

Abstract
We have established a physiologically relevant mechanism of CD4+ T cell-mediated neuroprotection involving axotomized wildtype (WT) mouse facial motoneurons (FMN) with significance in the treatment of amyotrophic lateral sclerosis (ALS), a fatal MN disease. Use of the transgenic mouse model of ALS involving expression of human mutant superoxide dismutase genes (SOD1G93A; abbreviated here as mSOD1) has accelerated basic ALS research. Superimposition of facial nerve axotomy (FNA) on the mSOD1 mouse during pre-symptomatic stages indicates that they behave like immunodeficient mice in terms of increased FMN loss and decreased functional recovery, through a mechanism that, paradoxically, is not inherent within the MN itself, but, instead, involves a defect in peripheral immune: CNS glial cell interactions. Our goal is to utilize our WT mouse model of immune-mediated neuroprotection after FNA as a template to elucidate how a malfunctioning peripheral immune system contributes to motoneuron cell loss in the mSOD1 mouse. This review will discuss potential immune defects in ALS, as well as provide an up-to-date understanding of how the CD4+ effector T cells provide neuroprotection to motoneurons through regulation of the central microglial and astrocytic response to injury. We will discuss an IL-10 cascade within the facial nucleus that requires a functional CD4+ T cell trigger for activation. The review will discuss the role of T cells in ALS, and our recent reconstitution experiments utilizing our model of T cell-mediated neuroprotection in WT vs mSOD1 mice after FNA. Identification of defects in neural:immune interactions could provide targets for therapeutic intervention in ALS.
Keywords: Motoneurons, Neuroprotection, Tcells, ALS, Neuroregeneration
Introduction
Over the past decade and a half, work from our laboratory has identified and characterized an immune-mediated model of endogenous neuroprotection following facial nerve axotomy in wild-type (WT) and immunodeficient recombinase-activating gene-2 knock-out (RAG-2−/−) mice lacking the adaptive arm of the immune system, while maintaining antigen-presenting cells (APC) of the innate arm of the immune system (Shinkai et al. 1992). In this review, we will present an up-to-date summary of our current understanding of how CD4+ T cells act to provide neuroprotection of mouse facial motoneurons (FMN) disconnected from their target musculature by complete transection of the facial nerve at its exit from the stylomastoid foramen (SMF).
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease involving progressive loss of motoneurons (MN), distal axonopathy, and paralysis of target muscle (Kennel et al. 1996; Rowland and Shneider 2001; Fischer et al. 2004; Hegedus et al. 2007; Park and Vincent 2008; Carrasco et al. 2010). Elegant work from multiple investigators has indicated that ALS pathophysiology involves a dysregulated immune response along with central neuroinflammation (Troost et al. 1992; O’Reilly et al. 1995; Graves et al. 2004; Henkel et al. 2004; Turner et al. 2004; Rafalowska et al. 2010; Sanagi et al. 2010; Mesnard et al. 2011). A widely used transgenic mouse model of ALS, involving the overexpression of human mutant superoxide dismutase-1 (SOD1G93A; abbreviated to mSOD1 in this review), develops disease pathology similar to that in familial and sporadic ALS patients (Gurney et al. 1994). Mariotti et al. demonstrated increased mSOD1 mouse FMN susceptibility to axotomy-induced cell death (Mariotti et al. 2002). The demonstration that axotomy increases cell loss in the animal model of ALS suggests that an additional central nervous system (CNS) pathology, beyond the initiating axonal die-back events, may contribute to differential axotomy-induced target deprivation responses. To explore the differences in response to axotomy by mSOD1 and WT mice, we extended the findings of Mariotti et al. (Mariotti et al. 2002) by superimposing facial nerve axotomy on pre-symptomatic mSOD1 mice and examining the molecular responses of both axotomized FMN and the surrounding microenvironment in the facial nucleus. In this review, we will summarize the use of axotomy as a tool to understand ALS pathogenesis in the mSOD1 mouse model.
Finally, we will discuss peripheral immune defects in the mSOD1 mouse model that prevent effective CD4+T cell activation/differentiation and outline the therapeutic potential of adoptive immuotherapy with neuroprotective mSOD1 T cells generated in vitro by facial nerve axotomy antigens. Identification of defects in neural:immune interactions could have an important impact because they can then be the target of therapeutic intervention in ALS.
CD4+ T Cell-Mediated Neuroprotection
For decades, the rodent facial nerve injury model has proven to be an ideal model in which to investigate mechanisms of neuroprotection and neuroregeneration (Moran and Graeber 2004). The Jones laboratory has examined facial nerve injury responses in hamster (Kinderman et al. 1998), mouse (Serpe et al. 1999), and rat (Foecking et al. 2012), and compared different locations of facial nerve damage as well (Sharma et al. 2009). One of the distinct advantages to the rodent facial nerve injury at its exit from the skull is that, while it produces a unilateral paralysis of the muscles of facial expression, vital functions that include breathing, swallowing, and locomotion are unaffected. In contrast to the relatively non-consequential facial nerve injury in animal models, loss of facial expression due to facial nerve injury in humans has both physical and psychological implications that result in significant restrictions for the affected individual. Thus, use of a simple animal model to understand neuroprotection and neurorepair could provide insights into therapeutic strategies designed to reduce disability and disfigurement associated with prolonged facial paralysis.
To explore neural:immune interactions in the context of neural injury and repair, we combined the mouse facial nerve injury model with a host of immunodeficient mouse models in order to investigate the impact of components of the immune system on neural repair. Initially, we began with two different immunodeficient mouse strains, the severe combined immunodeficiency and RAG2−/− models, which lack functional T and B cells, and explored the effects of removing the adaptive arm of the immune system on neuronal viability after target disconnection (Serpe et al. 2003). We discovered that, in the absence of T and B cells, FMN survival levels after axotomy at the SMF were decreased, but could be restored with adoptive transfer of whole splenocytes prior to axotomy (Serpe et al. 1999). Interestingly, following an extensive time course study (Serpe et al. 2000), we determined that a select percentage of FMN were vulnerable in the absence of T and B cells, whereas, approximately 50 % of axotomized FMN were resilient and did not die under immunodeficient conditions. Why some neurons within the same MN pool undergo a different fate when target-disconnected is unclear, but an important future area of research on MN cell death throughout the neuraxis.
Subsequently, a series of experiments was conducted to identify exactly which immune cell types contributed to FMN neuroprotection following facial nerve axotomy. These experiments utilized a host of relevant immunodeficient mouse models, including: CD4+T cell deficient, CD8+ T cell deficient, B cell deficient, interleukin-4 (IL-4) deficient, interferon-gamma (IFN-γ) deficient, STAT-4 and STAT-6 knock out (Byram et al. 2003; Serpe et al. 2003; DeBoy et al. 2006b; Deboy et al. 2006a) mice, to analyze the role of individual types of immune cells in neuroprotection. The collective results of those studies revealed that the CD4+ T cell is responsible for mediating the neuroprotective actions of the immune system on FMN survival after axotomy.
Historically, two types of CD4+ T effector cells, T helper type 2 (Th2) and T helper type 1 (Th1), producing anti-inflammatory (IL-4, IL-10) or pro-inflammatory (IFN-γ) cytokines, respectively, were described. In view of that literature, we explored which CD4+ T helper cell was responsible for FMN neuroprotection. The results of those studies indicate that the Th2, and not the Th1, cell subset is critical for FMN survival levels to equal WT after axotomy (Deboy et al. 2006a). These data are consistent with studies indicating that a shift toward the activation of a Th2 cell may delay neurodegenerative disease onset and/or inhibit disease progression (Reynolds et al. 2010; Yang et al. 2013). More recently, there has been the discovery of additional T cell subsets, including Tregulatory cells (Tregs), which may dampen the initiation and/or development of pro-inflammatory immune responses, and are considered to be anti-inflammatory. We characterized the phenotype of T cell subset development in lymphoid tissue following a unilateral facial nerve injury in the WT mouse and found, surprisingly, that all T cell subsets identified at the time did develop and expand in number in the draining lymph nodes, and were accompanied by cytokine increases (Xin et al. 2008). A future question will be to explore the impact of the other T cell subsets, both pro- and anti-inflammatory, on FMN survival after axotomy. At this point, we know that the Th1 cell, as an example of a pro-inflammatory cell, is not involved, and that the Th2 cell, as an example of an anti-inflammatory cell, is critical to achieve full neuroprotection. We have also examined CD4+ T cell subset involvement in functional recovery from facial paralysis in order to address the role of CD4+ T cells on the process of axonal elongation and nerve regeneration (Beahrs et al. 2010). Those studies utilized a series of immunodeficient mice in which impairment occurs in either the Th2 or the Th1 effector cell, and the results indicate that, while the Th2 cell is implicated selectively in neuronal viability, both the Th2 and Th1 cells with anti-and pro-inflammatory actions contribute to optimal functional recovery from axotomy-induced facial paralysis. These data argue that differential effects of the peripheral immune response to nerve injury occur in a location-dependent manner.
The results of our initial experiments utilizing a host of immunodeficient mice to examine FMN survival as well as functional recovery led next to the question of the site of the neural:immune interactions mediating the positive effects of immune cells on injured FMN. In 1998, Raivich et al. published a study demonstrating that T cells infiltrated the facial motor nucleus in WT mice between 7 and 21 days after peripheral axotomy (Raivich et al. 1998). Ha et al. (2007) also demonstrated a significant increase in CD4+T cells with the facial motor nucleus after peripheral axotomy. However, a substantial immune response at the site of injury, triggering Wallerian degeneration, has been a well-described, universal component of peripheral nerve injury (Brushart 2011). Thus, evidence existed within the literature in support of either a CNS or peripheral nervous system (PNS) site of action or both, with regard to the ability of CD4+ T cells to provide neuroprotection in the facial nerve injury paradigm. In order for naïve CD4+ T cells to differentiate into armed effector cells, such as the Th2 cell, and secrete appropriate cytokines, they must first be activated and then subsequently re-activated by the same antigen presented in the context of the major histocompatibility complex II (MHCII) molecule. In collaboration with Dr. Monica Carson, we created chimeric mice that were reciprocal in nature, i.e., APC lacking MHCII peripherally but retaining MHCII centrally or APC lacking MHCII centrally but retaining MHCII peripherally (Byram et al. 2004). These chimeric mice were utilized in a facial nerve injury study to determine the location of CD4+ T cell activation, with the results indicating a dual compartment model of CD4+ Tcell activation in the draining cervical lymph node after facial nerve axotomy at the SMF. First, the CD4+ T cell requires activation and differentiation in peripheral lymph tissue. Such peripheral activation is then followed by central re-activation by microglial cells in close association to FMN within the microenvironment in the facial motor nucleus.
With the determination that part of the mechanism by which CD4+ T cells exert a neuroprotective action involves interaction with microglial cells within the CNS, we next explored the question of how the CD4+ T cells are recruited into the facial nucleus. In a series of studies examining chemokine expression within the facial nucleus induced by peripheral nerve damage, along with the use of chemokine receptor-deficient mice, we established that Th2-associated chemokines are upregulated within the facial nucleus after injury (Wainwright et al. 2008, 2009a, b). Furthermore, chemokine receptor CCR3-deficient T cells failed to rescue FMN from axotomy-induced cell death, relative to WT CD4+ T cells (Wainwright et al. 2009c). In immunocytochemical experiments, Wainwright et al. (2009b) established that facial nerve axotomy induced the expression of CCL11, a Th2-associated chemokine, within astrocytes in the facial motor nucleus within the first 2 weeks after injury, and with a return to baseline by 30 days post-axotomy. Therefore, in addition to knowledge gained concerning microglial involvement in CD4+ T cell-mediated neuroprotection, it appears that the astrocyte, activated by axotomy in the microenvironment surrounding injured FMN cell bodies, is, along with the microglial cell, a key player in the ability of peripherally activated T cells to be recruited into the facial nucleus after axotomy.
We established that T cell recruitment into the facial nucleus through astrocytic production of appropriate chemokine, along with microglial-mediated T cell re-activation, are key elements in the mechanism by which T cell neuroprotection occurs. Therefore, we next tested the hypothesis that the reactivated CD4+ T cell produced neurotrophic or neuroprotective molecules within the facial nucleus, and that such T cell-secreted molecules were directly responsible for FMN rescue after axotomy. Research indicates brain-derived neurotrophic factor (BDNF) is expressed by mature resting CD4+ T cells (Kerschensteiner et al. 1999; Ziemssen et al. 2002). We found that supernatant from the draining lymph nodes 9 days after axotomy contained BDNF, and could be used to rescue FMN from axotomy-induced cell death when applied at the cut stump of the facial nerve and retrogradely transported back to the facial nucleus (Serpe et al. 2005). However, to our surprise, CD4+ T cells devoid of BDNF through cre-lox mouse technology were equally capable of FMN rescue as WT after axotomy (Xin et al. 2011).
Interleukin 10 (IL-10) is the phenotype-defining cytokine of type 1 regulatory T cells and is also produced by Th2 cells. IL-10 is a potent inhibitor of inflammation due to its ability to dampen Th1 cell activity and subsequent inflammatory cytokine production (Strle et al. 2001). Over the past decade, the beneficial effects of IL-10 have been shown in numerous neuroinflammatory disease models, including experimental encephalomyelitis, spinal cord injury, stroke, and Parkinson’s disease (Bethea et al. 1999; Brewer et al. 1999; Cua et al. 2001; Frenkel et al. 2005; Qian et al. 2006). Collectively, these findings indicate that IL-10 plays a role in regulation of CNS inflammation and perhaps viability of neurons under normal conditions. We examined the role of IL-10 in CD4+ T cell-mediated neuroprotection and discovered that both the CD4+ T cell and IL-10 are required for full FMN survival levels after axotomy (Xin et al. 2011). However, the T cell proved not to be the source of IL-10 (Xin et al. 2011).
Thus, the current working model of T cell-mediated neuroprotection after peripheral target disconnection is based upon the hypothesis that the centrally recruited CD4+ Tcell interacts with the microenvironment surrounding the injured FMN in such a way to induce an anti-inflammatory IL-10 cascade (Fig. 1). Our current hypothesis is that the microglial cell is the source of IL-10, and its release is dependent upon T cell-microglia communication. Once released into the environment, IL-10 could bind to astrocytes and modulate FMN indirectly and/or bind to FMN directly in order to exert its neuroprotective effects.
Fig. 1.
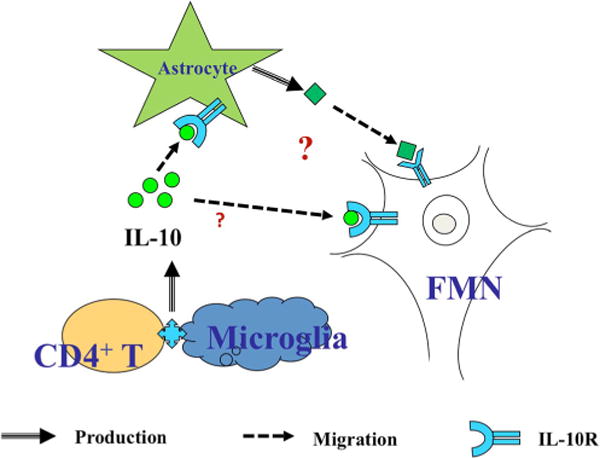
The current working model of T cell-mediated neuroprotection following peripheral target disconnection
Immune Defects in ALS
The earliest evidence suggesting a relationship between the immune system and ALS was that obtained from postmortem brain and spinal cord tissue from individuals with ALS. T cell infiltrates were found in the brain and/or spinal cord at not only end-stage disease in humans (Troost et al. 1989; Engelhardt et al. 1993), but also during disease progression in mSOD1 mice (Beers et al. 2008; Chiu et al. 2008). In the latter studies using transgenic mouse models lacking T cell activity, the rate of disease progression increased and survival time decreased. Importantly, these detrimental outcomes were prevented when the transgenic mice received an adoptive transfer of either WT or mSOD1 bone marrow cells. Both lines of evidence suggested that CD4+ T cells play a neuroprotective role in ALS. The exact nature of the neuroprotection has been investigated actively, with most evidence indicating a role for CD4+ T cells in the modulation of the microglial inflammatory response to neuronal injury (Troost et al. 1989; Beers et al. 2011). Since ALS is a neurodegenerative disease that involves MN loss, these important findings suggest that CD4+ T cells affect MN survival to regulate the ALS disease process.
The specific CD4+ T cell subset responsible for mediating the neuroprotective effect in ALS became an active area of investigation, with Foxp3+ T-regulatory (Treg) cells specifically a focus of attention. Some studies used the in vivo activation states of microglia to identify Treg-mediated effects on inflammation, including microglial anti-inflammatory (M2) versus pro-inflammatory (M1) responses (Zhao et al. 2013). More recently, using analyses at the molecular level, mSOD1 microglia were found to express neurodegeneration-specific genes that distinguished them from M1/M2 cells and correlated with the expression of T cell genes reflective of T cell-microglia interaction (Chiu et al. 2013). In ALS patients, the presence of Foxp3+ T-regulatory cells declined with disease progression (Henkel et al. 2013). In mice, during early stage disease, Treg cells increased in number, produced IL-4, and were associated with M2-mediated neuroprotection (Liao et al. 2012; Henkel et al. 2013), while Treg cells during late stage disease decreased in number, as did the level of neuroprotection (Troost et al. 1989; Beers et al. 2011). When immunodeficient mSOD1 mice were used, the adoptive transfer of CD4+Foxp3+ IL-4-producing T cells (Beers et al. 2011) or activated CD4+ CD25+ T cells (Banerjee et al. 2008) from either WT or mSOD1 mice promoted survival and decreased the rate of disease progression. However, the neuroprotective effect was not evident in mice adoptively transferred with naive CD4+ CD25− T cells in comparison to those mice receiving activated T cells (Banerjee et al. 2008) suggesting that naïve T cells may fail to become activated for neuroprotective potential when transferred to mSOD1 mice. When mSOD1 Treg cells were cultured with mSOD1 microglia, they suppressed microglial production of NOX2 and iNOS in an IL-4-dependent manner (Henkel et al. 2013), and thus could contribute to the slowly-progressing phase of the disease until their numbers decreased.
Epigenetic studies in immune cells of ALS patients have also shown that gene regulation may be altered in ALS. Changes in miRNA expression patterns in immune cells from ALS patients (Butovsky et al. 2012) and mSOD1 mice (Parisi et al. 2013) were linked to mSOD1 inflammation and disease progression, suggesting that a miRNA signature might be associated with ALS. Also along this line of changes occurring at the molecular level, one study identified specific epigenetic changes involving global methylation in postmortem ALS spinal cords, but not in blood (Figueroa-Romero et al. 2012), suggesting another mechanism that may be involved with ALS progression.
A role for other immune cells in ALS, including other CD4+ T cell subsets, remains under investigation. Few studies have focused on the role of the B cell, and these studies have produced varying results. One study reported that no changes in any antibody isotype occurred in ALS patients in comparison to normal controls (Rentzos et al. 2013), while another study reported changes in IgG and the level of immune complexes (Saleh et al. 2009). A much earlier study of humans with ALS found changes in the level of IgA and IgG, although these changes appeared to correlate with repeated infections in these patients and not with ALS disease progression (Hoffman et al. 1981). Thus, the role of B cells in the disease process is unclear. Also, a role for NKT cells was suggested when these cells were found in the spinal cords of mSOD1 mice and their presence was associated with a loss in neuroprotection, particularly when an inhibition of NKT activity delayed disease onset and increased survival (Finkelstein et al. 2011). Thus far, the majority of findings suggest a role for neuroprotective CD4+ Treg cells in ALS, and an unclear role for other immune cells.
Although a measurement of disease onset and progression in mSOD1 mice and humans suggested a role for immune cells in ALS, few studies have addressed the MN itself. For MN survival to occur in axotomized wild-type (WT) mice, Th2-like cells are required (Deboy et al. 2006a), and they must be activated in the periphery and reactivated for effector function in the brain (Byram et al. 2004). Another study suggested that CD4+CD25+ T cells may not be involved in MN survival (DeBoy et al. 2006b), although, as suggested above, they appear to play a beneficial role in disease progression and survival.
Use of Axotomy as Tool in Understanding ALS Pathogenesis
Contributing factors that influence the ability of an injured MN to survive include neuronal signals along with a tightly regulated microenviromental reaction involving CNS glial and immune cells. The rodent facial nucleus has a topography of subnuclear groups with distinct innervation patterns (Ashwell 1982). In a recent topographical mapping study (Canh et al. 2006), we analyzed the anatomical distribution pattern of FMN survival levels in 6 subnuclear groups within the WT mouse facial nucleus and unexpectedly found distinct differences in survival levels. While overall survival was around 85 %, there was essentially 100 % FMN survival in the ventromedial (VM), but only 70 % survival in the ventrolateral (VL), subnucleus. These results led to a molecular investigation of both the FMN injury response and the reaction of surrounding glial cells within the facial nucleus and in close proximity to the FMN cell bodies (Mesnard et al. 2010). Surprisingly, VM and VL FMN responded similarly to facial nerve injury and in a regenerative manner, despite differences in fate. In contrast, the surrounding glial responses between the 2 subnuclear regions were significantly different and support a causative role for glial and/or immune-derived molecules in directing the fate of VM and VL FMN subjected to the same injury.
In agreement with the literature (Mariotti et al. 2002), we found that FMN survival levels after facial nerve injury in pre-symptomatic mSOD1 were significantly decreased, relative to WT (Mesnard et al. 2011) and, interestingly, comparable to immunodeficient animals. Given the potential for immune dysfunction as a contributing factor to ALS disease onset/progression, the comparable reactions to axotomy by immunodeficient and mSOD1 mice was particularly striking. Axonal pathology, including motor axon withdrawal from neuromuscular junction, and the resulting axonal die-back have been show to precede symptom onset and MN loss in both humans and animal models (Fischer et al. 2004; Kano et al. 2012). The response to peripheral axotomy resembles the axonal die-back response, as both processes result in axonal disconnection from neuromuscular junctions in target muscle and after presynaptic stripping in the CNS. We, therefore, used axotomy as an investigative tool to delineate underlying molecular mechanisms that result in MN degeneration in pre-symptomatic mSOD1 mice (Mesnard et al. 2010, 2011, 2013; Haulcomb et al. 2014; Mesnard-Hoaglin et al. 2014). The superimposition of facial nerve axotomy on pre-symptomatic mice allowed us to experimentally produce simultaneous die-back of a select MN population at a set time and for a controlled length of time post-axotomy.
As with the different subnuclear FMN and despite differences in cell fate, mSOD1 FMN displayed a dynamic pro-survival/regeneration response phenotypic of WT injured MN (Mesnard et al. 2010). However, significant differences were revealed when the axotomy-induced gene expression response in the neuropil surrounding the injured FMN of pre-symptomatic mSOD1 was compared to WT. Collectively, the contrasting injury-induced responses of FMN vs the surrounding microenvironment within the facial nucleus suggests that there may be a dysregulation of nonneuronal cells following target disconnection that is pro-inflammatory in nature and perhaps contributory to the MN degeneration seen in the mSOD1 mouse model of ALS. Further use of laser capture microdissection to explore gene expression in the facial nucleus of mSOD1 mice, relative to WT, following axotomy-induced target disconnection provided evidence in support of a regenerative FMN phenotype surrounded by a pro-inflammatory environment with the CNS of mSOD1 mice (Mesnard et al. 2011; Haulcomb et al. 2014).
Given the results indicating similarities between mSOD1 and immunodeficient mouse FMN to axotomy, along with the dysregulation in nonneuronal cells close in proximity to the FMN cell bodies, we next began to explore the question of how might the immune system in the mSOD1 mouse be defective and whether axotomy could induce the production of neuroprotective CD4+ T cells as found in WT mice. Based upon the aforementioned studies, we hypothesized that a defective T cell population in the mSOD1 mouse could contribute to the immune dysfunction and MN cell death. To test this hypothesis, we conducted an extensive series of reconstitution experiments (Mesnard-Hoaglin et al. 2014) utilizing both immunodeficient and mSOD1 mouse models. To our surprise, the results of that study indicate that mSOD1 mouse CD4+ T cells ARE ABLE TO mediate neuroprotection after facial nerve axotomy in immunodeficient mice when isolated from a suppressive mSOD1 peripheral splenic microenvironment. We are currently testing how such a suppressive splenic microenvironment within the mSOD1 mouse might compromise neuroprotective CD4+T cell activation and/or differentiation.
Future Directions
No approach to date has been effective in providing meaningful immunotherapeutic impact for ALS patients, particularly with regard to reversing mortality levels associated with the disease. Thus, more immunology-focused studies are required to address the questions involving mechanism and potential therapeutic application. For example, what causes the activation and/or differentiation of a specific T cell subset during disease progression, and what factors influence the activation process? Are T cells activated before the disease process begins, but become refractive as the disease process progresses, thus lowering the potential neuroprotective effect of these cells? The future of research in this area will need to identify: 1) the specific antigen responsible for T cell activation, 2) key stages of immune cell involvement prior to disease development, and 3) immunotherapeutic transplant approaches that do not involve immunorejection.
Footnotes
Conflict of Interest The authors declare that they have no conflict of interest.
References
- Ashwell KW. The adult mouse facial nerve nucleus: morphology and musculotopic organization. J Anat. 1982;135:531–538. [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Mosley RL, Reynolds AD, Dhar A, Jackson-Lewis V, Gordon PH, Przedborski S, Gendelman HE. Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. PLoS ONE. 2008;3:e2740. doi: 10.1371/journal.pone.0002740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beahrs T, Tanzer L, Sanders VM, Jones KJ. Functional recovery and facial motoneuron survival are influenced by immunodeficiency in crush-axotomized mice. Exp Neurol. 2010;221:225–230. doi: 10.1016/j.expneurol.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Zhao W, Wang J, Appel SH. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A. 2008;105:15558–15563. doi: 10.1073/pnas.0807419105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Zhao W, Wang J, Huang A, Wen S, Liao B, Appel SH. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain. 2011;134:1293–1314. doi: 10.1093/brain/awr074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethea JR, Nagashima H, Acosta MC, Briceno C, Gomez F, Marcillo AE, Loor K, Green J, Dietrich WD. Systemically administered interleukin-10 reduces tumor necrosis factor-alpha production and significantly improves functional recovery following traumatic spinal cord injury in rats. J Neurotrauma. 1999;16:851–863. doi: 10.1089/neu.1999.16.851. [DOI] [PubMed] [Google Scholar]
- Brewer KL, Bethea JR, Yezierski RP. Neuroprotective effects of interleukin-10 following excitotoxic spinal cord injury. Exp Neurol. 1999;159:484–493. doi: 10.1006/exnr.1999.7173. [DOI] [PubMed] [Google Scholar]
- Brushart TM. Nerve repair. Oxford University Press; New York: 2011. [Google Scholar]
- Butovsky O, Siddiqui S, Gabriely G, Lanser AJ, Dake B, Murugaiyan G, Doykan CE, Wu PM, Gali RR, Iyer LK, Lawson R, Berry J, Krichevsky AM, Cudkowicz ME, Weiner HL. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest. 2012;122:3063–3087. doi: 10.1172/JCI62636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byram SC, Serpe CJ, Pruett SB, Sanders VM, Jones KJ. Natural killer cells do not mediate facial motoneuron survival after facial nerve transection. Brain Behav Immun. 2003;17:417–425. doi: 10.1016/s0889-1591(03)00089-8. [DOI] [PubMed] [Google Scholar]
- Byram SC, Carson MJ, DeBoy CA, Serpe CJ, Sanders VM, Jones KJ. CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. J Neurosci Off J Soc Neurosci. 2004;24:4333–4339. doi: 10.1523/JNEUROSCI.5276-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canh MY, Serpe CJ, Sanders V, Jones KJ. CD4(+) T cell-mediated facial motoneuron survival after injury: distribution pattern of cell death and rescue throughout the extent of the facial motor nucleus. J Neuroimmunol. 2006;181:93–99. doi: 10.1016/j.jneuroim.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Carrasco DI, Bichler EK, Seburn KL, Pinter MJ. Nerve terminal degeneration is independent of muscle fiber genotype in SOD1 mice. PLoS ONE. 2010;5:e9802. doi: 10.1371/journal.pone.0009802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Chen A, Zheng Y, Kosaras B, Tsiftsoglou SA, Vartanian TK, Brown RH, Jr, Carroll MC. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci U S A. 2008;105:17913–17918. doi: 10.1073/pnas.0804610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O’Keeffe S, Phatnani HP, Muratet M, Carroll MC, Levy S, Tavazoie S, Myers RM, Maniatis T. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013;4:385–401. doi: 10.1016/j.celrep.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cua DJ, Hutchins B, LaFace DM, Stohlman SA, Coffman RL. Central nervous system expression of IL-10 inhibits autoimmune encephalomyelitis. J Immunol. 2001;166:602–608. doi: 10.4049/jimmunol.166.1.602. [DOI] [PubMed] [Google Scholar]
- Deboy CA, Xin J, Byram SC, Serpe CJ, Sanders VM, Jones KJ. Immune-mediated neuroprotection of axotomized mouse facial motoneurons is dependent on the IL-4/STAT6 signaling pathway in CD4(+) T cells. Exp Neurol. 2006a;201:212–224. doi: 10.1016/j.expneurol.2006.04.028. [DOI] [PubMed] [Google Scholar]
- DeBoy CA, Byram SC, Serpe CJ, Wisuri D, Sanders VM, Jones KJ. CD4+CD25+ regulatory T cells and CD1-restricted NKT cells do not mediate facial motoneuron survival after axotomy. J Neuroimmunol. 2006b;176:34–38. doi: 10.1016/j.jneuroim.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Engelhardt JI, Tajti J, Appel SH. Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch Neurol. 1993;50:30–36. doi: 10.1001/archneur.1993.00540010026013. [DOI] [PubMed] [Google Scholar]
- Figueroa-Romero C, Hur J, Bender DE, Delaney CE, Cataldo MD, Smith AL, Yung R, Ruden DM, Callaghan BC, Feldman EL. Identification of epigenetically altered genes in sporadic amyotrophic lateral sclerosis. PLoS ONE. 2012;7:e52672. doi: 10.1371/journal.pone.0052672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein A, Kunis G, Seksenyan A, Ronen A, Berkutzki T, Azoulay D, Koronyo-Hamaoui M, Schwartz M. Abnormal changes in NKT cells, the IGF-1 axis, and liver pathology in an animal model of ALS. PLoS ONE. 2011;6:e22374. doi: 10.1371/journal.pone.0022374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Foecking EM, Fargo KN, Coughlin LM, Kim JT, Marzo SJ, Jones KJ. Single session of brief electrical stimulation immediately following crush injury enhances functional recovery of rat facial nerve. J Rehabil Res Dev. 2012;49:451–458. doi: 10.1682/jrrd.2011.03.0033. [DOI] [PubMed] [Google Scholar]
- Frenkel D, Huang Z, Maron R, Koldzic DN, Moskowitz MA, Weiner HL. Neuroprotection by IL-10-producing MOG CD4+ T cells following ischemic stroke. J Neurol Sci. 2005;233:125–132. doi: 10.1016/j.jns.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Graves MC, Fiala M, Dinglasan LA, Liu NQ, Sayre J, Chiappelli F, van Kooten C, Vinters HV. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotrophic lateral sclerosis and other motor neuron disorders: official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases. 2004;5:213–219. doi: 10.1080/14660820410020286. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Ha GK, Huang Z, Petitto JM. Prior facial motor neuron injury elicits endogenous T cell memory: relation to neuroregeneration. J Neuroimmunol. 2007;183:111–117. doi: 10.1016/j.jneuroim.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haulcomb MM, Mesnard NA, Batka RJ, Alexander TD, Sanders VM, Jones KJ. Axotomy-induced target disconnection promotes an additional death mechanism involved in motoneuron degeneration in amyotrophic lateral sclerosis transgenic mice. J Comp Neurol. 2014;522:2349–2376. doi: 10.1002/cne.23538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Gordon T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2007;28:154–164. doi: 10.1016/j.nbd.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Henkel JS, Engelhardt JI, Siklos L, Simpson EP, Kim SH, Pan T, Goodman JC, Siddique T, Beers DR, Appel SH. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol. 2004;55:221–235. doi: 10.1002/ana.10805. [DOI] [PubMed] [Google Scholar]
- Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE, Zhao W, Moore DH, Powell SZ, Appel SH. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med. 2013;5:64–79. doi: 10.1002/emmm.201201544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman PM, Robbins DS, Oldstone MB, Gibbs CJ, Jr, Gajdusek DC. Humoral immunity in Guamanians with amyotrophic lateral sclerosis and parkinsonism-dementia. Ann Neurol. 1981;10:193–196. doi: 10.1002/ana.410100210. [DOI] [PubMed] [Google Scholar]
- Kano O, Beers DR, Henkel JS, Appel SH. Peripheral nerve inflammation in ALS mice: cause or consequence. Neurology. 2012;78:833–835. doi: 10.1212/WNL.0b013e318249f776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennel PF, Finiels F, Revah F, Mallet J. Neuromuscular function impairment is not caused by motor neurone loss in FALS mice: an electromyographic study. Neuroreport. 1996;7:1427–1431. doi: 10.1097/00001756-199605310-00021. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner M, Gallmeier E, Behrens L, Leal VV, Misgeld T, Klinkert WE, Kolbeck R, Hoppe E, Oropeza-Wekerle RL, Bartke I, Stadelmann C, Lassmann H, Wekerle H, Hohlfeld R. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med. 1999;189:865–870. doi: 10.1084/jem.189.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinderman NB, Harrington CA, Drengler SM, Jones KJ. Ribosomal RNA transcriptional activation and processing in hamster facial motoneurons: effects of axotomy with or without exposure to testosterone. J Comp Neurol. 1998;401:205–216. [PubMed] [Google Scholar]
- Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol. 2012;237:147–152. doi: 10.1016/j.expneurol.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariotti R, Cristino L, Bressan C, Boscolo S, Bentivoglio M. Altered reaction of facial motoneurons to axonal damage in the presymptomatic phase of a murine model of familial amyotrophic lateral sclerosis. Neuroscience. 2002;115:331–335. doi: 10.1016/s0306-4522(02)00448-7. [DOI] [PubMed] [Google Scholar]
- Mesnard NA, Alexander TD, Sanders VM, Jones KJ. Use of laser microdissection in the investigation of facial motoneuron and neuropil molecular phenotypes after peripheral axotomy. Exp Neurol. 2010;225:94–103. doi: 10.1016/j.expneurol.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesnard NA, Sanders VM, Jones KJ. Differential gene expression in the axotomized facial motor nucleus of presymptomatic SOD1 mice. J Comp Neurol. 2011;519:3488–3506. doi: 10.1002/cne.22718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesnard NA, Haulcomb MM, Tanzer L, Sanders VM, Jones KJ. Delayed functional recovery in presymptomatic mSOD1 mice following facial nerve crush axotomy. J Neurodegener Regen. 2013;4:21–25. [PMC free article] [PubMed] [Google Scholar]
- Mesnard-Hoaglin NA, Xin J, Haulcomb MM, Batka RJ, Sanders VM, Jones KJ. SOD1(G93A) transgenic mouse CD4(+) T cells mediate neuroprotection after facial nerve axotomy when removed from a suppressive peripheral microenvironment. Brain Behav Immun. 2014;40:55–60. doi: 10.1016/j.bbi.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran LB, Graeber MB. The facial nerve axotomy model. Brain Res Brain Res Rev. 2004;44:154–178. doi: 10.1016/j.brainresrev.2003.11.004. [DOI] [PubMed] [Google Scholar]
- O’Reilly SA, Roedica J, Nagy D, Hallewell RA, Alderson K, Marklund SL, Kuby J, Kushner PD. Motor neuron-astrocyte interactions and levels of Cu, Zn superoxide dismutase in sporadic amyotrophic lateral sclerosis. Exp Neurol. 1995;131:203–210. doi: 10.1016/0014-4886(95)90042-x. [DOI] [PubMed] [Google Scholar]
- Parisi C, Arisi I, D’Ambrosi N, Storti AE, Brandi R, D’Onofrio M, Volonte C. Dysregulated microRNAs in amyotrophic lateral sclerosis microglia modulate genes linked to neuroinflammation. Cell Death Dis. 2013;4:e959. doi: 10.1038/cddis.2013.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KH, Vincent I. Presymptomatic biochemical changes in hindlimb muscle of G93A human Cu/Zn superoxide dismutase 1 transgenic mouse model of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2008;1782:462–468. doi: 10.1016/j.bbadis.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Block ML, Wei SJ, Lin CF, Reece J, Pang H, Wilson B, Hong JS, Flood PM. Interleukin-10 protects lipopolysaccharide-induced neurotoxicity in primary midbrain cultures by inhibiting the function of NADPH oxidase. J Pharmacol Exp Ther. 2006;319:44–52. doi: 10.1124/jpet.106.106351. [DOI] [PubMed] [Google Scholar]
- Rafalowska J, Dziewulska D, Gadamski R, Chrzanowska H, Modrzewska-Lewczuk M, Grieb P. Is the spinal cord motoneuron exclusively a target in ALS? Comparison between astroglial reactivity in a rat model of familial ALS and in human sporadic ALS cases. Neurol Res. 2010;32:867–872. doi: 10.1179/174313209X414542. [DOI] [PubMed] [Google Scholar]
- Raivich G, Jones LL, Kloss CU, Werner A, Neumann H, Kreutzberg GW. Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J Neurosci Off J Soc Neurosci. 1998;18:5804–5816. doi: 10.1523/JNEUROSCI.18-15-05804.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzos M, Evangelopoulos ME, Sereti E, Zouvelou V, Marmara S, Alexakis T, Evdokimidis I. Humoral immune activation in amyotrophic lateral sclerosis patients. Neurol Int. 2013;5:e3. doi: 10.4081/ni.2013.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson’s disease. J Immunol. 2010;184:2261–2271. doi: 10.4049/jimmunol.0901852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- Saleh IA, Zesiewicz T, Xie Y, Sullivan KL, Miller AM, Kuzmin-Nichols N, Sanberg PR, Garbuzova-Davis S. Evaluation of humoral immune response in adaptive immunity in ALS patients during disease progression. J Neuroimmunol. 2009;215:96–101. doi: 10.1016/j.jneuroim.2009.07.011. [DOI] [PubMed] [Google Scholar]
- Sanagi T, Yuasa S, Nakamura Y, Suzuki E, Aoki M, Warita H, Itoyama Y, Uchino S, Kohsaka S, Ohsawa K. Appearance of phagocytic microglia adjacent to motoneurons in spinal cord tissue from a pre-symptomatic transgenic rat model of amyotrophic lateral sclerosis. J Neurosci Res. 2010;88:2736–2746. doi: 10.1002/jnr.22424. [DOI] [PubMed] [Google Scholar]
- Serpe CJ, Kohm AP, Huppenbauer CB, Sanders VM, Jones KJ. Exacerbation of facial motoneuron loss after facial nerve transection in severe combined immunodeficient (scid) mice. J Neurosci Off J Soc Neurosci. 1999;19:RC7. doi: 10.1523/JNEUROSCI.19-11-j0004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpe CJ, Sanders VM, Jones KJ. Kinetics of facial motoneuron loss following facial nerve transection in severe combined immunodeficient mice. J Neurosci Res. 2000;62:273–278. doi: 10.1002/1097-4547(20001015)62:2<273::AID-JNR11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Serpe CJ, Coers S, Sanders VM, Jones KJ. CD4+ T, but not CD8+ or B, lymphocytes mediate facial motoneuron survival after facial nerve transection. Brain Behav Immun. 2003;17:393–402. doi: 10.1016/s0889-1591(03)00028-x. [DOI] [PubMed] [Google Scholar]
- Serpe CJ, Byram SC, Sanders VM, Jones KJ. Brain-derived neurotrophic factor supports facial motoneuron survival after facial nerve transection in immunodeficient mice. Brain Behav Immun. 2005;19:173–180. doi: 10.1016/j.bbi.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Sharma N, Cunningham K, Porter RG, Sr, Marzo SJ, Jones KJ, Foecking EM. Comparison of extratemporal and intratemporal facial nerve injury models. Laryngoscope. 2009;119:2324–2330. doi: 10.1002/lary.20627. [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall AM, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- Strle K, Zhou JH, Shen WH, Broussard SR, Johnson RW, Freund GG, Dantzer R, Kelley KW. Interleukin-10 in the brain. Crit Rev Immunol. 2001;21:427–449. [PubMed] [Google Scholar]
- Troost D, van den Oord JJ, de Jong JM, Swaab DF. Lymphocytic infiltration in the spinal cord of patients with amyotrophic lateral sclerosis. Clin Neuropathol. 1989;8:289–294. [PubMed] [Google Scholar]
- Troost D, Das PK, van den Oord JJ, Louwerse ES. Immunohistological alterations in muscle of patients with amyotrophic lateral sclerosis: mononuclear cell phenotypes and expression of MHC products. Clin Neuropathol. 1992;11:115–120. [PubMed] [Google Scholar]
- Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, Leigh PN, Banati RB. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C] (R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15:601–609. doi: 10.1016/j.nbd.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Wainwright DA, Xin J, Sanders VM, Jones KJ. Differential actions of pituitary adenylyl cyclase-activating polypeptide and interferon gamma on Th2- and Th1-associated chemokine expression in cultured murine microglia. J Neurodegener Regen. 2008;1:31–34. [PMC free article] [PubMed] [Google Scholar]
- Wainwright DA, Mesnard NA, Xin J, Sanders VM, Jones KJ. Effects of facial nerve axotomy on Th2-associated and Th1-associated chemokine mRNA expression in the facial motor nucleus of wild-type and presymptomatic SOD1 mice. J Neurodegener Regen. 2009a;2:39–44. [PMC free article] [PubMed] [Google Scholar]
- Wainwright DA, Xin J, Mesnard NA, Politis CM, Sanders VM, Jones KJ. Effects of facial nerve axotomy on Th2- and Th1-associated chemokine expression in the facial motor nucleus of wild-type and presymptomatic mSOD1 mice. J Neuroimmunol. 2009b;216:66–75. doi: 10.1016/j.jneuroim.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainwright DA, Xin J, Mesnard NA, Beahrs TR, Politis CM, Sanders VM, Jones KJ. Exacerbation of facial motoneuron loss after facial nerve axotomy in CCR3-deficient mice. ASN NEURO. 2009c;1:e00024. doi: 10.1042/AN20090017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin J, Wainwright DA, Serpe CJ, Sanders VM, Jones KJ. Phenotype of CD4+ T cell subsets that develop following mouse facial nerve axotomy. Brain Behav Immun. 2008;22:528–537. doi: 10.1016/j.bbi.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin J, Wainwright DA, Mesnard NA, Serpe CJ, Sanders VM, Jones KJ. IL-10 within the CNS is necessary for CD4+ T cells to mediate neuroprotection. Brain Behav Immun. 2011;25:820–829. doi: 10.1016/j.bbi.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang H, Xie Z, Wei L, Bi J. Systemic transplantation of human umbilical cord derived mesenchymal stem cells-educated T regulatory cells improved the impaired cognition in AbetaPPswe/PS1dE9 transgenic mice. PLoS ONE. 2013;8:e69129. doi: 10.1371/journal.pone.0069129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Beers DR, Appel SH. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J NeuroImmune Pharmacol Off J Soc NeuroImmune Pharmacol. 2013;8:888–899. doi: 10.1007/s11481-013-9489-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemssen T, Kumpfel T, Klinkert WE, Neuhaus O, Hohlfeld R. Glatiramer acetate-specific T-helper 1- and 2-type cell lines produce BDNF: implications for multiple sclerosis therapy. Brain-derived neurotrophic factor. Brain. 2002;125:2381–2391. doi: 10.1093/brain/awf252. [DOI] [PubMed] [Google Scholar]