

Abstract
Regulation of many biological processes in eukaryotes involves distant communication between the regulatory DNA sequences (e.g. enhancers) and their targets over the DNA regions organized in chromatin. However previously developed methods for analysis of communication in chromatin in vitro are artifact-prone and/or do not allow analysis of communication on physiologically relevant, saturated arrays of nucleosomes. Here we describe a method for quantitative analysis of the rate of distant communication in cis on saturated arrays of nucleosomes capable of forming the 30-nm chromatin fibers in vitro.
Keywords: Chromatin, nucleosome, enhancer, promoter, transcription
1. Introduction
Many DNA transactions in eukaryotic nuclei involve protein-mediated interactions between two or more DNA sites widely separated along DNA that is organized into chromatin. Such processes most often are accompanied by direct interaction between proteins bound at the enhancer and target promoter with accompanying formation of large chromatin loops that includes the intervening DNA in vivo (1–6). Therefore efficient enhancer action over a distance critically depends on structural and dynamic communication properties of chromatin that are largely unknown.
Several experimental approaches for analysis of distant enhancer-promoter communication have been developed recently. The FLP DNA recombination assay was employed for measuring communication over 74-bp to 15-kb distances in chromatin in vivo (7). However applicability of this method for studies of communication in chromatin in vitro has not been evaluated. Furthermore, communication between the DNA sequences required for recombination and regulation over a distance may occur by different mechanisms. Thus, recombination can occur between DNA sequences positioned within different, sometimes widely spaced domains of chromatin while enhancer-promoter communication is largely limited by a single chromatin loop (8).
DNA ligation-circularization assay was used as an alternative method for analysis of intramolecular communication over a distance on DNA (9–11) and in chromatin in vitro (12). However extensive internucleosomal interactions strongly complicate interpretation of the experiments (Y.S.P., data not shown). Furthermore, the ligation-circularization assay can be applied only to linear DNA molecules and therefore communication in chromatin cannot be studied using supercoiled DNA templates.
Recently we have developed an experimental assay allowing quantitative analysis of the rate of distant communication between a bacterial transcription regulatory element (enhancer) and its target (promoter) in chromatin (13, 14). The use of the bacterial experimental system is dictated by the low efficiency of eukaryotic RNA polymerase II-dependent in vitro transcription systems (15) and inconsistency of eukaryotic enhancer action over a distance in vitro (16, 17). At the same time, bacterial transcriptional enhancers can work efficiently over a large distance (up to at least 5 kb) both in vivo and in vitro (18–20). Moreover, pro- and eukaryotic transcriptional enhancers share many key properties, such as the looping mechanism of enhancer-promoter communication (21). The mechanism of action of bacterial transcriptional enhancers has been extensively studied using the glnAp2 promoter of Escherichia coli as a model (18, 19). Transcriptional activity of glnAp2 promoter is entirely controled by the NtrC-dependent, σ54-dependent transcriptional enhancer (20, 22, 23). The enhancer is activated by the NtrC protein, which is phosphorylated by the NtrB protein kinase (24, 25). When phosphorylated, enhancer-bound NtrC interacts with the Eσ54 holoenzyme and stimulates conversion of the closed (RPclosed) to the open (RPopen) initiation complex (20, 22, 26–29). During this direct enhancer-promoter interaction, intervening DNA is transiently looped out (30, 31).
This experimental system is relatively simple, highly efficient, and is very well studied. Transcription is strongly (>100-fold) stimulated by the enhancer and the mechanism of communication can be analyzed both in vitro and in vivo. Activity of the promoter itself in this system does not depend on the level of negative DNA supercoiling (Y.S.P., unpublished data) allowing analysis of communication properties of linear, relaxed or supercoiled DNA and chromatin templates. Using this experimental technique, the mechanisms of distant communication on histone-free DNA (18, 19) and in chromatin (13, 14) have been studied. In our previous studies of communication in chromatin sub-saturated arrays of randomly positioned nucleosomes were utilized (32, 33). However nucleosomes under physiologically relevant conditions are organized in regularly spaced, saturated arrays of nucleosomes forming higher-order chromatin structure (the 30-nm fibers (34, 35)). Here we describe an experimental approach that allows analysis of communication properties of structurally defined, saturated arrays of precisely positioned nucleosomes capable of 30-nm chromatin fiber formation.
2. Materials
2.1. Buffers and Reagents
TAE buffer (1×): 40 mM Tris-HCl (pH 7.6), 31.2 mM Acetic Acid, 1 mM EDTA.
TBE buffer (0.5×): 89 mM Tris-HCl (pH 8.3), 89 mM Boric Acid, 1 mM EDTA.
Chromatin Reconstitution Buffer A: 1 M NaCl, 10 mM Tris-HCl (pH 7.5), 0.2 mM EDTA, 0.1% NP40 and 5 mM β-mercaptoethanol.
Chromatin Reconstitution Buffer B: 10 mM Tris-HCl (pH 7.5), 0.2 mM EDTA, 0.1% NP40 and 5 mM β-mercaptoethanol.
NEBuffer #2 (1×, New England Biolabs): 50 mM NaCl, 10 mM Tris-HCl (pH 7.9), 10 mM MgCl2, 1 mM DTT.
ThermoPol Buffer (1×, New Englad Biolabs): 20 mM Tris-HCl (pH 8.8), 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4, 0.1% Triton-X100.
Transcription buffer (1×): 50 mM Tris-Ac (pH 8.0), 100 mM KAc, 8 mM Mg(Ac)2, 27 mM NH4Ac, 0.7% PEG-8000 and 0.2 mM DTT.
2.2. Native Electrophoresis of Reconstituted Chromatin
Native agarose gel: 1.2% agarose, 20 mM HEPES-Na (pH 8.0), 0.2 mM EDTA, 5% glycerol.
Running buffer: 20 mM HEPES-Na (pH 8.0), 0.2 mM EDTA.
Electrophoresis was carried out in a vertical apparatus between two glass plates using 1 mm spacers at 120 volts for approximately 1.5 hours until the bromphenol blue front reached the bottom of the gel.
2.3. Denaturing PAGE of Purified DNA and RNA
8% denaturing polyacrylamide gel (19:1) containing 0.5× TBE buffer and 8 M urea was prepared.
Running buffer: 0.5× TBE buffer.
Electrophoresis was carried out in a vertical apparatus between two 20 × 30 cm glass plates using 0.4 mm spacers at 2000 volts and no more than 50 watts for approximately 1 hour until the bromphenol blue front reached the bottom of the gel.
3. Methods
3.1. Design of the DNA Template for Analysis of Distant Communication
Assembly of saturated arrays is impossible using the experimental techniques developed earlier because randomly positioned nucleosomes can assemble on the enhancer and promoter and completely block these DNA elements from binding of the proteins when the level of chromatin assembly approaches saturation (32). To prevent nucleosome assembly on the enhancer and promoter and to form saturated arrays of precisely positioned nucleosomes on the spacer DNA we have employed a high-affinity histone-binding, nucleosome positioning (601) sequence (13, 14). The 601 sequence support formation of precisely positioned arrays of nucleosomes that can form the 30-nm chromatin fibers in the absence of linker histones H1/H5 (35, 36). Furthermore, if reconstitution of the arrays is conducted in the presence of an excess of competitor DNA and a limited supply of histone octamers, nucleosomes can form on the 601 sequences with a high preference (37) leaving the enhancer and promoter histone-free and accessible to corresponding DNA-binding proteins.
The overall experimental approach for the transcriptional analysis of distant enhancer-promoter communication in chromatin is outlined in Fig. 1B.
The pYP05 plasmid (Fig. 1B (19, 38), see Note 1) used as template for chromatin reconstitution and in vitro transcription was purified using the QIAfilter Plasmid Maxi Kit (Qiagen)
Figure 1.
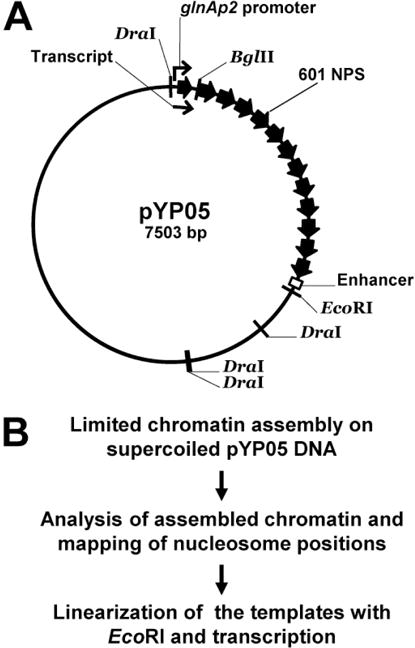
An experimental approach for the transcriptional analysis of distant enhancer-promoter communication in chromatin. A. Restriction map of the pYP05 template. Positions of the enhancer, promoter and 13 high-affinity nucleosome positioning 601 sequences (601 NPS) are indicated. 35 bp of the promoter-proximal NPS is truncated from its promoter-distal end and is replaced by bacteriophage T7 transcription terminator site, limiting the length of the transcript to 176 nucleotides. The length of the internucleosomal spacer DNA between each two NPSs is 29 bp. B. The experimental approach.
3.2. Chromatin Assembly on Supercoiled DNA
Chromatin reconstitution was conducted on negatively supercoiled pYP05 plasmid at different mass ratios of donor chromatin to template DNA (1:4, 1:2, 1:1) by a modified transfer method using continuous dialysis from 1 M NaCl (39, 40). Long chromatin from chicken erythrocytes was used as a donor of wild type core histone octamers.
Reconstitution of chromatin was conducted at 10 nM (50 μg/ml) DNA concentration and 12.5 μg/ml, 25 μg/ml or 50 μg/ml of donor chromatin (1:4, 1:2 or 1:1 chromatin/DNA mass ratios, respectively) to achieve sub-saturated, saturated and over-saturated levels of chromatin assembly, respectively.
DNA and donor chromatin were mixed in reconstitution buffer A in a total volume of 120 μl, transferred into a small dialysis bag (Spectra/Por, MWCO 12-14000) and placed into a glass bottle containing 100 ml of reconstitution buffer A.
The bottle was then tightly sealed and connected to the gradient maker, the inner beaker was filled with 500 ml of buffer A and the outer beaker contained 500 ml of buffer B.
Dialysis was performed at 4°C in cold room in gradient maker and on magnetic stirrer overnight at flow rate of 1 ml/min created by peristaltic pump.
The vast majority (>90%) of the nucleosomes on the saturated and oversaturated reconstituted chromatin samples are positioned on the NPSs (see Note 2).
3.3. Characterization of the Reconstituted Chromatin in a Native Gel
In a native gel DNA-protein complexes remain intact. The mobility in the gel is determined by the charge, mass and shape of the complexes.
For this experiment 5′-radioactively labeled ApoI/EcoRI-restriction fragment of pYP05 plasmid (Fig. 1A) was used. It comprises the promoter and enhancer separated by 13 NPSs.
After nucleosome assembly (see Note 3) chromatin samples were analyzed by native agarose gel electrophoresis.
After the electrophoresis the gel was transferred to Whatman 3MM paper, covered with polyethylene wrap and dried for 20 minutes at 50°C and then for 20 minutes at 80°C.
The dried gel was exposed overnight to PhosphorScreen (Perkin Elmer) and the screen was scanned on Cyclone PhosphorImager (Perkin Elmer, Fig. 2).
Figure 2.
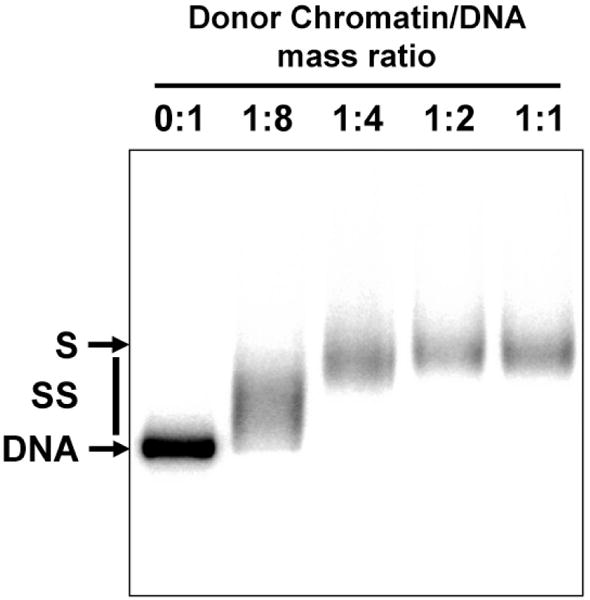
Analysis of chromatin templates by native PAGE. Chromatin was assembled on the radioactively labeled 2.5 kb ApoI/EcoRI pYP05 fragment at the indicated mass ratios of donor chromatin to template DNA. Chromatin assembly results in a progressive decrease of the mobility of the complexes in the gel. Chromatin reconstituted at 1:2 ratio of the donor-chromatin to DNA is saturated (S), since further increase in the ratio up to 1:1 does not result in further shift of the corresponding band. Saturated chromatin assembled at 1:2 ratio was used in the transcription experiment (Fig. 5).
3.4. Characterization of Reconstituted Chromatin using a Restriction Enzyme Sensitivity Assay
This is a simple and fast assay to evaluate DNA occupancy by nucleosomes. The assay is based on the observation that chromatin assembly results in strong protection of nucleosome-covered DNA from digestion with restriction enzymes (41, 42). The cutting sites should be chosen to generate DNA fragments of different lengths to facilitate their separation in the agarose gel and analysis of the gel. In the case presented below the sites were chosen in the NPS-free region of the plasmid to evaluate the extent of undesired nucleosome assembly. However a similar technique could be applied to any DNA region on the plasmid (32, 33).
750 ng of DNA or nucleosomal templates (20 μg/ml) were incubated in the presence of an excess of DraI and BglII restriction endonucleases (10 units each) in the NEBuffer #2 at 37°C for 2 hours.
DNA was purified by phenol/chloroform extraction followed by ethanol precipitation and analyzed by electrophoresis in 1% agarose/TAE gel. The intensities of the bands in the gel were quantified using the OptiQuant software (Perkin Elmer, see Note 4).
The intensities of the bands corresponding to the final products of digestion are decreased as the efficiency of chromatin assembly is increased (Fig. 3). The extent of chromatin assembly is directly proportional to the decrease in the intensity of the bands. However even on saturated chromatin (1:2, Fig. 3) the analyzed restriction sites remain largely accessible suggesting (in combination with the data shown in Fig. 2) that nucleosomes are poorly formed on plasmid DNA regions that do not contain NPSs.
Figure 3.
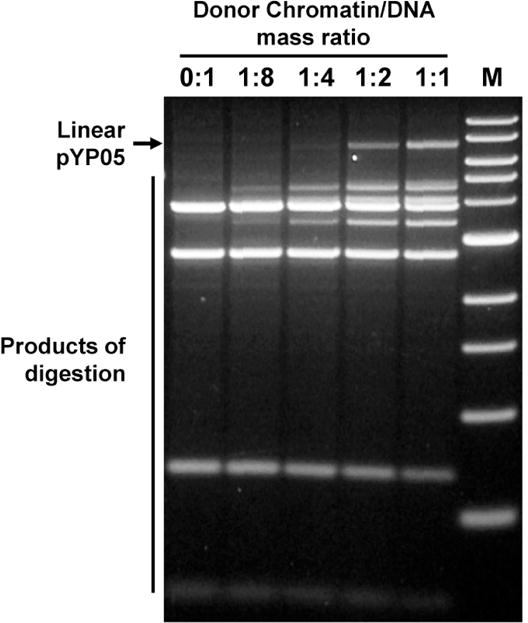
Characterization of chromatin templates using restriction enzyme sensitivity assay. Chromatin was assembled on supercoiled pYP05 plasmid and digested with an excess of restriction enzymes DraI and BglII. Then DNA was purified and analyzed in 1% agarose gel. Nucleosomes protect DNA from digestion with restriction enzymes. The restriction sites are localized beyond the NPSs (Fig. 1B); therefore the increase of donor-chromatin/DNA ratio results in progressively better protection of the template DNA from the enzymes, but this protection is still minimal even at highest ratio of donor chromatin to DNA (1:1), indicating that the majority of nucleosomes were formed predominantly on the desired NPSs. Only a small fraction (<5%) of all nucleosomes formed on the plasmid occupy the NPS-free regions of the pYP05 plasmid. M – 1-kb DNA ladder (New England Biolabs).
3.5. Characterization of Reconstituted Chromatin using a Restriction Enzyme Sensitivity Assay with Primer Extension
This is a more advanced and involved version of the restriction enzyme sensitivity assay (see 3.4) allowing more straightforward, quantitative and high-resolution analysis of nucleosome occupancy and positioning on polynucleosomal templates. Overall experimental approach is illustrated on Fig. 4A.
Chromatin samples were digested with an excess of restriction enzymes AluI, MspI or ScaI (Fig. 4B), followed by phenol/chloroform extraction and ethanol precipitation of DNA.
Purified DNA fragments obtained after AluI, MspI or ScaI digestion were further digested with EcoRI restriction enzyme (Fig. 1B) to set the 3′-end; the 5′-end is set by the primer (Fig. 4B).
EcoRI-digested DNA was subjected to primer annealing (Fig. 4B, see Note 5). The annealed primer was extended with Taq DNA polymerase (New England Biolabs) in 1× ThermoPol buffer and conditions recommended by manufacturer.
The products of extension were purified by phenol/chloroform extraction, ethanol precipitation and analyzed by a denaturing PAGE.
The distribution of the bands in the gel (Fig. 4C) reflects the sensitivity of each restriction site to corresponding restriction enzyme in chromatin and allows quantitative analysis of nucleosome occupancy (see Note 6).
Figure 4.
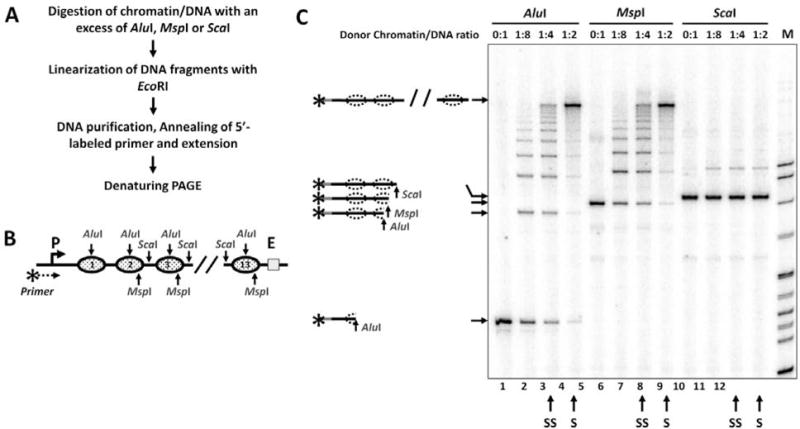
Analysis of chromatin templates using a restriction digestion sensitivity assay with primer extension. A. The experimental approach. B. Schematic diagram of the fragment of pYP05 plasmid used for in vitro chromatin reconstitution. Nucleosome positioning sequences (NPS) are shown by solid ovals. Restriction enzyme sites within the NPSs are shown by arrows. AluI and MspI sites are localized within NPS, ScaI sites - in the middle of NPS-separating spacer DNA. MspI and ScaI sites are missing from the first NPS and the spacer DNA respectively. C. Analysis of the products of primer extension by a denaturing PAGE. The increase in the level of chromatin assembly results in progressively better protection of the templates from AluI and MspI, but not from ScaI restriction enzyme. Possible products of digestion are shown on the right. S/SS: Saturated/sub-saturated chromatin samples.
3.6. Analysis of Communication in Chromatin using a Single-round Transcriptional Assay In Vitro
In vitro transcription assay was optimized for maximal utilization of the chromatin templates and was used previously to measure the rate of enhancer-promoter (E-P) communication (Fig. 5A (33, 43)). Physical E-P interaction is the rate-limiting step during transcription (19, 33). E-P communication and direct interaction of corresponding bound protein are accompanied by looping of the intervening DNA (30, 31) and are required for conversion of the closed (RPclosed) to the open initiation complex (RPopen) (20, 24–28). The RPopen formed at the promoter is stable in the presence of heparin; however its formation de novo is strongly inhibited (44). Heparin also disrupts nucleosomes, so that they are present only during initiation step, but not during elongation. Therefore when E-P communication is aloowed for a limited time (1 min) the overall efficiency of transcription conducted in the presence of heparin can serve as a direct and quantitative measure of the rate of E-P communication (see Note 7 (43, 45)).
All templates were linearized at the EcoRI site (Fig. 1A), and the closed initiation complexes (RPclosed) were formed in 50-μl aliquots in 1× transcription buffer (TB) at 1 nM chromatin concentration and 10 nM core RNA polymerase, 300 nM σ54, 120 nM NtrC, and 400 nM NtrB for 15 min at 37°C.
5 μl of 40 mM ATP in 1× TB were added to the reaction volume to 4 mM final ATP concentration, and the reaction was incubated at 37°C for 1 more minute to form the open, elongation-competent initiation complex (RPopen).
To start elongation and to limit it to a single round (prevent further formation of the RPopen) a mixture of all four ribonucleotide-triphospates (4 mM each) in 1× TB with 2.5 μCi of [α-32P]-GTP (3000 Ci/mmol) and 2 mg/ml heparin was added to the reaction.
The reaction was continued at 37°C for 15 minutes and terminated with an equal volume of phenol/chloroform (1:1).
The samples were precipitated with ethanol, dissolved in formamide-containing loading solution, denatured at 95°C for 5 minutes, cooled on ice and separated by denaturing PAGE.
The gel was transferred to Whatman 3MM paper, covered with polyethylene wrap and dried for 30 minutes at 80°C.
PhosphorScreen (Perkin Elmer) was placed above the dried gel, exposed overnight and scanned on Cyclone PhosphorImager (Perkin Elmer). The data was quantified using the OptiQuant software (Fig. 5B, see Note 8).
Figure 5.
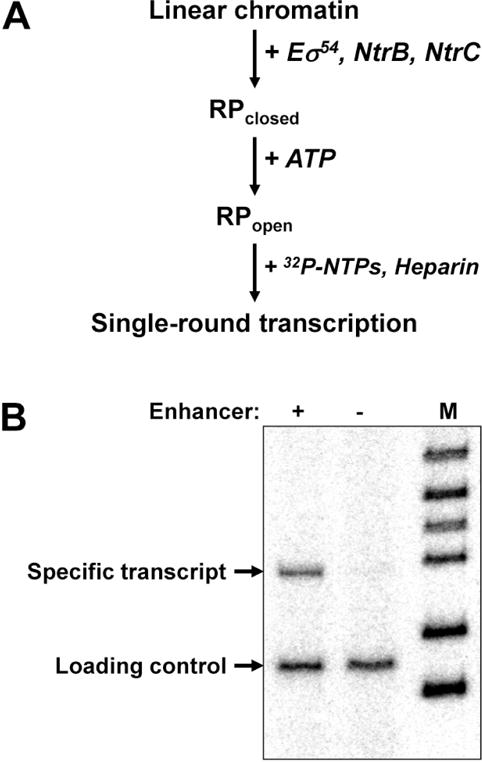
Analysis of enhancer-dependent glnAp2 promoter activation in chromatin using the single-round transcription assay. A. The experimental approach. Eσ54: RNA polymerase holoenzyme that recognizes the glnAp2 promoter. NtrB: protein kinase that phosphorylates NtrC (transcription activator that after the phosphorylation interacts with the enhancer and activates transcription). RPclosed and RPopen – closed and open initiation complexes, respectively. B. Transcription of enhancer-containing and enhancer-less saturated chromatin templates. The 3.9-kb HindIII/PstI fragment of the pYP05 plasmid containing (+) the enhancer or 3.0-kb EcoRV fragment missing it (−) were assembled into saturated arrays of nucleosomes and transcribed using the single-round transcription assay. Chromatin structure supports efficient transcription that is almost entirely enhancer-dependent. M – end-labeled pBR322-MspI digest. End-labeled DNA fragment was added to the reactions as a loading control.
Acknowledgments
This work was supported by NSF (0549593) and NIH (GM58650) grants to V.M.S. We would like to thank Dr. T. J. Richmond for providing the 12×177-601 plasmid containing array of twelve 601 NPSs.
Footnotes
The pYP05 plasmid contains an NtrC-dependent enhancer that strongly activates the glnAp2 promoter over 2.5 kb distance (19, 38). It contains thirteen strong 601 nucleosome positioning sequences (NPS) between the enhancer and promoter. The remaining portion of the plasmid does not contain any DNA sequences having high affinity to histones and therefore serves as a “sink” for the excess of histones or histone octamers during reconstitution. Thus chromatin assembly on the plasmid can be conducted either using purified histones or donor chromatin; in both cases the presence of DNA “sink” guarantees complete occupancy of NPSs and minimal promoter/enhancer blockage.
After reconstitution of the saturated arrays of 601 nucleosomes only one to two randomly positioned nucleosomes are formed on the DNA region of the plasmid that does not contain NPSs (see Figs 2 and 3; Y.S.P., data not shown). Use of regularly spaced nuclesomal templates allows structural interpretation of the obtained data because the X-ray structures of nucleosomes and the 30-nm fibers formed on the templates have been solved (36, 46, 47).
Chromatin assembly on the linear fragment was conducted using the same protocol as in the case of assembly on supercoiled DNA (part 3.2). Presence of an excess of donor chromatin in the reaction does not result in binding of an excessive amounts of core histones when chromatin is reconstituted by histone transfer from donor chromatin (Fig. 2). However reconstitution from purified histones (40) has to be conducted much more carefully because even small excess of purified histones can result in complete occupancy of the promoter and enhancer in the absence of DNA “sink” (see Note 1). In this case, a non-specific competitor DNA with random sequence can be added during reconstitution to serve as DNA “sink”.
The loading was adjusted to guarantee that intensities of the bands are in linear range of the measuring device. When needed, the standards (DNA fragments with known concentrations) were separated in the same gel.
The primer (5′-gaatttcgagggcatgataacgccttttaggg-3′) is localized immediately upstream of the promoter (Fig. 4B) and was 5′-end-labeled with γ[32]-ATP (Perkin Elmer) by polynucleotide kinase (NEB) according to the manufacturer protocol.
In the particular example (Fig. 4C), DNA is completely sensitive to each of the restriction enzymes (lanes 1, 5 and 9) indicating the absence of nucleosomes. In sub-saturated chromatin all bands on the gel have similar intensities after AluI or MspI digestion (lanes 3 and 7) indicating that in each chromatin sample on average one of the 13 AluI or 12 MspI sites is accessible to the enzymes cutting within NPSs. Thus on average one of the 13 nucleosomes is missing in sub-saturated chromatin. In saturated chromatin all NPSs are >95% protected from AluI and/or MspI digestion (lanes 4 and 8) indicating >95% nucleosome occupancy of each site. At the same time, non-NPS, internucleosomal linker DNA regions are fully sensitive to ScaI both in sub-saturated and saturated chromatin (lanes 11 and 12) indicating that nucleosomes preferentially occupy NPSs (less than 5% of nucleosomes are formed on the linker DNA regions).
Enhancer-promoter communication in chromatin occurs only in cis (32) allowing analysis of the same intramolecular interactions that occur in vivo. It depends on the presence of all functional components involved in enhancer action on histone-free DNA (32, 33).
The rates of enhancer-promoter communication on DNA, supercoiled or relaxed chromatin templates can be measured quantitatively using the single-round transcription assay. In this case, communication is also initiated by adding ATP after pre-formation of the RPclosed and NtrC-DNA complexes. This approach is similar to the one described in section 3.6., but ATP should be added for various time intervals (0, 1, 2, 4, 8, 16 or 32 minutes) to allow E-P communication for different times. Then t1/2 for the rates of communication can be calculated.
References
- 1.de Laat W, Grosveld F. Spatial organization of gene expression: the active chromatin hub. Chromosome Res. 2003;11:447–459. doi: 10.1023/a:1024922626726. [DOI] [PubMed] [Google Scholar]
- 2.Dean A. Chromatin remodelling and the interaction between enhancers and promoters in the beta-globin locus. Brief Funct Genomic Proteomic. 2004;2:344–354. doi: 10.1093/bfgp/2.4.344. [DOI] [PubMed] [Google Scholar]
- 3.Tsytsykova AV, Rajsbaum R, Falvo JV, Ligeiro F, Neely SR, Goldfeld AE. Activation-dependent intrachromosomal interactions formed by the TNF gene promoter and two distal enhancers. Proc Natl Acad Sci USA. 2007;104:16850–16855. doi: 10.1073/pnas.0708210104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carter D, Chakalova L, Osborne CS, Dai YF, Fraser P. Long-range chromatin regulatory interactions in vivo. Nat Genet. 2002;32:623–626. doi: 10.1038/ng1051. [DOI] [PubMed] [Google Scholar]
- 5.Kooren J, Palstra RJ, Klous P, Splinter E, von Lindern M, Grosveld F, de Laat W. Beta-globin active chromatin Hub formation in differentiating erythroid cells and in p45 NF-E2 knock-out mice. J Biol Chem. 2007;282:16544–16552. doi: 10.1074/jbc.M701159200. [DOI] [PubMed] [Google Scholar]
- 6.Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, Grosveld F, Galjart N, de Laat W. CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev. 2006;20:2349–2354. doi: 10.1101/gad.399506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ringrose L, Chabanis S, Angrand PO, Woodroofe C, Stewart AF. Quantitative comparison of DNA looping in vitro and in vivo: chromatin increases effective DNA flexibility at short distances. EMBO J. 1999;18:6630–6641. doi: 10.1093/emboj/18.23.6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.West AG, Fraser P. Remote control of gene transcription. Hum Mol Genet. 2005;14(Spec No 1):R101–111. doi: 10.1093/hmg/ddi104. [DOI] [PubMed] [Google Scholar]
- 9.Shore D, Langowski J, Baldwin RL. DNA flexibility studied by covalent closure of short fragments into circles. Proc Natl Acad Sci USA. 1981;78:4833–4837. doi: 10.1073/pnas.78.8.4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cloutier TE, Widom J. Spontaneous sharp bending of double-stranded DNA. Mol Cell. 2004;14:355–362. doi: 10.1016/s1097-2765(04)00210-2. [DOI] [PubMed] [Google Scholar]
- 11.Crothers DM, Drak J, Kahn JD, Levene SD. DNA bending, flexibility, and helical repeat by cyclization kinetics. Methods Enzymol. 1992;212:3–29. doi: 10.1016/0076-6879(92)12003-9. [DOI] [PubMed] [Google Scholar]
- 12.Stein A, Dalal Y, Fleury TJ. Circle ligation of in vitro assembled chromatin indicates a highly flexible structure. Nucleic Acids Res. 2002;30:5103–5109. doi: 10.1093/nar/gkf671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 14.Thastrom A, Lowary PT, Widlund HR, Cao H, Kubista M, Widom J. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J Mol Biol. 1999;288:213–229. doi: 10.1006/jmbi.1999.2686. [DOI] [PubMed] [Google Scholar]
- 15.Knezetic JA, Jacob GA, Luse DS. Assembly of RNA polymerase II preinitiation complexes before assembly of nucleosomes allows efficient initiation of transcription on nucleosomal templates. Mol Cell Biol. 1988;8:3114–3121. doi: 10.1128/mcb.8.8.3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laybourn PJ, Kadonaga JT. Threshold phenomena and long-distance activation of transcription by RNA polymerase II. Science. 1992;257:1682–1685. doi: 10.1126/science.1388287. [DOI] [PubMed] [Google Scholar]
- 17.Ptashne M, Gann AA. Activators and targets. Nature. 1990;346:329–331. doi: 10.1038/346329a0. [DOI] [PubMed] [Google Scholar]
- 18.Bondarenko VA, Jiang YI, Studitsky VM. Rationally designed insulator-like elements can block enhancer action in vitro. EMBO J. 2003;22:4728–4737. doi: 10.1093/emboj/cdg468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Bondarenko V, Ninfa A, Studitsky VM. DNA supercoiling allows enhancer action over a large distance. Proc Natl Acad Sci USA. 2001;98:14883–14888. doi: 10.1073/pnas.261477898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Popham DL, Szeto D, Keener J, Kustu S. Function of a bacterial activator protein that binds to transcriptional enhancers. Science. 1989;243:629–635. doi: 10.1126/science.2563595. [DOI] [PubMed] [Google Scholar]
- 21.Bondarenko VA, Liu YV, Jiang YI, Studitsky VM. Communication over a large distance: enhancers and insulators. Biochem Cell Biol. 2003;81:241–251. doi: 10.1139/o03-051. [DOI] [PubMed] [Google Scholar]
- 22.Sasse-Dwight S, Gralla JD. Probing the Escherichia coli glnALG upstream activation mechanism in vivo. Proc Natl Acad Sci USA. 1988;85:8934–8938. doi: 10.1073/pnas.85.23.8934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ninfa AJ, Reitzer LJ, Magasanik B. Initiation of transcription at the bacterial glnAp2 promoter by purified E. coli components is facilitated by enhancers. Cell. 1987;50:1039–1046. doi: 10.1016/0092-8674(87)90170-x. [DOI] [PubMed] [Google Scholar]
- 24.Ninfa AJ, Magasanik B. Covalent modification of the glnG product, NRI, by the glnL product, NRII, regulates the transcription of the glnALG operon in Escherichia coli. Proc Natl Acad Sci USA. 1986;83:5909–5913. doi: 10.1073/pnas.83.16.5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keener J, Kustu S. Protein kinase and phosphoprotein phosphatase activities of nitrogen regulatory proteins NTRB and NTRC of enteric bacteria: roles of the conserved amino-terminal domain of NTRC. Proc Natl Acad Sci USA. 1988;85:4976–4980. doi: 10.1073/pnas.85.14.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porter SC, North AK, Wedel AB, Kustu S. Oligomerization of NTRC at the glnA enhancer is required for transcriptional activation. Genes Dev. 1993;7:2258–2273. doi: 10.1101/gad.7.11.2258. [DOI] [PubMed] [Google Scholar]
- 27.Wedel A, Kustu S. The bacterial enhancer-binding protein NTRC is a molecular machine: ATP hydrolysis is coupled to transcriptional activation. Genes Dev. 1995;9:2042–2052. doi: 10.1101/gad.9.16.2042. [DOI] [PubMed] [Google Scholar]
- 28.Wyman C, Rombel I, North AK, Bustamante C, Kustu S. Unusual oligomerization required for activity of NtrC, a bacterial enhancer-binding protein. Science. 1997;275:1658–1661. doi: 10.1126/science.275.5306.1658. [DOI] [PubMed] [Google Scholar]
- 29.Buck M, Cannon W. Activator-independent formation of a closed complex between sigma 54-holoenzyme and nifH and nifU promoters of Klebsiella pneumoniae. Mol Microbiol. 1992;6:1625–1630. doi: 10.1111/j.1365-2958.1992.tb00887.x. [DOI] [PubMed] [Google Scholar]
- 30.Su W, Porter S, Kustu S, Echols H. DNA-looping and enhancer activity: association between DNA-bound NtrC activator and RNA polymerase at the bacterial glnA promoter. Proc Natl Acad Sci USA. 1990;87:5504–5508. doi: 10.1073/pnas.87.14.5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rippe K, Guthold M, von Hippel PH, Bustamante C. Transcriptional activation via DNA-looping: visualization of intermediates in the activation pathway of E. coli RNA polymerase × sigma 54 holoenzyme by scanning force microscopy. J Mol Biol. 1997;270:125–138. doi: 10.1006/jmbi.1997.1079. [DOI] [PubMed] [Google Scholar]
- 32.Rubtsov MA, Polikanov YS, Bondarenko VA, Wang YH, Studitsky VM. Chromatin structure can strongly facilitate enhancer action over a distance. Proc Natl Acad Sci USA. 2006;103:17690–17695. doi: 10.1073/pnas.0603819103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polikanov YS, Rubtsov MA, Studitsky VM. Biochemical analysis of enhancer-promoter communication in chromatin. Methods. 2007;41:250–258. doi: 10.1016/j.ymeth.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas JO, Butler PJ. Changes in chromatin folding in solution. J Mol Biol. 1980;144:89–93. doi: 10.1016/0022-2836(80)90268-5. [DOI] [PubMed] [Google Scholar]
- 35.Dorigo B, Schalch T, Kulangara A, Duda S, Schroeder RR, Richmond TJ. Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science. 2004;306:1571–1573. doi: 10.1126/science.1103124. [DOI] [PubMed] [Google Scholar]
- 36.Schalch T, Duda S, Sargent DF, Richmond TJ. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005;436:138–141. doi: 10.1038/nature03686. [DOI] [PubMed] [Google Scholar]
- 37.Huynh VA, Robinson PJ, Rhodes D. A method for the in vitro reconstitution of a defined “30 nm” chromatin fibre containing stoichiometric amounts of the linker histone. J Mol Biol. 2005;345:957–968. doi: 10.1016/j.jmb.2004.10.075. [DOI] [PubMed] [Google Scholar]
- 38.Bondarenko V, Liu Y, Ninfa A, Studitsky VM. Action of prokaryotic enhancer over a distance does not require continued presence of promoter-bound sigma54 subunit. Nucleic Acids Res. 2002;30:636–642. doi: 10.1093/nar/30.3.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walter W, Kireeva ML, Tchernajenko V, Kashlev M, Studitsky VM. Assay of the fate of the nucleosome during transcription by RNA polymerase II. Methods Enzymol. 2003;371:564–577. doi: 10.1016/S0076-6879(03)71042-8. [DOI] [PubMed] [Google Scholar]
- 40.Walter W, Studitsky VM. Construction, analysis, and transcription of model nucleosomal templates. Methods. 2004;33:18–24. doi: 10.1016/j.ymeth.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 41.Studitsky VM. Preparation and analysis of positioned nucleosomes. Methods Mol Biol. 1999;119:17–26. doi: 10.1385/1-59259-681-9:17. [DOI] [PubMed] [Google Scholar]
- 42.Polach KJ, Widom J. Restriction enzymes as probes of nucleosome stability and dynamics. Methods Enzymol. 1999;304:278–298. doi: 10.1016/s0076-6879(99)04017-3. [DOI] [PubMed] [Google Scholar]
- 43.Bondarenko V, Liu YV, Ninfa AJ, Studitsky VM. Assay of prokaryotic enhancer activity over a distance in vitro. Methods Enzymol. 2003;370:324–337. doi: 10.1016/S0076-6879(03)70029-9. [DOI] [PubMed] [Google Scholar]
- 44.Feng J, Goss TJ, Bender RA, Ninfa AJ. Activation of transcription initiation from the nac promoter of Klebsiella aerogenes. J Bacteriol. 1995;177:5523–5534. doi: 10.1128/jb.177.19.5523-5534.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Polikanov YS, Bondarenko VA, Tchernaenko V, Jiang YI, Lutter LC, Vologodskii A, Studitsky VM. Probability of the site juxtaposition determines the rate of protein-mediated DNA looping. Biophys J. 2007;93:2726–2731. doi: 10.1529/biophysj.107.111245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 47.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J Mol Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]