

Abstract
Brain edema, the first stage of intracranial hypertension, has been associated with poor prognosis and increased mortality after acute brain injury, such as ischemic stroke, intracranial hemorrhage (ICH), and traumatic brain injury (TBI). The acute brain injury often initiates release of many molecules, including glutamate, adenosine, thrombin, oxyhemoglobin, cytokines, reactive oxygen species (ROS), damage associated molecular pattern molecules (DAMPs), and others. Most of those molecules activate Src family kinases (SFKs), a family of proto-oncogenic non-receptor tyrosine kinases, resulting in blood-brain barrier (BBB) disruption and brain edema at the acute stage after brain injury. However, SFKs also contributes to BBB self-repair and brain edema resolution in the chronic stage that follows brain injury. In this review we summarize possible pathways through which SFKs are implicated in both brain edema formation and its eventual resolution.
Introduction
Brain edema occurs when a cerebral blood vessel is blocked or ruptured following ischemic stroke, intracerebral hemorrhage (ICH), traumatic brain injury (TBI) and other neurological diseases [1–3]. There are two main categories of brain edema, namely cytotoxic (cellular) edema and vasogenic (extracellular) edema [4]. In cytotoxic edema, the blood-brain barrier (BBB) remains intact, but there is essentially a compartment shift of water from the extracellular to the intracellular compartment, with no increase of brain water content or rise in ICP. Though it does not require BBB disruption, cytotoxic brain edema changes cellular metabolism and eventually damages BBB after brain injury. By contrast, vasogenic edema requires BBB disruption, allowing fluid (i.e., circulating blood) to accumulate in the extracellular space in brain parenchyma and will increase ICP [4]. It is generally thought that cytotoxic edema is dominant immediately following ischemic stroke [5], while vasogenic edema is dominant at the acute stage after TBI [4]. However, cytotoxic and vasogenic edema usually combine when brain injury progresses into the chronic phase in which a characteristic breakdown of BBB occurs no matter what type of edema was first in the acute stage post brain injury [6]. Therefore, maintenance of BBB integrity has become a focus of recent research to prevent brain edema and improve outcomes of acute brain injury.
Brain edema has been associated with high mortality, mostly because it can induce rapid increase in intracranial pressure (ICP), which leads to compression of blood vessels, reduced tissue blood flow, reduced oxygenation and shifts tissue down pressure gradients (herniations) that may crush vital brain centers and eventually cause respiratory or heart failure [4]. An aggressive treatment for raised ICP can reduce mortality and improve outcome [7, 8], though ICP control alone (i.e. osmotherapy) may be insufficient to benefit long-term recovery after brain injury [9]. This is probably because osmotherapy is unable to block the release of many toxic molecules that follow acute brain injury, such as glutamate, adenosine, oxyhemoglobin, thrombin, cytokines, reactive oxygen species (ROS), damage associated molecular pattern molecules (DAMPs) and others [10–40]. These molecules mediate BBB disruption and brain edema through multiple ligand-receptor pathways. Since brain edema might occur via many parallel pathways, blocking just one or two of these pathways may not be clinically effective in treating human brain injury [16].
Src family kinases (SFKs), a family of proto-oncogenic, non-receptor tyrosine kinases, include nine family members: c-Src, Fyn, Yes, Yrk, Lyn, Fgr, Hck, Blk and Lck [41–43]. They can be activated by many trans-membrane receptors, such as adhesion receptors, tyrosine kinase receptors, G protein-coupled receptors, cytokine receptors, and others [44]. This makes SFKs a point of convergence for many molecules, and targeting SFKs has potential to prevent disruption of BBB components (i.e., endothelial cells, astrocytes, pericytes, neurons, tight junctions, and others) and block brain edema via modulating their multiple downstream targets, such as NMDA receptors [45–50], mitogen-activated protein kinases (MAPKs) [51–57], and cyclin-dependent kinases (Cdks) [58–62]. Many studies have demonstrated that acute administration of SFK inhibitors (e.g., PP1, PP2) attenuates BBB breakdown and prevents brain edema after acute brain injury [18–20, 63–66]. However, delayed and chronic administration of PP2 prevents the BBB self-repair and lengthens the period to resolve the edema in the recovery stage after brain injury [20]. These suggest SFKs may play dual roles in both brain edema formation and resolution during the different stages following acute brain injury (Figure 1).
Figure 1.
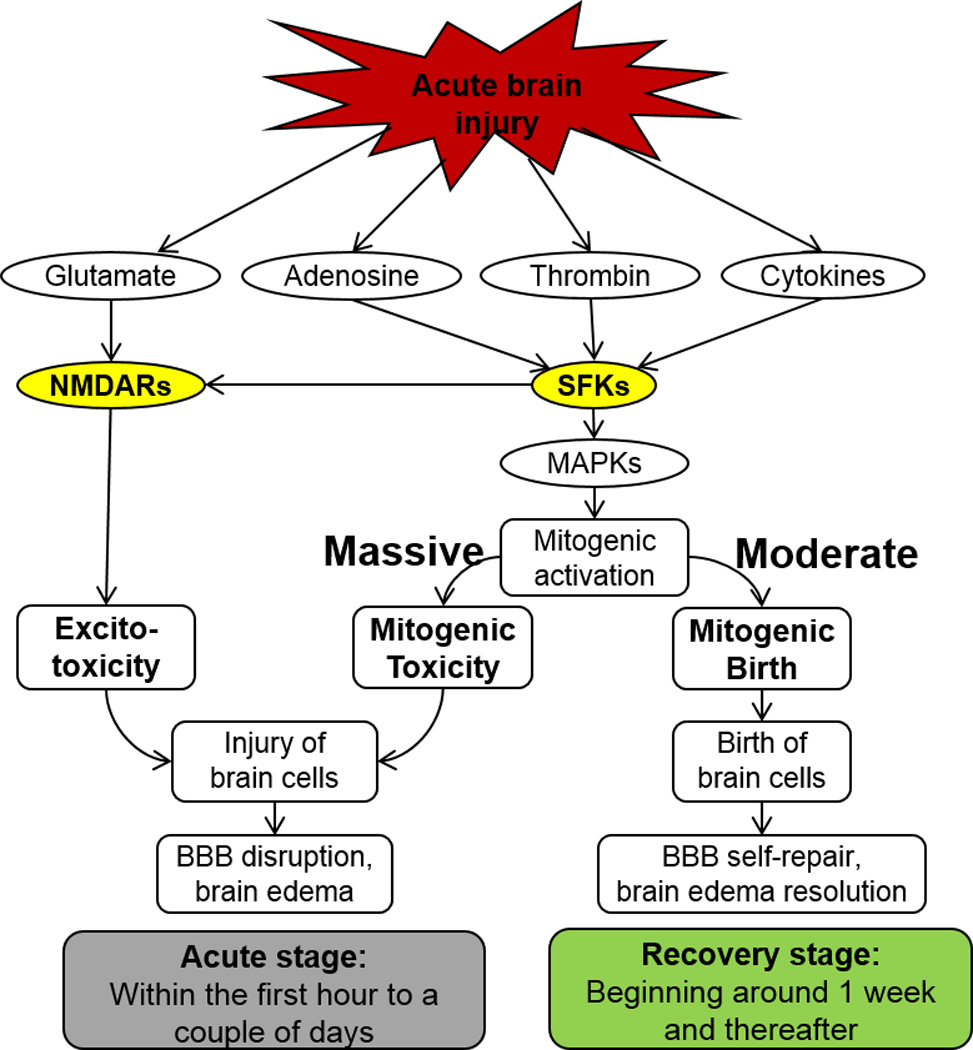
Activation of SFKs results in BBB disruption and brain edema formation in the acute stage, but leads to BBB self-repair and brain edema resolution in the recovery stage after acute brain injury, such as ICH, TBI and ischemic stroke.
Tissue specificity, structure, activity and functions of SFKs
Several SFK family members (c-Src, Fyn, Yes, Yrk) are ubiquitously expressed, whereas others (Lyn, Fgr, Hck, Blk, Lck) are generally found in brain and hematopoietic cells [47, 67–72]. In adult mice, Fyn and c-Src mRNA expression is highest in hippocampal neurons [73, 74]. Importantly, one tissue can express multiple SFK members, for example, Src, Fyn, Yes, and Lck have been examined in brain [47, 67–72], and the different SFK family members are often found to compensate for one another [75].
Structurally, SFK family members share a conserved domain structure consisting of consecutive SH3 (polyproline type II helix for protein-protein interaction), SH2 (phosphotyrosine recognition), and SH1 (tyrosine kinase catalytic activity) [43]. They also contain a membrane-targeting region at their N-terminus that is followed by a unique domain of 50–70 residues, which is divergent among family members [43]. Although it still incompletely clear, Src activity is regulated by tyrosine phosphorylation at two sites (one is at Tyr416 in the SH1 domain, the other at Tyr527 in the short C-terminal tail), but with opposing effects. While phosphorylation at Tyr416 activates Src, phosphorylation at Tyr527 results in inactivation [72, 76].
Under normal physiological conditions, SFKs are implicated in the regulation of embryonic development, cell growth, cellular differentiation and inflammatory responses [74, 77–79]. SFKs can initiate negative feedback to prevent their sustained activation via recruitment of inhibitory factor C-terminal Src (Csk) [80]. The feedback loop consists of SFK activation leading to phosphorylation of Csk binding protein (Cbp), and the phosphorylated Cbp targets Csk to SFKs and promotes inhibitory Csk phosphorylation of SFKs [81].
Due to mutations in SFKs or Csk, aberrant activation of SFKs can occur in cancers, and the abnormal SFK signaling contributes to many aspects of tumor development, including proliferation, survival, adhesion, migration, invasion, as well as metastasis [82–84]. Thus, it is likely that targeting SFKs may be a promising therapeutic approach for cancer, as SFK antagonists have been tested and well tolerated in cancer clinical trials [85–88].
Recently, we and others have demonstrated a new function of SFKs in acute brain injury, that is, transient activation of SFKs associated with BBB disruption, brain edema and spatial memory deficits following experimental ICH (intracerebroventricular fresh blood or thrombin model), TBI (lateral fluid percussion (LFP) model), and stroke (middle cerebral artery occlusion (MCAO) model) [18, 19, 63–66].
SFK activation, excitotoxicity, BBB disruption and brain edema
Following acute brain injury (i.e., ICH, TBI, ischemic stroke), there occurs a transient increase of glucose utilization and local cerebral blood flow [53, 89, 90], presumably because of the actions of glutamate in blood at the time of brain injury. This was supported by findings that glucose hypermetabolism could be blocked by antagonists of NMDA and AMPA receptors [53, 90]. However, glutamate alone could not explain the hypermetabolism since glutamate injected directly into brain does not produce hypermetabolism [53]. This suggests that acute brain injury affects NMDA receptors in some way to make them more sensitive to glutamate in order to mediate brain injury and/or hypermetabolism.
A large number of studies have revealed that the molecules released following acute brain injury (e.g. adenosine, thrombin, cytokines) can activate SFKs [10–32], and SFKs directly bind NMDA receptors and modulate their activity [45–50]. Our data show that either an NMDA receptor inhibitor (MK801) or an SFK inhibitor (PP2) is able to prevent brain edema and improve behavioral outcomes after intracerebroventricular injection of thrombin in rats [19]. Therefore, it is plausible that SFKs and NMDA receptors are coupling to mediate calcium overload, glucose hypermetabolism and brain edema after acute brain injury.
SFK activation, mitogenic signaling, brain edema formation and resolution
SFKs can be activated by many trans-membrane receptors, such as adhesion receptors, tyrosine kinase receptors, G protein-coupled receptors, cytokine receptors, and others [44]. This unique feature of SFKs makes them a point of convergence for many toxic molecules that are released after brain injury [10–40]. Most of those molecules are abruptly released and reach peak concentrations within a couple of hours to a day after brain injury. In the acute stage over-activated SFK mitogenic signaling causes neurons to enter the cell cycle and die, and damages astroctyes and endothelial cells via MAPKs or CdKs [14, 19, 20, 51–62]. The disruption of BBB components increase BBB permeability, resulting in brain edema after acute brain injury.
Within about a day after acute brain injury, the molecules resolve gradually, and the disease progresses to a recovery stage of brain injury. The restored moderate SFK/mitogenic signaling leads to birth of new endothelial cells, astrocytes and other cells that mediate BBB self-repair and brain edema resolution. Recent studies suggest that a number of stem cells exist throughout the mammalian brain, and some of these are associated with vascular niches [91]. Such stem cells could serve as a source of newborn endothelial cells, astrocytes and other cells of the neurovascular unit that would play a major role in re-establishing the BBB after brain injury [92].
Though SFK inhibitors prevent toxicity signaling at the acute phase after ICH, they also block cellular proliferation of stem cells to delay and prolong BBB self-repair [55, 56, 93, 94]. This may provide at least a partial explanation for the findings that: (1) acute single administration of SFK inhibitors (PP2, 1mg/kg, i.p. immediately after ICH) can attenuate the intracerebroventricular injection (i.c.v.) of thrombin-induced BBB disruption and brain edema [20, 52]; (2) and that delayed and chronic administration of SFK inhibitor (PP2, 1 mg/kg, i.p. daily, day 2 through 6) prevents thrombin-induced BBB repair and brain edema resolution in rats [20, 52].
Additionally, SFKs also activate hypoxia-inducible factors (HIFs) that can increase BBB permeability for brain edema formation or promote angiogenesis for brain edema resolution after brain injury through expression of aquaporins (AQPs), matrix metalloproteinases (MMPs), vascular endothelial growth factor (VEGF), BBB proteins (i.e., occluding), and others [95–97]. Interactions and cross-talk with these and other molecules and pathways add complexity to timing and development of appropriate treatment strategies involving the SFKs.
Future directions
Future studies need to address exactly which specific SFK members found in brain (e.g., Src, Fyn, Lck and Yrk) mediate edema following acute brain injury. In view that SFKs also play critical roles in brain edema resolution, the therapeutic time window of SFK inhibition should be studied for treating edema following acute brain injury and avoid the potential side effects caused by chronic inhibition of SFKs. A nanoparticle-based siRNA transfection system can be used for knockdown of individual SFK genes, as it allows transient knockdown of target genes, high efficiency of in vivo siRNA delivery, high specificity for gene targets, low cytotoxicity [98, 99], and is approved by the FDA for human use [100–103].
Acknowledgments
Disclosure: The authors acknowledge the support of AHA Beginning Grant-in-Aid 12BGIA12060381 and NIH grant NS054652.
References
- 1.Marmarou A. Pathophysiology of traumatic brain edema: current concepts. Acta Neurochir Suppl. 2003;86:7–10. doi: 10.1007/978-3-7091-0651-8_2. [DOI] [PubMed] [Google Scholar]
- 2.Thiex R, Tsirka SE. Brain edema after intracerebral hemorrhage: mechanisms, treatment options, management strategies, and operative indications. Neurosurg Focus. 2007;22:E6. doi: 10.3171/foc.2007.22.5.7. [DOI] [PubMed] [Google Scholar]
- 3.Kasner SE, Demchuk AM, Berrouschot J, Schmutzhard E, Harms L, et al. Predictors of fatal brain edema in massive hemispheric ischemic stroke. Stroke. 2001;32:2117–2123. doi: 10.1161/hs0901.095719. [DOI] [PubMed] [Google Scholar]
- 4.Donkin JJ, Vink R. Mechanisms of cerebral edema in traumatic brain injury: therapeutic developments. Curr Opin Neurol. 2010;23:293–299. doi: 10.1097/WCO.0b013e328337f451. [DOI] [PubMed] [Google Scholar]
- 5.Rosenberg GA. Ischemic brain edema. Prog Cardiovasc Dis. 1999;42:209–216. doi: 10.1016/s0033-0620(99)70003-4. [DOI] [PubMed] [Google Scholar]
- 6.Iencean SM. Brain edema -- a new classification. Med Hypotheses. 2003;61:106–109. doi: 10.1016/s0306-9877(03)00127-0. [DOI] [PubMed] [Google Scholar]
- 7.Sadaka F, Veremakis C. Therapeutic hypothermia for the management of intracranial hypertension in severe traumatic brain injury: a systematic review. Brain Inj. 2012;26:899–908. doi: 10.3109/02699052.2012.661120. [DOI] [PubMed] [Google Scholar]
- 8.Pitfield AF, Carroll AB, Kissoon N. Emergency management of increased intracranial pressure. Pediatr Emerg Care. 2012;28:200–204. doi: 10.1097/PEC.0b013e318243fb72. quiz 205-207. [DOI] [PubMed] [Google Scholar]
- 9.Sandsmark DK, Sheth KN. Management of increased intracranial pressure. Curr Treat Options Neurol. 2014;16:272. doi: 10.1007/s11940-013-0272-3. [DOI] [PubMed] [Google Scholar]
- 10.Prins M, Greco T, Alexander D, Giza CC. The pathophysiology of traumatic brain injury at a glance. Dis Model Mech. 2013;6:1307–1315. doi: 10.1242/dmm.011585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Namjoshi DR, Good C, Cheng WH, Panenka W, Richards D, et al. Towards clinical management of traumatic brain injury: a review of models and mechanisms from a biomechanical perspective. Dis Model Mech. 2013;6:1325–1338. doi: 10.1242/dmm.011320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaetz M. The neurophysiology of brain injury. Clin Neurophysiol. 2004;115:4–18. doi: 10.1016/s1388-2457(03)00258-x. [DOI] [PubMed] [Google Scholar]
- 13.Perel P, Roberts I, Bouamra O, Woodford M, Mooney J, et al. Intracranial bleeding in patients with traumatic brain injury: a prognostic study. BMC Emerg Med. 2009;9:15. doi: 10.1186/1471-227X-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu D, Sharp FR, Van KC, Ander BP, Ghiasvand R, et al. Inhibition of Src Family Kinases Protects Hippocampal Neurons and Improves Cognitive Function after Traumatic Brain Injury. J Neurotrauma. 2014;31:1268–1276. doi: 10.1089/neu.2013.3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.NINDS. Traumatic Brain Injury: Hope Through Research. 2013 http://www.ninds.nih.gov/disorders/tbi/detail_tbi.htm.
- 16.Ray SK, Dixon CE, Banik NL. Molecular mechanisms in the pathogenesis of traumatic brain injury. Histol Histopathol. 2002;17:1137–1152. doi: 10.14670/HH-17.1137. [DOI] [PubMed] [Google Scholar]
- 17.Xi G, Reiser G, Keep RF. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? J Neurochem. 2003;84:3–9. doi: 10.1046/j.1471-4159.2003.01268.x. [DOI] [PubMed] [Google Scholar]
- 18.Sharp F, Liu DZ, Zhan X, Ander BP. Intracerebral hemorrhage injury mechanisms: glutamate neurotoxicity, thrombin, and Src. Acta Neurochir Suppl. 2008;105:43–46. doi: 10.1007/978-3-211-09469-3_9. [DOI] [PubMed] [Google Scholar]
- 19.Liu DZ, Cheng XY, Ander BP, Xu H, Davis RR, et al. Src kinase inhibition decreases thrombin-induced injury and cell cycle re-entry in striatal neurons. Neurobiol Dis. 2008;30:201–211. doi: 10.1016/j.nbd.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu DZ, Ander BP, Xu H, Shen Y, Kaur P, et al. Blood-brain barrier breakdown and repair by Src after thrombin-induced injury. Ann Neurol. 2010;67:526–533. doi: 10.1002/ana.21924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11:720–731. doi: 10.1016/S1474-4422(12)70104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yao X, Balamurugan P, Arvey A, Leslie C, Zhang L. Heme controls the regulation of protein tyrosine kinases Jak2 and Src. Biochem Biophys Res Commun. 2010;403:30–35. doi: 10.1016/j.bbrc.2010.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corcoran A, Cotter TG. Redox regulation of protein kinases. FEBS J. 2013;280:1944–1965. doi: 10.1111/febs.12224. [DOI] [PubMed] [Google Scholar]
- 24.Giannoni E, Chiarugi P. Redox Circuitries Driving Src Regulation. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2013.5525. [DOI] [PubMed] [Google Scholar]
- 25.Johnson P, Cross JL. Tyrosine phosphorylation in immune cells: direct and indirect effects on toll-like receptor-induced proinflammatory cytokine production. Crit Rev Immunol. 2009;29:347–367. doi: 10.1615/critrevimmunol.v29.i4.50. [DOI] [PubMed] [Google Scholar]
- 26.Cabodi S, Di Stefano P, Leal Mdel P, Tinnirello A, Bisaro B, et al. Integrins and signal transduction. Adv Exp Med Biol. 2010;674:43–54. doi: 10.1007/978-1-4419-6066-5_5. [DOI] [PubMed] [Google Scholar]
- 27.Page TH, Smolinska M, Gillespie J, Urbaniak AM, Foxwell BM. Tyrosine kinases and inflammatory signalling. Curr Mol Med. 2009;9:69–85. doi: 10.2174/156652409787314507. [DOI] [PubMed] [Google Scholar]
- 28.Hou CH, Fong YC, Tang CH. HMGB-1 induces IL-6 production in human synovial fibroblasts through c-Src, Akt and NF-kappaB pathways. J Cell Physiol. 2011;226:2006–2015. doi: 10.1002/jcp.22541. [DOI] [PubMed] [Google Scholar]
- 29.Banerjee S, de Freitas A, Friggeri A, Zmijewski JW, Liu G, et al. Intracellular HMGB1 negatively regulates efferocytosis. J Immunol. 2011;187:4686–4694. doi: 10.4049/jimmunol.1101500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Musumeci D, Roviello GN, Montesarchio D. An overview on HMGB1 inhibitors as potential therapeutic agents in HMGB1-related pathologies. Pharmacol Ther. 2014;141:347–357. doi: 10.1016/j.pharmthera.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Ibrahim ZA, Armour CL, Phipps S, Sukkar MB. RAGE and TLRs: relatives, friends or neighbours? Mol Immunol. 2013;56:739–744. doi: 10.1016/j.molimm.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 32.Zhong C, Zhao X, Van KC, Bzdega T, Smyth A, et al. NAAG peptidase inhibitor increases dialysate NAAG and reduces glutamate, aspartate and GABA levels in the dorsal hippocampus following fluid percussion injury in the rat. J Neurochem. 2006;97:1015–1025. doi: 10.1111/j.1471-4159.2006.03786.x. [DOI] [PubMed] [Google Scholar]
- 33.Hua Y, Keep RF, Hoff JT, Xi G. Brain injury after intracerebral hemorrhage: the role of thrombin and iron. Stroke. 2007;38:759–762. doi: 10.1161/01.STR.0000247868.97078.10. [DOI] [PubMed] [Google Scholar]
- 34.Matz PG, Fujimura M, Lewen A, Morita-Fujimura Y, Chan PH. Increased cytochrome c-mediated DNA fragmentation and cell death in manganese-superoxide dismutase-deficient mice after exposure to subarachnoid hemolysate. Stroke. 2001;32:506–515. doi: 10.1161/01.str.32.2.506. [DOI] [PubMed] [Google Scholar]
- 35.Wu J, Hua Y, Keep RF, Schallert T, Hoff JT, et al. Oxidative brain injury from extravasated erythrocytes after intracerebral hemorrhage. Brain Res. 2002;953:45–52. doi: 10.1016/s0006-8993(02)03268-7. [DOI] [PubMed] [Google Scholar]
- 36.Jung KH, Chu K, Jeong SW, Han SY, Lee ST, et al. HMG-CoA reductase inhibitor, atorvastatin, promotes sensorimotor recovery, suppressing acute inflammatory reaction after experimental intracerebral hemorrhage. Stroke. 2004;35:1744–1749. doi: 10.1161/01.STR.0000131270.45822.85. [DOI] [PubMed] [Google Scholar]
- 37.Dziedzic T, Bartus S, Klimkowicz A, Motyl M, Slowik A, et al. Intracerebral hemorrhage triggers interleukin-6 and interleukin-10 release in blood. Stroke. 2002;33:2334–2335. doi: 10.1161/01.str.0000027211.73567.fa. [DOI] [PubMed] [Google Scholar]
- 38.Rincon F, Mayer SA. Novel therapies for intracerebral hemorrhage. Curr Opin Crit Care. 2004;10:94–100. doi: 10.1097/00075198-200404000-00003. [DOI] [PubMed] [Google Scholar]
- 39.Castillo J, Davalos A, Alvarez-Sabin J, Pumar JM, Leira R, et al. Molecular signatures of brain injury after intracerebral hemorrhage. Neurology. 2002;58:624–629. doi: 10.1212/wnl.58.4.624. [DOI] [PubMed] [Google Scholar]
- 40.Mayne M, Ni W, Yan HJ, Xue M, Johnston JB, et al. Antisense oligodeoxynucleotide inhibition of tumor necrosis factor-alpha expression is neuroprotective after intracerebral hemorrhage. Stroke. 2001;32:240–248. doi: 10.1161/01.str.32.1.240. [DOI] [PubMed] [Google Scholar]
- 41.Oda H, Kumar S, Howley PM. Regulation of the Src family tyrosine kinase Blk through E6AP-mediated ubiquitination. Proc Natl Acad Sci U S A. 1999;96:9557–9562. doi: 10.1073/pnas.96.17.9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biscardi JS, Ishizawar RC, Silva CM, Parsons SJ. Tyrosine kinase signalling in breast cancer: epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Res. 2000;2:203–210. doi: 10.1186/bcr55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23:7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 44.Tatosyan AG, Mizenina OA. Kinases of the Src family: structure and functions. Biochemistry (Mosc) 2000;65:49–58. [PubMed] [Google Scholar]
- 45.Groveman BR, Feng S, Fang XQ, Pflueger M, Lin SX, et al. The regulation of N-methyl-D-aspartate receptors by Src kinase. FEBS J. 2012;279:20–28. doi: 10.1111/j.1742-4658.2011.08413.x. [DOI] [PubMed] [Google Scholar]
- 46.Yu XM, Askalan R, Keil GJ, 2nd, Salter MW. NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]
- 47.Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- 48.Trepanier CH, Jackson MF, MacDonald JF. Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J. 2012;279:12–19. doi: 10.1111/j.1742-4658.2011.08391.x. [DOI] [PubMed] [Google Scholar]
- 49.Choi UB, Xiao S, Wollmuth LP, Bowen ME. Effect of Src kinase phosphorylation on disordered C-terminal domain of N-methyl-D-aspartic acid (NMDA) receptor subunit GluN2B protein. J Biol Chem. 2011;286:29904–29912. doi: 10.1074/jbc.M111.258897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu DZ, Ander BP. Cell cycle phase transitions: signposts for aberrant cell cycle reentry in dying mature neurons. Journal of Cytology & Histology. 2011;2:5. [Google Scholar]
- 52.Liu DZ, Sharp FR. The dual role of SRC kinases in intracerebral hemorrhage. Acta Neurochir Suppl. 2011;111:77–81. doi: 10.1007/978-3-7091-0693-8_13. [DOI] [PubMed] [Google Scholar]
- 53.Ardizzone TD, Lu A, Wagner KR, Tang Y, Ran R, et al. Glutamate receptor blockade attenuates glucose hypermetabolism in perihematomal brain after experimental intracerebral hemorrhage in rat. Stroke. 2004;35:2587–2591. doi: 10.1161/01.STR.0000143451.14228.ff. [DOI] [PubMed] [Google Scholar]
- 54.Copani A, Nicoletti F. Cell-cycle mechanisms and neuronal cell death. New York: Kluwer Academic/Plenum; 2005. [Google Scholar]
- 55.Liu DZ, Ander BP. Cell cycle inhibition without disruption of neurogenesis is a strategy for treatment of aberrant cell cycle diseases: an update. ScientificWorldJournal. 2012;2012:491737. doi: 10.1100/2012/491737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu DZ, Ander BP, Sharp FR. Cell cycle inhibition without disruption of neurogenesis is a strategy for treatment of central nervous system diseases. Neurobiol Dis. 2010;37:549–557. doi: 10.1016/j.nbd.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rodriguez PL, Sahay S, Olabisi OO, Whitehead IP. ROCK I-mediated activation of NF-kappaB by RhoB. Cell Signal. 2007;19:2361–2369. doi: 10.1016/j.cellsig.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, et al. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128:269–280. doi: 10.1016/j.cell.2006.11.047. [DOI] [PubMed] [Google Scholar]
- 59.Kasahara K, Nakayama Y, Nakazato Y, Ikeda K, Kuga T, et al. Src signaling regulates completion of abscission in cytokinesis through ERK/MAPK activation at the midbody. J Biol Chem. 2007;282:5327–5339. doi: 10.1074/jbc.M608396200. [DOI] [PubMed] [Google Scholar]
- 60.Liu Z, Falola J, Zhu X, Gu Y, Kim LT, et al. Antiproliferative effects of Src inhibition on medullary thyroid cancer. J Clin Endocrinol Metab. 2004;89:3503–3509. doi: 10.1210/jc.2003-031917. [DOI] [PubMed] [Google Scholar]
- 61.Mishra R, Wang Y, Simonson MS. Cell cycle signaling by endothelin-1 requires Src nonreceptor protein tyrosine kinase. Mol Pharmacol. 2005;67:2049–2056. doi: 10.1124/mol.104.010546. [DOI] [PubMed] [Google Scholar]
- 62.Taylor SJ, Shalloway D. The cell cycle and c-Src. Curr Opin Genet Dev. 1993;3:26–34. doi: 10.1016/s0959-437x(05)80337-5. [DOI] [PubMed] [Google Scholar]
- 63.Ardizzone TD, Zhan X, Ander BP, Sharp FR. SRC kinase inhibition improves acute outcomes after experimental intracerebral hemorrhage. Stroke. 2007;38:1621–1625. doi: 10.1161/STROKEAHA.106.478966. [DOI] [PubMed] [Google Scholar]
- 64.Park Y, Luo T, Zhang F, Liu C, Bramlett HM, et al. Downregulation of Src-kinase and glutamate-receptor phosphorylation after traumatic brain injury. J Cereb Blood Flow Metab. 2013;33:1642–1649. doi: 10.1038/jcbfm.2013.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bai Y, Xu G, Xu M, Li Q, Qin X. Inhibition of Src phosphorylation reduces damage to the blood-brain barrier following transient focal cerebral ischemia in rats. Int J Mol Med. 2014;34:1473–1482. doi: 10.3892/ijmm.2014.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kusaka G, Ishikawa M, Nanda A, Granger DN, Zhang JH. Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2004;24:916–925. doi: 10.1097/01.WCB.0000125886.48838.7E. [DOI] [PubMed] [Google Scholar]
- 67.Morse WR, Whitesides JG, 3rd, LaMantia AS, Maness PF. p59fyn and pp60c-src modulate axonal guidance in the developing mouse olfactory pathway. J Neurobiol. 1998;36:53–63. doi: 10.1002/(sici)1097-4695(199807)36:1<53::aid-neu5>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 68.Encinas M, Tansey MG, Tsui-Pierchala BA, Comella JX, Milbrandt J, et al. c-Src is required for glial cell line-derived neurotrophic factor (GDNF) family ligand-mediated neuronal survival via a phosphatidylinositol-3 kinase (PI-3K)-dependent pathway. J Neurosci. 2001;21:1464–1472. doi: 10.1523/JNEUROSCI.21-05-01464.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sperber BR, Boyle-Walsh EA, Engleka MJ, Gadue P, Peterson AC, et al. A unique role for Fyn in CNS myelination. J Neurosci. 2001;21:2039–2047. doi: 10.1523/JNEUROSCI.21-06-02039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heidinger V, Manzerra P, Wang XQ, Strasser U, Yu SP, et al. Metabotropic glutamate receptor 1-induced upregulation of NMDA receptor current: mediation through the Pyk2/Src-family kinase pathway in cortical neurons. J Neurosci. 2002;22:5452–5461. doi: 10.1523/JNEUROSCI.22-13-05452.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rouer E. Neuronal isoforms of Src, Fyn and Lck tyrosine kinases: A specific role for p56lckN in neuron protection. C R Biol. 2010;333:1–10. doi: 10.1016/j.crvi.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 72.Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–7909. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]
- 73.Umemori H, Wanaka A, Kato H, Takeuchi M, Tohyama M, et al. Specific expressions of Fyn and Lyn, lymphocyte antigen receptor-associated tyrosine kinases, in the central nervous system. Brain Res Mol Brain Res. 1992;16:303–310. doi: 10.1016/0169-328x(92)90239-8. [DOI] [PubMed] [Google Scholar]
- 74.Ross CA, Wright GE, Resh MD, Pearson RC, Snyder SH. Brain-specific src oncogene mRNA mapped in rat brain by in situ hybridization. Proc Natl Acad Sci U S A. 1988;85:9831–9835. doi: 10.1073/pnas.85.24.9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stein PL, Vogel H, Soriano P. Combined deficiencies of Src, Fyn, and Yes tyrosine kinases in mutant mice. Genes Dev. 1994;8:1999–2007. doi: 10.1101/gad.8.17.1999. [DOI] [PubMed] [Google Scholar]
- 76.Hunter T. A tail of two src's: mutatis mutandis. Cell. 1987;49:1–4. doi: 10.1016/0092-8674(87)90745-8. [DOI] [PubMed] [Google Scholar]
- 77.Okutani D, Lodyga M, Han B, Liu M. Src protein tyrosine kinase family and acute inflammatory responses. Am J Physiol Lung Cell Mol Physiol. 2006;291:L129–L141. doi: 10.1152/ajplung.00261.2005. [DOI] [PubMed] [Google Scholar]
- 78.Salmond RJ, Filby A, Qureshi I, Caserta S, Zamoyska R. T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunol Rev. 2009;228:9–22. doi: 10.1111/j.1600-065X.2008.00745.x. [DOI] [PubMed] [Google Scholar]
- 79.Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004;23:7990–8000. doi: 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- 80.Place AT, Chen Z, Bakhshi FR, Liu G, O'Bryan JP, et al. Cooperative role of caveolin-1 and C-terminal Src kinase binding protein in C-terminal Src kinase-mediated negative regulation of c-Src. Mol Pharmacol. 2011;80:665–672. doi: 10.1124/mol.111.073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kaimachnikov NP, Kholodenko BN. Toggle switches, pulses and oscillations are intrinsic properties of the Src activation/deactivation cycle. FEBS J. 2009;276:4102–4118. doi: 10.1111/j.1742-4658.2009.07117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang S, Yu D. Targeting Src family kinases in anti-cancer therapies: turning promise into triumph. Trends Pharmacol Sci. 2012;33:122–128. doi: 10.1016/j.tips.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huveldt D, Lewis-Tuffin LJ, Carlson BL, Schroeder MA, Rodriguez F, et al. Targeting Src family kinases inhibits bevacizumab-induced glioma cell invasion. PLoS One. 2013;8:e56505. doi: 10.1371/journal.pone.0056505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Park SI, Zhang J, Phillips KA, Araujo JC, Najjar AM, et al. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 2008;68:3323–3333. doi: 10.1158/0008-5472.CAN-07-2997. [DOI] [PubMed] [Google Scholar]
- 85.Herold CI, Chadaram V, Peterson BL, Marcom PK, Hopkins J, et al. Phase II Trial of Dasatinib in Patients with Metastatic Breast Cancer Using Real-Time Pharmacodynamic Tissue Biomarkers of Src Inhibition to Escalate Dosing. Clin Cancer Res. 2011;17:6061–6070. doi: 10.1158/1078-0432.CCR-11-1071. [DOI] [PubMed] [Google Scholar]
- 86.Gucalp A, Sparano JA, Caravelli J, Santamauro J, Patil S, et al. Phase II Trial of Saracatinib (AZD0530), an Oral SRC-inhibitor for the Treatment of Patients with Hormone Receptor-negative Metastatic Breast Cancer. Clin Breast Cancer. 2011 doi: 10.1016/j.clbc.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anbalagan M, Carrier L, Glodowski S, Hangauer D, Shan B, et al. KX-01, a novel Src kinase inhibitor directed toward the peptide substrate site, synergizes with tamoxifen in estrogen receptor alpha positive breast cancer. Breast Cancer Res Treat. 2011 doi: 10.1007/s10549-011-1513-3. [DOI] [PubMed] [Google Scholar]
- 88.Fujisaka Y, Onozawa Y, Kurata T, Yasui H, Goto I, et al. First report of the safety, tolerability, and pharmacokinetics of the Src kinase inhibitor saracatinib (AZD0530) in Japanese patients with advanced solid tumours. Invest New Drugs. 2013;31:108–114. doi: 10.1007/s10637-012-9809-7. [DOI] [PubMed] [Google Scholar]
- 89.Jiang XB, Ohno K, Qian L, Tominaga B, Kuroiwa T, et al. Changes in local cerebral blood flow, glucose utilization, and mitochondrial function following traumatic brain injury in rats. Neurol Med Chir (Tokyo) 2000;40:16–28. doi: 10.2176/nmc.40.16. discussion 28-19. [DOI] [PubMed] [Google Scholar]
- 90.Simon R, Shiraishi K. N-methyl-D-aspartate antagonist reduces stroke size and regional glucose metabolism. Ann Neurol. 1990;27:606–611. doi: 10.1002/ana.410270604. [DOI] [PubMed] [Google Scholar]
- 91.Ohab JJ, Fleming S, Blesch A, Carmichael ST. A neurovascular niche for neurogenesis after stroke. J Neurosci. 2006;26:13007–13016. doi: 10.1523/JNEUROSCI.4323-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425:479–494. doi: 10.1002/1096-9861(20001002)425:4<479::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 93.Bernabeu R, Sharp FR. NMDA and AMPA/kainate glutamate receptors modulate dentate neurogenesis and CA3 synapsin-I in normal and ischemic hippocampus. J Cereb Blood Flow Metab. 2000;20:1669–1680. doi: 10.1097/00004647-200012000-00006. [DOI] [PubMed] [Google Scholar]
- 94.Liu J, Solway K, Messing RO, Sharp FR. Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J Neurosci. 1998;18:7768–7778. doi: 10.1523/JNEUROSCI.18-19-07768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Higashida T, Kreipke CW, Rafols JA, Peng C, Schafer S, et al. The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg. 2011;114:92–101. doi: 10.3171/2010.6.JNS10207. [DOI] [PubMed] [Google Scholar]
- 96.Karni R, Dor Y, Keshet E, Meyuhas O, Levitzki A. Activated pp60c-Src leads to elevated hypoxia-inducible factor (HIF)-1alpha expression under normoxia. J Biol Chem. 2002;277:42919–42925. doi: 10.1074/jbc.M206141200. [DOI] [PubMed] [Google Scholar]
- 97.Madri JA. Modeling the neurovascular niche: implications for recovery from CNS injury. J Physiol Pharmacol. 2009;60(Suppl 4):95–104. [PubMed] [Google Scholar]
- 98.Hannon GJ. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 99.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, et al. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 100.de Fougerolles A, Vornlocher HP, Maraganore J, Lieberman J. Interfering with disease: a progress report on siRNA-based therapeutics. Nat Rev Drug Discov. 2007;6:443–453. doi: 10.1038/nrd2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Eifler AC, Thaxton CS. Nanoparticle therapeutics: FDA approval, clinical trials, regulatory pathways, and case study. Methods Mol Biol. 2011;726:325–338. doi: 10.1007/978-1-61779-052-2_21. [DOI] [PubMed] [Google Scholar]
- 102.Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm. 2009;6:659–668. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- 103.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]