

Abstract
α-Allenols are attractive and versatile compounds whose preparation can be a nontrivial task. In this Letter, we provide a method for the prompt synthesis of substituted α-allenols via a catalytic cross-coupling reaction which makes use of a nontoxic and cost-effective iron catalyst. The catalyst loading is typically as low as 1–5 mol %. The mild reaction conditions (−20 °C) and the short reaction time (15 min) allow for the presence of a variety of functional groups. Moreover, the reaction was shown to be scalable up to gram-scale and the propargyl substrates are readily accessible by a one-pot synthesis.
Keywords: iron catalysis, cross coupling, allenol, allenyl acetate, Grignard reagents
Transition metal-catalyzed cross-coupling reactions have been established as one of the most important tools in organic chemistry for the formation of C–C bonds of all orders, as well as C-heteroatom bonds. The dominant metals in this field are palladium,1 copper,2 and nickel,3 whereas other transition metals such as iron are less explored. However, iron is an inexpensive, nontoxic and environmentally benign transition metal that in the past few years has gained new impetus.4,5 It has also been found that iron works well for cross coupling of sp3-substrates including both nucleophilic and electrophilic reaction partners.6
For several years our research group has been interested in allene-chemistry, in particular, oxidative palladium-catalyzed carbocyclizations of enallenes,7 allenynes,8 arylallenes,9 and aza-enallenes10 as well as the dynamic kinetic resolution of allenols.11 Recently our research group published an efficient one-pot method for the enzyme- and ruthenium-catalyzed enantioselective transformation of α-allenols into 2,3-dihydrofurans. The reaction proceeds with excellent enantioselectivity by an enzymatic kinetic resolution and a subsequent ruthenium-catalyzed cycloisomerization.12 α-Allenols are also desired substrates for many other transformations such as the gold-,13 silver-,14 ruthenium-,15 and palladium-catalyzed16 cyclization or the palladium-catalyzed dibromination.17 They are also important building-blocks in total synthesis of natural products, such as β-carboline alkaloids (−)-isocyclocapitelline and (−)-isochrysotricine18 or amphidinolide polyketide alkaloids amphidinolide X and Y.19
α-Allenols are conventionally synthesized by the Crabbé reaction,20 which for terminal allenes is reliable but can be very substrate dependent for internal allenes.21 The latter procedure commonly involves the semistoichiometric use of metals such as copper21b or cadmium.22 However, α-allenols, tetra-substituted with respect to the allene moiety are not accessible via the Crabbé reaction. These types of α-allenols have to be prepared via transition metal-catalyzed cross-coupling, mostly via copper catalysis, utilizing alkynyl oxiranes23 or dioxolanones24 as substrates. In the case of propargyl oxiranes, there is also an iron-catalyzed variant.23d However, the preparation of these highly reactive substrates involves a multistep route.19
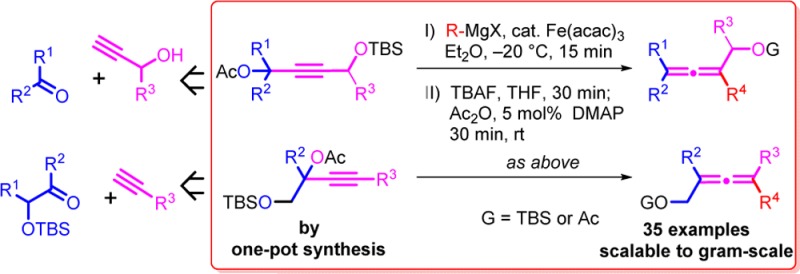
In light of the extensive use of α-allenols and the limited amount of methods for their preparation, there is a demand for novel and complementary methods. On the basis of our previous work,5 we now present a novel synthesis of highly substituted α-allenols via iron-catalyzed cross-coupling of propargyl substrates with Grignard reagents (Figure 1).
Figure 1.
Iron-catalyzed cross-coupling reaction for the synthesis of highly substituted α-allenols. Typical reaction conditions: 1 equiv of substrate (0.1 M) and 1 mol % Fe(acac)3 in Et2O treated with 1.25 equiv of R4 MgX for 5 min at −20 °C and stirred for 10 min.
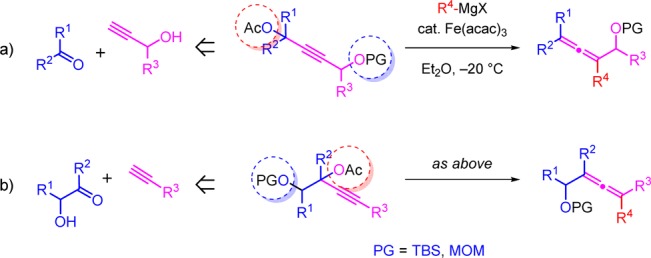
The reaction conditions are typically mild (ether, – 20 °C, 15 min), and the reaction requires a low catalyst loading of 1–5 mol % of Fe(acac)3. With this strategy, the hydroxyl group can be introduced on the opposite side of the alkyne to that of the acetate leaving group (Figure 1a) or on the same side as the acetate leaving group (Figure 1b). The approach therefore allows for the synthesis of a wide variety of differently substituted allenols (Figure 1). The propargyl substrates are in turn prepared in one pot from propargyl alcohols or terminal acetylenes and aldehydes or ketones.25
The obvious benefit of the herein described iron-catalyzed synthesis is the simple accessibility of the starting materials, the little preparative effort, the cost effectiveness, the nontoxic catalyst, and the mild conditions, allowing the swift preparation of a wide range of substituted allenols.
We started our investigations with the propargyl substrates 1 bearing the protected hydroxyl group on the opposite side of the alkyne to that of the acetate leaving group (Table 1).
Table 1. Preparation of α-Allenols from Propargyl Substrates with Acetate and Protected Hydroxyl on Opposite Sides of the Alkynea.
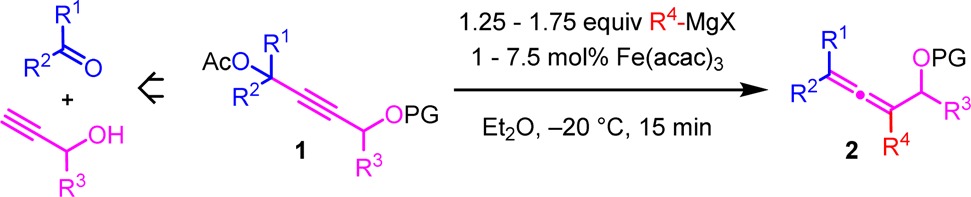
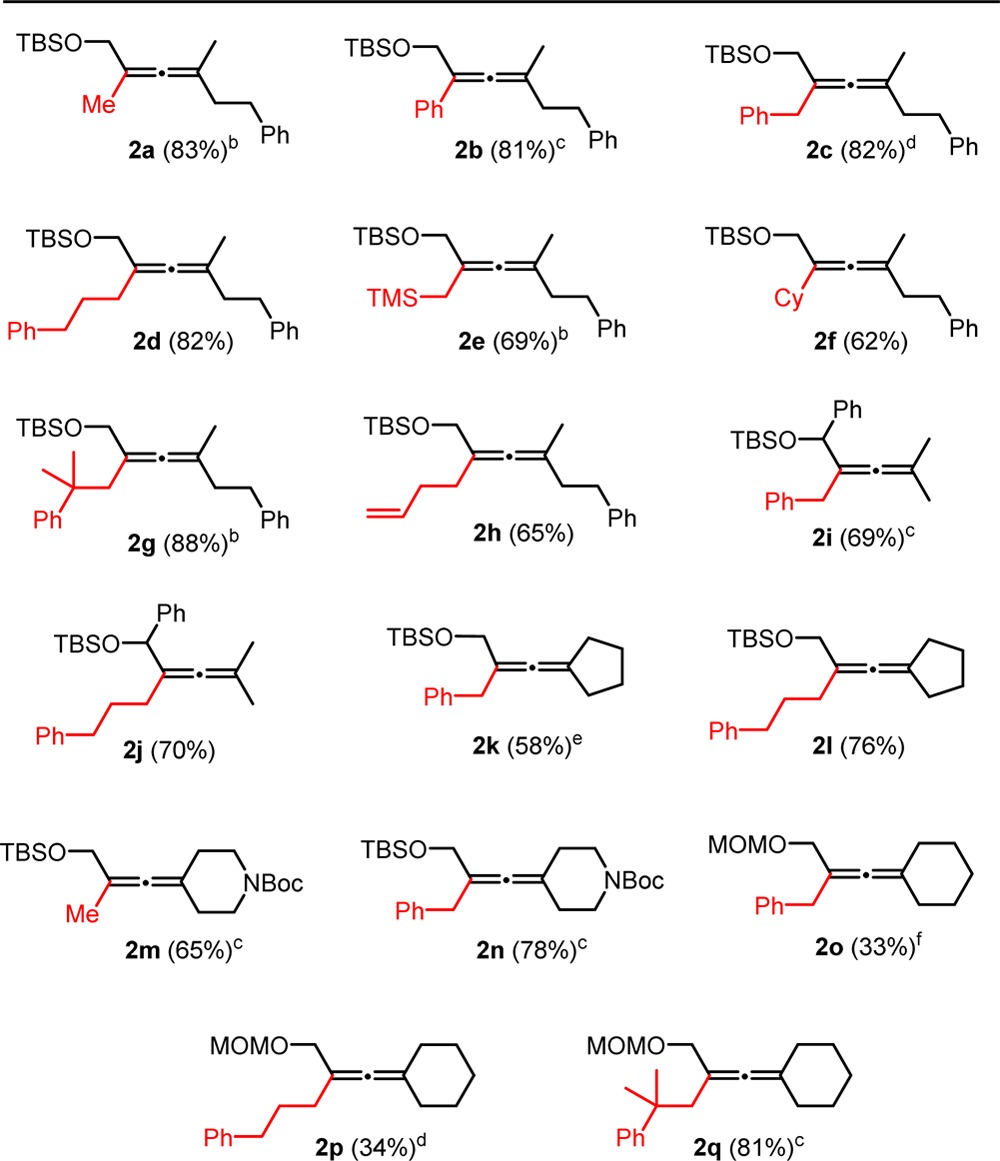
Isolated yields. Conditions: 0.1 M solution of substrate 1 (1 equiv), R4 MgX (1.25 equiv), and 1 mol % of Fe(acac)3 in Et2O, addition of R4 MgX for 5 and 10 min reaction time.
R4 MgX (1.5 equiv), 5 mol % of Fe(acac)3.
R4 MgX (1.75 equiv), 7.5 mol % of Fe(acac)3.
R4 MgX (1.75 equiv), 5 mol % of Fe(acac)3.
5 mol % of Fe(acac)3.
R4 MgX (2 equiv), 7.5 mol % of Fe(acac)3.
As protecting group, a silyl moiety, tert-butyldimethylsilyl (TBS) was chosen because of its orthogonal reactivity to most other functional groups. The less-hindered trimethylsilyl (TMS) as protecting group was unsuitable for the one-pot preparation of the substrate as well as the allene formation. In this way, the otherwise acid-sensitive26 allenols can be easily formed in situ and for example be directly transformed to acetates (vide infra). The Grignard reagents can be divided into two main categories: those with and those without β-hydrogens. The β-H-containing magnesium organyls require only 1 mol % of Fe(acac)3, whereas the others require 5 mol % or more of the catalyst. This difference is most likely due to different mechanistic pathways, as previous reports suggest.6c To reflect the two categories of Grignard reagents, mainly benzyl and (3-phenylpropyl)magnesium halides have been utilized, which also add enough molecular weight to the final product to avoid volatility issues. The yields are generally high, and it is interesting to note that sterically congested Grignard reagents such as (2-methyl-2-phenylpropyl)magnesium chloride give the highest yields (Table 1: 2g and 2q). The mild conditions tolerate functional groups such as silyl ethers, carbamates, and acetales (Table 1: 2a–2n, 2m and 2n, 2o–2q), as well as a variety of Grignard reagents such as methyl, alkyl, phenyl, benzyl, homoallyl and (trimethylsilyl)methyl magnesium organyls (Table 1 e.g.: 2m, 2f, 2b, 2c, 2h, 2e). The low yield in case of the MOM-derivatives 2o and 2p are attributed to a moderate conversion of around 60 percent.
Another variation is when substrates have the protected hydroxyl group on the same side of the alkyne as that of the leaving group (Table 2: 3). This not only extends the scope of accessible products but also enables the preparations of allenols with a less-substituted allene moiety (Table 2: 4a–4d, 4g, and 4h). For tri- and tetrasubstituted allenes, the yields are comparable to those in Table 1. However, the yields rapidly decrease when going to less-substituted allenes. To explore the tolerance toward halogens, a chloride was introduced in the substrate. The chlorinated substrate showed good tolerance toward magnesium organyls lacking β-hydrogens (Table 1: 4e). However, in the case of β-hydrogen-containing magnesium organyls, the chloro substrate proceeded with moderate yield (Table 2: 4f).
Table 2. Allenols from Propargyl Substrates with Acetate and Protected Hydroxyl on the Same Side of the Alkynea.

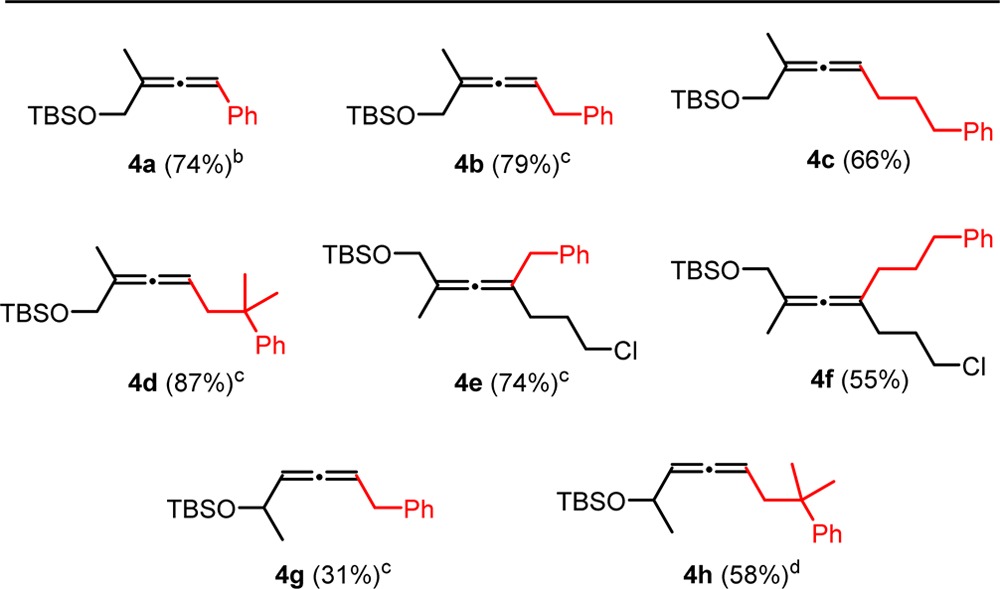
Isolated yields. Conditions: 0.1 M solution of substrate 3 (1 equiv), R4 MgX (1.25 equiv), and 1 mol % of Fe(acac)3 in Et2O, addition of R4 MgX for 5 and 10 min reaction time.
R4 MgX (1.75 equiv), 7.5 mol % of Fe(acac)3.
5 mol % of Fe(acac)3.
R4 MgX (2.0 equiv), 5 mol % of Fe(acac)3.
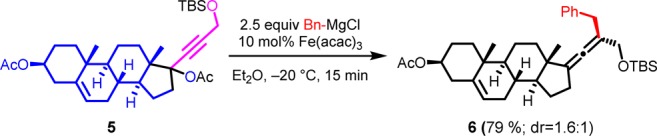
The iron-catalyzed cross-coupling reaction was also successfully applied to more complex molecules such as steroids, leading to a steroidal allene in good yield (Scheme 1). This strategy shows potential to be used in order to gain rapid access to a variety of functionalized steroidal compounds interesting to medicinal chemistry or as precursors for further transformations.
Scheme 1. Functionalization of Steroidal Compounds.
The protocol was extended to the transformation of the silyl protected alcohol into an acetate, which in turn is a valuable substrate for transition metal-catalyzed transformations. This transformation can be achieved in one step via a Lewis acid-catalyzed deprotection-esterification sequence with acetic anhydride (Ac2O). However, this sequence did not show very high yields and was expected to be incompatible with Lewis acid labile groups such as carbamates. Hence, we decided to choose the two-step route by deprotection of the crude allenol with tetrabutylammonium fluoride (TBAF) and subsequent esterification by Ac2O catalyzed by the Steglich-base (DMAP), subsequent to the cross-coupling reaction, without purification of the intermediate TBS-ether nor the alcohol (Table 3). Furthermore, to demonstrate the scalability of the herein described iron-catalyzed synthesis of substituted allenols and subsequent transformation to acetates, the substrates have been used on gram-scale, which worked well for both type of substrates (7 and 8, Table 3). The procedure allows the presence of ester or carbamate functions to give 10e or 10f, respectively (Table 3) and show good yields except for the butyl-substituted allene which might in this particular case be mainly attributed to the higher volatility of this compound (8b, Table 3).
Table 3. Gram-Scale Synthesis of Allenyl Acetates from Propargyl Substratesa.
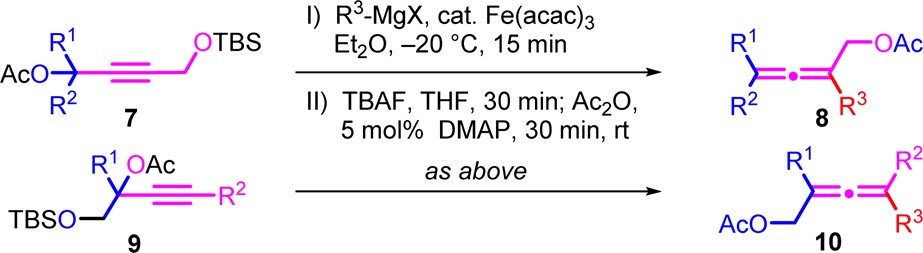
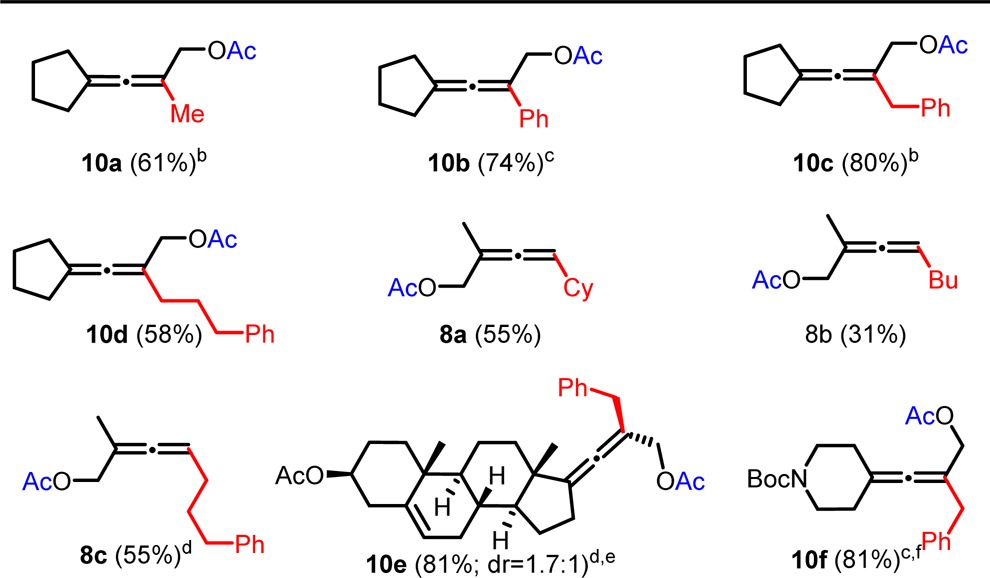
Isolated yields. Conditions: 0.1 M solution of substrate 7 or 9 (1.25–2.25 g, 1 equiv), R4 MgX (1.25 equiv), and 1 mol % of Fe(acac)3 in Et2O, addition of R4 MgX for 5 and 10 min reaction time.
5 mol % of Fe(acac)3, R4 MgX (1.5 equiv).
7.5 mol % of Fe(acac)3, R4 MgX (1.75 equiv).
half gram-scale.
10 mol % of Fe(acac)3, BnMgX (2.5 equiv).
by 2 steps.
In conclusion, we have developed a mild and facile synthesis of highly substituted α-allenols by the utility of a nontoxic and cost-effective iron catalyst with loadings as low as 1 mol % at a reaction temperature of only −20 °C. The iron-catalyzed synthesis of allenols can easily be scaled up to gram-scale, it shows convenient functional group tolerance, and it allows for the employment of a variety of Grignard reagents. In addition, more complex molecules such as steroids can be readily functionalized, which is of interest in medicinal chemistry.
Acknowledgments
Financial support from the European Research Council (ERC AdG 247014), the Swedish Research Council (621-2013-4653), and the Berzelii Centre EXSELENT is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.6b02114.
Experimental procedures and compound characterization data for all new compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Kapdi A. R.; Prajapati D. RSC Adv. 2014, 4, 41245. 10.1039/C4RA07895K. [DOI] [Google Scholar]; b Nicolaou K. C.; Bulger P. G.; Sarlah D. Angew. Chem., Int. Ed. 2005, 44, 4442. 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]; c Johansson Seechurn C. C. C.; Kitching M. O.; Colacot T. J.; Snieckus V. Angew. Chem., Int. Ed. 2012, 51, 5062. 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- a Beletskaya I. P.; Cheprakov A. V. Coord. Chem. Rev. 2004, 248, 2337. 10.1016/j.ccr.2004.09.014. [DOI] [Google Scholar]; b Evano G.; Theunissen C.; Pradal A. Nat. Prod. Rep. 2013, 30, 1467. 10.1039/c3np70071b. [DOI] [PubMed] [Google Scholar]
- a Standley E. A.; Tasker S. Z.; Jensen K. L.; Jamison T. F. Acc. Chem. Res. 2015, 48, 1503. 10.1021/acs.accounts.5b00064. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tasker S. Z.; Standley E. A.; Jamison T. F. Nature 2014, 509, 299. 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cornella J.; Edwards J. T.; Qin T.; Kawamura S.; Wang J.; Pan C.-M.; Gianatassio R.; Schmidt M.; Eastgate M. D.; Baran P. S. J. Am. Chem. Soc. 2016, 138, 2174. 10.1021/jacs.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sherry B. D.; Fürstner A. Acc. Chem. Res. 2008, 41, 1500. 10.1021/ar800039x. [DOI] [PubMed] [Google Scholar]; b Correa A.; Mancheño O. G.; Bolm C. Chem. Soc. Rev. 2008, 37, 1108. 10.1039/b801794h. [DOI] [PubMed] [Google Scholar]; c Legros J.; Figadère B. Nat. Prod. Rep. 2015, 32, 1541. 10.1039/C5NP00059A. [DOI] [PubMed] [Google Scholar]; d Bedford R. B. Acc. Chem. Res. 2015, 48, 1485. 10.1021/acs.accounts.5b00042. [DOI] [PubMed] [Google Scholar]; e Nakamura E.; Hatakeyama T.; Ito S.; Ishizuka K.; Ilies L.; Nakamura M.. Iron-Catalyzed Cross-Coupling Reactions. In Organic Reactions, Vol. 83; Denmark S. E., Ed.; John Wiley & Sons: Hoboken, NJ, 2014; pp 1–210. [Google Scholar]
- Kessler S. N.; Bäckvall J.-E. Angew. Chem., Int. Ed. 2016, 55, 3734. 10.1002/anie.201511139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fürstner A.; Leitner A.; Méndez M.; Krause H. J. Am. Chem. Soc. 2002, 124, 13856. 10.1021/ja027190t. [DOI] [PubMed] [Google Scholar]; b Nakamura M.; Matsuo K.; Ito S.; Nakamura E. J. Am. Chem. Soc. 2004, 126, 3686. 10.1021/ja049744t. [DOI] [PubMed] [Google Scholar]; c Fürstner A.; Martin R.; Krause H.; Seidel G.; Goddard R.; Lehmann C. W. J. Am. Chem. Soc. 2008, 130, 8773. 10.1021/ja801466t. [DOI] [PubMed] [Google Scholar]; d Adams C. J.; Bedford R. B.; Carter E.; Gower N. J.; Haddow M. F.; Harvey J. N.; Huwe M.; Cartes M. Á.; Mansell S. M.; Mendo-za C.; Murphy D. M.; Neeve E. C.; Nunn J. J. Am. Chem. Soc. 2012, 134, 10333. 10.1021/ja303250t. [DOI] [PubMed] [Google Scholar]; e Jin M.; Adak L.; Nakamura M. J. Am. Chem. Soc. 2015, 137, 7128. 10.1021/jacs.5b02277. [DOI] [PubMed] [Google Scholar]; f Gärtner D.; Stein A. L.; Grupe S.; Arp J.; Jacobi von Wangelin A. Angew. Chem., Int. Ed. 2015, 54, 10545. 10.1002/anie.201504524. [DOI] [PubMed] [Google Scholar]; g Bekhradnia A.; Norrby P.-O. Dalton Trans. 2015, 44, 3959. 10.1039/C4DT03491K. [DOI] [PubMed] [Google Scholar]
- a Franzén J.; Löfstedt J.; Dorange I.; Bäckvall J.-E. J. Am. Chem. Soc. 2002, 124, 11246. 10.1021/ja026150m. [DOI] [PubMed] [Google Scholar]; b Jiang T.; Bartholomeyzik T.; Mazuela J.; Willersinn J.; Bäckvall J.-E. Angew. Chem., Int. Ed. 2015, 54, 6024. 10.1002/anie.201501048. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhu C.; Yang B.; Jiang T.; Bäckvall J.-E. Angew. Chem., Int. Ed. 2015, 54, 9066. 10.1002/anie.201502924. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zhu C.; Yang B.; Bäckvall J.-E. J. Am. Chem. Soc. 2015, 137, 11868. 10.1021/jacs.5b06828. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Bartholomeyzik T.; Bäckvall J.-E. Angew. Chem., Int. Ed. 2013, 52, 6283–6287. 10.1002/anie.201301167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazuela J.; Banerjee D.; Bäckvall J.-E. J. Am. Chem. Soc. 2015, 137, 9559. 10.1021/jacs.5b06068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson A. K. Å.; Bäckvall J.-E. Angew. Chem., Int. Ed. 2010, 49, 4624. 10.1002/anie.201000726. [DOI] [PubMed] [Google Scholar]
- Deska J.; del Pozo Ochoa C.; Bäckvall J.-E. Chem. - Eur. J. 2010, 16, 4447. 10.1002/chem.201000301. [DOI] [PubMed] [Google Scholar]
- Yang B.; Zhu C.; Qiu Y.; Bäckvall J.-E. Angew. Chem., Int. Ed. 2016, 55, 5568. 10.1002/anie.201601505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause N.; Winter C. Chem. Rev. 2011, 111, 1994. 10.1021/cr1004088. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zheng K.; Hong R. J. Am. Chem. Soc. 2012, 134, 4096. 10.1021/ja300453u. [DOI] [PubMed] [Google Scholar]
- a Yoneda E.; Zhang S.-W.; Zhou D.-Y.; Onitsuka K.; Takahashi S. J. Org. Chem. 2003, 68, 8571. 10.1021/jo0350615. [DOI] [PubMed] [Google Scholar]; b Yoneda E.; Kaneko T.; Zhang S.-W.; Onitsuka K.; Takahashi S. Org. Lett. 2000, 2, 441. 10.1021/ol990377d. [DOI] [PubMed] [Google Scholar]
- Ma S.; Zhao S. J. Am. Chem. Soc. 1999, 121, 7943. 10.1021/ja9904888. [DOI] [Google Scholar]
- Horváth A.; Bäckvall J.-E. J. Org. Chem. 2001, 66, 8120. 10.1021/jo015950x. [DOI] [PubMed] [Google Scholar]
- Volz F.; Krause N. Org. Biomol. Chem. 2007, 5, 1519. 10.1039/B703995F. [DOI] [PubMed] [Google Scholar]
- Fürstner A.; Kattnig E.; Lepage O. J. Am. Chem. Soc. 2006, 128, 9194. 10.1021/ja061918e. [DOI] [PubMed] [Google Scholar]
- a Rona P.; Crabbé P. J. Am. Chem. Soc. 1968, 90, 4733. 10.1021/ja01019a043. [DOI] [Google Scholar]; b Rona P.; Crabbé P. J. Am. Chem. Soc. 1969, 91, 3289. 10.1021/ja01040a033. [DOI] [Google Scholar]
- a Yu S.; Ma S. Chem. Commun. 2011, 47, 5384. 10.1039/c0cc05640e. [DOI] [PubMed] [Google Scholar]; b Kuang J.; Luo H.; Ma S. Adv. Synth. Catal. 2012, 354, 933. 10.1002/adsc.201100772. [DOI] [Google Scholar]
- Tang X.; Zhu C.; Cao T.; Kuang J.; Lin W.; Ni S.; Zhang J.; Ma S. Nat. Commun. 2013, 4, 2450. 10.1038/ncomms3450. [DOI] [PubMed] [Google Scholar]
- a Herr R. W.; Wieland D. M.; Johnson C. R. J. Am. Chem. Soc. 1970, 92, 3813. 10.1021/ja00715a060. [DOI] [Google Scholar]; b de Montellano P. R. O. J. Chem. Soc., Chem. Commun. 1973, (19), 709. 10.1039/c39730000709. [DOI] [Google Scholar]; c Bertozzi F.; Crotti P.; Macchia F.; Pineschi M.; Arnold A.; Ferin-ga B. L. Tetrahedron Lett. 1999, 40, 4893. 10.1016/S0040-4039(99)00905-3. [DOI] [Google Scholar]; d Fürstner A.; Méndez M. Angew. Chem., Int. Ed. 2003, 42, 5355. 10.1002/anie.200352441. [DOI] [PubMed] [Google Scholar]
- Tang X.; Woodward S.; Krause N. Eur. J. Org. Chem. 2009, 2009, 2836. 10.1002/ejoc.200900226. [DOI] [Google Scholar]
- In Figure 1: for (a), ketones are commercially available; for (b), the aldehydes and ketones were prepared by one or two steps (see SI).
- Alcaide B.; Almendros P.; Cembellín S.; Martínez del Campo T. Adv. Synth. Catal. 2015, 357, 1070. 10.1002/adsc.201400928. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.