

Acetylation-mediated inactivation of MnSOD triggers activation of prosurvival AKT signaling in uterine smooth muscle tumors.
Keywords: ROS, MnSOD, leiomyoma, AKT, fibroids
Abstract
AKT signaling promotes cell growth and survival and is often dysregulated via multiple mechanisms in different types of cancer, including uterine leiomyomas (ULMs). ULMs are highly prevalent fibrotic tumors that arise from the smooth muscular layer of the uterus, the myometrium (MM). ULMs pose a major public health issue because they can cause severe morbidity and poor pregnancy outcomes. We investigate the mechanisms driving ULM growth and survival via aberrant activation of AKT. We demonstrate that an acetylation-mediated impairment of the manganese superoxide dismutase (MnSOD) activity is prevalent in ULM cells compared to the normal-matched MM from the same patients. This impairment increases the levels of superoxide and oxidative stress, which activate AKT via oxidative inactivation of the phosphatase and tensin homolog deleted on chromosome 10 (PTEN). Redox activation of AKT promotes ULM cell survival under conditions of moderate but persistent oxidative stress that are compatible with ULM’s prooxidative microenvironment. Moreover, because of impaired MnSOD activity, ULM cells are sensitive to high levels of reactive oxygen species (ROS) and superoxide-generating compounds, resulting in decreased ULM cell viability. On the contrary, MM cells with functional MnSOD are more resistant to high levels of oxidants. This study demonstrates a causative role of acetylation-mediated MnSOD dysfunction in activating prosurvival AKT signaling in ULMs. The specific AKT and redox states of ULM cells provide a potential novel therapeutic rationale to selectively target ULM cells because of their defective ROS-scavenging system.
INTRODUCTION
Abnormal cellular growth is a common neoplastic event in smooth muscle tissues, giving rise to tumors that escape normal growth control mechanisms and survive despite restrictive conditions (1). Uterine leiomyomas (ULMs), also called uterine fibroids, are benign smooth muscle tumors arising from the myometrium (MM) that grow to sizes that can exceed 10 cm in diameter and cause extreme morbidity in women, including vaginal bleeding, anemia, and poor pregnancy outcomes (2, 3). ULMs occur in approximately 70% of reproductive-age women and are the leading cause of hysterectomy in the United States (4, 5), resulting in a financial burden of $5.9 billion to $34.4 billion per year in health-related costs (6).
The pathophysiology of ULM has been relatively understudied. Previous findings from our group and others revealed that ULMs, unlike parental MM tissues, are characterized by abnormal activation of the AKT pathway (7, 8), a widely recognized critical regulator of tumor initiation and maintenance (9, 10). The biological relevance of the AKT pathway in ULM was demonstrated in our previous studies when, upon inhibition of AKT, multiple downstream effects, including decreased cell proliferation and viability, increased autophagy and death, and disruption of mitochondrial structures were observed in ULM cells (8, 11). However, the causes of AKT overactivation in ULM are unclear. In other systems, the AKT pathway was shown to be highly sensitive to variations of the intracellular redox state (12–14).
ULMs are hypoxic in nature and, as in many disease states, low oxygen content and oxidative stress are closely associated (15). In ULMs, oxidative stress biomarkers, such as NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) oxidase 4 (NOX4) (16) and 8-hydroxy-2′-deoxyguanosine (8-OH-dG) (17), have been shown to be increased compared to MM. Oxidative stress, which is characterized by elevated levels of reactive oxygen species (ROS), causes cellular damage. However, low to moderate levels of ROS play a critical role in fine-tuning normal cellular functions, such as proliferation, differentiation, and metabolic adaptation, because they are important second messengers that ensure cell homeostasis and mediate adaptation to stress (18–20). In ULM cells, it has been shown that ROS are important mediators of platelet-derived growth factor– and epidermal growth factor–dependent activation of mitogen-activated protein kinase signaling, thus promoting cell proliferation (21). Mitochondria are the major source and target of ROS, and to preserve their integrity under conditions of oxidative stress, cells have evolved sophisticated mechanisms to limit excessive ROS (22). In the mitochondria, the manganese superoxide dismutase (MnSOD; also known as SOD2) rapidly reacts with the superoxide anion (O2•−), converting it into hydrogen peroxide (H2O2), which, in turn, diffuses across membranes and acts as a signaling molecule or is eliminated by catalase and other peroxidases in the form of water (23, 24). MnSOD is the primary ROS-scavenging enzyme of the cell and the only antioxidant enzyme indispensable for maintaining normal cell development and function (22, 23), and besides being a protective enzyme, it is also a regulator of intracellular redox signaling by controlling the O2•−/H2O2 ratio. In cellular systems where the antioxidant machinery is impaired, a shift toward O2•− has shown to create a favorable environment for cell transformation (25, 26). Recent mechanistic insights have demonstrated that the function of MnSOD is controlled by acetylation of specific and evolutionarily conserved lysine residue(s), suggesting that regulation of its activity via acetylation may play a key role in mitochondrial function (27). Hyperacetylation of MnSOD leads to loss of its enzymatic activity, resulting in increased O2•− levels (28, 29).
Given the key role of the AKT pathway in regulating mitochondrial dynamics and the evidence that AKT inhibition causes damage of mitochondria in ULM (8), we investigated the function of the primary ROS mitochondrial scavenger MnSOD and its possible involvement in activating AKT in ULM. Here, we demonstrate an interplay between inactivating MnSOD acetylation and activation of redox-sensitive AKT signaling in promoting ULM survival and a role for ROS in determining ULM cell fate.
RESULTS
An acetylation-mediated alteration of MnSOD activity determines the unique redox profile of ULM
Alterations in redox homeostasis are generally associated with changes in the expression or activity of antioxidant enzymes, such as mitochondrial MnSOD (30). It has previously been shown that acetylation of MnSOD at lysine (K) 122 (MnSOD K122-Ac) leads to inactivation of MnSOD, increasing mitochondrial O2•− levels (29). To investigate whether MnSOD was intrinsically acetylated in ULM, a tissue microarray (TMA) containing a cohort of matched human MM and ULM tissue cores from 60 hysterectomy specimens was immunostained with an antibody to MnSOD K122-Ac (Fig. 1A). For each hysterectomy specimen, two tissue cores were derived from MM and three were derived from ULM. Immunoreactivities for MnSOD K122-Ac and total MnSOD were scored for intensity on a four-point scale by a pathologist, with 0 being negative, 1 being weak, 2 being moderate, and 3 being strong. The frequency distribution of each staining score (0 to 3) was calculated for MnSOD K122-Ac and total MnSOD antibodies, and data are presented in Fig. 1B. No significant change in immunoreactivity for total MnSOD was observed between MM and ULM specimens, whereas immunoscores for MnSOD K122-Ac were significantly higher in ULM compared to normal MM (P = 0.0009). Moreover, 53.8% of ULM tissue cores displayed the strongest immunointensity [score, 3 (strong)] for MnSOD K122-Ac compared to normal-matched MM (30.2%) (Fig. 1B). Sirtuin 3 (SIRT3) is known to interact with and deacetylate MnSOD, increasing its dismutating activity (28, 29). To investigate a possible role of SIRT3 in promoting MnSOD hyperacetylation in ULM, its expression was analyzed by staining the TMA with an antibody against SIRT3. As shown in fig. S1A, the χ2 test revealed a significant difference between MM and ULM, in that SIRT3 expression was slightly increased in ULM tissues.
Fig. 1. ULMs exhibit increased levels of acetylated MnSOD and 3-NO as well as decreased MnSOD activity.
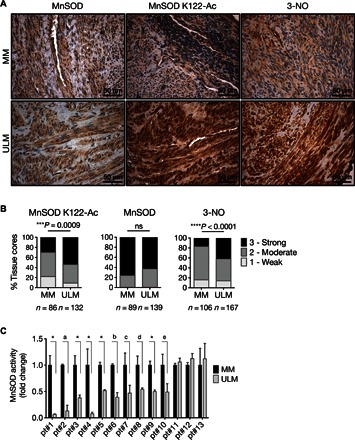
(A) Representative images of the TMA containing 60 matched MM and ULM specimens immunostained with MnSOD K122-Ac, MnSOD, and 3-NO antibodies. For each human specimen, two tissue cores were derived from MM and three were derived from ULM. (B) Score frequency distribution of TMA immunostaining. The intensity of well-preserved matched MM/ULM tissue cores was scored numerically as 0 (negative), 1 (weak), 2 (moderate), or 3 (strong). Statistically significant differences between normal MM and ULM were evaluated using a χ2 test (***P = 0.0009, ****P < 0.0001; ns, not significant). (C) MnSOD activity was assessed in 13 untreated patient-derived MM and matched ULM cells. Each data point represents the means ± SD of a quadruplicate measurement. Data are represented as fold change to untreated MM cells for each patient (*P < 0.0001; aP = 0.0183; bP = 0.0046; cP = 0.0008; dP = 0.0025; eP = 0.0035; paired t test).
To assess whether MnSOD acetylation in ULM was associated with protein damage caused by increased reactive oxygen/nitrogen species, the TMA was immunostained with 3-nitrotyrosine (3-NO) antibody (Fig. 1A). 3-NO is a well-established biomarker of oxidative protein damage by peroxynitrite (ONOO−), a highly reactive free radical derived from the reaction of intracellularly cogenerated O2•− and nitric oxide (NO) (31). In ULM tissue cores, significant levels of 3-NO correlated with high levels of MnSOD K122-Ac (P < 0.0001). The strongest immunointensity for 3-NO (score, 3) was detected in 42.5% of ULM tissue against 17% of normal MM (Fig. 1B). Higher 3-NO expression was also associated with increased levels of inducible nitric oxide synthase (iNOS) in ULM (fig. S1B; P < 0.0001). iNOS generates large amounts of NO in response to a variety of stimuli, including inflammation, hypoxia, and steroid hormones, and its activity is often increased in gynecologic disorders (32, 33). The increases in 3-NO and iNOS expression in ULM suggest that high levels of NO and O2•− are simultaneously generated in ULM, and these high levels are likely due to augmented iNOS and impaired MnSOD activity, respectively.
Next, MnSOD activity was assessed in patient-derived primary ULM cells and normal-matched MM cells. Of 13 cases, 10 demonstrated significantly decreased MnSOD activity in ULM cells compared to normal-matched MM cells (Fig. 1C). Together, these results suggest that ULMs are characterized by an acetylation-mediated impairment of MnSOD activity compared to healthy MM cells.
Mitochondrial ROS derived from dysfunctional acetylated MnSOD lead to AKT activation in ULM cells
ULM tissues displayed higher levels of MnSOD K122-Ac than normal MM (Fig. 1, A and B). We observed these differences in cultured patient-derived primary cells as well. Western blot analyses revealed that in most of the samples analyzed, ULM cells have higher levels of MnSOD K122-Ac compared to MM cells, and this corresponded to higher levels of phospho-AKT(S473) (pAKT). On the other hand, MM cells with lower MnSOD K122-Ac and higher total MnSOD content had lower levels of pAKT (Fig. 2A and fig. S2A). In the three patient-derived MM and ULM cells (pt#11 to pt#13 from Fig. 1C) where no changes in MnSOD activity were detected, we did not observe significant variations in pAKT levels (fig. S2B).
Fig. 2. Mitochondrial ROS derived from dysfunctional acetylated MnSOD lead to AKT activation in ULM cells.
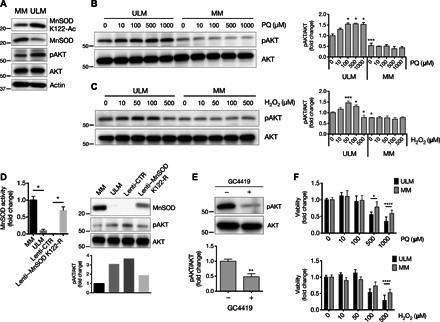
(A) Levels of MnSOD K122-Ac, MnSOD, and pAKT(S473) (pAKT) proteins from cell lysates of untreated MM and ULM cells. pan-AKT (AKT) and actin were used as loading controls. (B and C) ULM and MM cells were exposed to increasing concentrations of PQ or H2O2 for 6 hours, and cellular protein extracts were analyzed by Western blot for pAKT(S473) and AKT. A representative Western blot and the means ± SD of densitometric quantification of three independent experiments are shown [(B) *P < 0.05, ***P < 0.001 versus PQ 0 μM ULM; (C) *P < 0.05, ***P < 0.001 versus H2O2 0 μM ULM; one-way analysis of variance (ANOVA), n = 3]. (D) ULM cells were transiently infected with a lenti-CTR or a lenti–MnSOD K122-R. MnSOD activity (*P = 0.0286; unpaired t test) as well as MnSOD and pAKT protein levels were analyzed, and corresponding densitometric analysis is shown. Data are representative of three patients. (E) ULM cells were treated with 10 μM GC4419 for 6 hours, and pAKT levels were analyzed. AKT was used as loading control. Corresponding densitometric analysis of three independent experiments is shown as means ± SD (**P = 0.0045; unpaired t test, n = 3). (F) ULM and MM cells were treated for 24 hours with increasing doses of PQ or H2O2, and cell viability was measured using WST-1 assay (*P < 0.05, ****P < 0.0001; one-way ANOVA). Results are expressed as means ± SD from three independent experiments (n = 3).
Because the AKT pathway is redox-sensitive and ULMs are characterized by a reduced capacity of detoxifying ROS due to aberrant MnSOD activity, we next assessed pAKT levels upon oxidative stimulation in ULM versus MM cells. Cells were treated with paraquat (PQ) or H2O2 for 6 hours. PQ is used to specifically generate O2•− when analyzing oxidative stress (34). In ULM cells, PQ induced pAKT levels at all the concentrations used (Fig. 2B). Similarly, H2O2 increased pAKT levels at all doses (with a maximum increase at 50 μM) except at 500 μM (Fig. 2C). No changes in pAKT expression were detected in MM cells with PQ or H2O2.
To further investigate the role of dysfunctional MnSOD in activating the AKT pathway in ULMs, primary ULM cells from different patients were infected with a control lentivirus (lenti-CTR) or a mutant MnSOD K122-R lentivirus (lenti–MnSOD K122-R), and pAKT levels were analyzed. It has previously been demonstrated that substitution of lysine (K) 122 with arginine (R) results in a constitutively active dominant-positive form of MnSOD that mimics deacetylation (29). Infection of ULM cells with lenti–MnSOD K122-R led to a significant increase in both MnSOD activity and protein levels as well as a decrease in pAKT expression in ULM cells, supporting the idea that mitochondrial ROS derived from dysfunctional acetylated MnSOD are capable of activating AKT in ULM (Fig. 2D and fig. S3). This was further corroborated by the fact that pAKT levels were reduced following treatment with the MnSOD mimetic GC4419, a small molecule that selectively detoxifies O2•− (Fig. 2E).
Because ULM and MM cells displayed a different ROS detoxification potential, we then investigated the sensitivity of those cells to oxidative stimuli. Exposure of ULM and MM cells to increasing doses of PQ or H2O2 for 24 hours showed lower cell viability in ULM than in MM cells (Fig. 2F), demonstrating an increased sensitivity to oxidative insults in ULM cells.
ULM cells feature aberrant MnSOD acetylation and AKT activation under both normoxic and hypoxic conditions
ULMs are solid tumors featuring a hypoxic environment unlike the normal uterine MM tissues (35, 36). We investigated whether low oxygen content may contribute to the aberrant acetylation of MnSOD and activation of AKT in ULM. First, we observed that ULM cells grown in hypoxia (2% O2) had higher levels of mitochondrial O2•− compared to the normoxic counterpart (20% O2), as shown by increased MitoSOX staining (Fig. 3A). Also, Western blotting analysis revealed that hypoxia [verified by the stabilization of hypoxia-inducible factor–1α (HIF-1α)] further augmented the expression of pAKT. Under hypoxia, MnSOD K122-Ac levels were comparable to those of ULM cells grown in normoxia and around twofold higher compared to MM (Fig. 3B). Accordingly, we found that MnSOD activity in ULM cells grown in hypoxia was still lower than that in control MM cells and that the MnSOD mimetic GC4419 simultaneously increased the capacity of ULM cells to dismutate O2•− and reduced pAKT levels under hypoxic conditions as shown before under normoxic conditions (Fig. 3, C and D).
Fig. 3. ULM cells are characterized by aberrant MnSOD acetylation and AKT activation under normoxic and hypoxic conditions.
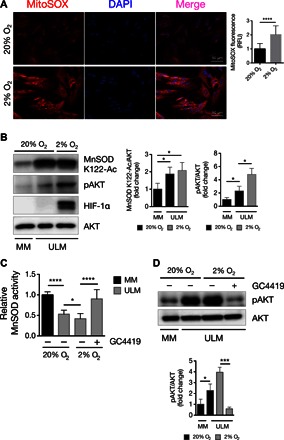
(A) MitoSOX Red was used to assess mitochondrial superoxide levels in ULM cells under normoxic (20% O2) and hypoxic (2% O2) conditions after 48 hours of incubation time. MitoSOX fluorescence was quantified by analyzing the fluorescence intensity of more than 10 cells for each condition using ImageJ software (****P < 0.0001; one-way ANOVA, n = 3 independent experiments ± SD). RFU, relative fluorescence unit. (B) ULM cells were grown under normoxia and hypoxia for 48 hours, and levels of MnSOD K122-Ac and pAKT(S473) were analyzed. MM cells grown in normoxia were used as control. Hypoxia was verified by detection of HIF-1α. Equal loading was confirmed by anti-AKT. Corresponding densitometric analysis of three independent experiments is shown as means ± SD (*P < 0.05; one-way ANOVA, n = 3). (C and D) MM and ULM cells were grown under both normoxic and hypoxic atmosphere for a total of 48 hours. Twenty-four hours before harvesting, the MnSOD mimetic GC4419 (10 μM) was added to ULM cells. (C) MnSOD activity was assessed as previously described. Data are expressed as means ± SD from three independent experiments (n = 3). (D) pAKT and AKT levels were analyzed by Western blotting, and densitometric analysis of three independent experiments is shown as means ± SD (*P < 0.05, ***P < 0.001, ****P < 0.0001; one-way ANOVA, n = 3).
Inactive MnSOD promotes AKT activation via mitochondrial ROS–dependent PTEN oxidation
ROS induce oxidative posttranslational modifications that can negatively affect the structure and function of many proteins, including the phosphatase and tensin homolog deleted on chromosome 10 (PTEN), a major negative regulator of AKT signaling (37). Reversible oxidation of PTEN by H2O2 results in its inactivation and translocation to the nucleus (38, 39). To investigate whether increased pAKT expression in ULM was due to upstream inactivation of PTEN, we first measured PTEN activity in MM and ULM cells, assessing the conversion rate of its substrate, phosphatidylinositol 3,4,5-trisphosphate (PIP3), to phosphatidylinositol 4,5-bisphosphate. PTEN activity was detectable at very low levels in four of five patient-derived ULM cells, whereas MM cells displayed significantly higher levels of PTEN activity (Fig. 4A).
Fig. 4. PTEN activity is reduced in ULM cells as a result of superoxide-dependent oxidation.
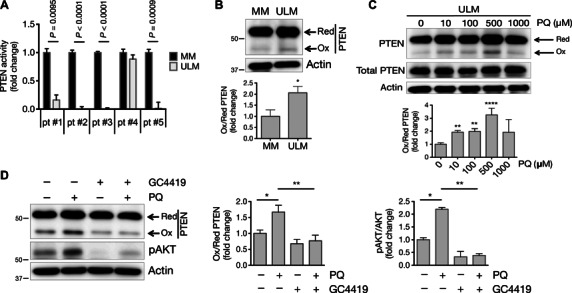
(A) PTEN phosphatase activity was measured in immunoprecipitated PTEN samples from five patient-derived MM and ULM cells using PIP3 as the substrate. Data are represented as fold change to untreated MM cells (paired t test). (B) Protein extracts from untreated MM and ULM cells were analyzed under nonreducing conditions and immunoblotted with anti-PTEN antibody (*P = 0.0162; unpaired t test). (C) ULM cells were treated for 6 hours with 10, 100, 500, and 1000 μM PQ in serum-free medium, and protein extracts were analyzed as described above for oxidized and total PTEN (**P < 0.01, ****P < 0.0001; one-way ANOVA). (D) ULM cells were treated with 100 μM PQ, 10 μM GC4419, or a combination of both drugs for 6 hours. Protein extracts were analyzed under nonreducing conditions and immunoblotted with anti-PTEN antibody (*P < 0.05, **P < 0.01; one-way ANOVA) or under reducing conditions and immunoblotted with pAKT antibody (*P < 0.05, **P < 0.01; one-way ANOVA). For all Western blots in (B) to (D), one representative blot and corresponding densitometric analysis of three independent experiments are shown as means ± SD (n = 3). Anti-actin antibody was used as loading control.
In ULM, increased MnSOD K122-Ac and 3-NO staining indicated enhanced oxidative stress and O2•− formation. To determine whether endogenous ROS were capable of oxidizing and inactivating PTEN while increasing pAKT, basal levels of oxidized PTEN were analyzed in untreated MM and ULM cells. Oxidation of PTEN induces the appearance of a higher-mobility (oxidized) form of PTEN on an SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gel under nonreducing conditions (40). Accumulation of the oxidized form of PTEN was observed at higher levels in protein extracts from ULM cells compared to MM cells (Fig. 4B). Moreover, treatment of ULM cells with increasing concentrations of PQ induced a further accumulation of the oxidized form of PTEN, usually peaking at 500 μM (Fig. 4C). No increase in oxidized PTEN was detectable in MM cells under the same conditions (fig. S4A). Consistently, immunofluorescent staining showed increased nuclear accumulation of PTEN in ULM cells following treatment with 100 μM PQ compared to untreated ULM cells (fig. S4, B to D). Quantitation of the nuclear/cytoplasmic ratio of PTEN cellular distribution further confirmed a significantly increased nuclear localization of PTEN upon PQ treatment (fig. S4E).
To assess whether acetylated MnSOD was responsible for PTEN oxidation due to increased O2•− and oxidative stress, ULM cells were treated with PQ together with the MnSOD mimetic agent GC4419. Treatment of ULM cells with the combination of 100 μM PQ and 10 μM GC4419 reduced both the appearance of the oxidized form of PTEN and pAKT levels induced by PQ alone (Fig. 4D). Together, these results suggest that dysfunctional acetylated MnSOD sustains AKT activation through mitochondrial ROS–dependent oxidation of PTEN in ULM.
MK-2206 effectively triggers death of ULM cells through simultaneous AKT inhibition and increase of superoxide
The potential involvement of the AKT pathway in regulating ULM growth and survival has previously been reported by Sefton et al. (8) and Hoekstra et al. (11). MK-2206 is a selective allosteric pan-AKT inhibitor that leads to multiple downstream effects depending on the dose and/or duration of the treatment. Treatment of ULM cells with increasing concentrations of MK-2206 effectively decreased pAKT protein levels at all the concentrations used (fig. S5A). In agreement with previous findings, 25 μM MK-2206 reduced viability of ULM cells (fig. S5B) despite the fact that there is no increase in cleaved caspases (8). Down-regulation of all AKT isoforms by specific small interfering RNA (siRNA) transfection did not affect ULM cell viability (fig. S5C). Given the recent evidence that the MK-2206 compound promotes ROS generation (41, 42) and disrupts mitochondrial structures (8), we sought to determine whether MK-2206 specifically induced O2•−, causing ULM cell death. We observed that treatment of ULM cells with increasing concentrations of MK-2206 (from 1 to 10 to 25 μM) resulted in a dose-dependent increase in O2•− levels (Fig. 5, A and B), unlike silencing of AKT, which did not alter O2•− levels (fig. S5D). High doses of MK-2206 increased superoxide generation in MM cells as well (fig. S5E). Reduced glutathione (GSH) is another ROS-scavenging molecule that is oxidized [to GSSG (oxidized GSH)] and is one of the first lines of defense against oxidative stress in mammalian cells (43). We found that MK-2206 decreased the levels of the reduced form of GSH in a dose-dependent manner (Fig. 5C), which is suggestive of GSH oxidation by ROS, because its levels were inversely proportional to MK-2206–induced O2•− levels (Fig. 5, A and B). The GSH synthesis inhibitor buthionine sulfoximine (BSO) and the antioxidant N-acetyl-l-cysteine (NAC) were used as negative and positive controls, respectively. Treatment of ULM cells with the MnSOD mimetic GC4419 inhibited MK-2206–induced increase of O2•− in ULM cells (Fig. 5, A and B). Moreover, we found that MK-2206–induced cytotoxicity was completely reversed by GC4419 in ULM and MM cells. Although viability of MM cells decreased with 25 μM MK-2206, the decrease observed in ULM cells was greater (Fig. 5D). These results suggest that the AKT inhibitor MK-2206 promotes superoxide-mediated toxicity in the background of inactive AKT.
Fig. 5. Treatment with the AKT inhibitor MK-2206 leads to superoxide generation in ULM cells.
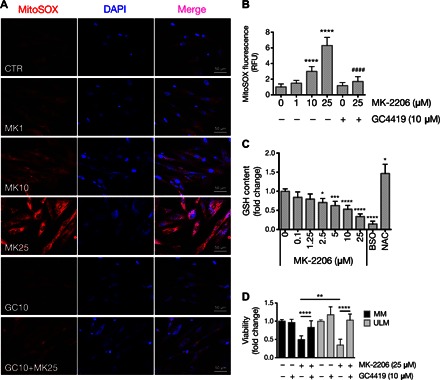
(A) Mitochondrial superoxide levels were assessed in ULM cells using MitoSOX Red. ULM cells were treated with vehicle (CTR), various concentrations of MK-2206 (MK; 1, 10, and 25 μM) and 10 μM GC4419 (GC) alone or with 25 μM MK-2206 (GC+MK25) for 6 hours in serum-free media. Representative pictures from three independent experiments are shown. (B) MitoSOX fluorescence was quantified by analyzing the fluorescence intensity of more than 10 cells for each condition using ImageJ software (****P < 0.0001 versus 0 μM MK-2205; ####P < 0.0001 versus 25 μM MK-2205; one-way ANOVA, n = 3 independent experiments ± SD). (C) GSH content was quantified using GSH-Glo reagent. ULM cells were treated at the indicated concentrations of MK-2206 for 24 hours. BSO (10 μM) and 50 μM NAC were used as negative and positive controls, respectively [*P < 0.05, ***P < 0.001, ****P < 0.0001 versus vehicle (dimethyl sulfoxide); one-way ANOVA, n = 3 independent experiments ± SD]. (D) MM and ULM cells were treated with 25 μM MK-2206 alone or in combination with 10 μM GC4419 for 24 hours. Cell viability was determined using WST-1 (**P < 0.01, ****P < 0.0001; one-way ANOVA, n = 3 independent experiments ± SD).
AKT serves as a survival factor in the prooxidative environment of ULM
To further demonstrate the idea that ROS are cytotoxic in ULM when AKT is inactive or, in other words, the idea that AKT serves as a survival factor for ULM under high oxidative stress, AKT was silenced using siRNA (fig. S6A) in ULM cells that were consequently treated with increasing concentrations of H2O2. H2O2 led to a progressive decrease of ULM cell viability compared to control siRNA (siCTR) cells treated with the same doses of H2O2 (Fig. 6A). Moreover, viability of MM cells under the same conditions was unaffected (Fig. 6B and fig. S6B). Together, these results suggest that AKT serves as a survival factor in the prooxidative environment occurring in ULM.
Fig. 6. AKT protects ULM cells from oxidative-induced damage.
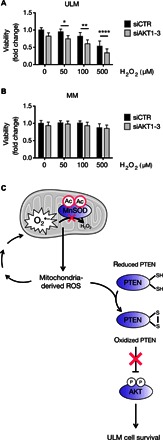
(A and B) AKT1, AKT2, and AKT3 were silenced in ULM and MM cells by reverse transfection using siAKT1, siAKT2, and siAKT3. Following AKT knockdown, 50, 100, or 500 μM H2O2 was added to ULM and MM cells for 6 hours, and cell viability was determined using WST-1. Data are shown as means ± SD from three independent experiments (*P < 0.05, **P < 0.01, ****P < 0.0001; one-way ANOVA, n = 3). (C) Proposed working model for the interplay between dysfunctional MnSOD and activation of the AKT pathway and its effects on ULM cell survival. In ULM cells, acetylation of MnSOD impairs its activity, leading to the increase of mitochondrial ROS, which, in turn, activate AKT through oxidative inactivation of PTEN and promote cell survival in the prooxidative ULM microenvironment. The mitochondrion in the figure was taken from the Servier Medical Art database (http://servier.com/Powerpoint-image-bank).
DISCUSSION
Oxidative stress plays a role in the pathophysiology of several gynecologic diseases, including ULM. Previous studies showed that multiple oxidative stress biomarkers, such as NOX4 (16), 8-OH-dG (17), and malonaldehyde (44), as well as serum oxidative stress markers (45), are increased in ULM. Moreover, unlike immortalized MM cells, activity of both catalase and extracellular SOD3 is significantly reduced in immortalized ULM cells (46). To date, our study is the most comprehensive study to investigate the redox regulatory system in exclusively patient-derived ULM and MM specimens and, thus, significantly furthers our understanding of the redox biology of ULM. We have demonstrated that patient-derived primary ULM cells are inherently characterized by an acetylation-mediated impairment of MnSOD activity compared to healthy MM cells. In ULM, inactivating acetylation of MnSOD increases the levels of mitochondrial ROS, leading to a state of persistent oxidative stress that activates AKT through oxidative inactivation of PTEN. Redox-sensitive AKT activation in turn promotes cell survival in the prooxidative ULM microenvironment (Fig. 6C). Therefore, our results point to a potential key signaling role of mitochondrial ROS and mild oxidative stress in the biology of ULM. Because of the dysfunctional MnSOD activity, ULM cells also feature a defective response to high ROS concentrations. In contrast to MM cells, whose viability is less affected by exposure to oxidants, ULM cells that are deficient in detoxifying ROS are very sensitive to high doses of O2•−-generating compounds.
Several reports suggest that moderate increases in the levels of ROS, especially O2•−, can affect numerous aspects of tumor initiation and progression (47, 48). Oxidative stress can result from defects in the cellular antioxidant response, of which MnSOD is a major component (30, 49). Tao et al. (29) demonstrated that the enzymatic function of MnSOD is regulated by lysine acetylation. They found that, in Sirt3−/− mouse embryonic fibroblasts, increased acetylation of MnSOD at lysine 122 (MnSOD K122-Ac) resulted in the inactivation of MnSOD and the subsequent increment of O2•− levels, promoting a tumor-permissive environment. We found that MnSOD K122-Ac was highly expressed in ~60% of ULM human tissues compared to the matched MM tissues analyzed, and this coincided with a decreased activity of MnSOD in ~70% of the patient-derived ULM cells compared to the normal-matched MM cells. We also showed that, in ULM, 3-NO and iNOS were accumulated in a similar fashion to the K122-acetylated inactive form of MnSOD (~60%). 3-NO is a well-known biomarker of oxidative stress (31) that is formed from the reaction between tyrosine residues of proteins and ONOO−, a damaging oxidant whose production depends on the availability of mitochondrial O2•− and NO. Therefore, high levels of MnSOD K122-Ac, 3-NO, and iNOS suggested a correlation between inactivating MnSOD acetylation and establishment of a prooxidative milieu in ULM that could be ascribed to increased mitochondrial O2•− levels. Up-regulation of NOX4 was also found in ULM compared to MM (16). It has previously been reported that mitochondrial O2•− derived from MnSOD deficiency increases the activity of extramitochondrial NADPH oxidase, the major source of cytosolic O2•−, via a feed-forward mechanism (50–52). This leads to a vicious cycle of ROS-induced ROS release between mitochondrial and cytosolic sources of O2•− that synergistically fosters oxidative stress and may contribute to the pathogenesis of many tumor types, including ULM. Our study demonstrates the importance of acetylated MnSOD in a pathological context. The reasons for increased acetylated MnSOD in ULM are unknown. To date, no acetylases that acetylate MnSOD have been identified (53). It is known that mitochondrial SIRT3 plays a role in MnSOD deacetylation (28, 29), and members of the sirtuin deacetylase family were shown to be dysregulated in cancer (54). We did not detect lower SIRT3 expression in ULM, but rather an increase, suggesting that augmented acetylation of MnSOD is not likely due to a reduced deacetylation of MnSOD by SIRT3 but due to other unknown mechanisms. The increased acetylation of MnSOD may occur in instances of metabolic reprogramming. Cancer cells often exhibit an altered metabolism that is characterized by a shift from oxidative phosphorylation to glycolysis, also known as the Warburg effect, and is associated with cancer cell growth and survival (55). The link between metabolic reprogramming and protein acetylation has been previously documented and includes the lactate-mediated increase of intracellular pH and the glycolytic increase in the production of acetyl–coenzyme A, which is the substrate used by acetyltransferases to acetylate proteins (56–58). ULMs appear to have features associated with the Warburg effect, such as increased hypoxia, acetylation, and augmented ROS. Whether metabolic reprogramming has occurred in ULM and whether this increases acetylation of MnSOD or other key proteins will require further investigation.
It has been shown that ULMs are locally hypoxic with abnormal vasculature, and it is thought that low oxygen levels may contribute to ULM pathogenesis and growth (35, 36). We found that, under hypoxic conditions, ULM cells exhibited increased levels of pAKT compared to the normoxic counterpart as well as higher levels of acetylated MnSOD and lower MnSOD activity than normal MM cells. Moreover, rescuing MnSOD activity prevents pAKT accumulation in hypoxia as similarly shown in normoxia. These data show that a low-oxygen environment further promotes oxidative stress and AKT dysregulation in ULM.
In our study, we demonstrated that AKT activation occurs via oxidative inactivation of PTEN in ULM and suggested that this is driven by MnSOD deficiency. It is known that PTEN is sensitive to variations of the intracellular redox state (38, 39, 59). In our model, aberrant MnSOD activity leads to increased levels of mitochondrial superoxide that may directly inactivate PTEN if released to the cytoplasm. Multiple studies showed that superoxide can be released from the mitochondria toward the cytosol via voltage-dependent anion channels under conditions of elevated mitochondrial oxidative stress (60–62). However, because superoxide is a short-lived radical species, indirect mechanisms of superoxide-dependent PTEN oxidation cannot be excluded. For example, Dikalov’s group (51, 52) showed that reduced MnSOD activity leads to increased H2O2 release from the mitochondria, which can trigger PTEN oxidation. A similar mechanism may occur in ULM, resulting in acetylated MnSOD–dependent oxidation of PTEN and ultimately AKT activation. Moreover, although we believe that mitochondria-derived ROS are largely responsible for PTEN oxidation, other mechanisms, such as increased growth factor receptor signaling, cannot be ruled out in inactivating PTEN, as previously shown by other groups (63, 64).
Our findings suggest that mitochondrial ROS play an important role as a survival factor in ULM through increased AKT activation. Although AKT is localized in the membrane and cytosol, nuclear localization (65) and localization in the mitochondria have been reported (66). Under oxidative stress conditions, cytosolic AKT can translocate into the mitochondria to preserve mitochondrial integrity and provide cytoprotection (66–68). It would be interesting to investigate whether AKT localizes to the mitochondria in ULM cells to preserve their integrity following oxidative stimulation.
Together, we have provided new insights into the pathophysiology of ULM that go beyond the comparative expression analysis of redox targets in MM versus ULM. We have demonstrated the mechanisms by which ULM cells have adapted to survive in otherwise suboptimal conditions that are very different from those of the normal tissue from which they arise, the MM. Using exclusively human tissues and patient-derived primary cells, we have found an acetylation-mediated impairment of the activity of a critical ROS-sensing protein, MnSOD, which promotes a mitochondrial ROS–mediated increase in oxidative stress. Dysfunctional MnSOD activity not only increases oxidative stress to levels that promote survival through activation of the AKT pathway but also renders ULM cells to be more vulnerable to further prooxidative stimulation. Not all fibroids exhibited high MnSOD K122-Ac and reduced MnSOD activity, and not all matched patient samples showed a correlation between MnSOD K122-Ac and pAKT levels. The biological reasons for these differences remain unknown, but future investigation on the influence of the most prominent mutations that occur in ULM (such as in the MED12, HMGA2, and FH genes) on the oxidative stress–mediated activation of AKT would be of interest. The differential response to ROS between ULM and MM cells has translational potential and paves the way for possible redox-modulating therapeutic strategies that can selectively target ULM while minimizing the effects on normal MM that has a functional ROS-scavenging system.
MATERIALS AND METHODS
Collection of tissue samples and culture of primary cells
Human ULM and MM tissues were collected from premenopausal women (age range, 30 to 52 years) undergoing hysterectomy or myomectomy at the Northwestern University Prentice Women’s Hospital (Chicago, Illinois), according to an International Review Board–approved protocol. At the moment of the surgery, subjects included in the study were not taking hormonal contraceptives or a gonadotropin-releasing hormone antagonist or agonist for at least 3 months. Informed consent was obtained from all the patients participating in the study. Primary ULM and MM cells were isolated and cultured as previously described (69) and passaged up to two times before they were discarded. Primary cells were cultivated in Dulbecco’s modified Eagle’s medium/nutrient Ham’s Mixture F-12 (DMEM-F12) 1:1 containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C and 5% CO2 atmosphere. For experiments in hypoxia, cells were placed in a humidified incubator at 37°C under a 2% O2, 5% CO2, and balance N2 atmosphere for 48 hours.
Lentiviral transduction
A lenti-CTR and a lenti–MnSOD K122-R were provided by D. Gius. ULM cells at 60% confluence were transiently infected with lenti-CTR and lenti–MnSOD K122-R (107 copies/ml for both) in the presence of polybrene (8 μg/ml). After 2 days of infection, the culture medium of infected cells was replaced with fresh DMEM-F12 containing 10% FBS and 1% penicillin-streptomycin, and cells were cultured for an additional 2 days. Cells were then harvested, and protein extracts were prepared for both MnSOD activity assay and Western blot analysis.
Chemicals
GC4419 was provided by D. Gius and Galera Therapeutics Inc. MK-2206 was provided by Merck Sharp & Dohme Corp. and the National Cancer Institute, National Institutes of Health. PQ, H2O2, and NAC were purchased from Sigma-Aldrich, and BSO was purchased from Cayman Chemical.
Immunohistochemical staining of TMAs
Immunostaining of TMA was performed on an automated system by the Robert H. Lurie Comprehensive Cancer Center’s Pathology Core Facility. Anti-MnSOD (NBP2-20535; Novus Biologicals), anti–MnSOD K122-Ac (NCI-156, clone ID 33; Epitomics), anti–3-NO (06–284; EMD Millipore), iNOS (PA3-030A; Thermo Fisher Scientific), and SIRT3 (ARP32389_P050; Aviva Systems Biology) antibodies were used at 1:200, 1:250, 1:500, 1:1000, and 1:250 dilutions, respectively. Primary antibodies were detected using a standard anti-rabbit secondary antibody followed by 3,3′-diaminobenzidine revelation (Dako).
MnSOD activity assay
MnSOD activity was measured using the Superoxide Dismutase Assay Kit (Cayman Chemical) following the manufacturer’s instructions for specific quantitation of mitochondrial MnSOD activity. Addition of 2 mM KCN (Sigma-Aldrich) (Cu-ZnSOD/SOD1 and FeSOD/SOD3 inhibitor) to the assay allowed specific detection of MnSOD activity. Absorbance was read at 450 nm using a Cytation 3 Cell Imaging Multi-Mode Reader (BioTek).
SDS-PAGE and Western blotting
Protein lysates were extracted from MM and ULM cells using M-PER mammalian protein extraction reagent (Thermo Fisher Scientific) with protease and phosphatase inhibitors (Sigma-Aldrich). Protein concentration was determined using Bradford reagent (Sigma-Aldrich). Equal amount of proteins were subjected to SDS-PAGE and subsequently transferred to polyvinylidene difluoride (PVDF) membranes. Immunoblotting was performed using the following primary antibodies: rabbit anti-pAKT(S473) (#9271; Cell Signaling Technology), rabbit anti–pan-AKT (#4691; Cell Signaling Technology), rabbit anti-AKT1 (#2932; Cell Signaling Technology), rabbit anti-AKT2 (#2964; Cell Signaling Technology), rabbit anti-PTEN (#9188; Cell Signaling Technology), rabbit anti-MnSOD (NBP2-20535; Novus Biologicals), rabbit anti–MnSOD K122-Ac (NCI-156 Clone ID 33; Epitomics), and mouse anti-actin (A1972; Sigma-Aldrich). Secondary antibodies were horseradish peroxidase (HRP)–labeled anti-mouse or anti-rabbit (Bio-Rad). Chemiluminescence was detected by adding a chemiluminescent HRP substrate (Thermo Fisher Scientific) and measured with a Fujifilm LAS-3000 Imager.
WST-1 viability assay
ULM and/or MM cells were cultured in 96-well plates in complete DMEM-F12 1:1 medium until 80 to 90% confluence. At the end of treatments, WST-1 (BioVision) was added to each well (1:10 dilution) for 2 hours at 37°C, and absorbance was read at 420 nm with reference wavelength set at 650 nm. Data were analyzed following the manufacturer’s manual.
PTEN activity assay
MM and ULM cells were cultured in 100-mm dishes in complete DMEM-F12 1:1 medium until 80 to 90% confluence. Cells were lysed in ice-cold 25 mM tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 1 mM EDTA, and 5% glycerol lysis buffer. PTEN (500 μg) was immunoprecipitated from whole-cell lysates with 8 μl of rabbit anti-PTEN antibody (Cell Signaling Technologies) under agitation at 4°C overnight. The antigen-antibody complex was recovered with 30 μl of protein A Sepharose beads (Sigma-Aldrich) after 3 hours with agitation at 4°C, and then the beads were washed twice in lysis buffer, washed once in PTEN reaction buffer [25 mM tris-HCl (pH 7.4), 140 mM NaCl, 2.7 mM KCl, and 10 mM DTT], and resuspended in 80 μl of PTEN reaction buffer. PTEN phosphatase activity was measured using the Malachite Green Assay Kit (Echelon Biosciences) and PIP3 as substrate according to the manufacturer’s instructions.
Detection of oxidized PTEN
MM and ULM cells were cultured in 60-mm dishes in complete DMEM-F12 1:1 medium until 80 to 90% confluence. Untreated cells or cells treated with PQ at the indicated concentrations for 6 hours were washed twice with phosphate-buffered saline (PBS)/EDTA, scraped in PBS/EDTA, and collected by centrifugation. Cell pellets were washed with PBS and resuspended in 200 μl of lysis buffer [100 mM tris-HCl (pH 6.8), 2% SDS, and 40 mM N-ethylmaleimide] as described by Connor et al. (40). Equal amounts of protein were loaded on a nonreducing SDS-PAGE gel (8% polyacrylamide) and electroblotted onto a PVDF membrane, which was incubated first with anti-PTEN (Cell Signaling Technologies) and then with anti-rabbit secondary antibody conjugated to HRP (Bio-Rad). Chemiluminescence was detected by adding a chemiluminescent HRP substrate (Thermo Fisher Scientific) and measured with a Fujifilm LAS-3000 Imager.
Immunofluorescence staining for oxidized PTEN
ULM cells were grown on glass coverslips in a 12-well plate in complete DMEM-F12 1:1 medium until 70% confluence. After treatment with PQ at the indicated concentrations for 6 hours, cells were washed three times with PBS, fixed with 4% paraformaldehyde (PFA), permeabilized with 0.1% Triton X-100 in PBS, and blocked with 5% bovine serum albumin in PBS for 1 hour. Cells were then incubated with anti-PTEN antibody (1:200; Cell Signaling Technologies) overnight at 4°C. Anti-rabbit secondary antibody Alexa Fluor 488 (Thermo Fisher Scientific) was then added for 1 hour at room temperature. Coverslips were then mounted onto glass slides using ProLong Gold Antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific) as counterstain to visualize the nuclei. Images were taken using a Leica DM5000 B microscope.
PTEN nuclear/cytoplasmic ratio
The nuclear and cytoplasmic distribution of PTEN was analyzed using ImageJ software and R. Details about the procedure can be found in the Supplementary Materials. The mean nuclear/cytoplasmic PTEN signal ratio of more than 10 cells per condition was measured using ImageJ software. Average ratios and SDs from independent experiments were plotted using GraphPad Prism software.
Superoxide anion detection
ULM cells were grown on glass coverslips in a 12-well plate in complete DMEM-F12 1:1 medium until 70% confluence. Cells were then washed twice with Hanks’ balanced salt solution (HBSS) Ca-Mg and preincubated with 5 μM MitoSOX Red (Thermo Fisher Scientific) in HBSS Ca-Mg for 20 min at 37°C in the dark. MitoSOX Red allows the selective visualization of O2•− generated in the mitochondria because it is rapidly oxidized by O2•− only. After treatments, the dye was removed, and the cells were washed three times with HBSS Ca-Mg and fixed with 4% PFA. Coverslips were then mounted onto glass slides using ProLong Gold Antifade reagent with DAPI (Thermo Fisher Scientific) as counterstaining to visualize the nuclei. Images were taken using a Leica DM5000 B microscope.
Relative fluorescence unit quantification
The relative fluorescence units of individual cells were quantified using ImageJ software. Corrected total cell fluorescence units were determined using the following formula: integrated density of selected cell − (area of selected cell × mean integrated density of background readings).
GSH measurement
ULM cells were cultured in a 96-well white plate in complete DMEM-F12 1:1 medium until 80 to 90% confluence. At the end of treatments, GSH content was measured using the GSH-Glo Glutathione Assay (Promega Corporation) according to the supplier’s instructions. Luminescence was read using a luminometer plate reader (Cytation 3 Cell Imaging Multi-Mode Reader, BioTek).
AKT knockdown, RNA isolation, and RT-PCR
AKT1, AKT2, and AKT3 were silenced in ULM or MM cells by reverse transfection using siAKT1, siAKT2, and siAKT3 ON-TARGET plus SMARTpool (GE Dharmacon) and Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. A nontargeting siCTR (GE Dharmacon) was used in parallel. Cells were harvested for immunoblotting or reverse transcription polymerase chain reaction (RT-PCR) after 72 hours from transfection.
RNA was isolated from ULM cells using TRIzol Reagent (Thermo Fisher Scientific) and reverse-transcribed with M-MLV Reverse Transcriptase (Thermo Fisher Scientific) following the manufacturer’s instructions. AKT3 TaqMan gene expression assay was purchased from Thermo Fisher Scientific (the supplier did not provide the primers’ sequence). RT-PCR was performed using TaqMan Gene Expression Master Mix (Thermo Fisher Scientific) on a QuantStudio 12K Flex RT-PCR system (Thermo Fisher Scientific). 18S was used as housekeeping gene, and relative mRNA levels were calculated using the 2−ΔΔCt method. Each data point is the average of three replicates.
Statistical analysis
GraphPad Prism software was used for statistical analysis. According to the experimental design, χ2 test, unpaired t test, paired t test, or one-way ANOVA was performed. Statistical analysis on fold change data was performed after log transformation of the data to obtain a more normalized distribution. Data from each patient were considered as an independent experiment.
Supplementary Material
Acknowledgments
We thank D. R. Principe and B. Shmaltuyeva for assistance with immunohistochemical staining, S. S. Malpani and S. A. Kujawa for providing the tissue samples for this study, Y. Zhu for providing the lenti-CTR and lenti–MnSOD K122-R viruses, and D. Fantini for insightful discussions and editorial assistance in writing the manuscript. Funding: This work was supported by NIH grant NICHD P01 HD057877. Author contributions: V.V. and J.J.K. designed research. V.V. performed research. D.G. and J.-J.W. contributed reagents and analytic tools. V.V., J.-J.W., and J.J.K. analyzed data. V.V., D.G., D.C., S.E.B., J.J.W., and J.J.K. wrote and reviewed the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/11/e1601132/DC1
fig. S1. SIRT3 and iNOS protein levels in ULM.
fig. S2. Differential expression of MnSOD K122-Ac, MnSOD, and pAKT in MM and ULM cells.
fig. S3. Overexpression of MnSOD reduces pAKT levels in ULM cells from multiple patients.
fig. S4. PQ causes PTEN nuclear translocation in ULM cells.
fig. S5. Different effects of MK-2206 and AKT silencing on ULM cell viability and superoxide generation.
fig. S6. AKT silencing in ULM and MM cells.
REFERENCES AND NOTES
- 1.Hanahan D., Weinberg R. A., The hallmarks of cancer. Cell 100, 57–70 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Peddada S. D., Laughlin S. K., Miner K., Guyon J.-P., Haneke K., Vahdat H. L., Semelka R. C., Kowalik A., Armao D., Davis B., Baird D. D., Growth of uterine leiomyomata among premenopausal black and white women. Proc. Natl. Acad. Sci. U.S.A. 105, 19887–19892 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallach E. E., Vlahos N. F., Uterine myomas: An overview of development, clinical features, and management. Obstet. Gynecol. 104, 393–406 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Bulun S. E., Uterine fibroids. N. Engl. J. Med. 369, 1344–1355 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Catherino W. H., Parrott E., Segars J., Proceedings from the National Institute of Child Health and Human Development conference on the Uterine Fibroid Research Update Workshop. Fertil. Steril. 95, 9–12 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cardozo E. R., Clark A. D., Banks N. K., Henne M. B., Stegmann B. J., Segars J. H., The estimated annual cost of uterine leiomyomata in the United States. Am. J. Obstet. Gynecol. 206, 211.e1–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kovács K. A., Lengyel F., Környei J. L., Vértes Z., Szabó I., Sümegi B., Vértes M., Differential expression of Akt/protein kinase B, Bcl-2 and Bax proteins in human leiomyoma and myometrium. J. Steroid Biochem. Mol. Biol. 87, 233–240 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Sefton E. C., Qiang W., Serna V., Kurita T., Wei J.-J., Chakravarti D., Kim J. J., MK-2206, an AKT inhibitor, promotes caspase-independent cell death and inhibits leiomyoma growth. Endocrinology 154, 4046–4057 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altomare D. A., Testa J. R., Perturbations of the AKT signaling pathway in human cancer. Oncogene 24, 7455–7464 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Vivanco I., Sawyers C. L., The phosphatidylinositol 3-Kinase–AKT pathway in human cancer. Nat. Rev. Cancer 2, 489–501 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Hoekstra A. V., Sefton E. C., Berry E., Lu Z., Hardt J., Marsh E., Yin P., Clardy J., Chakravarti D., Bulun S., Kim J. J., Progestins activate the AKT pathway in leiomyoma cells and promote survival. J. Clin. Endocrinol. Metab. 94, 1768–1774 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leslie N. R., The redox regulation of PI 3-kinase–dependent signaling. Antioxid. Redox Signal. 8, 1765–1774 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Pelicano H., Xu R.-h., Du M., Feng L., Sasaki R., Carew J. S., Hu Y., Ramdas L., Hu L., Keating M. J., Zhang W., Plunkett W., Huang P., Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J. Cell Biol. 175, 913–923 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nogueira V., Park Y., Chen C.-C., Xu P.-Z., Chen M.-L., Tonic I., Unterman T., Hay N., Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 14, 458–470 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell E. L., Emerling B. M., Chandel N. S., Mitochondrial regulation of oxygen sensing. Mitochondrion 5, 322–332 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Fletcher N. M., Saed M. G., Abuanzeh S., Abu-Soud H. M., Al-Hendy A., Diamond M. P., Saed G. M., Nicotinamide adenine dinucleotide phosphate oxidase is differentially regulated in normal myometrium versus leiomyoma. Reprod. Sci. 21, 1145–1152 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foksinski M., Kotzbach R., Szymanski W., Olinski R., The level of typical biomarker of oxidative stress 8-hydroxy-2′-deoxyguanosine is higher in uterine myomas than in control tissues and correlates with the size of the tumor. Free Radic. Biol. Med. 29, 597–601 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Bolisetty S., Jaimes E. A., Mitochondria and reactive oxygen species: Physiology and pathophysiology. Int. J. Mol. Sci. 14, 6306–6344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sena L. A., Chandel N. S., Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landriscina M., Maddalena F., Laudiero G., Esposito F., Adaptation to oxidative stress, chemoresistance, and cell survival. Antioxid. Redox Signal. 11, 2701–2716 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Mesquita F. S., Dyer S. N., Heinrich D. A., Bulun S. E., Marsh E. E., Nowak R. A., Reactive oxygen species mediate mitogenic growth factor signaling pathways in human leiomyoma smooth muscle cells. Biol. Reprod. 82, 341–351 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holley A. K., Bakthavatchalu V., Velez-Roman J. M., St. Clair D. K., Manganese superoxide dismutase: Guardian of the powerhouse. Int. J. Mol. Sci. 12, 7114–7162 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fridovich I., Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 64, 97–112 (1995). [DOI] [PubMed] [Google Scholar]
- 24.Miriyala S., Spasojevic I., Tovmasyan A., Salvemini D., Vujaskovic Z., St. Clair D., Batinic-Haberle I., Manganese superoxide dismutase, MnSOD and its mimics. Biochim. Biophys. Acta 1822, 794–814 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buettner G. R., Superoxide dismutase in redox biology: The roles of superoxide and hydrogen peroxide. Anticancer Agents Med Chem. 11, 341–346 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pervaiz S., Clement M.-V., Superoxide anion: Oncogenic reactive oxygen species?. Int. J. Biochem. Cell Biol. 39, 1297–1304 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Ozden O., Park S.-H., Kim H.-S., Jiang H., Coleman M. C., Spitz D. R., Gius D., Acetylation of MnSOD directs enzymatic activity responding to cellular nutrient status or oxidative stress. Aging 3, 102–107 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y., Zhang J., Lin Y., Lei Q., Guan K. L., Zhao S., Xiong Y., Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 12, 534–541 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tao R., Daniel Pennington J., Ozden O., Park S.-H., Jiang H., Kim H.-S., Robb Flynn C., Hill S., Hayes McDonald W., Olivier A. K., Spitz D. R., Gius D., Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 40, 893–904 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Candas D., Li J. J., MnSOD in oxidative stress response-potential regulation via mitochondrial protein influx. Antioxid. Redox Signal. 20, 1599–1617 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Darwish R. S., Amiridze N., Aarabi B., Nitrotyrosine as an oxidative stress marker: Evidence for involvement in neurologic outcome in human traumatic brain injury. J. Trauma 63, 439–442 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Lechner M., Lirk P., Rieder J., Inducible nitric oxide synthase (iNOS) in tumor biology: The two sides of the same coin. Semin. Cancer Biol. 15, 277–289 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Thomsen L. L., Lawton F. G., Knowles R. G., Beesley J. E., Riveros-Moreno V., Moncada S., Nitric oxide synthase activity in human gynecological cancer. Cancer Res. 54, 1352–1354 (1994). [PubMed] [Google Scholar]
- 34.Cochemé H. M., Murphy M. P., Complex I is the major site of mitochondrial superoxide production by paraquat. J. Biol. Chem. 283, 1786–1798 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Mayer A., Höckel M., Wree A., Leo C., Horn L.-C., Vaupel P., Lack of hypoxic response in uterine leiomyomas despite severe tissue hypoxia. Cancer Res. 68, 4719–4726 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Tal R., Segars J. H., The role of angiogenic factors in fibroid pathogenesis: Potential implications for future therapy. Hum. Reprod. Update 20, 194–216 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X., Jiang X., Post-translational regulation of PTEN. Oncogene 27, 5454–5463 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Lee S.-R., Yang K.-S., Kwon J., Lee C., Jeong W., Rhee S. G., Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 277, 20336–20342 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Chang C.-J., Mulholland D. J., Valamehr B., Mosessian S., Sellers W. R., Wu H., PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression. Mol. Cell. Biol. 28, 3281–3289 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Connor K. M., Subbaram S., Regan K. J., Nelson K. K., Mazurkiewicz J. E., Bartholomew P. J., Aplin A. E., Tai Y.-T., Aguirre-Ghiso J., Flores S. C., Melendez J. A., Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J. Biol. Chem. 280, 16916–16924 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Quast S.-A., Berger A., Eberle J., ROS-dependent phosphorylation of Bax by wortmannin sensitizes melanoma cells for TRAIL-induced apoptosis. Cell Death Dis. 4, e839 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu X., Lu Z., Qiang W., Vidimar V., Kong B., Kim J. J., Wei J.-J., Inactivation of AKT induces cellular senescence in uterine leiomyoma. Endocrinology 155, 1510–1519 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Forman H. J., Zhang H., Rinna A., Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 30, 1–12 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hou P., Zhao L., Li Y., Luo F., Wang S., Song J., Bai J., Comparative expression of thioredoxin-1 in uterine leiomyomas and myometrium. Mol. Hum. Reprod. 20, 148–154 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Santulli P., Borghese B., Lemaréchal H., Leconte M., Millischer A.-E., Batteux F., Chapron C., Borderie D., Increased serum oxidative stress markers in women with uterine leiomyoma. PLOS ONE 8, e72069 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fletcher N. M., Saed M. G., Abu-Soud H. M., Al-Hendy A., Diamond M. P., Saed G. M., Uterine fibroids are characterized by an impaired antioxidant cellular system: Potential role of hypoxia in the pathophysiology of uterine fibroids. J. Assist. Reprod. Genet. 30, 969–974 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Storz P., Reactive oxygen species in tumor progression. Front. Biosci. 10, 1881–1896 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Trachootham D., Zhou Y., Zhang H., Demizu Y., Chen Z., Pelicano H., Chiao P. J., Achanta G., Arlinghaus R. B., Liu J., Huang P., Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell 10, 241–252 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Cairns R. A., Harris I. S., Mak T. W., Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Dikalov S., Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 51, 1289–1301 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dikalov S. I., Nazarewicz R. R., Bikineyeva A., Hilenski L., Lassègue B., Griendling K. K., Harrison D. G., Dikalova A. E., Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid. Redox Signal. 20, 281–294 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dikalova A. E., Bikineyeva A. T., Budzyn K., Nazarewicz R. R., McCann L., Lewis W., Harrison D. G., Dikalov S. I., Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 107, 106–116 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiong Y., Guan K.-L., Mechanistic insights into the regulation of metabolic enzymes by acetylation. J. Cell Biol. 198, 155–164 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roth M., Chen W. Y., Sorting out functions of sirtuins in cancer. Oncogene 33, 1609–1620 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Warburg O., On the origin of cancer cells. Science 123, 309–314 (1956). [DOI] [PubMed] [Google Scholar]
- 56.Cluntun A. A., Huang H., Dai L., Liu X., Zhao Y., Locasale J. W., The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer Metab. 3, 10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Donohoe D. R., Collins L. B., Wali A., Bigler R., Sun W., Bultman S. J., The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 48, 612–626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wellen K. E., Hatzivassiliou G., Sachdeva U. M., Bui T. V., Cross J. R., Thompson C. B., ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leslie N. R., Bennett D., Lindsay Y. E., Stewart H., Gray A., Downes C. P., Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 22, 5501–5510 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han D., Antunes F., Canali R., Rettori D., Cadenas E., Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 278, 5557–5563 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Kokoszka J. E., Coskun P., Esposito L. A., Wallace D. C., Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc. Natl. Acad. Sci. U.S.A. 98, 2278–2283 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lustgarten M. S., Bhattacharya A., Muller F. L., Jang Y. C., Shimizu T., Shirasawa T., Richardson A., Remmena H. S. V., Complex I generated, mitochondrial matrix-directed superoxide is released from the mitochondria through voltage dependent anion channels. Biochem. Biophys. Res. Commun. 422, 515–521 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kwon J., Lee S.-R., Yang K.-S., Ahn Y., Kim Y. J., Stadtman E. R., Rhee S. G., Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. U.S.A. 101, 16419–16424 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peng L., Wen Y., Han Y., Wei A., Shi G., Mizuguchi M., Lee P., Hernando E., Mittal K., Wei J.-J., Expression of insulin-like growth factors (IGFs) and IGF signaling: Molecular complexity in uterine leiomyomas. Fertil. Steril. 91, 2664–2675 (2009). [DOI] [PubMed] [Google Scholar]
- 65.Xuan Nguyen T. L., Choi J. W., Lee S. B., Ye K., Woo S.-D., Lee K.-H., Ahna J.-Y., Akt phosphorylation is essential for nuclear translocation and retention in NGF-stimulated PC12 cells. Biochem. Biophys. Res. Commun. 349, 789–798 (2006). [DOI] [PubMed] [Google Scholar]
- 66.Bijur G. N., Jope R. S., Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J. Neurochem. 87, 1427–1435 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miyamoto S., Murphy A. N., Brown J. H., Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 15, 521–529 (2008). [DOI] [PubMed] [Google Scholar]
- 68.Tapodi A., Debreceni B., Hanto K., Bognar Z., Wittmann I., Gallyas F. Jr, Varbiro G., Sumegi B., Pivotal role of Akt activation in mitochondrial protection and cell survival by poly(ADP-ribose)polymerase-1 inhibition in oxidative stress. J. Biol. Chem. 280, 35767–35775 (2005). [DOI] [PubMed] [Google Scholar]
- 69.Yin P., Lin Z., Cheng Y.-H., Marsh E. E., Utsunomiya H., Ishikawa H., Xue Q., Reierstad S., Innes J., Thung S., Kim J. J., Xu E., Bulun S. E., Progesterone receptor regulates Bcl-2 gene expression through direct binding to its promoter region in uterine leiomyoma cells. J. Clin. Endocrinol. Metab. 92, 4459–4466 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/11/e1601132/DC1
fig. S1. SIRT3 and iNOS protein levels in ULM.
fig. S2. Differential expression of MnSOD K122-Ac, MnSOD, and pAKT in MM and ULM cells.
fig. S3. Overexpression of MnSOD reduces pAKT levels in ULM cells from multiple patients.
fig. S4. PQ causes PTEN nuclear translocation in ULM cells.
fig. S5. Different effects of MK-2206 and AKT silencing on ULM cell viability and superoxide generation.
fig. S6. AKT silencing in ULM and MM cells.