


Abstract
Gene transcription is regulated mainly by transcription factors (TFs). ENCODE and Roadmap Epigenomics provide global binding profiles of TFs, which can be used to identify regulatory regions. To this end we implemented a method to systematically construct cell-type and species-specific maps of regulatory regions and TF–TF interactions. We illustrated the approach by developing maps for five human cell-lines and two other species. We detected ∼144k putative regulatory regions among the human cell-lines, with the majority of them being ∼300 bp. We found ∼20k putative regulatory elements in the ENCODE heterochromatic domains suggesting a large regulatory potential in the regions presumed transcriptionally silent. Among the most significant TF interactions identified in the heterochromatic regions were CTCF and the cohesin complex, which is in agreement with previous reports. Finally, we investigated the enrichment of the obtained putative regulatory regions in the 3D chromatin domains. More than 90% of the regions were discovered in the 3D contacting domains. We found a significant enrichment of GWAS SNPs in the putative regulatory regions. These significant enrichments provide evidence that the regulatory regions play a crucial role in the genomic structural stability. Additionally, we generated maps of putative regulatory regions for prostate and colorectal cancer human cell-lines.
INTRODUCTION
Recent genomic technologies have demonstrated that the functional DNA is not only formed by genes, but also by a sizeable fraction of the non-coding sequences (1,2). Consequently, two large consortia (3,4) were set to prioritize the identification, interpretation and interconnection of all the genomic elements related to genomic transcription regulation, including transcribed sequences and gene regulatory elements. Chromatin immunoprecipitation (ChIP) followed by high-throughput sequencing (ChIP-seq) is nowadays a commonly used technique to identify transcription factor binding sites (TFBS) and locations of histone modifications (HM) on a genome-wide scale (5). Such marks have been identified in various locations throughout the genome including gene coding and non-coding regions (6). The ENCODE (4,6) and NIH Roadmap Epigenomics (3) projects focused on producing of comprehensive, publicly available data and analysis of such data sets.
The vast majority of the human genome (∼98%) consists of non-coding regions and a surprisingly large fraction has been proven to have a functional regulatory role containing promoters, enhancers, Locus Control Regions (LCR), insulators and silencers (7). Promoters, enhancers and LCR are associated with gene expression activation, while insulators and silencers are associated with gene expression repression (8).
The main focus of the research performed using ChIP-seq data has been targeting specific biological issues such as promoter and enhancer annotation marks (9), genome accessibility (10), HM functionality (11), nucleosome positioning (12), exon inclusion (13) etc. Additionally, various research projects have focused on applying machine learning techniques on such data, in order to reveal genomic marks that can characterize and annotate multiple genome-wide phenomena (14–16).
Several pipelines and software tools have been established to detect TFBSs and their motifs throughout the genome (17). However, very few methods have been developed to detect transcription regulatory regions genome-wide (18–21). These methods require large data pre-processing and do not dynamically integrate public or custom data collections into their pipelines since they use prior knowledge to report regulatory regions, such as TF binding motifs. While TF binding is largely affected by sequence specific motifs, recruiting these motifs in a regulatory region detection pipeline may lead to overfitting due to ignoring the sequence-independent TF–TF interactions. Specific synergistic operations are also very important in defining the entire regulatory landscape.
Here, we aimed to provide sets of genomic loci of significant importance to gene regulation and to facilitate their experimental investigation in a TFBS-data-driven, cell-line-specific and species-specific manner. To this end, we recruited the well-established notion that clusters of TFs mark putative regulatory regions (22–24) and developed tfNet, an algorithm that constructs genomic regions from sets of ChIP-seq data. tfNet builds clusters of genomic signals, based on their distance, that constitute putative regulatory regions. The output consists of two types of results: (i) genomic-region data sets, and (ii) genomic signals interaction networks. Using public ChIP-seq data we detected large sets of putative cell-specific regulatory regions in five human cell lines and in a collection of cell lines of two cancer types. Additionally, we detected maps of putative regulatory regions in five Mus musculus (M. musculus) cell lines and in four developmental stages of Drosophila melanogaster (D. melanogaster). A subset of the identified genomic regions has been validated experimentally to test for transcriptional activity. More importantly, we investigated the role of the proposed regions in the genomic regulatory mechanism, especially in the less studied heterochromatin, and the three dimensional structure of the genome. We discovered that the majority of the regions were located in open chromatin and intersected with the ENCODE genomic annotations. The participation of the putative regulatory regions in the formation and the body of the genomic 3D looping structure was confirmed computationally in over 90% of the cases. With such a large enrichment we could map a large number of the identified regulatory regions in the 3D genomic space. The putative regulatory regions are publically available at http://bioinf.icm.uu.se/tfnet.php.
MATERIALS AND METHODS
Identification of the regulatory landscape
tfNet assigns clusters of consecutive TFBSs located within a predefined distance to regulatory regions. The details of tfNet are provided in Supplementary Note S1 and Supplementary Figure S1. We applied tfNet to identify putative regulatory regions for five human cell lines (GM12878, H1-hESC, HeLa-S3, HepG2 and K562). We employed TF ChIP-seq and DNaseI Hypersensitivity data sets from ENCODE for the selected cell lines (Supplementary Table S1) (4). We merged the overlapping TFBSs originating from different replicates of the same TF into single peaks in order to avoid artefacts and misleading TF interactions (multiple peaks of the same TF originating from different replicates binding the same genomic loci) using the function mergeBed of bedtools (25). Next, we ran tfNet to detect regulatory regions for each cell line. The distance threshold between consecutive peak summits (dm) was set to 300 bp, which was greater than the distance between 87% of the input TFBS data (Supplementary Figure S2). We considered only those regulatory regions harbouring at least 2 peaks of different sources (regions containing at least two different TFs or those containing a TF and a DNaseI signal). The tfNet tool is freely accessible through http://figshare.com/articles/tfNet_manual/1408532 and the generated putative regulatory regions for all five cell lines are made publically available on http://bioinf.icm.uu.se/tfnet.php.
TF interaction detection
In order to detect TF–TF interactions we constructed three regulatory networks: co-occurring, neighbouring and overlapping. The co-occurring network models the interactions between TFs that appeared to bind (co-occurred) in the same regulatory regions. The neighbouring network stands to model the interactions of TFs whose ChIP-seq peak summits are located 20–60 bp away from each other. The overlapping network models the interactions of TFs that have summit pairs within 20 bp. These chosen distance values are empirical and may be sensitive towards data resolution or systematic technical differences among various ChIP-seq experiments. Here, we aim at providing potential interactions of TFs to be subjected to further investigation. Only statistically significant TF interactions were reported for the generated models. P-values for the neighbouring and the overlapping TF pairs were calculated using the hypergeometric distribution since each peak may have at most one succeeding neighbour (Supplementary Equation S1A). Significant interactions in the co-occurring pairs were calculated using the binomial distribution since one peak can be used to construct several pair connections with replacement (Supplementary Equation S1B). Stringent multiple-test correction (Bonferroni) was applied.
Regulatory region annotation
We investigated the genomic context where the putative regulatory regions were located using the GENCODEv23 data set (26) for all five cell lines (Supplementary Table S1). We first converted the genomic coordinates from hg38 to hg19 genome assembly, removed all the pseudogenes and the “to be experimentally confirmed" genes, and we constructed promoters for each gene spanning ±1.5 kb from the transcription start site (TSS). Finally, we extracted all exons and introns from the gene annotation data set and intersected the locations of the putative regulatory regions with the promoters, the exons and the introns.
We merged the 12 ChromHMM annotation classes proposed by ENCODE into 7 (enhancers, promoters, heterochromatin, repetitive, repressed, transcribed and insulators) (Supplementary Table S2). Next, we intersected the putative regulatory regions with the annotated regions for the available cell lines (GM12878, H1-hESC, HepG2 and K562). ChromHMM has not annotated HeLa-S3 hence its putative regulatory regions could not be characterized. To avoid artefacts, we considered as robust the unique annotations that overlapped with a putative regulatory region by more than 20% and for the rest we introduced a new annotation class named “Mixed".
Enrichment of GWAS SNPs in regulatory regions
Single nucleotide polymorphisms (SNPs) reported in the GWAS catalogue were downloaded from the European Bioinformatics Institute and converted to the hg19 genome assembly. The enrichment of SNPs located within the putative regulatory regions of HepG2, K562 and GM12878 were compared to those enriched in randomly generated regions in the corresponding cell lines. The shuffleBed feature of bedtools (25) was used to obtain a random set of regulatory regions with similar properties to those of the original set. This process was repeated 10 000 times and a P-value was calculated according to a one sided T-test.
We compared the enrichment of SNPs associated with selected terms (liver for HepG2; lymphoma and blood for GM12878 and K562) in the putative regulatory regions and the random sets using the statistical T-test to check for enrichment of SNPs related to a particular disease or trait.
Analysis of regulatory regions in the three dimensional space
Employing the genomic signals (peaks) (27) of regions brought to close proximity by the three dimensional conformation, we investigated all types of annotated putative regulatory regions for contacting domains. We used data for the cell lines common between Rao et al., 2014 (27) and our analysis (GM12878, K562 and HeLa-S3) to map interactions of the annotated putative regulatory regions. We intersected the putative regulatory regions with the contacting domains of the corresponding cell line and we created pairs of interactions between putative regulatory regions marking the upstream and downstream contacting domains. In cases when more than one putative regulatory region was intersecting with the same contacting domain we assumed a complete interaction graph. In cases when no putative regulatory region intersected with the contacting domain we marked this domain as “Unknown". As a result we obtained maps of pairs of annotated putative regulatory region interactions.
From the same data set of Rao et al., 2014 (27) we extracted all the formed genomic loops, excluding the contacting domains and we studied the participation of our putative regulatory regions in the 3D loops. As the starting position of a looping domain we set the end of the upstream contacting domain and as the end position of the looping domain we set the start of the downstream contacting domain. Next, we intersected all the putative regulatory regions that did not intersect with any contacting domain in order to obtain those located in the genomic loops. The putative regulatory regions not located in contacting or looping domains were marked as “Out".
RESULTS
A regulatory map of the genome
We developed a fast parallel platform-independent computational tool called tfNet for detecting regulatory regions on a genome wide scale from a collection of TFBSs. tfNet offers a range of features to the users to adapt the results to their specific research interests. The resulting set of regions is provided in BED file format and it may be reused by other computational tools and visualized in genome browsers. tfNet also identifies and reports networks of significant TF–TF interactions for the detected putative regulatory regions. The interactions are reported in three types of networks based on the TF binding proximity. The co-occurring networks refer to TFs located within the same regulatory regions. The neighbouring networks indicate TFs appearing in sequences. The overlapping networks that the tool reports are a reflection of the potential competition for the same binding site (antagonism) or formation of protein–protein complexes that result in one direct and one indirect DNA binding (tethering). Clearly, these networks may be sensitive to technical biases or resolution of ChIP-seq data.
We detected whole-genome regulatory maps for five human cell lines (GM12878, H1-hESC, HepG2, K562 and HeLa-S3). The total number of the obtained putative regulatory regions appeared to be correlated to the total number of TFBSs available for each cell line (Supplementary Table S3; Supplementary Figures S3 and S4). For cell lines with a large number of ChIP-seq-ed TFs (GM12878, H1-hESC, HepG2 and K562) our algorithm resulted in a larger number of putative regulatory regions (Figure 1A and B). On average 76% of the putative regulatory regions intersected with DNaseI peaks (Supplementary Figure S3). This was in agreement with previous findings (14) and suggested that a large number of the putative regulatory regions that were located within open chromatin domains are potentially functional.
Figure 1.

(A) Annotation of the putative regulatory regions for each of the five cell lines according to their proximity to GENCODEv23 genes (26). (B) Annotation of putative regulatory regions according to the merged annotations from the ChromHMM data set (cf. Materials and Methods). The regions that did not intersect with any of the ChromHMM annotations are marked as “Unannotated". We lack ChromHMM annotations for HeLa-S3. (C) Pair-wise comparison of gene expression differences among ChromHMM heterochromatic, insulator and promoter putative regulatory regions located in physical promoters (Supplementary Note S4). The Y-axis represents the number of physical gene promoters intersecting with heterochromatic, insulator or promoter putative regulatory regions in three different cell lines. The P-value shows the statistically significant difference (Wilcoxon rank-sum test) between gene expression levels in heterochromatin, insulator and promoters according to ChromHMM. “ns" denotes that there was no statistical significance between the gene expression levels. (D) Biological validation of a subset of the proposed regulatory regions by tfNet. The information in the X-axis contains the GWAS reference SNP IDs (rs) for the SNPs located within the regulatory regions and the ChromHMM annotation. The Y-axis shows the relative luciferase activity for each tested region. P-values are calculated between the control and each corresponding tested region (Mann–Whitney U test). “ns" denotes that there was no statistical significance between the tested region and the control.
In order to confirm the robustness of the algorithm and the generated maps, we recursively detected regions from randomly generated TFBS data sets. We observed that the regulatory regions detected using the experimentally derived data contained more TFBSs than those generated using the randomized data. Similarly, the number of the regulatory regions and their genome coverage were orders of magnitude lower than those obtained from the synthetic data (Supplementary Figure S4). We also investigated the overlap between the obtained putative regulatory regions with a set of manually curated TFBSs that is used as a ChIP-seq benchmark data set (28). On average more than 86% of the true positive benchmark peaks overlapped with the putative regulatory regions (Supplementary Table S4).
We experimentally validated the regulatory function of a selected subset of regions using the luciferase assays (Supplementary Note S3). The selected regions contained SNPs associated to liver diseases within the TFBSs. The luminescence ratios obtained for the four experimental samples were significantly higher (Mann–Whitney U test P-value < 0.05) for plasmids containing the putative regulatory regions than the controls, indicating that they are active regulatory elements (Figure 1D; Supplementary Table S5).
Genome-wide association studies have associated thousands of SNPs to hundreds of complex traits and common diseases (29). The majority of these SNPs map to non-protein coding sequences (30). Using our map of putative regulatory regions defined in GM12878, K562 and HepG2, we found a significant enrichment of GWAS SNPs (P-value < 10−3 T-test from Monte Carlo simulations). Since our identified regulatory maps are cell type specific we could search for enrichment of particular traits or diseases. The regulatory regions in GM12878 showed a significant enrichment of lymphoma-related SNPs (P-value < 10−3). While the regulatory regions in K562 showed a significant enrichment of blood-related traits (P-value < 10−3) and finally liver-related traits were significantly enriched in the regulatory regions of HepG2 (P-value = 2 × 10−3).
To show the utility of tfNet on diverse ChIP-seq experiments we collected binding sites for 117 TFs of the LoVo cell line in colorectal cancer. We also generated the binding sites for 34 TFs from 135 ChIP-seq experiments curated from 29 independent studies of different prostate cancer cell lines (Supplementary Note S2; Supplementary Table S6). In both cases tfNet mapped ∼120K putative regulatory regions and revealed several TF–TF interactions (Supplementary Figures S5 and S6).
Additionally, we used tfNet to generate regulatory maps of M. musculus and D. melanogaster (Supplementary Table S7). For M. musculus we ran tfNet for five different cell types (C2C12, CH12.LX, ES-E14, MEL and myocyte). We discovered a large number of putative regulatory regions and statistically significant TF–TF interactions within the identified regions (Supplementary Figure S7A; Supplementary Figure S8). For D. melanogaster we constructed regulatory regions for various developmental stages (Supplementary Note S2). We detected a large number of putative regulatory regions and several strong TF–TF interactions for each developmental stage (Supplementary Figure S7B; Supplementary Figure S9). Moreover, by combining TFBSs of all developmental stages for D. melanogaster we constructed a full map of putative regulatory regions and detected a large number of statistically significant TF interactions (Supplementary Figure S7B; Supplementary Figure S10). This map recovered ∼83% of the known D. melanogaster cis regulatory modules (31) (Supplementary Figure S7C). These case-studies, additionally to showing that our hypothesis was valid, proved that the approach is species-independent in generating putative regulatory-region landscapes.
Annotation of the putative regulatory regions
As it was expected, we observed a significant and similar number of putative regulatory regions harbouring promoters (±1.5 kb from gene TSS) among cell lines (∼19%), while the number of putative regulatory regions located in the exonic components was lower (∼10%). Introns and intergenic regions, that are distal regulatory candidates, appeared to contain the largest number of putative regulatory regions (∼39% and ∼32%, respectively) (Figure 1A).
In the next step we investigated the types of putative regulatory regions retrieved for the cell lines where chromatin-state annotation using hidden Markov model combinations of chromatin modification patterns (ChromHMM) annotations were available. The majority of the regions (99.8%) where annotated by ChromHMM annotations, of these 13.8% were labelled with more than one annotation; marked as “Mixed" (Figure 1B). These findings indicated that the putative regulatory regions show distinct histone modification mark patterns suggesting a robust region annotation. As expected, enhancers and promoters appeared to be among the overrepresented regulatory region annotations. Together they covered a significant part of the total number of the regulatory regions (44%) where clusters of TFBSs appeared (Figure 1B). Both genomic regions’ annotations have been extensively studied and characterized for being marked by specific genomic signals and by participating in a wide range of genomic interactions. Additionally, heterochromatin, insulators, repetitive and repressed annotations appeared to cover on average 32% of the putative regulatory regions. Interestingly, transcribed regions, previously noted to be marked by specific histone modifications (14) and depleted from DNaseI accessibility, were observed to host a substantial number (10%) of putative regulatory regions (Figure 1B).
Heterochromatic elements from ChromHMM annotations are associated to nuclear lamina and lack histone chromatin marks (14). The number of TF marks that we observed in such elements was comparable to that of enhancers and promoters (Figure 1B), which may indicate regulatory activity of the heterochromatic regions.
Investigating physical promoters, ±1.5 kb from TSSs of genes, revealed at least 40- and 51-fold higher abundancy of ChromHMM promoters compared to heterochromatic or insulator putative regulatory regions, respectively (Figure 1C). Genes with ChromHMM promoter domains were expressed at least 12- and 4-fold higher than those with heterochromatic (P-value < 10−3) and insulator domains (P-value < 10−3), respectively. Generally, the average expression of genes with proximal ChromHMM heterochromatic regions was very low (RPKM ∼3.9). Additionally to that, the putative regulatory regions that did not show any significant relative luciferase activity was a heterochromatic region (Figure 1D). These findings suggest that regulatory elements harbouring complexes of TFs in heterochromatic regions may act over large distances as activators and/or silencers.
Frequent TFs and TF–TF interactions in heterochromatic regulatory regions
We investigated TFs that were abundant in heterochromatic regions and focused on the most significant TF interactions appearing at the weighted TF networks (Figure 2A–H; Supplementary Figures S11–S15). These networks demonstrate TF interactions occurring in heterochromatic putative regulatory regions and rely on the absolute distances between TFBSs, hence the quality of the ChIP-seq data may affect the information they present. In order to avoid potential biases towards active regulatory regions and to explore the cell-line-specific TF–TF interactions, we regenerated the results after excluding DNaseI hypersensitive sites from the region detection pipeline.
Figure 2.

Heatmap networks modelling the significant TF–TF interactions in putative regulatory regions of heterochromatin annotation. The colour intensity in each cell represents the TF–TF interaction significance for each network type in the corresponding cell line. The shown interactions are between (A) CTCF-RAD21-SMC3, (B) NRSF-SIX5-ZNF143, (C) BACH1-MAFF-MAFK, (D) CJUN-FOSL1-JUND, (E) USF1-USF2, (F) ARF2-BATF-NFIC-RUNX3, (G) HNF4A-HNF4G and (H) FOXA1-FOXA2.
CTCF has recently been extensively linked to pioneering the three dimensional conformation of the genome (27,32). It has also been characterized as a TF with unique properties that bridges the gap between the nuclear architecture and the genomic expression through coupling with the cohesin complex (33,34). Here, we observed that CTCF and a major component of the cohesin complex RAD21 were the most frequent TFs that occurred in the heterochromatic putative regulatory regions (Table 1). CTCF, RAD21 and additionally SMC3 were among the TFs interacting strongly in GM12878, HepG2 and K562 (Figure 2A; Supplementary Figures S11 and S13).
Table 1. Abundance of TFBSs participating in putative regulatory regions of heterochromatic annotation for four examined cell lines and for each TF network model (co-occurring, overlapping and neighbouring). The percentages and the colour code demonstrate the ratio of the TFBSs participating in heterochromatin compared to the total number of input TFBSs.
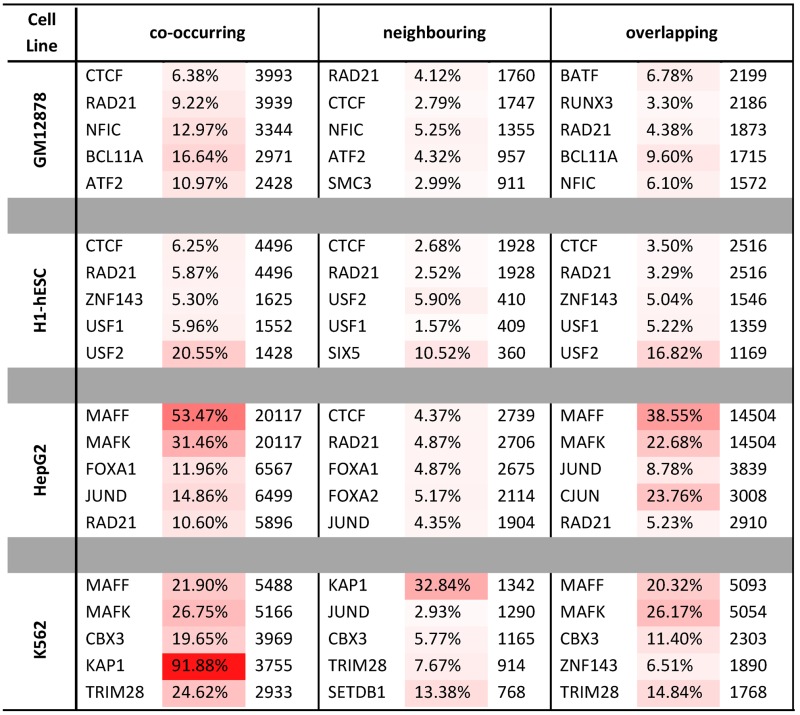
|
There is a plethora of statistically significant interactions between CTCF, RAD21 and SMC3 in all the networks and cell lines (Figure 2A; Supplementary Figures S11–S15). Although the stoichiometric ratio of the CTCF-cohesin interactions is fixed across cell types, the SMC3 binding sites in some of the interactions were depleted in GM12878 and K562. This observation indicated that differences in data quality and resolution across different cell types affect the observed interaction patterns, since the cohesin complex cannot be formed in absence of SMC3 (35). The presence of significant CTCF-cohesin interactions in heterochromatic putative regulatory regions across different cell types indicated functionality of these regions.
In H1-hESC the zinc finger protein ZNF143 was enriched in heterochromatic regions when compared to the other cell lines (Table 1). The plethora of binding sites of ZNF143 suggested another dimension, additional to its already known functionality in cell cycle regulation (36), proliferation (37) and apoptosis (38) in embryonic stem cells. Additionally, its binding sites overlapped significantly with NRSF and SIX5 (Figure 2B; Supplementary Figures S12B and S14). The homologous nuclear proteins MAFF and MAFK showed high occupancy of the heterochromatic putative regulatory regions in H1-hESC, HepG2 and K562 (Table 1) and very strong interactions, confirming the previous studies uncovering their cooperative action (Figure 2C, Supplementary Figure S11) (39–41). Specifically, we detected strong interactions in HepG2 and K562 for the overlapping model (Figure 2C; Supplementary Figures S12B and S15) pointing out antagonism or tethering. Additionally, the TFBSs of MAFK appeared to overlap significantly with those of BACH1 in H1-hESC, a finding that agrees with their role in transcription activation and repression (Figure 2C; Supplementary Figures S12B and S15) (42).
JUND is a member of the AP-1 transcription factor complex that is considered to act both as an onco-suppressor and as an oncogenic driver (43). We also saw it to be one of the most frequent TFs in heterochromatic regions, mainly in HepG2 and K562 cells (Table 1). The molecular function of JUND has not been accurately defined. However, current research suggests that it negatively affects cell proliferation (44). These findings were in agreement with our results, since we did not observe JUND in GM12878 and H1-hESC, while we did observe it in the other two cell lines originating from cancers. Even more convincing were the results of significant interactions that occurred between JUND and two other members of the AP-1 complex, FOSL1 and c-JUN. JUND overlapped significantly with c-JUN in HepG2 and K562 (Figure 2D; Supplementary Figures S12B and S15), and it co-occurred significantly with FOSL1 in K562 (Figure 2D; Supplementary Figures S11 and S13). Additionally, we observed that USF1 and USF2 despite binding infrequently to the heterochromatic regions (Table 1), except from H1-hESC they appeared to cooperate significantly (Figure 2D; Supplementary Figures S11 and S13). USF1 and USF2 have been previously associated with familial combined hyperlipidaemia (45), the metabolic syndrome (46) and to higher risk of cardiovascular disease (47). In our study, they appeared to overlap significantly in the heterochromatic regions of all cell lines (Figure 2E; Supplementary Figures S12B and S14). The extent of the overlap between these two TFs suggested antagonism or tethering between USF1 and USF2. This offers a complementary evidence in support of our previous findings that have detected USF1 and USF2 at protein coding gene promoters (48).
BATF occurred repeatedly in the heterochromatic regions of GM12878 (Table 1). BATF is a TF known to cooperate with RUNX3 in regulation of vital CD8+ effector T cells (49). Here, we observed that BATF had a plethora of binding sites and cooperated with NFIC in heterochromatin regions, even though the latter was not among the five most frequent TFs in the heterochromatin of GM12878. Moreover, NFIC appeared to be significantly correlated with RUNX3 and even more significantly with ATF2 (Figure 2F; Supplementary Figures S13–S15).
The homodimers HNF4α and HNF4γ (50) that have been reported to coordinate gene expression (51) appeared to have a significant overlap of binding sites in HepG2 (Figure 2G; Supplementary Figures S13–S15). In the same cell line, FOXA1 and FOXA2 that are known for their extensive homology in their DNA binding domain (52), participated significantly in the heterochromatin domains (Figure 2H; Supplementary Figures S13–S15). They are known for controlling liver tissue development, regulation of liver specific genes (53,54) and have been proposed to share DNA binding motifs. In both cases the statistical significance of the overlap of the binding sites suggested antagonism or tethering for the aforementioned pairs of TFs (Figure 2G–H; Supplementary Figures S13–S15).
Here we observed that the two most informative networks were the overlapping (Supplementary Figure S12B) and the co-occurring (Supplementary Figure S11). The neighbouring network (Supplementary Figure S12A) appeared to be depleted of interactions. Hence, in addition to a general intuition of the TF interactions the generated networks can offer detailed information about the detected TF interactions.
Interactions between regulatory regions in three-dimensional space
Finally, we investigated the involvement of the putative regulatory regions in the three dimensional genome architecture employing annotated interactions from the recently published results of Rao et al., 2014 (27). We examined the participation of the identified putative regulatory regions in the contacting domains that constitute the basis for the loop formation and the looping domains (as defined by Rao et al., 2014 (27)). The identified regions covered 84%, 91% and 93% of the contacting domains for GM12878, K562 and HeLa-S3, respectively (Supplementary Figure S16) and the majority of the chromatin loops contained putative regulatory regions in both the upstream and the downstream domains suggesting a strong regulatory effect on the formation of the three dimensional genome structure. On average only 10% of the total number of the contacting domains did not contain any of the regions (Figure 3 red ribbon).
Figure 3.

Participation of the putative regulatory regions (including DNaseI) in interacting domains for (A) GM12878 and (B) K562 (27,57). Region annotations are shown outside the circles. The percentages show the participation of regulatory regions of each annotation. The numbers between the inner and the outer circle represent the amount of putative regulatory regions of a specific annotation interacting with other annotated regions. Putative regulatory regions participating in multiple interactions have been counted multiple times while the numbers in pink stand for the actual amount of putative regulatory regions detected by tfNet. The colour code for the putative regulatory region annotations is the same as in Figure 1B. The thickness of the ribbons shows the number of interacting regions of each annotation. The arks of the innermost circle denote the edges of the corresponding ribbon. (C) Enrichment of GWAS SNPs in putative regulatory regions of interacting domains. In the first track, the blue-box clusters represent ChIP-seq peaks constituting regulatory regions located in the chromatin interacting domains. The lines show the three-dimensional interactions between the upstream and the two downstream domains (Supplementary Table S8). The GWAS SNPs enriched in the regulatory regions are shown in the second track. The red bars are harboured by regions within the interacting domains while those in blue harboured by the nearby regions. In the third track enrichment of histone modification and DNaseI signals are shown. In the final track the ENSEMBL genes close to the looping domains are shown. The arrows show the transcription direction.
Next we constructed the coordinates of the chromatin loops from the coordinates of their boundaries (contacting domains). Within the loops we observed a large participation of the putative regulatory regions. The proposed putative regulatory regions were present by 68%, 58% and 36% in the looping domains of GM12878, K562 and HeLa-S3, respectively (Supplementary Figure S17). On average ∼65% of all the putative regulatory regions associated with gene activation and transcription were enriched in the loops while insulators showed a lower level of enrichment (∼44%). On the other hand, more than half of the overall putative heterochromatic regulatory regions (∼65%) were detected within such loops indicating a regulatory role. Taking together these results and the current findings of Heidari et al., 2014 (55) we hypothesized that the putative regulatory regions located within genomic loops participate in the regulation of genes within these loops. For the unannotated cell line, HeLa-S3, we lacked annotation information hence we could not derive any conclusion (Supplementary Figure S17B).
Insulators, defined by CTCF marks, contributed most to the loop formation. This is in agreement with the suggestion that CTCF and the two major components of the cohesin complex, RAD21 and SMC3, anchor the majority of the interacting domains (27,55). More importantly, a large number of the domain interactions occurred between insulators which is in agreement with the property of CTCF to bridge distal genomic regions (Figure 3A and B). Furthermore, our results suggested a large number of distal interactions between promoters, enhancers and putative regulatory regions of multiple annotations (Mixed) (Figure 3A and B). This implies the involvement of other types of genomic loci in forming genomic loops in addition to insulators. The majority of the HeLa-S3 interactions were between distal regions (intronic and intergenic). Moreover, putative regulatory regions located within physical gene promoters appeared to also participate in the distal interactions mechanism. This, in addition to the observed interactions between promoters and distal regions suggested cooperative gene regulation (Supplementary Figure S18).
Rao et al., 2014 showed that the promoter–enhancer interactions constitute a major part of the interacting domains (Figure 3A and B). Here, we also observed that the participation of putative regulatory regions annotated as heterochromatic was poor (Figure 3A and B). Nevertheless, 75% and 56% of the heterochromatic putative regulatory regions were present within the looping domains of GM12878 and K562, respectively (Supplementary Figure S17). Taken together this evidence demonstrated that heterochromatin is not as silent and inactive as originally assumed by classical biology. Our data indicated that regulatory regions lacking histone modification signals may play an important role in the gene expression regulation. Yet a large number of regulatory regions remains to be characterized, as well as the genes they act on.
The role of GWAS SNPs in the 3D genome conformation
The majority of the SNPs identified through GWAS are located in non-coding regions which have made their characterization challenging. Integration of regulatory regions with genomic interactions allows us to characterize these SNPs by mapping the regulatory regions where the SNPs are harboured by contacting domains. In total, we mapped 46 GWAS SNPs to putative regulatory regions harboured by contacting domains. Figure 3C shows enrichment of blood-related GWAS SNPs in putative regulatory regions of K562. Specifically, four GWAS SNPs were enriched in putative regulatory regions in chromosome 11. These regulatory regions except for harbouring SNPs and containing large clusters of TFs were also part of the chromatin three-dimensional formation of two genomic loops (Figure 3C). Additionally, we detected five other SNPs that were located within putative regulatory regions and genomic loops. One of these SNPs, rs174548, has been recently reported to participate in the regulation of cis/trans-18:2 by FADS1 and FADS2 genes (56).
Most of the SNPs detected in this specific region have been associated to red blood cell fatty acids levels, and they are also located nearby genomic loci encapsulating the fatty acid desaturase genes. Based on this finding we investigated if any of the four SNPs located in putative regulatory regions within the interacting domain of K562 was enriched in putative regulatory regions of HepG2. We discovered that rs174541 which is located in the intergenic region downstream the FADS1 and FEN1 genes, and upstream the TMEM258 gene, and rs174538 which is located in the 5-prime UTR variant of the TMEM258 gene were present in HepG2 regions. The map of histone modifications, the presence of several TFBSs and the enrichment of GWAS SNPs near the genes FADS1, FADS2, FADS3, FEN1 and TEM258 may provide further explanations of the role of these SNPs. Our data suggested that in addition to their previously known role (56), these SNPs may also affect the functionality of their distant interacting domains.
DISCUSSION
In this study we validated our hypothesis that co-localized clusters of TFs can accurately define putative regulatory regions in a genome-wide scale. Additionally, we demonstrated the species-independency and the cell-line-specificity of the algorithm. Moreover, we manifested the adaptability of the tool and its ability to efficiently visualize the results.
TF-clusters are candidate regulatory regions of various functionalities, e.g. promoters, enhancers and insulators. We attempted to annotate the detected putative regulatory regions based on the physical gene locations and based on the machine learning annotations provided by ENCODE (ChromHMM). We observed an extensive overlap with both ChromHMM annotations and DNaseI peaks, suggesting that our findings were indeed functional. We also observed a significant enrichment of GWAS SNPs. Surprisingly, a large number of putative regulatory regions was detected in DNA compartments depleted of any histone modification signal, characterized as heterochromatin by ChromHMM. The latter suggested a regulatory function of genomic regions located in “silent" DNA domains.
In addition to the regulatory region detection functionality, our algorithm sheds light onto the interactions of the TFs participating in the formation of the regions. Specifically, we took advantage of the detected TF interactions and we constructed three types of statistically significant TF-interaction networks, co-occurring, neighbouring and overlapping. Studying the networks we identified the binding preferences of TFs into putative regulatory regions. For example, we observed that MAFF and MAFK preferred binding to the same regulatory regions and they did appear to interact with each other. Furthermore, peaks of HNF4α-HNF4γ and of FOXA1-FOXA2 appeared to bind on the exact same DNA locations, suggesting antagonism or tethering. We were able to identify differences in TF interactions among cell lines. Provided that there will be enough data, we believe that this study may be extended towards investigating the differences in TF interactions among different tissues or even different species.
Next, we investigated the participation of the detected putative regulatory regions in the formation of the 3D genomic loops. We detected a range of putative regulatory regions located within the looping domains that regulate genes of similar expression patterns. We were also able to identify the majority of the regulatory regions that interacted with each other to construct the loops. Insulators, or CTCF-bound regions, appeared to be the most frequent regions in this mechanism. However, the regulatory regions annotated as heterochromatic did not appear to participate largely in the formation of looping domains, suggesting an unknown but promising regulatory functionality.
Finally, we searched for GWAS SNPs located in the putative regulatory regions participating in the three dimensional genome conformation and we detected 46 SNPs that were harboured in these regions for K562. The majority of the investigated SNPs have been reported to affect the levels of fatty acids in blood cells. Here, we suggest that they are also closely related to affect the bridging of distal loci.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
AstraZeneca [to K.D.]; Uppsala University [to H.M.U., M.C., C.W. and J.K.]; Foundation of Polish Science, International PhD Projects (MPD) program [to M.K.]; Institute of Computer Science, Polish Academy of Sciences [to M.J.D. and J.K.]; Swedish Research Council [to M.C. and C.W.]; Swedish Diabetes Foundation [to C.W. and M.C.]; Diabetes Wellness Network Sweden [to C.W. and M.C.]; Family Ernfors Fund [to C.W. and M.C.]; eSSence project [to J.K.]; Polish Ministry of Science and Higher Education [N301 239536 to J.K.]; National Science Centre [DEC-2015/16/W/NZ2/00314 to J.K. and M.D]. Funding for open access charge: Uppsala University.
Conflict of interest statement. None declared.
REFERENCES
- 1.Heard E., Tishkoff S., Todd J.A., Vidal M., Wagner G.P., Wang J., Weigel D., Young R. Ten years of genetics and genomics: what have we achieved and where are we heading. Nat. Rev. Genet. 2010;11:723–733. doi: 10.1038/nrg2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Consortium E.P. The ENCODE (ENCyclopedia Of DNA Elements) project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein B.E., Stamatoyannopoulos J.A., Costello J.F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M.A., Beaudet A.L., Ecker J.R., et al. The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 6.Consortium E.P., Birney E., Stamatoyannopoulos J.A., Dutta A., Guigo R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thurman R.E., Day N., Noble W.S., Stamatoyannopoulos J.A. Identification of higher-order functional domains in the human ENCODE regions. Genome Res. 2007;17:917–927. doi: 10.1101/gr.6081407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maston G.A., Evans S.K., Green M.R. Transcriptional regulatory elements in the human genome. Ann. Rev. Genomics Hum. Genet. 2006;7:29–59. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- 9.Rajagopal N., Xie W., Li Y., Wagner U., Wang W., Stamatoyannopoulos J., Ernst J., Kellis M., Ren B. RFECS: A random-forest based algorithm for enhancer identification from chromatin state. PLoS Comput. Biol. 2013;9:e1002968. doi: 10.1371/journal.pcbi.1002968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B., et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S.A., Flynn R.A., Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersson R., Enroth S., Rada-Iglesias A., Wadelius C., Komorowski J. Nucleosomes are well positioned in exons and carry characteristic histone modifications. Genome Res. 2009;19:1732–1741. doi: 10.1101/gr.092353.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enroth S., Bornelov S., Wadelius C., Komorowski J. Combinations of histone modifications mark exon inclusion levels. PloS One. 2012;7:e29911. doi: 10.1371/journal.pone.0029911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M., et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffman M.M., Ernst J., Wilder S.P., Kundaje A., Harris R.S., Libbrecht M., Giardine B., Ellenbogen P.M., Bilmes J.A., Birney E., et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013;41:827–841. doi: 10.1093/nar/gks1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yip K.Y., Cheng C., Bhardwaj N., Brown J.B., Leng J., Kundaje A., Rozowsky J., Birney E., Bickel P., Snyder M., et al. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol. 2012;13:R48. doi: 10.1186/gb-2012-13-9-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran N.T., Huang C.H. A survey of motif finding Web tools for detecting binding site motifs in ChIP-Seq data. Biol. Direct. 2014;9:1–22. doi: 10.1186/1745-6150-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitington T., Frith M.C., Johnson J., Bailey T.L. Inferring transcription factor complexes from ChIP-seq data. Nucleic Acids Res. 2011;39:e98. doi: 10.1093/nar/gkr341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun H., Guns T., Fierro A.C., Thorrez L., Nijssen S., Marchal K. Unveiling combinatorial regulation through the combination of ChIP information and in silico cis-regulatory module detection. Nucleic Acids Res. 2012;40:e90. doi: 10.1093/nar/gks237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen G., Zhou Q. Searching ChIP-seq genomic islands for combinatorial regulatory codes in mouse embryonic stem cells. BMC Genomics. 2011;12:1–18. doi: 10.1186/1471-2164-12-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niu M., Tabari E.S., Su Z. De novo prediction of cis-regulatory elements and modules through integrative analysis of a large number of ChIP datasets. BMC Genomics. 2014;15:1–20. doi: 10.1186/1471-2164-15-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerstein M.B., Lu Z.J., Van Nostrand E.L., Cheng C., Arshinoff B.I., Liu T., Yip K.Y., Robilotto R., Rechtsteiner A., Ikegami K., et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moorman C., Sun L.V., Wang J., de Wit E., Talhout W., Ward L.D., Greil F., Lu X.J., White K.P., Bussemaker H.J., et al. Hotspots of transcription factor colocalization in the genome of Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 2006;103:12027–12032. doi: 10.1073/pnas.0605003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan J., Enge M., Whitington T., Dave K., Liu J., Sur I., Schmierer B., Jolma A., Kivioja T., Taipale M., et al. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell. 2013;154:801–813. doi: 10.1016/j.cell.2013.07.034. [DOI] [PubMed] [Google Scholar]
- 25.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S., et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rye M.B., Sætrom P., Drabløs F. A manually curated ChIP-seq benchmark demonstrates room for improvement in current peak-finder programs. Nucleic Acids Res. 2010;39:e25. doi: 10.1093/nar/gkq1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L., et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gallo S.M., Gerrard D.T., Miner D., Simich M., Des Soye B., Bergman C.M., Halfon M.S. REDfly v3.0: Toward a comprehensive database of transcriptional regulatory elements in Drosophila. Nucleic Acids Res. 2011;39:D118–D123. doi: 10.1093/nar/gkq999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Handoko L., Xu H., Li G., Ngan C.Y., Chew E., Schnapp M., Lee C.W., Ye C., Ping J.L., Mulawadi F., et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat. Genet. 2011;43:630–638. doi: 10.1038/ng.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanyal A., Lajoie B.R., Jain G., Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paredes S.H., Melgar M.F., Sethupathy P. Promoter-proximal CCCTC-factor binding is associated with an increase in the transcriptional pausing index. Bioinformatics. 2013;29:1485–1487. doi: 10.1093/bioinformatics/bts596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuin J., Dixon J.R., van der Reijden M.I., Ye Z., Kolovos P., Brouwer R.W., van de Corput M.P., van de Werken H.J., Knoch T.A., van I.W.F., et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. U.S.A. 2014;111:996–1001. doi: 10.1073/pnas.1317788111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Myslinski E., Gerard M.A., Krol A., Carbon P. Transcription of the human cell cycle regulated BUB1B gene requires hStaf/ZNF143. Nucleic Acids Res. 2007;35:3453–3464. doi: 10.1093/nar/gkm239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Izumi H., Wakasugi T., Shimajiri S., Tanimoto A., Sasaguri Y., Kashiwagi E., Yasuniwa Y., Akiyama M., Han B., Wu Y., et al. Role of ZNF143 in tumor growth through transcriptional regulation of DNA replication and cell-cycle-associated genes. Cancer Sci. 2010;101:2538–2545. doi: 10.1111/j.1349-7006.2010.01725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu W., Chen Z., Zhang H., Wang Y., Luo Y., Huang P. ZNF143 transcription factor mediates cell survival through upregulation of the GPX1 activity in the mitochondrial respiratory dysfunction. Cell Death Dis. 2012;3:e422. doi: 10.1038/cddis.2012.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kannan M.B., Solovieva V., Blank V. The small MAF transcription factors MAFF, MAFG and MAFK: current knowledge and perspectives. Biochim. Biophys. Acta. 2012;1823:1841–1846. doi: 10.1016/j.bbamcr.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 40.Shimohata H., Yoh K., Fujita A., Morito N., Ojima M., Tanaka H., Hirayama K., Kobayashi M., Kudo T., Yamagata K., et al. MafA-deficient and beta cell-specific MafK-overexpressing hybrid transgenic mice develop human-like severe diabetic nephropathy. Biochem. Biophys. Res. Commun. 2009;389:235–240. doi: 10.1016/j.bbrc.2009.08.124. [DOI] [PubMed] [Google Scholar]
- 41.Menegazzo L., Albiero M., Avogaro A., Fadini G.P. Endothelial progenitor cells in diabetes mellitus. BioFactors. 2012;38:194–202. doi: 10.1002/biof.1016. [DOI] [PubMed] [Google Scholar]
- 42.Oyake T., Itoh K., Motohashi H., Hayashi N., Hoshino H., Nishizawa M., Yamamoto M., Igarashi K. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell. Biol. 1996;16:6083–6095. doi: 10.1128/mcb.16.11.6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eferl R., Wagner E.F. AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 44.Greenblatt M.B., Shim J.H., Glimcher L.H. Mitogen-activated protein kinase pathways in osteoblasts. Annu. Rev. Cell Dev. Biol. 2013;29:63–79. doi: 10.1146/annurev-cellbio-101512-122347. [DOI] [PubMed] [Google Scholar]
- 45.Pajukanta P., Lilja H.E., Sinsheimer J.S., Cantor R.M., Lusis A.J., Gentile M., Duan X.J., Soro-Paavonen A., Naukkarinen J., Saarela J., et al. Familial combined hyperlipidemia is associated with upstream transcription factor 1 (USF1) Nat. Genet. 2004;36:371–376. doi: 10.1038/ng1320. [DOI] [PubMed] [Google Scholar]
- 46.Ng H.H., Robert F., Young R.A., Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 47.Komulainen K., Alanne M., Auro K., Kilpikari R., Pajukanta P., Saarela J., Ellonen P., Salminen K., Kulathinal S., Kuulasmaa K., et al. Risk alleles of USF1 gene predict cardiovascular disease of women in two prospective studies. PLoS Genet. 2006;2:e69. doi: 10.1371/journal.pgen.0020069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rada-Iglesias A., Ameur A., Kapranov P., Enroth S., Komorowski J., Gingeras T.R., Wadelius C. Whole-genome maps of USF1 and USF2 binding and histone H3 acetylation reveal new aspects of promoter structure and candidate genes for common human disorders. Genome Res. 2008;18:380–392. doi: 10.1101/gr.6880908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kurachi M., Barnitz R.A., Yosef N., Odorizzi P.M., DiIorio M.A., Lemieux M.E., Yates K., Godec J., Klatt M.G., Regev A., et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat. Immunol. 2014;15:373–383. doi: 10.1038/ni.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bogan A.A., Dallas-Yang Q., Ruse M.D., Jr, Maeda Y., Jiang G., Nepomuceno L., Scanlan T.S., Cohen F.E., Sladek F.M. Analysis of protein dimerization and ligand binding of orphan receptor HNF4alpha. J. Mol. Biol. 2000;302:831–851. doi: 10.1006/jmbi.2000.4099. [DOI] [PubMed] [Google Scholar]
- 51.Archer A., Sauvaget D., Chauffeton V., Bouchet P.E., Chambaz J., Pincon-Raymond M., Cardot P., Ribeiro A., Lacasa M. Intestinal apolipoprotein A-IV gene transcription is controlled by two hormone-responsive elements: A role for hepatic nuclear factor-4 isoforms. Mol. Endocrinol. 2005;19:2320–2334. doi: 10.1210/me.2004-0462. [DOI] [PubMed] [Google Scholar]
- 52.Clark K.L., Halay E.D., Lai E., Burley S.K. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature. 1993;364:412–420. doi: 10.1038/364412a0. [DOI] [PubMed] [Google Scholar]
- 53.Lee C.S., Friedman J.R., Fulmer J.T., Kaestner K.H. The initiation of liver development is dependent on Foxa transcription factors. Nature. 2005;435:944–947. doi: 10.1038/nature03649. [DOI] [PubMed] [Google Scholar]
- 54.Schrem H., Klempnauer J., Borlak J. Liver-enriched transcription factors in liver function and development. Part I: the hepatocyte nuclear factor network and liver-specific gene expression. Pharmacol. Rev. 2002;54:129–158. doi: 10.1124/pr.54.1.129. [DOI] [PubMed] [Google Scholar]
- 55.Heidari N., Phanstiel D.H., He C., Grubert F., Jahanbani F., Kasowski M., Zhang M.Q., Snyder M.P. Genome-wide map of regulatory interactions in the human genome. Genome Res. 2014;24:1905–1917. doi: 10.1101/gr.176586.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith C.E., Follis J.L., Nettleton J.A., Foy M., Wu J.H., Ma Y., Tanaka T., Manichakul A.W., Wu H., Chu A.Y., et al. Dietary fatty acids modulate associations between genetic variants and circulating fatty acids in plasma and erythrocyte membranes: Meta-analysis of nine studies in the CHARGE consortium. Mol. Nutr. Food Res. 2015;59:1373–1383. doi: 10.1002/mnfr.201400734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.