

Abstract
New technologies can make previously invisible phenomena visible. Nowhere is this more obvious than in the field of light microscopy. Beginning with the observation of “animalcules” by Antonie van Leeuwenhoek, when he figured out how to achieve high magnification by shaping lenses, microscopy has advanced to this day by a continued march of discoveries driven by technical innovations. Recent advances in single-molecule-based technologies have achieved unprecedented resolution, and were the basis of the Nobel prize in Chemistry in 2014. In this article, we focus on developments in camera technologies and associated image processing that have been a major driver of technical innovations in light microscopy. We describe five types of developments in camera technology: video-based analog contrast enhancement, charge-coupled devices (CCDs), intensified sensors, electron multiplying gain, and scientific complementary metal-oxide-semiconductor cameras, which, together, have had major impacts in light microscopy.
Video-Based Analog Contrast Enhancement
Technology for television broadcasts became available during the 1930s. The early developers of this technology were well aware of the potential usefulness of microscopy. Vladimir Zworykin, a Russian-born scientist who made pivotal contributions to the development of the television, in his landmark paper wrote, “Wide possibilities appear in application of such tubes in many fields as a substitute for the human eye, or for the observation of phenomena at present completely hidden from the eye, as in the case of the ultraviolet microscope” (Zworykin, 1934/1997). Over the following decades, while television technology became more mature and more widely accessible, its use in microscopes often appeared to be limited to classroom demonstrations. Shinya Inoué, a Japanese scientist whose decisive contributions to the field of cytoskeleton dynamics included ground-breaking new microscope technologies, described “the exciting displays of a giant amoeba, twice my height, crawling up on the auditorium screen at Princeton as I participated in a demonstration of the RCA projection video system to which we had coupled a phase-contrast microscope” (Inoué, 1986).
While teaching a summer optical microscopy course at the Marine Biological Laboratory in Woods Hole, Robert Day Allen, his wife, Nina Strömgen Allen, and Jeff Travis attached a video camera to a microscope setup for differential interference contrast (DIC), and discovered that sub-resolution structures could be made visible that were not visible when using the eye-pieces or on film (Davidson, 2015). To view samples by eye, it was necessary to reduce the light intensity and to close down the iris diaphragm to increase apparent contrast, at the expense of reduced resolution. The video equipment enabled the use of a fully opened iris diaphragm and all available light, because one could now change the brightness and contrast and display the full resolution of the optical system on the television screen (Allen et al., 1981).
Independently, yet at a similar time, Shinya Inoué used video cameras with auto-gain and auto-black level controls, which helped to improve the quality of both polarization and DIC microscope images (Inoué, 1981). The Allens subsequently explored the use of frame memory to store a background image and continuously subtract it from a live video stream, further refining image quality (Allen and Allen, 1983). In practice, this background image was collected by slightly defocusing the microscope, accumulating many images, and averaging them (see Fig. 1 and Salmon, 1995). These new technologies enabled visualization of transport of vesicles in the squid giant axon (Allen et al., 1982), followed by visualization of such transport in extracts of the squid giant axon (Brady et al., 1985; Vale et al., 1985a), leading to the development of microscopy-based assays for motor activity (Fig. 2) and subsequent purification of the motor protein, kinesin (Vale et al., 1985b, c).
Figure 1.
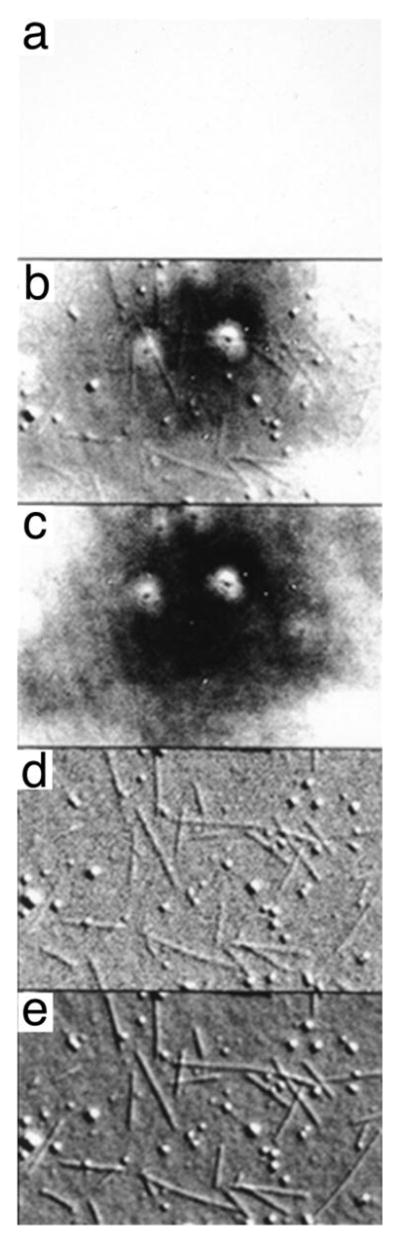
Example of the work flow in video-enhanced differential interference contrast (DIC) microscopy. The sample consists of taxol-stablized microtubules attached to a coverslip. A Hamamatsu C2400 Newvicon camera and Argus 10 digital image processor (Hamamatsu Photonics, Inc., Bridgewater, NJ) were used. (a) Raw image, as seen through the eyepiece, exhibiting very low contrast. (b) Video image after analog contrast enhancement. (c) Background image created by defocusing the microscope. (d) Video image after background subtraction, addition of a bias gray level, and contrast enhancement. (e) Video image after averaging four frames to reduce noise. (Reproduced, with modifications, from fig. 3 of Salmon, E. D., 1995, Trends Cell Biol.5: 154 –158, with permission from Elsevier.)
Figure 2.
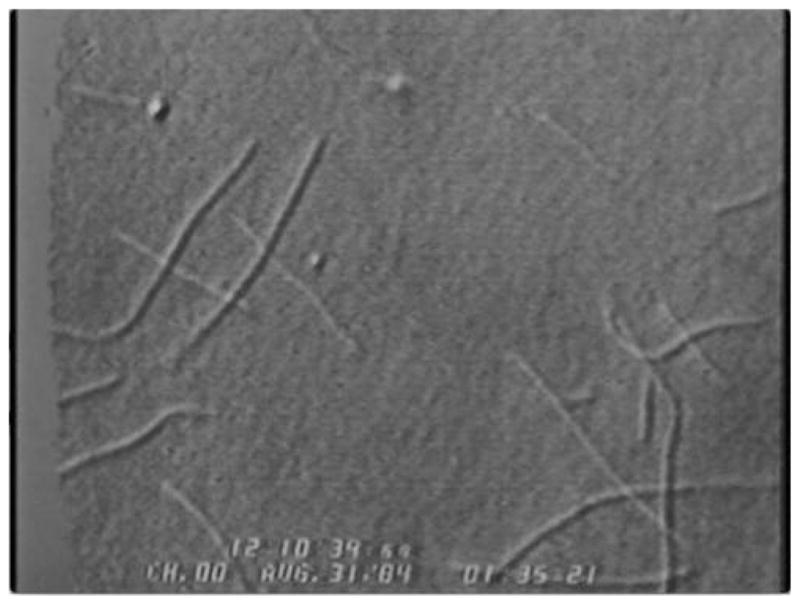
Differential interference contrast (DIC) image of microtu-bules moving on a glass coverslip in the presence of soluble extract from the squid giant axon. Shown is a still image of a video clip obtained with a Newvicon video camera, using brightness-contrast optimization and background subtraction, as detailed in the main text (see also Vale et al., 1985c).
Until the development of the charge-coupled device (see next section), video cameras consisted of vacuum cathode ray tubes with a photosensitive front plate (target). These devices operated by scanning the cathode ray over the target while the resulting current varied with the amount of light “seen” by the scanned position. The analog signal (which most often followed the National Television System Committee (NTSC) standard of 30 frames per second and 525 lines) was displayed on a video screen. To store the data, either a picture of the screen was taken with a photo camera and silver emulsion-based film, or the video signal was stored on tape. Over the years, the design of both video cameras and recording equipment improved dramatically; cost reduction was driven by development for the consumer market. (In fact, to this day microscope camera technology development benefits enormously from industrial development aimed at the consumer electronics market.) However, the physical design of a vacuum tube and video tape recording device places limits on the minimum size possible, an important consideration motivating development of alternative technologies. Initially, digital processing units for background subtraction and averaging at video rate were home-built; but, later, commercial versions became available. The last widely used analog, a vacuum tube-based system used in microscopes, was probably the Hamamatsu Newvicon camera in combination with the Hamamatsu Argus image processor (Hamamatsu Photonics, Inc., Bridge-water, NJ; see Fig. 1 for an example of the workflow typically used with that equipment).
CCD Technology
Development of charge-coupled devices (CCDs) was driven by the desire to reduce the physical size of video cameras used in television stations to encourage their use by the consumer market. The CCD was invented in 1969 by Willard Boyle and George E. Smith at AT&T’s Bell Labs, an invention for which they received a half-share of the Nobel Prize in Physics in 2009. The essence of the invention was its ability to transfer charge from one area storage capacitor on the surface of a semiconductor to the next. In a CCD camera, these capacitors are exposed to light and they collect photoelectrons through the photoelectric effect––in which a photon is converted into an electron. By shifting these charges along the surface of the detector to a readout node, the content of each storage capacitor can be determined and translated into a voltage that is equivalent to the amount of light originally hitting that area.
Various companies were instrumental in the development of the CCD, including Sony (for a short history of CCD camera development within Sony, see Sony Corporate Info, 2016) and Fairchild (Kodak developed the first digital camera based on a Fairchild 100 × 100-pixel sensor in 1975). These efforts quickly led to the wide availability of consumer-grade CCD cameras.
Even after CCD sensors had become technically superior to the older, tube-based sensors, it took time for everyone to transition from the tube-based camera technology and associated hardware, such as matching video recorders and image analyzers. One of the first descriptions of the use of a CCD sensor on a microscope can be found in Roos and Brady (1982), who attached a 1728-pixel CCD (which appears to be a linear CCD) to a standard microscope setup for Nomarski-type DIC microscopy. The authors built their own circuitry to convert the analog signals from the CCD to digital signals, which were stored in a micro-computer with 192 KB of memory. They were able to measure the length of sarcomeres in isolated heart cells at a time resolution of ~6 ms and a spatial precision of ~50 nm. Shinya Inoué’s book, Video Microscopy (1986), mentions CCDs as an alternative to tube-based sensors, but clearly there were very few CCDs in use on microscopes at that time. Remarkably, one of the first attempts to use deconvolution, an image-processing technique to remove out-of-focus “blur,” used photographic images that were digitized by a densitometer rather than a camera (Agard and Sedat, 1980). It was only in 1987 that the same group published a paper on the use of a CCD camera (Hiraoka et al., 1987). In that publication, the work of John A. Connor at AT&T’s Bell Labs is mentioned as the first use of the CCD in microscope imaging. Connor (1986) used a 320 × 512-pixel CCD camera from Photo-metrics (Tucson, AZ) for calcium imaging, using the fluorescent dye, Fura-2, as a reporter in rat embryonic nerve cells. Hiraoka et al. (1987) highlighted the advantages of the CCD technology, which included extraordinary sensitivity and numerical accuracy, and noted its main downside, slow speed. At the time, the readout speed was 50,000 pixels/s, and readout of the 1024 × 600-pixel camera took 13 s (speeds of 20 MHz are now commonplace, and would result in a 31-ms readout time for this particular camera).
One of the advantages of CCD cameras was their ability to expose the array for a defined amount of time. Rather than frame-averaging multiple short-exposure images in a digital video buffer (as was needed with a tube-based sensor), one could simply accumulate charge on the chip itself. This allowed for the recording of very sensitive measurements of dim, non-moving samples. As a result, CCD cameras became the detector of choice for (fixed) fluorescent samples during the 1990s.
Because CCDs are built using silicon, they inherit its excellent photon absorption properties at visible wavelengths. A very important parameter of camera performance is the fraction of light (photons) hitting the sensor that is converted into signal (in this case, photoelectrons). Hence, the quantum efficiency (QE) is expressed as the percentage of photoelectrons resulting from a given number of photons hitting the sensor. Even though the QE of crystalline silicon itself can approach 100%, the overlying electrodes and other structures reduce light absorption and, therefore, QE, especially at lower wavelengths (i.e., towards the blue range of the spectrum). One trick to increase the QE is to turn the sensor around so that its back faces the light source and to etch the silicon to a thin layer (10 –15 μm). Such back-thinned CCD sensors can have a QE of ~95% at certain wavelengths. The QE of charge-coupled devices tends to peak at wavelengths of around 550 nm, and drop off towards the red, because photons at wavelengths of 1100 nm are not energetic enough to elicit an electron in the silicon. Other tricks have been employed to improve the QE and its spectral properties, such as coating with fluorescent plastics to enhance the QE at lower wavelengths, changing the electrode material, or using micro-lenses that focus light on the most sensitive parts of the sensor. The Sony ICX285 sensor, which is still in use today, uses micro-lenses, achieving a QE of about 65% from ~450 –550 nm.
Concomitant with the advent of the CCD camera in microscope imaging was the widespread availability of desktop computers. Computers not only provided a means for digital storage of images, but also enabled image processing and analysis. Even though these desktop computers at first were unable to keep up with the data generated by a video rate camera (for many years, it was normal to store video on tape and digitize only sections of relevance), they were ideal for storage of images from the relatively slow CCD cameras. For data to enter the computer, analog-to-digital conversion (AD) is needed. AD conversion used to be a complicated step that took place in dedicated hardware or, later, in a frame grabber board or device, but nowadays it is often carried out in the camera itself (which provides digitized output). The influence of computers on microscopy cannot be overstated. Not only are computers now the main recording device, they also enable image reconstruction approaches––such as deconvolution, structured illumination, and super-resolution microscopy––that use the raw images to create realistic models for the microscopic object with resolutions that can be far greater than the original data. These models (or the raw data) can be viewed in many different ways, such as through 3D reconstructions, which are impossible to generate without computers. Importantly, computers also greatly facilitate extraction of quantitative information from the microscope data.
Intensified Sensors
The development of image intensifiers, which amplify the brightness of the image before it reaches the sensor or eye, started early in the twentieth century. These devices consisted of a photocathode that converted photons into electrons, followed by an electron amplification mechanism, and, finally, a layer that converted electrons back into an image. The earliest image intensifiers were developed in the 1930s by Gilles Holst, who was working for Philips in the Netherlands (Morton, 1964). His intensifier consisted of a photocathode upon which an image was projected in close proximity to a fluorescent screen. A voltage differential of several thousand volts accelerated electrons emitted from the photocathode, directing them onto a phosphor screen. The high-energy electrons each produced many photons in the phosphor screen, thereby amplifying the signal. By cascading intensifiers, the signal can be intensified significantly. This concept was behind the so-called Gen I image-intensifiers.
The material of the photocathode determines the wavelength detected by the intensifier. Military applications required high sensitivity at infrared wavelengths, driving much of the early intensifier development; however, intensifiers can be built for other wavelengths, including X-rays.
Most intensifier designs over the last forty years or so (i.e., Gen II and beyond) include a micro-channel plate consisting of a bundle of thousands of small glass fibers bordered at the entrance and exit by nickel chrome electrodes. A high-voltage differential between the electrodes accelerates electrons into the glass fibers, and collisions with the wall elicit many more electrons, multiplying electrons coming from the photocathode. Finally, the amplified electrons from the micro-channel plate are projected onto a phosphor screen.
The sensitivity of an intensifier is ultimately determined by the quantum efficiency (QE) of the photocathode, and––despite decades of developments––it still lags significantly behind the QE of silicon-based sensors (i.e., the QE of the photocathode of a modern intensified camera peaks at around 50% in the visible region).
Intensifiers must be coupled to a camera in order to record images. For instance, intensifiers were placed in front of vidicon tubes either by fiber-optic coupling or by using lens systems in intensified vidicon cameras. Alternatively, intensifiers were built into the vacuum imaging tube itself. Probably the most well-known implementation of such a design is the silicon-intensifier target (SIT) camera. In this design, electrons from the photocathode are accelerated onto a silicon target made up of p-n junction silicon diodes. Each high-energy electron generates a large number of electron-hole pairs, which are subsequently detected by a scanning electron beam that generates the signal current. The SIT camera had a sensitivity that was several hundredfold higher than that of standard vidicon tubes (Inoué, 1986). SIT cameras were a common instrument in the 1980s and 1990s for low-light imaging. For instance, our lab used a SIT camera for imaging of sliding microtubules, using dark-field microscopy (e.g., Vale and Toyoshima, 1988) and other low-light applications such as fluorescence imaging. The John W. Sedat group used SIT cameras, at least for some time during the transition from film to CCD cameras, in their work on determining the spatial organization of DNA in the Drosophila nucleus (Gruenbaum et al., 1984).
When charge-coupled devices began to replace vidicon tubes as the sensor of choice, intensifiers were coupled to CCD or complementary metal-oxide-semiconductor (CMOS) sensors, either with a lens or by using fiber-optic bonding between the phosphor plate and the solid-state sensor. Such “ICCD” cameras can be quite compact and produce images from very low-light scenes at astonishing rates (for current commercial offerings, see, e.g., Stanford Photonics, 2016 and Andor, 2016a).
Intensified cameras played an important role at the beginning of single-molecule-imaging experimentation. Toshio Yanagida’s group performed the first published imaging of single fluorescent molecules in solution at room temperature. They visualized individual, fluorescently labeled myosin molecules as well as the turnover of individual ATP molecules, using total internal reflection microscopy, an ISIT camera (consisting of an intensifier in front of a SIT), and an ICCD camera (Funatsu et al., 1995). Until the advent of the electron multiplying charge-coupled device (EMCCD; see next section), ICCD cameras were the detector of choice for imaging of single fluorescent molecules. For instance, ICCD cameras were used to visualize single kinesin motors moving on axonemes (Vale et al., 1996), the blinking of green fluorescent protein (GFP) molecules (Dickson et al., 1997), and in demonstrating that F1-ATPase is a rotational motor that takes 120-degree steps (Yasuda et al., 1998).
Since the gain of an ICCD depends on the voltage differential between entrance and exit of the micro-channel plate, it can be modulated at extremely high rates (i.e., MHz rates). This gating not only provides an “electronic shutter,” but also can be used in more interesting ways. For example, the lifetime of a fluorescent dye (i.e., the time between absorption of a photon and emission of fluorescence) can be determined by modulating both the excitation light source and the detector gain. It can be appreciated that the emitted fluorescence will be delayed with respect to the excitation light, and that the amount of delay depends on the lifetime of the dye. By gating the detector at various phase delays with respect to the excitation light, signals with varying intensity will be obtained, from which the fluorescent lifetime can be deduced. This frequency-domain fluorescence lifetime imaging (FLIM) can be executed in wide-field mode, using an ICCD as a detector. FLIM is often used for measurement of Foerster energy transfer (FRET) between two dyes, which can be used as a proxy for the distance between the dyes. By using carefully designed probes, researchers have visualized cellular processes such as epidermal growth factor phosphorylation (Wouters and Bastiaens, 1999) and Rho GTPase activity (Hinde et al., 2013).
Electron Multiplying CCD Cameras
Despite the unprecedented sensitivity of intensified CCD cameras, which enable observation of single photons with relative ease, this technology has a number of drawbacks. These include the small linear range of the sensor (often no greater than a factor of 10), relatively low quantum efficiency (even the latest-generation ICCD cameras have a maximal QE of 50%), spreading of the signal due to the coupling of the intensifier to a CCD in an ICCD, and the possibility of irreversible sensor damage by high-light intensities, which can happen easily and at great financial cost. The signal spread was so significant that researchers were using non-amplified, traditional CCD cameras rather than ICCDs to obtain maximal localization precision in single-molecule experiments (see, e.g., Yildiz et al., 2003), despite the much longer readout times needed to collect sufficient signal above the readout (read) noise. Clearly, there was a need for CCD cameras with greater spatial precision, lower effective read noise, higher readout speeds, and a much higher damage threshold.
In 2001, both the British company, e2v technologies (Chelmsford, UK), and Texas Instruments (Dallas, TX) launched a new chip design that amplified the signal on the chip before reaching the readout amplifier, rather than using an external image intensifier. This on-chip amplification is carried out by an extra row of silicon “pixels,” through which all charge is transferred before reaching the readout amplifier. The well-to-well transfer in this special register is driven by a relatively high voltage, resulting in occasional amplification of electrons through a process called “impact ionization.” This process provides the transferred electrons with enough kinetic energy to knock an electron out of the silicon from the next well. Repeating this amplification in many wells (the highly successful e2v chip CCD97 has 536 elements in the amplification register) leads to very high effective amplification gains. Although the relation between voltage and gain is non-linear, the gain versus voltage curve has been calibrated by the manufacturer in modern electron multiplying (EM) CCD cameras, so that the end user can set a desired gain rather than a voltage. EM gain enables readout of the CCD at a much higher speed and read noise than normal, because the signal is amplified before readout. For instance, when the CCD readout noise is 30 e− (i.e., 30 electrons of noise per pixel) and a 100-fold EM gain is used, the effective read noise is 0.3 e−, using the unrealistic assumption that the EM gain itself does not introduce noise (read noise below 1 e− is negligible).
Amplification is never noise-free and several additional noise factors need to be considered when using EM gain. (For a thorough discussion of noise sources, see Andor, 2016b). Dark noise, or the occasional spontaneous accumulation of an electron in a CCD well, now becomes significant, since every “dark noise electron” will also be increased by EM amplification. Some impact ionization events take place during the normal charge transfers on the CCD. These “spurious charge” events are of no concern in a standard CCD, since they disappear in the noise floor dominated by readout noise, but they do become an issue when using EM gain. EM amplification itself is a stochastic process, and has noise characteristics very similar to that of the Poisson distributed photon shot noise, resulting in a noise factor (representing the additional noise over the noise expected from noise-free amplification) equal to √2, or ~1.41. Therefore, it was proposed that one can think of EM amplification as being noise-free but reducing the input signal by a factor of two, or halving the QE (Pawley, 2006).
Very quickly after their initial release around 2001, EMCCDs became the camera of choice for fluorescent, single-molecule detection. The most popular detector was the back-thinned EMCCD from e2v technologies, which has a QE reaching 95% in some parts of the spectrum and 512 × 512 × 16 μm-square pixels; through a frame transfer architecture, it can run continuously at ~30 frames per second (fps) full frame. One of the first applications of this technology in biological imaging was by the Jim Spudich group at Stanford University, who used the speed and sensitive detection offered by EMCCD cameras to image the mechanism of movement of the molecular motor protein, myosin VI. They showed that both actin-binding sites (heads) of this dimeric motor protein take 72-nm steps and that the heads move in succession, strongly suggesting a hand-over-hand displacement mechanism (Ökten et al., 2004).
One of the most spectacular contributions made possible by EMCCD cameras was the development of super-resolution localization microscopy. For many years, single-molecule imaging experiments had shown that it was possible to localize a single fluorescent emitter with high resolution, in principle limited only by the amount of photons detected. However, to image biological structures with high fidelity, one needs to image many single molecules, whose projections on the camera (the point spread function) overlap. As William E. Moerner and Eric Betzig explained in the 1990s, as long as one can separate the emission of single molecules based on any physical criteria, such as time or wavelength, it is possible to uniquely localize many single molecules within a diffraction-limited volume. Several groups implemented this idea in 2006, using blinking of fluorophores, either photo-activatable GFPs, in the case of photo-activated localization microscopy (PALM; Betzig et al., 2006) and fluorescence PALM (fPALM; Hess et al., 2006), or small fluorescent molecules, as in stochastic optical reconstruction microscopy (STORM; Rust et al., 2006). Clearly, the development and availability of fluorophores with the desired properties was essential for these advances (which is why the Nobel prize in Chemistry was awarded in 2014 to Moerner and Betzig, as well as Stefan W. Hell, who used non-camera based approaches to achieve super-resolution microscopy images). But successful implementation of super-resolution localization microscopy was greatly aided by EMCCD camera technology, which allowed the detection of single molecules with high sensitivity and low background, and at high speeds (the Betzig, Xiaowei Zhuang, and Samuel T. Hess groups all used EMCCD cameras in their work).
Other microscope-imaging modalities that operate at very low light levels have also greatly benefited from the use of EMCCD cameras. Most notably, spinning disk confocal microscopy is aided enormously by EMCCD cameras, since that microscopy enables visualization of the biological process of interest at lower light exposure of the sample and at higher speed than possible with a normal CCD. EMCCD-based imaging reduces photobleaching and photodamage of the live sample compared to CCDs, and offers better spatial resolution and larger linear dynamic range than do intensified CCD cameras. Hence, EMCCDs have largely replaced other cameras as the sensor of choice for spinning disk confocal microscopes (e.g., Thorn, 2010 and Oreopoulos et al., 2013).
Scientific CMOS Cameras
Charge-coupled device (CCD) technology is based on shifting charge between potential wells with high accuracy and the use of a single, or very few, readout amplifiers. (Note: It is possible to attach a unique readout amplifier to a subsection of the CCD, resulting in so-called multi-tap CCD sensors. But application of this approach has been limited in research microscopy). In active-pixel sensor (APS) architecture, each pixel contains not only a photodetector, but also an amplifier composed of transistors located adjacent to the photosensitive area of the pixel. These APS sensors are built using complementary metal-oxide-semiconductor (CMOS) technology, and are referred to as CMOS sensors. Because of their low cost, CMOS sensors were used for a long time in consumer-grade devices such as web cameras, cell phones, and digital cameras. However, they were considered far too noisy for use in scientific imaging, since every pixel contains its own amplifier, each slightly different from the other. Moreover, the transistors take up space on the chip that is not photosensitive, a problem that can be partially overcome by the use of micro-lenses to focus the light onto the photosensitive area of the sensor. Two developments, however, made CMOS cameras viable for microscopy imaging. First, Fairchild Imaging (Fairchild Imaging, 2016) improved the design of the CMOS sensor so that low read noise (around 1 electron per pixel) and high quantum efficiency (current sensors can reach 82% QE) became possible. These new sensors were named sCMOS (scientific CMOS). Second, the availability of field-programmable gate arrays (FPGAs), which are integrated circuits that can be configured after they have been produced (i.e., they can be used as custom-designed chips, cost much less because only the software has to be written. No new hardware needs to be designed). All current sCMOS cameras contain FPGAs that execute blemish corrections, such as reducing hot pixels and noisy pixels, and linearize the output of pixels, in addition to performing other functions, such as binning (pooling) of pixels. More and more image-processing functions are being integrated into these FPGAs. For instance, the latest sCMOS camera
Remarkably, sCMOS cameras can run at very high speeds (100 frames per s for a ~5-megapixel sensor), have desirable pixel sizes (the standard is 6.5 μm2, which matches the resolution provided by the often used 100 × 1.4-na objective lens; see Maddox et al., 2003 for an explanation), and cost significantly less than electron multiplying CCD (EMCCD) cameras. These features led to the rapid adoption of these cameras, even though the early models still had obvious defects, such as uneven dark image, non-linear pixel response (most pronounced around pixel value 2048 due to the use of separate digital-to-analog converters for the low- and high-intensity ranges), and the rolling shutter mode, which causes the exposure to start and end at varying time points across the chip (up to 10 ms apart). The speed, combined with large pixel number and low-read noise, makes sCMOS cameras highly versatile for most types of microscopy. In practice, however, EMCCDs still offer an advantage under extremely low-light conditions, such as is often encountered in spinning disk confocal microscopy (Fig. 3; Oreopoulos et al., 2013). However, super-resolution microscopy can make good use of the larger field of view and higher speed of sCMOS cameras, resulting in much faster data acquisition of larger areas. For example, acquisition of reconstructed super-resolution images at a rate of 32 per s using sCMOS cameras has been demonstrated (Huang et al., 2013). Another application that has greatly benefited from sCMOS cameras is light sheet fluorescence microscopy, in which objects ranging in size from single cells to small animals are illuminated sideways, such that only the area to be imaged is illuminated, greatly reducing phototoxicity. The large field of view, low-read noise, and high speed of sCMOS cameras has, for instance, made it possible to image calcium signaling in 80% of the neurons of a zebrafish embryo at 0.8 Hz (Ahrens et al., 2013). A recent development is lattice light sheet microscopy, which uses a very thin sheet and allows for imaging of individual cells at high resolution for extended periods of time. Lattice light sheet microscopes use sCMOS cameras because of their high speed, low-read noise, and large field of view (Chen et al., 2015). New forms and improvements in light sheet microscopy will occur in the next several years, and make significant contributions to the understanding of biological systems.
Figure 3.

Comparison of the electron multiplying charge-coupled device (EMCCD) and scientific complementary metal-oxide-semiconductor (sCMOS) cameras for use with a spinning disk confocal microscope. Adjacent cells were imaged, making use of either an EMCCD or sCMOS camera, using different tube lenses such that the pixel size in the image plane was the same for both (16-μm pixels for the EMCCD, with a 125-mm tube lens; 6.5-μm pixels for the sCMOS, with a 50-mm tube lens). (Reprinted from fig. 9.8, panel b, of Oreopoulos, J., et al., 2013, Methods Cell Biol. 123: 153–175, with permission from Elsevier.) from Photometrics can execute a complicated noise reduction algorithm on the FPGA of the camera before the image reaches the computer.
Summary and Outlook
Microscope imaging has progressed from the written recording of qualitative observations to a quantitative technique with permanent records of images. This leap was made possible through the emergence of highly quantitative, sensitive, and fast cameras, as well as computers, which make it possible to capture, display, and analyze the data generated by the cameras. It is safe to say that, despite notable improvements in microscope optics, progress in microscopy over the last three decades has been largely driven by sensors and analysis of digital images; structured illumination, super-resolution, lifetime, and light sheet microscopy, to name a few, would have been impossible without fast quantitative sensors and computers. The development of camera technologies was propelled by the interests of the military, the consumer market, and researchers, who have benefited from the much larger economic influence of the other groups.
Camera technology has become very impressive, pushing closer and closer to the theoretical limits. The newest sCMOS cameras have an effective read noise of about 1 e− and high linearity over a range spanning almost 4 orders of magnitude; they can acquire 5 million pixels at a rate of 100 fps and have a maximal QE of 82%. Although there is still room for improvement, these cameras enable sensitive imaging at the single-molecule level, probe biochemistry in living cells, and image organs or whole organisms at a fast rate and high resolution in three dimensions. The biggest challenge is to make sense of the enormous amount of data generated. At maximum rate, sCMOS cameras produce data at close to 1 GB/s, or 3.6 TB/h, making it a challenge to even store on computer disk, let alone analyze the images with reasonable throughput. Reducing raw data to information useful for researchers, both by extracting quantitative measurements from raw data and by visualization (especially in 3D), is increasingly becoming a bottleneck and an area where improvements and innovations could have profound effects on obtaining new biological insights. We expect that this data reduction will occur more and more “upstream,” close to the sensor itself, and that data analysis and data reduction will become more integrated with data acquisition. The aforementioned noise filters built into the new Photometrics sCMOS camera, as well as recently released software by Andor that can transfer data directly from the camera to the computer’s graphical processing unit (GPU) for fast analysis, foreshadow such developments. Whereas researchers now still consider the entire acquired image as the de facto data that needs to be archived, we may well transition to a workflow in which images are quickly reduced to the measurements of interest (using well-described, open, and reproducible procedures), either by computer processing in the camera itself or in closely connected computational units. Developments in this area will open new possibilities for the ways in which scientists visualize and analyze biological samples.
Acknowledgments
We thank Ted Salmon for inspiring stories, and John Oreopoulos, as well as members of our laboratory for reading the manuscript and providing helpful suggestions. Related work in our laboratory on software integration of cameras with microscopes was supported by NIH grant no. R01EB007187.
Abbreviations
- AD
analog-to-digital conversion
- APS
active pixel sensor
- CCD
charge-coupled device
- CMOS
complementary metal-oxide-semiconductor
- DIC
differential interference contrast
- EMCCD
electron multiplying charge-coupled device
- FLIM
fluorescence lifetime imaging microscopy
- fps
frames per second
- FRET
Foerster resonance energy transfer
- GFPs
green fluorescent proteins
- PALM
photoactivated localization microscopy
- QE
quantum efficiency
- sCMOS
scientific complementary metal-oxide-semiconductor
- SIT
silicon-intensifier target
- STORM
stochastic optical reconstruction microscopy
Literature Cited
- Agard DA, Sedat JW. Three-dimensional analysis of biological specimens utilizing image processing techniques. In: Elliott DA, editor. Proceedings of the Conference on Applications of Digital Image Processing to Astronomy, Vol. 0264, SPIE Proceedings; Bellingham, WA: Society of Photo-optical Engineers, Jet Propulsion Laboratory; 1980. pp. 110–117. [Google Scholar]
- Ahrens MB, Orger MB, Robson DN, Li JM, Keller PJ. Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat Methods. 2013;10:413– 420. doi: 10.1038/nmeth.2434. [DOI] [PubMed] [Google Scholar]
- Allen RD, Allen NS. Video-enhanced microscopy with a computer frame memory. J Microsc. 1983;129:3–17. doi: 10.1111/j.1365-2818.1983.tb04157.x. [DOI] [PubMed] [Google Scholar]
- Allen RD, Allen NS, Travis JL. Video-enhanced contrast, differential interference contrast (AVEC-DIC) microscopy: a new method capable of analyzing microtubule-related motility in the reticulopodial network of Allogromia laticollaris. Cell Motil. 1981;1:291–302. doi: 10.1002/cm.970010303. [DOI] [PubMed] [Google Scholar]
- Allen RD, Metuzals J, Tasaki I, Brady ST, Gilbert SP. Fast axonal transport in squid giant axon. Science. 1982;218:1127–1129. doi: 10.1126/science.6183744. [DOI] [PubMed] [Google Scholar]
- Andor . Intensified Camera Series [Online] Andor Technology, Ltd; Belfast, Northern Ireland: 2016a. Available: http://www.andor.com/scientific-cameras/intensified-camera-series. [2016, January 5] [Google Scholar]
- Andor . CCD, EMCCD and ICCD Comparisons [Online] Andor Technology, Ltd; Belfast, Northern Ireland: 2016b. Available: http://www.andor.com/learning-academy/ccd,-emccd-and-iccd-comparisons-difference-between-the-sensors [2016, January 5] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nano-meter resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Brady ST, Lasek RJ, Allen RD. Video microscopy of fast axonal transport in extruded axoplasm: a new model for study of molecular mechanisms. Cell Motil. 1985;5:81–101. doi: 10.1002/cm.970050203. [DOI] [PubMed] [Google Scholar]
- Chen BC, Legant WR, Wang K, Shao L, Milkie DE, Davidson MW, Janetopoulos C, Wu XS, Hammer JA, III, Liu Z, et al. Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science. 2015;346:1257998. doi: 10.1126/science.1257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JA. Digital imaging of free calcium changes and of spatial gradients in growing processes in single, mammalian central nervous system cells. Proc Natl Acad Sci USA. 1986;83:6179– 6183. doi: 10.1073/pnas.83.16.6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson MW. Pioneers in optics: Robert Day Allen. Microsc Today. 2015;23:58–59. [Google Scholar]
- Dickson RM, Cubitt AB, Tsien RY, Moerner WE. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature. 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- Fairchild Imaging. sCMOS [Online] Fairchild Imaging; San Jose, CA: 2016. Available: http://fairchildimaging.com [2016, January 5] [Google Scholar]
- Funatsu T, Harada Y, Tokunaga M, Saito K, Yanagida T. Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution. Nature. 1995;374:555–559. doi: 10.1038/374555a0. [DOI] [PubMed] [Google Scholar]
- Gruenbaum Y, Hochstrasser M, Mathog D, Saumweber H, Agard DA, Sedat JW. Spatial organization of the Drosophila nucleus: a three-dimensional cytogenetic study. J Cell Sci Suppl. 1984;1:223–234. doi: 10.1242/jcs.1984.supplement_1.14. [DOI] [PubMed] [Google Scholar]
- Hess ST, Girirajan TPK, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258– 4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinde E, Digman MA, Hahn KM, Gratton E. Millisecond spatiotemporal dynamics of FRET biosensors by the pair correlation function and the phasor approach to FLIM. Proc Natl Acad Sci USA. 2013;110:135–140. doi: 10.1073/pnas.1211882110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka Y, Sedat JW, Agard DA. The use of a charge-coupled device for quantitative optical microscopy of biological structures. Science. 1987;238:36– 41. doi: 10.1126/science.3116667. [DOI] [PubMed] [Google Scholar]
- Huang F, Hartwich TMP, Rivera-Molina FE, Lin Y, Duim WC, Long JJ, Uchil PD, Myers JR, Baird MA, Mothes W, et al. Video-rate nanoscopy using sCMOS camera-specific single-molecule localization algorithms. Nat Methods. 2013;10:653– 658. doi: 10.1038/nmeth.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoué S. Video image processing greatly enhances contrast, quality, and speed in polarization-based microscopy. J Cell Biol. 1981;89:346–356. doi: 10.1083/jcb.89.2.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoué S. Video Microscopy. Plenum Press; New York: 1986. [Google Scholar]
- Maddox PS, Moree B, Canman JC, Salmon ED. Spinning disk confocal microscope system for rapid high-resolution, multimode, fluorescence speckle microscopy and green fluorescent protein imaging in living cells. Methods Enzymol. 2003;360:597– 617. doi: 10.1016/s0076-6879(03)60130-8. [DOI] [PubMed] [Google Scholar]
- Morton GA. Image intensifiers and the scotoscope. Appl Optics. 1964;3:651– 672. [Google Scholar]
- Ökten Z, Churchman LS, Rock RS, Spudich JA. Myosin VI walks hand-over-hand along actin. Nat Struct Mol Biol. 2004;11:884– 887. doi: 10.1038/nsmb815. [DOI] [PubMed] [Google Scholar]
- Oreopoulos J, Berman R, Browne M. Spinning-disk confocal microscopy: present technology and future trends. Methods Cell Biol. 2013;123:153–175. doi: 10.1016/B978-0-12-420138-5.00009-4. [DOI] [PubMed] [Google Scholar]
- Pawley J, editor. Handbook Of Biological Confocal Microscopy. 3. Springer; Boston: 2006. [Google Scholar]
- Roos KP, Brady AJ. Individual sarcomere length determination from isolated cardiac cells using high-resolution optical microscopy and digital image processing. Biophys J. 1982;40:233–244. doi: 10.1016/S0006-3495(82)84478-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–796. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon ED. VE-DIC light microscopy and the discovery of kinesin. Trends Cell Biol. 1995;5:154–158. doi: 10.1016/s0962-8924(00)88979-5. [DOI] [PubMed] [Google Scholar]
- Sony Corporate Info. Determination drove the development of the CCD “electronic eye” [Online] 2016 Available: http://www.sony.net/SonyInfo/CorporateInfo/History/SonyHistory/2-11.html [2016, January 5]
- Stanford Photonics, Inc. Electronic imaging technologies [Online] Stanford Photonics, Inc; Palo Alto, CA: 2016. Available: http://www.stanfordphotonics.com [2016, January 5] [Google Scholar]
- Thorn K. Spinning-disk confocal microscopy of yeast. Methods Enzymol. 2010;470:581– 602. doi: 10.1016/S0076-6879(10)70023-9. [DOI] [PubMed] [Google Scholar]
- Vale RD, Toyoshima YY. Rotation and translocation of microtubules in vitro induced by dyneins from Tetrahymena cilia. Cell. 1988;52:459– 469. doi: 10.1016/s0092-8674(88)80038-2. [DOI] [PubMed] [Google Scholar]
- Vale RD, Schnapp BJ, Reese TS, Sheetz MP. Movement of organelles along filaments dissociated from the axoplasm of the squid giant axon. Cell. 1985a;40:449– 454. doi: 10.1016/0092-8674(85)90159-x. [DOI] [PubMed] [Google Scholar]
- Vale RD, Schnapp BJ, Reese TS, Sheetz MP. Organelle, bead, and microtubule translocations promoted by soluble factors from the squid giant axon. Cell. 1985b;40:559–569. doi: 10.1016/0092-8674(85)90204-1. [DOI] [PubMed] [Google Scholar]
- Vale RD, Reese TS, Sheetz MP. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985c;42:39–50. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Funatsu T, Pierce DW, Romberg L, Harada Y, Yanagida T. Direct observation of single kinesin molecules moving along microtubules. Nature. 1996;380:451– 453. doi: 10.1038/380451a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters FS, Bastiaens PIH. Fluorescence lifetime imaging of receptor tyrosine kinase activity in cells. Curr Biol. 1999;9:1127–1130. doi: 10.1016/s0960-9822(99)80484-9. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Noji H, Kinosita K, Jr, Yoshida M. F1- ATPase is a highly efficient molecular motor that rotates with discrete 120° steps. Cell. 1998;93:1117–1124. doi: 10.1016/s0092-8674(00)81456-7. [DOI] [PubMed] [Google Scholar]
- Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 2003;300:2061–2065. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]
- Zworykin VK. The iconoscope: a modern version of the electric eye. Proc IEEE. 1997;85:1327–1333. (Reprinted from the Proceedings of the Institute of Radio Engineers 1934; 22: 16 –32) [Google Scholar]