectral identification rate. Bioinformatics 2010, 26, i399–i406. 10.1093/bioinformatics/btq185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez R. S.; Sánchez S. S. Characterization of pax1, pax9, and uncx sclerotomal genes during Xenopus laevis embryogenesis. Dev. Dyn. 2013, 242, 572–579. 10.1002/dvdy.23945. [DOI] [PubMed] [Google Scholar]
- Li S.; Rousseau D. ATAD3, a vital membrane bound mitochondrial ATPase involved in tumor progression. J. Bioenerg. Biomembr. 2012, 44, 189–197. 10.1007/s10863-012-9424-5. [DOI] [PubMed] [Google Scholar]
- Mouradov D.; et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014, 74, 3238–3247. 10.1158/0008-5472.CAN-14-0013. [DOI] [PubMed] [Google Scholar]
- Paik Y.-K.; Hancock W. S. Uniting ENCODE with genome-wide proteomics. Nat. Biotechnol. 2012, 30, 1065–1067. 10.1038/nbt.2416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.