

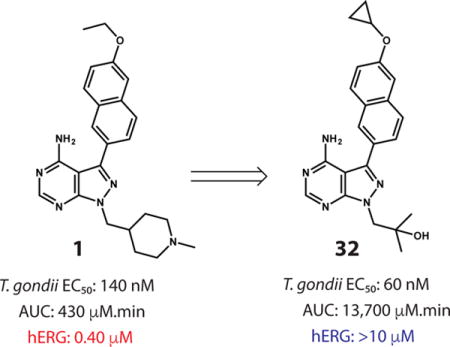
Abstract
New therapies are needed for the treatment of toxoplasmosis, which is a disease caused by the protozoan parasite Toxoplasma gondii. To this end, we previously developed a potent and selective inhibitor (compound 1) of Toxoplasma gondii calcium-dependent protein kinase 1 (TgCDPK1) that possesses anti-toxoplasmosis activity in vitro and in vivo. Unfortunately, 1 has potent human Ether-à-go-go-Related Gene (hERG) inhibitory activity, associated with long Q-T syndrome, and, consequently, presents a cardiotoxicity risk. Here, we describe the identification of an optimized TgCDPK1 inhibitor 32, which does not have a hERG liability and possesses a favorable pharmacokinetic profile in small and large animals. 32 is CNS-penetrant and highly effective in acute and latent mouse models of T. gondii infection, significantly reducing the amount of parasite in the brain, spleen, and peritoneal fluid and reducing brain cysts by >85%. These properties make 32 a promising lead for the development of a new anti-toxoplasmosis therapy.
Graphical abstract
Introduction
Toxoplasmosis is an infectious disease that results from the infection of the protozoan parasite Toxoplasma gondii.1 While cats are the definitive host of T. gondii, this zoonotic parasite is estimated to be one of the most prevalent in humans, with approximately 30–50% of the world’s population being seropositive.2 T. gondii infections of humans can be caused by the consumption of undercooked meat containing tissue cysts or by the accidental ingestion or inhalation of oocysts from cat feces.1,3 T. gondii parasites rapidly proliferate during acute infection before being controlled by the immune system in immunocompetent individuals.4 However, despite this robust immune response, the T. gondii parasite is not eliminated. Instead, the parasite transforms into a slowly dividing cellular stage called the bradyzoite, which cluster into tissue cysts mainly located in the brain, eyes, and striated muscle.5 Tissue cysts can persist for the lifetime of the host; periodically rupturing and releasing tachyzoites that can again rapidly replicate and reform cysts. While in immunocompetent individuals toxoplasmosis is usually asymptomatic and recovery occurs in the absence of antibiotic treatment, T. gondii infection can sometimes trigger severe retinal diseases, which has been recently recognized to be particularly prevalent in regions of South America.6,7 When T. gondii infection occurs during pregnancy, vertical transmission can ensue, resulting in birth defects or miscarriage.8 For individuals with compromised immune systems, active T. gondii infection primarily leads to encephalitis or chorioretinitis, but may cause disease in other organs as well.9
Current first-line therapy for toxoplasmosis involves treatment with a combination of pyramethamine and sulfadiazine, which inhibit the folate pathway in T. gondii.10 However, this therapeutic regimen requires long dosing periods and can be limited by rash, leukopenia, nephrotoxicity, teratogenicity, and kernicterus in newborns.11 There is a longstanding need for better toxoplasmosis drugs, especially therapeutic agents that are non-toxic, nonteratogenic, and with simple dosing profiles.
In order to develop new therapies for the treatment of toxoplasmosis, we and others have explored strategies that involve inhibiting enzymatic components of calcium-dependent cellular processes in the parasite, which are necessary for invasion into and egress from host cells. T. gondii calcium-dependent protein kinase 1 (TgCDPK1) is a particularly promising target due to its role in gliding and motility.12 Gliding and motility are necessary for T. gondii invasion and egress, and as T. gondii is an obligate intracellular parasite that requires these processes to proliferate, inhibition of TgCDPK1 represents a promising new anti-parasitic strategy. Indeed, genetic or pharmacological disruption of TgCDPK1 function blocks parasite growth in vitro and is able to prevent and treat established T. gondii infections in mice.13–20
We previously developed a number of highly promising pyrazolopyrimidine (PP)-based ATP-competitive inhibitors that are able to potently block the enzymatic activity of TgCDPK1. These selective TgCDPK1 inhibitors are able to block the invasion of T. gondii into host cells, preventing proliferation of the parasite.15,19,20 Recently, we have demonstrated that the TgCDPK1 inhibitor 1 (1294) is an orally available inhibitor that possesses anti-toxoplasmosis activity in vitro and in vivo, with no apparent signs of toxicity.15 Furthermore, 1 was able to block vertical transmission of Neospora caninum in a pregnant mouse model, and since N. caninum is closely related to T. gondii, it speaks to a potential therapeutic application for toxoplasmosis in pregnancy.21,22 Unfortunately, we have found that 1 has potent human Ether-à-go-go-Related Gene (hERG) inhibitory activity23–associated with long Q-T syndrome–and, thus, presents a significant cardiotoxicity risk in humans.24 Here, we describe the identification of optimized PP-based TgCDPK1 inhibitor 32, which does not have an hERG liability but possesses the favorable anti-parasitic activity of lead compound 1.
Results and Discussion
Molecular Design and Synthesis
TgCDPK1 contains a C-terminal calcium-binding domain of 4 EF-hands (CDPK activation domain) linked to an N-terminal kinase domain. The ATP-binding cleft of TgCDPK1, which is the site of interaction with PP-based inhibitors, is similar in structure to mammalian protein kinases with the exception that it contains a rare glycine residue at the gatekeeper position. The starting point for this study was compound 1, which is based on a PP scaffold that forms two hydrogen-bonding interactions with the hinge region of TgCDPK1 (Figure 1). The C-3 (R1) position of 1 is derivatized with a bulky 6-ethoxy-2-naphthyl group that occupies a hydrophobic pocket adjacent to TgCDPK1’s glycine gatekeeper residue. As glycine is a very uncommon gatekeeper residue, the space occupied by the 6-ethoxy-2-naphthyl group provides a means to obtain high selectivity over mammalian kinases, which often have larger gatekeepers that constrain access to the hydrophobic back pocket. The 6-alkoxy-2-naphthyl substitution pattern appears to be optimal for complementing the hydrophobic gatekeeper pocket of TgCDPK1 and enhancing overall inhibitor selectivity. At the N-1 position, 1 contains an N-methyl-4-piperidinemethylene group, directed toward the sugar pocket. This basic group increases potency for TgCDPK1 and, in combination with 6-alkoxy-2-naphthyl substituents at the C-3(R1) position, the 4-piperidinemethyl group greatly enhances kinase selectivity.25
Figure 1.
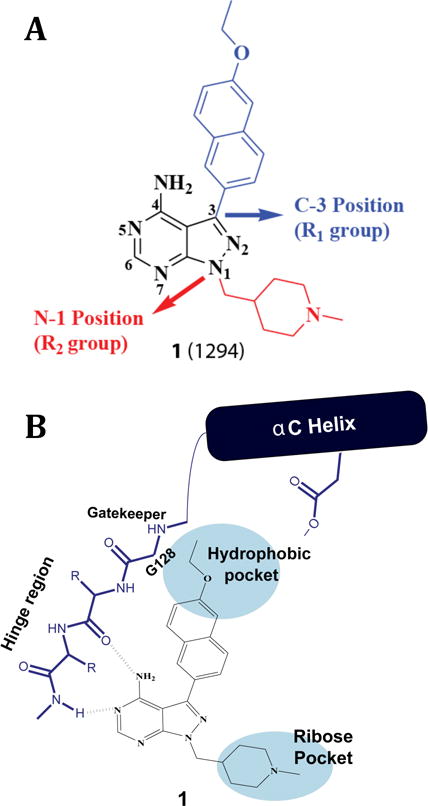
Inhibitor 1. (A) The chemical structure of compound 1. Pyrazolopyrimidine scaffold numbering and sites of inhibitor derivatization (R1 and R2) are shown. (B) Schematic of how 1 interacts with the ATP-binding site of TgCDPK1.
Several synthetic strategies were used to generate derivatives of 1. The synthetic route shown in Scheme 1 was used to generate inhibitors that contain a 6-ethoxy-2-naphthyl group at the R1 position and various 4-piperidinemethylene or 3-azetidinemethylene derivatives at the R2 position (Table 1). Inhibitors that contain an N-methyl-4-piperidinemethylene R2 group and various 6-alkoxy-2-naphthyl or 6-alkoxy-2-quinolinyl groups at the R1 position were generated by either alkylation of t-butyl-4-((4-amino-3-(6-hydroxynaphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)piperidine-1-carboxylate, followed by Boc deprotection and reductive alkylation, or by palladium-catalyzed Suzuki-Miyaura couplings between appropriate aryl boronic acids and boronate esters and t-butyl-4-((4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)piperidine-1-carboxylate, followed by Boc deprotection and reductive alkylation (Scheme 2). Boronic esters and boronic acids that are not commercially available were prepared from aryl bromides using standard methods.26 Compounds containing variable groups at the R2 position were generated by alkylating 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine with alkyl halides, alkyl mesylates, or expoxides followed by Suzuki-Miyaura coupling to R1-boronic acids or boronate pinacol esters (Scheme 3).
Scheme 1.
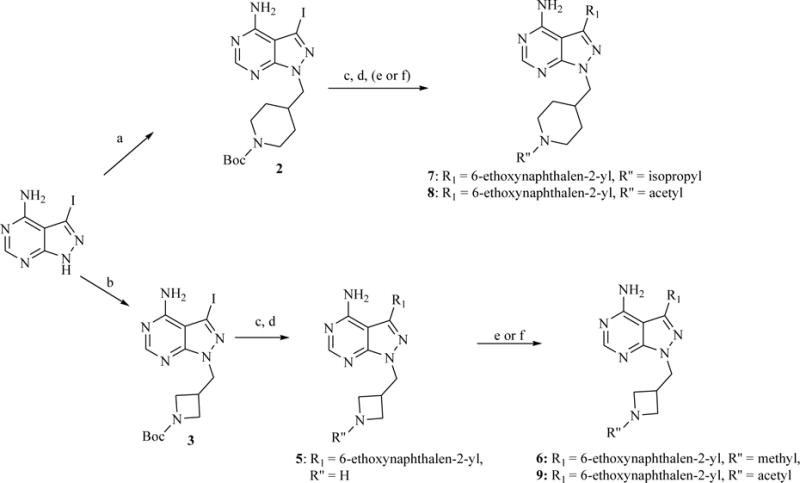
a) N-Boc-4-piperidinemethanol mesylate, Cs2CO3, DMF, 80 °C; b) N-Boc-3-azetidinemethanol mesylate, Cs2CO3, DMF, 80 °C; c) Na2CO3, Pd(PPh3)4, 6-ethoxy-2-naphthaleneboronic acid, H2O/DME, 80 °C (microwave); d) TFA/CH2Cl2 (1:1); e) sodium methoxide in MeOH, then 2% AcOH, formaldehyde or acetone, NaBH3CN; f) Ac2O, TEA, DMF.
Table 1.
Enzymatic TgCDPK1 and Src inhibition (IC50), hERG inhibition (IC50), HepG2 and CRL-8155 growth inhibition (GI50) and T. gondii cellular assay (EC50) results for compounds with variable R2 substructures (1–9). IC50 and GI50 values are the averages of at least three assays. T. gondii cellular assay EC50 values are the average of at least two runs that were performed in triplicate.
| |||||||
|---|---|---|---|---|---|---|---|
| R2 | IC50 (μM)
|
human cells, GI50 (μM)
|
EC50 (μM)
|
||||
| TgCDPK1 | Src | hERG | HepG2 | CRL-8155 | T. gondii | ||
| 1 |
|
0.0029 | >10 | 0.40 | >10 | >10 | 0.14 |
| 5 |
|
0.0032 | >10 | 1.7 | >10 | >10 | 0.283 |
| 6 |
|
0.0060 | >10 | 1.4 | >10 | >10 | 0.41 |
| 7 |
|
0.010 | >10 | 0.30 | >10 | >10 | 0.51 |
| 8 |
|
0.011 | >10 | >10 | >10 | >10 | 0.22 |
| 9 |
|
0.004 | 1.1 | >10 | >10 | >10 | 0.48 |
Scheme 2.
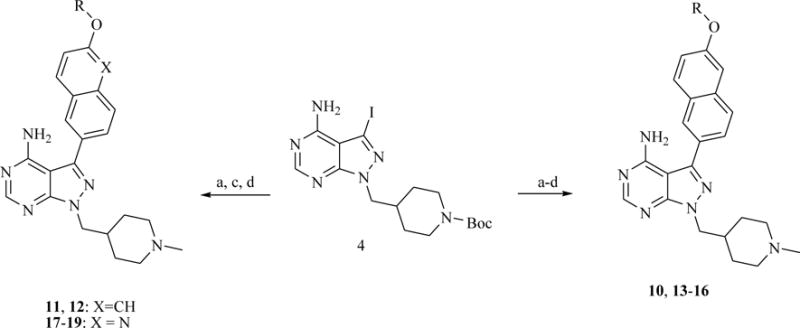
a) aryl boronate ester, K3PO4/Na2CO3, PdCl2(dppf), 1,4-Dioxane:H2O or DME:H2O, 80 °C (microwave); b) alkyl halide or epoxide, NaH2PO4:K2CO3 (1:1), DMF, 60 °C; c) TFA/CH2Cl2 (1:1); d) sodium methoxide in MeOH, then 2% AcOH, formaldehyde, NaBH3CN. Compound 4 was synthesized according to a previously reported procedure.19
Scheme 3.
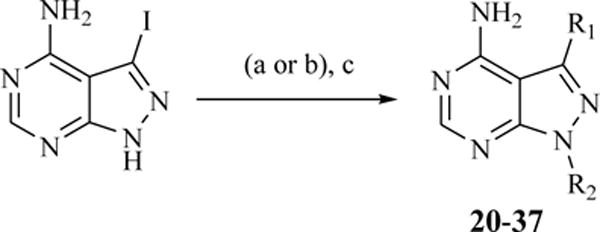
a) alkyl halide or alkyl mesylate, Cs2CO3, DMF, 80 °C; b) epoxide, NaH2PO4:K2CO3 (1:1), DMF, 80 °C; c) aryl boronate ester, K3PO4 or Na2CO3, PdCl2(dppf) or Pd(PPh3)4, 1,4-Dioxane:H2O or DME:H2O, 80 °C (microwave).
Inhibitor Testing Cascade
A multi-assay testing cascade was used to determine if newly generated analogs demonstrated superior anti-parasitic properties compared to compound 1. Compounds were first tested for the ability to block the activity of recombinant TgCDPK1 using a luminescence-based assay that measures the consumption of ATP in the presence of the peptide substrate Syntide 2.19 All assays were performed in the presence of 2 mM CaCl2 and at an ATP concentration–10 μM–equal to TgCDPK1’s Michaelis constant (Km). Compounds of sufficient in vitro enzyme inhibitory activity were then tested for their ability to block the invasion and growth of parasites into human foreskin fibroblast cells using a previously described T. gondii cellular assay.19 Inhibitors with comparable in vitro enzymatic and cellular potencies as 1, were next screened for selectivity in two assays. To determine selectivity for TgCDPK1 over mammalian kinases, compounds were tested against the tyrosine kinase Src, which is representative of the most likely off targets of pyrazolopyrimidine-based inhibitors that contain a large aryl group at the C-3 (R1) position due to its small threonine gatekeeper residue.27 General off target toxicity was determined by testing for growth inhibition (GI50) of two mammalian cell lines, HepG2 (liver) and CRL-8155 (lymphoblast). As elimination of hERG activity was a major goal of this study, all compounds with favorable profiles were tested for hERG activity using a previously described assay.23,28,29
In generating analogs of 1, we felt that it would be important to retain a 6-alkoxynaphthalen-2-yl group at the C-3 (R1) position. In addition, due to the unexpected contribution of the 4-piperidinemethyl group to inhibitor potency and selectivity,25 compounds that contain a similar substituent at the R2 position were focused on first. Results for compounds containing a fixed 6-ethoxynaphthyl at the R1 position and various 4-piperidinemethyl or 3-azetidinemethyl groups at the R2 position are shown in Table 1. Increasing the size of the N-alkyl group on the 4-piperidinemethyl moiety (7) resulted in slightly reduced target potency and did not diminish hERG inhibition. Replacing the 1-methylpiperidin-4-ylmethyl group of 1 with either a 3-azetidinemethyl (5) or a 1-methyl-3-azetidinemethyl (6) group resulted in potent inhibition of TgCDPK1, albeit with attenuated activity against parasite proliferation, but only a slight decrease in hERG inhibition. However, an analog containing an acetylated 4-piperidinemethyl group (inhibitor 8) displayed acceptable levels of inhibition of the TgCDPK1 enzyme and parasite proliferation with no hERG activity. Inhibitor 9, which contains a 1-acetyl-3-azetidinemethylene group instead of a 1-acetyl-4-piperidinemethyl moiety, also lacks hERG activity but possesses unacceptably low activity in the T. gondii cellular assay. These results are consistent with the observed correlation between inhibitor basicity and hERG inhibition.30–32
We next explored whether hERG inhibition could be reduced in analogs of 1 that maintain the 1-methyl-4-piperidinemethyl group by modifying the N-1 position (R2) of the pyrazolopyrimidine scaffold (Table 2). For the 6-alkoxynaphthalen-2-yl series, inhibitors containing cycloalkyloxy groups larger than ethyl (10–12) were potent TgCDPK1 inhibitors and were of reasonable potency in the T. gondii cellular assay. However, the hERG activity of these compounds was comparable to 1. Unfortunately, introduction of more polar groups at the 6 position of the naphthyl ring (13–16), which reduces overall inhibitor lipophilicity and would be expected to diminish hERG activity, led to less potent inhibition of TgCDPK1 and subsequent reduced potency in the T. gondii cellular assay. A more favorable inhibitor profile was obtained by replacing the 6-ethoxy-2-naphthyl group of 1 with a 2-ethoxyquinolin-6-yl moiety (17). Inhibitor 17 showed comparable levels of TgCDPK1 enzyme inhibition and efficacy in the invasion assay as 1, with no detectable Src inhibition or mammalian cell toxicity. Compound 17 also possesses 25-fold lower activity against hERG than 1. However, replacing the ethyl group of 17 with more hydrophobic substituents, like trifluoroethyl (18) or cyclopropyl (19), led to unacceptable levels of hERG inhibition (Table 2).
Table 2.
Enzymatic TgCDPK1 and Src inhibition (IC50), hERG inhibition (IC50), HepG2 and CRL-8155 growth inhibition (GI50) and T. gondii cellular assay (EC50) results for compounds with variable R1 substructures (10–19). IC50 and GI50 values are the averages of at least three assays. T. gondii cellular assay EC50 values are the average of at least two runs that were performed in triplicate. Compounds that did not show an IC50 < 0.010 μM for TgCDPK1 or an IC50 > 5 μM for hERG were de-prioritized for further testing.
| |||||||
|---|---|---|---|---|---|---|---|
| R1 | IC50 (μM)
|
human cells, GI50 (μM)
|
EC50 (μM)
|
||||
| TgCDPK1 | Src | hERG | HepG2 | CRL-8155 | T. gondii | ||
| 1 |
|
0.0029 | >10 | 0.40 | >10 | >10 | 0.14 |
| 10 |
|
0.0040 | >10 | 0.90 | >10 | >10 | 0.61 |
| 11 |
|
0.0025 | >10 | 0.20 | >10 | >10 | 0.32 |
| 12 |
|
0.0015 | >10 | 0.37 | – | >10 | 0.12 |
| 13 |
|
0.015 | >10 | – | >10 | >10 | 0.71 |
| 14 |
|
0.010 | >10 | – | >10 | >10 | – |
| 15 |
|
0.046 | – | – | – | – | – |
| 16 |
|
0.024 | >10 | – | >10 | >10 | – |
| 17 |
|
0.0061 | >10 | ~10 | >10 | >10 | 0.27 |
| 18 |
|
0.0022 | – | 0.50 | – | – | 0.33 |
| 19 |
|
0.0016 | >10 | 1.7 | >10 | – | 0.098 |
–Not tested
Finally, we investigated whether the cyclic amine substituent at the R2 position of 1 can be replaced with another group that confers similar potency, selectivity, and solubility. We have previously shown that inhibitors possessing R2 groups that are connected to the N-1 position of the PP scaffold through a methylene linkage demonstrate the highest degree of selectivity for TgCDPK1 over mammalian kinases with small gatekeepers, like the tyrosine kinases Src and Abl.25 Therefore, a panel of analogs with variable R2 groups, which extend into the ribose-binding pocket of the ATP-binding site was generated (Table 3). In general, inhibitors of this class are potent inhibitors of the TgCDPK1 enzyme and efficacious in the T. gondii cellular assay. Furthermore, all of the inhibitors in Table 3 are highly selective (>150-fold) for TgCDPK1 over Src. Consistent with basicity at the R2 position playing a major role in hERG inhibition, inhibitor 20, which contains a dimethylamino group at the R2 position, has a similar level of hERG activity as 1. Compounds 21–23, which all contain an isobutyl group at the R2 position, are potent inhibitors of the TgCDPK1 enzyme and are at least as effective as 1 against the parasite. Importantly, these compounds are >3500-fold selective for TgCDPK1 over Src, demonstrate no detectable growth inhibition of HepG2 and CRL-8155 cells, and lack hERG activity. Compounds 24–29, which all contain a cycloalkyl ether group linked to the N-1 (R2) position through a methylene, are also effective inhibitors of TgCDPK1 and parasite proliferation. All of these compounds, except 28, have an IC50 >10 μM for Src activity and do not inhibit the growth of mammalian cells. Furthermore, all of the inhibitors in this series, except 24 (IC50 = 1.9 μM), have an IC50 of greater than 10 μM for hERG activity. Inhibitors 30–36, which either contain a 2-methylpropan-2-ol or a 2,2-dimethylpropan-1-ol group at the R2 position also possess desirable profiles: potent TgCDPK1 enzyme inhibition, effective inhibition of parasite proliferation, and no detectable off-target inhibition or toxicity.
Table 3.
Enzymatic TgCDPK1 and Src inhibition (IC50), hERG inhibition (IC50), HepG2 and CRL-8155 growth inhibition (GI50) and T. gondii cellular assay (EC50) results for compounds with variable R1 and R2 substructures (20–37). IC50 and GI50 values are the averages of at least three assays. T. gondii cellular assay EC50 values are the average of at least two runs that were performed in triplicate.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| R2 | R1 | IC50 (μM)
|
human cells, GI50 (μM)
|
EC50 (μM)
|
||||
| TgCDPK1 | Src | hERG | HepG2 | CRL-8155 | T. gondii | |||
|
|
1 |
|
0.0029 | >10 | 0.40 | >10 | >10 | 0.14 |
|
|
||||||||
|
|
20 |
|
0.0045 | >10 | 0.082 | >10 | >10 | 0.26 |
|
|
||||||||
|
|
21 |
|
0.00080 | >10 | >10 | >10 | >10 | 0.12 |
| 22 |
|
0.0011 | 3.7 | >10 | – | >10 | 0.071 | |
| 23 |
|
0.0012 | >10 | >10 | >10 | >10 | 0.090 | |
|
|
||||||||
|
|
24 |
|
0.0021 | >10 | 1.9 | >10 | >10 | 0.045 |
| 25 |
|
0.0018 | >10 | >10 | >10 | >10 | 0.21 | |
|
|
||||||||
|
|
26 |
|
0.0020 | >10 | >10 | >10 | >10 | 0.076 |
| 27 |
|
0.0020 | >10 | >10 | >10 | >10 | 0.078 | |
| 28 |
|
0.0024 | 0.37 | >10 | >10 | >10 | 0.077 | |
| 29 |
|
0.0018 | >10 | >10 | >10 | >10 | 0.081 | |
|
|
||||||||
|
|
30 |
|
0.0056 | >10 | – | >10 | >10 | 0.75 |
| 31 |
|
0.0033 | >10 | >10 | >10 | >10 | 0.17 | |
| 32 |
|
0.001 | >10 | >10 | >10 | >10 | 0.060 | |
| 33 |
|
0.002 | >10 | >10 | >10 | >10 | 0.16 | |
|
|
||||||||
|
|
34 |
|
0.00090 | 3.1 | >10 | >10 | >10 | 0.056 |
| 35 |
|
0.0013 | 7.4 | >10 | >10 | >10 | 0.018 | |
| 36 |
|
0.017 | >10 | – | >10 | >10 | 0.70 | |
|
|
||||||||
|
|
37 |
|
0.094 | >10 | – | >10 | >10 | – |
–Not tested
Pharmacokinetic Profiling of Potent and Selective TgCDPK1 Inhibitors
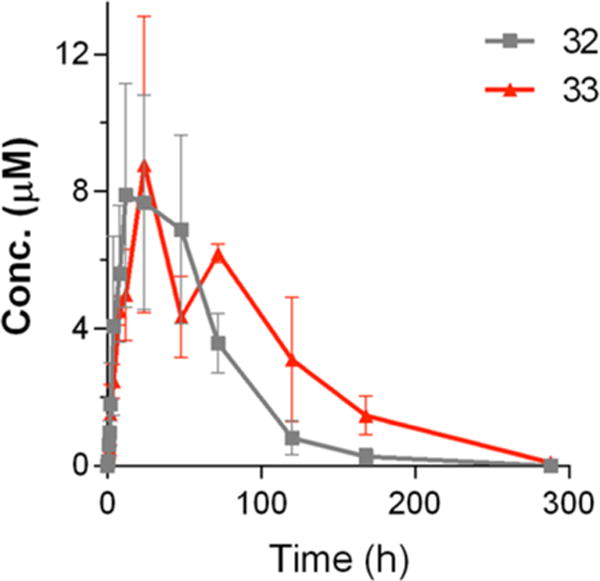
Based on the abilities of a number of inhibitors to potently block T. gondii proliferation in mammalian cells, while demonstrating little or no off-target toxicity or hERG inhibition, we next prioritized compounds based on their solubility in water and pharmacokinetic (PK) properties (Table 4). The aqueous solubility of inhibitors 8, 17, 29, 31–34, and 35 was determined at pH = 6.5. The initial PK profiles of these inhibitors were determined after a single 10 mg/kg oral dose in three Balb/c mice, with sampling conducted at the time points indicated in the Experimental Section. As a reference, after a 10 mg/kg per os (PO) dose, lead inhibitor 1 demonstrated a maximum concentration (Cmax) of 0.75 ±0.15 μM and total exposure (area under the plasma concentrations versus time curve, AUC) of 430 ±84 μM·min. Inhibitors 29, 34, and 35 were poorly soluble (<20 μM) relative to 1 (solubility = 82 μM) and all three compounds had lower exposures and higher clearances than 1. Inhibitors 8 and 17 demonstrated better solubility but also had a lower exposure and higher clearance than 1, which is likely due to the more efficient metabolism of these compounds. In contrast, inhibitors 31–33, which all contain a 2-methylpropan-2-ol at the R2 position, were highly soluble and 32 and 33 reached maximum serum concentrations (Cmax) >10-fold higher and total exposure >30-fold higher than 1 (Table 4 and Figure 2). Compound 31 was absorbed rapidly with a Tmax of 50 min and a Cmax nearly 10-fold higher than 1. Yet, the overall exposure of 31 was only twice as high as 1 and much lower than 32 and 33. Based on this mouse pharmacokinetic data, compounds 32 and 33 were selected for further pharmacokinetic and tolerability studies.
Table 4.
Solubility (in phosphase-buffered saline, pH= 6.5) and PK properties of the most potent and selective TgCDPK1 inhibitors dosed at 10 mg/kg PO to mice.
| Solubility (μM) | Tmax (min) | Cmax (μM) | AUCo->inf (μM.min) | Half life (min) | clearance (mL/min) | |
|---|---|---|---|---|---|---|
| 1 | 82 | 120 | 0.75 ±0.15 | 430 ±58 | 233 ±26 | 0.7 |
| 8 | 48 | 50 | 0.89 ±0.17 | 94 ±51 | 68 ±64 | 2.5 |
| 17 | 54 | 200 | 0.26 ±0.04 | 128 ± 53 | 223 ±182 | 1.8 |
| 29 | 17 | 30 | 0.88 ±0.23 | 67 ±60 | 50 ±73 | 110 |
| 31 | >100 | 50 | 5.2 ±1.0 | 850 ± 411 | 114 ±115 | 0.2 |
| 32 | >100 | 320 | 13 ±1 | 13700 ±1500 | 1190 ±510 | 0.2 |
| 33 | >100 | 560 | 7.8 ±1.4 | 16600 ±4300 | 1110 ±400 | 0.3 |
| 34 | 6.2 | 40 | 0.39 ±0.16 | 31 ±8 | 40 ±5 | 6.7 |
| 35 | 8.0 | 30 | 3.9 ±1.1 | 3.9 ±1.1 | 41 ±21 | 21 |
Figure 2.
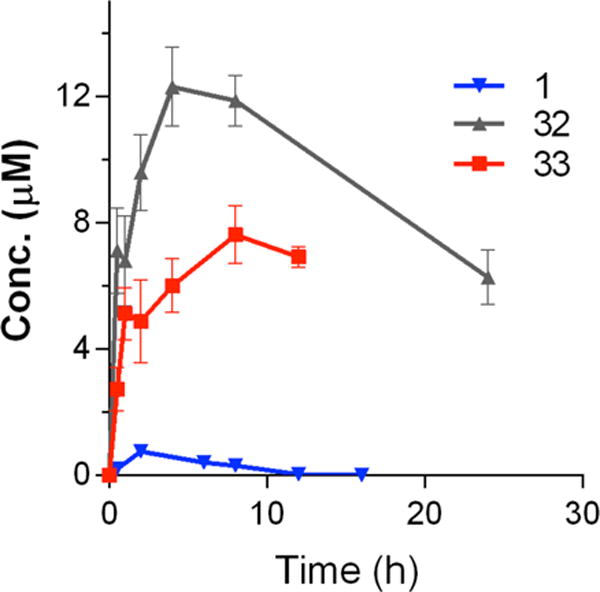
Comparison of oral PK for compounds 32 and 33 in mice. Both compounds were dosed at 10 mg/kg, PO.
Dose Finding and Tolerability of 32 and 33
Based on the PK parameters of 32 and 33 following single dose administration to mice, these compounds were evaluated for tolerability following escalated dosing. Both compounds were initially dosed at 10 mg/kg on day one. As no observable adverse effects were detected in mice dosed with either 32 or 33, the next dose was increased to 50 mg/kg on day 4, followed by a final dose of 100 mg/kg on day 8. Mice showed no overt signs of toxicity or weight loss over the 10-day observation period. Next, the plasma protein binding of both compounds was evaluated. Both compounds were highly protein bound in mouse plasma with plasma protein binding of 96% for 32 and 88% for 33. Based on the protein binding values the plasma concentrations required to maintain unbound plasma exposure above the TgCDPK1 EC50 are 1.33 μM for 33 and 1.5 μM for 32. Using these target concentrations and the single dose PK data, a multiple dosing regimen with a loading dose of 20 mg/kg of 32 and 33 followed by a 5 mg/kg daily dose for five days was evaluated. Blood was collected at multiple time points to determine plasma concentrations over the course of treatment (Figure 3). Both compounds demonstrated excellent exposure through the 24-hour dosing interval with the trough concentrations remaining >1.5 μM throughout the study. The measured concentrations following multiple dosing were similar to those predicted from the single dose PK studies in mice (Figure 3). Neither compound showed any evident signs of toxicity compared to mice dosed with vehicle only. Cardiac puncture blood collection was performed at the end of the study for complete blood count and serum biochemical profiles. All results were reported by Phoenix Central Laboratory to be within a normal range for species and age.
Figure 3.
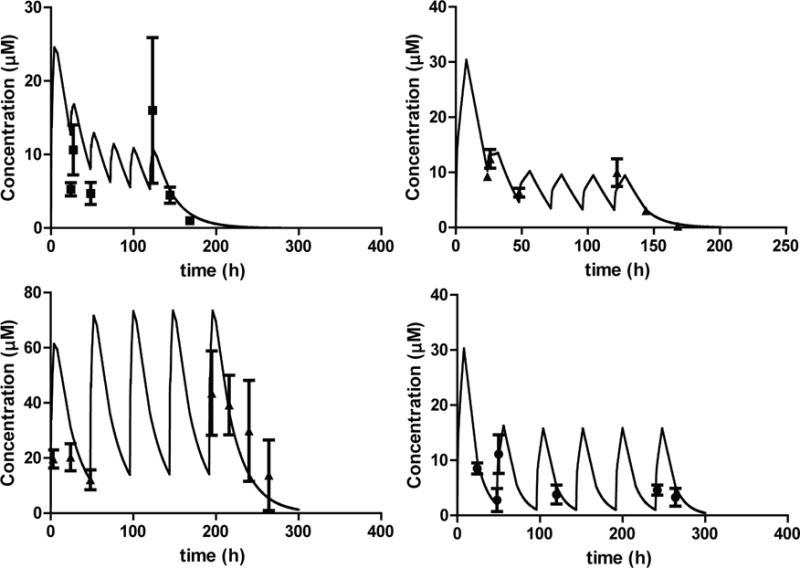
Multiple dose PK studies with compound 32 and 33. The dosing regimens were simulated based on the disposition data following single oral doses to mice (lines) and the observed plasma concentrations (dots) compared to the simulation. (top left panel) 32 was dosed at 20 mg/kg on day 1 and then 5 mg/kg every 24 hours for 5 doses. (bottom left panel) 32 was dosed at 50 mg/kg every 48 hours for 5 doses. (top right panel) 33 was dosed at 20 mg/kg on day 1 and then 5 mg/kg every 24 hours for 5 doses. (bottom right panel) 33 was dosed at 20 mg/kg on day 1 and then 10 mg/kg every 48 hours for 5 doses. Each dot is a mean of 3 mice with standard deviation.
In a second multiple dose study compound 32 was administered at 50 mg/kg PO every other day for 5 doses and compound 33 was administered with a 20 mg/kg loading dose followed by a 10 mg/kg maintenance dose every other day for 5 doses. The exposure to 33 with this dosing regimen was similar to the one observed with daily dosing of the lower dose and the plasma concentration at various time points were well predicted based on the single dose PK results. With compound 32 plasma concentrations remained >10 μM for the entire duration of the study (Figure 3) with maximum concentrations reaching 43 μM. The exposure to compound 32 following the last 50 mg/kg dose was higher than predicted from single dose studies with longer half-life suggesting possible saturation of metabolism at this dose.
Based on the favorable single and multiple dose PK and the lack of observable toxicity, compounds 32 and 33 were evaluated for tolerability and PK characteristics in rats following intravenous (IV) and PO dosing. Following IV administration to rats both 32 and 33 displayed biphasic kinetics with compound 33 reaching distribution equilibrium more rapidly than 32 (Figure 4). Compound 32 distributed approximately to total body water with a volume of distribution at steady state (Vss) of 0.9 L/kg. The distribution of 33 was more extensive with a Vss of 5.8 L/kg. Both compounds had very low systemic clearances in rats but, in contrast to mice, the clearance of 33 was 4-fold higher than that of 32. The elimination half-lives of both compounds were acceptable for multiple dosing regimens, 9.6 and 13 hours for 32 and 33, respectively (Figure 4).
Figure 4.
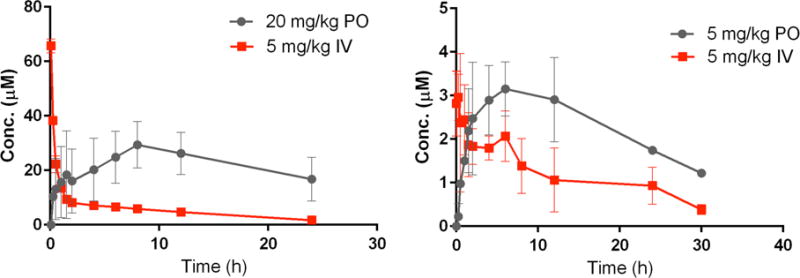
Plasma concentration time curves for compound 32 (left) and 33 (right) following IV and PO dosing to rats.
Following oral administration to rats both compounds showed slow absorption with absorption phase continuing for 12 hours after oral dosing. The plasma concentrations over the 24-hour period after PO dosing exceeded those observed after IV dosing (Figure 4) suggesting that both 32 and 33 had essentially complete bioavailability in rats. The apparent bioavailability of these compounds was greatly improved compared to that of compound 1 (46%).
Because it is important that anti-toxoplasmosis therapies are able to prevent reactivation of parasites within tissue cysts, which largely reside in the central nervous system (CNS), the distribution of compounds 32 and 33 to the brain was next determined. To do this, the distribution of 32 and 33 into mouse brain (n=3) at one hour after intraperitoneal dosing of 5 mg/kg was measured. The mean concentration of compound 32 at one hour in brain was 1.2 ±0.5 μM, a concentration well above the TgCDPK1 EC50. The corresponding plasma concentration of 32 was 4.1 ±1.1 μM resulting in a brain to plasma concentration ratio of 32 of 0.33 ±0.22. This brain penetration was comparable to 1 (0.31). In accordance with the larger distribution volume of compound 33, it demonstrated a greater brain to plasma concentration ratio (1.65 ±0.84). Both the brain and plasma concentration of 33, 1.90 ±0.24 μM and 1.23 ±0.76 μM, respectively, were above the TgCDPK1 EC50. As both 32 and 33 provide adequate exposure in CNS, they are both excellent candidates for animal efficacy models of toxoplasmosis.
Selectivity Profiling of 32 and 33
Due to the impressive PK properties of 32 and 33, these inhibitors were profiled further. To demonstrate that TgCDPK1 is the kinase target of these compounds in T. gondii, compounds 32 and 33 were tested against parasitic cell lines overexpressing the Gly128Met TgCDPK1 gatekeeper mutant or wildtype (wt) TgCDPK1. Expression of the Gly128Met gatekeeper mutant of TgCDPK1, but not wtTgCDPK1, makes T. gondii highly resistant to PP-based inhibitors that contain 6-alkoxynaphthalen-2-yl groups at the R1 position.19,33 Both 32 and 33 show a dramatic loss in potency against parasites overexpressing the Gly128Met gatekeeper mutant relative to the parent RH strain and to T. gondii overexpressing wild type TgCDPK1 (Figure 5), which is consistent with TgCDPK1 being the primary target through which these inhibitors exert their anti-parasitic effects.
Figure 5.
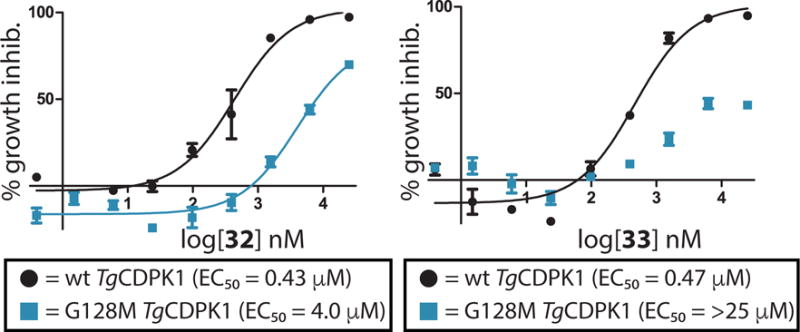
EC50 curves of inhibitors 32 (left) and 33 (right) for T. gondii over-expressing either wild type (wt) TgCDPK1 (black circles) or a drug resistant G128M TgCDPK1 mutant (blue squares). All experiments were performed in triplicate.
32 and 33 were further profiled for any mammalian kinase off targets using a panel of 80 human kinases representing different subfamilies of the kinome tree with a fluorescence-based competition assay (Supporting Information Table 1).17 78 of the 80 mammalian kinases tested have an IC50 >1.5 μM (>1500-fold selective for TgCDPK1) for 32. For the two kinases–PKCν (PKD3) and MEK1–that have sub-micromolar IC50 values, compound 32 is a >120-fold and >900-fold less potent inhibitor than for TgCDPK1, respectively. Compound 33 appears to be slightly more selective than compound 32. Indeed, it demonstrated an IC50 value of greater than 5 μM (>2500-fold selective) for 79 of the kinases tested, with only PKCν (PKD3) demonstrating a sub-micromolar (IC50 = 0.280 μM; 140-fold selective) IC50 value.
Structure of the TgCDPK1·33 Complex
Previously, we demonstrated that certain substituents at the R2 position of the pyrazolopyrimidine scaffold enhance inhibitor potency and selectivity for TgCDPK1.25 While 1 has an IC50 of 2.9 nM for TgCDPK1 and is >3000-fold selective for TgCDPK1 over the mammalian tyrosine kinase Src, the R2 isopropyl analog of 1 has an IC50 of 5.0 nM for TgCDPK1 and is only 76-fold selective for TgCDPK1 over Src.19,25 The 2-methylpropan-2-ol R2 group of inhibitors 32 and 33 seem to have a similar effect on selectivity as the N-methyl-4-piperidinemethylene group of 1. For example, the R2 isopropyl analog of inhibitor 33 is only 40–fold selective for TgCDPK1 over Src, while 33, which contains a 2-methylpropan-2-ol R2 group, is >5000-fold selective. To gain a better understanding of the contribution of the 2-methylpropan-2-ol R2 group to inhibitor binding, a co-crystal structure of 33 bound to TgCDPK1 in the calcium-free inactive form was determined (Figure 6). As expected, 33 occupies the ATP-binding cleft of TgCDPK1, with the pyrazolopyrimidine scaffold making the same hydrophobic and hydrogen-bonding contacts as the adenine ring of ATP. In addition, the 2-cyclopropyloxyquinolin-6-yl R1 group of 33 is directed towards the Gly128 gatekeeper residue of TgCDPK1, with the quinoline moiety occupying the enlarged hydrophobic pocket that is available due to the lack of a side chain at this position. Side chains of residues in helix-αC and the fourth β-strand of the N-terminal lobe form hydrophobic interactions with the 2-cyclopropyloxy group, most likely contributing to the favorable potencies of inhibitors containing this substituent. The R2 substituent of 33 occupies the ribose-binding pocket of TgCDPK1, with the tertiary alcohol of the R2 substituent directed towards the phosphate-binding loop (P-loop), which caps the ATP-binding site. Superposition of the TgCDPK1·33 complex with the co-crystal structure of TgCDPK1 bound to a close analog of 1 shows that the pyrazolopyrimidine scaffolds and R1 substituents of these inhibitors are well aligned within the ATP-binding site. However, the R2 groups of both inhibitors adopt very different orientations. While the 2-methylpropan-2-ol R2 group of 33 is oriented towards the P-loop of TgCDPK1, the larger 4-piperidinemethylene group of the 1 analog is directed out of the ATP-binding site (Figure 6C) and forms an electrostatic interaction with the side chain of Glu155, located on the surface of the C-terminal lobe. Therefore, it appears that different interactions in the ribose pocket can be exploited by R2 substituents to confer high selectivity for TgCDPK1.
Figure 6.
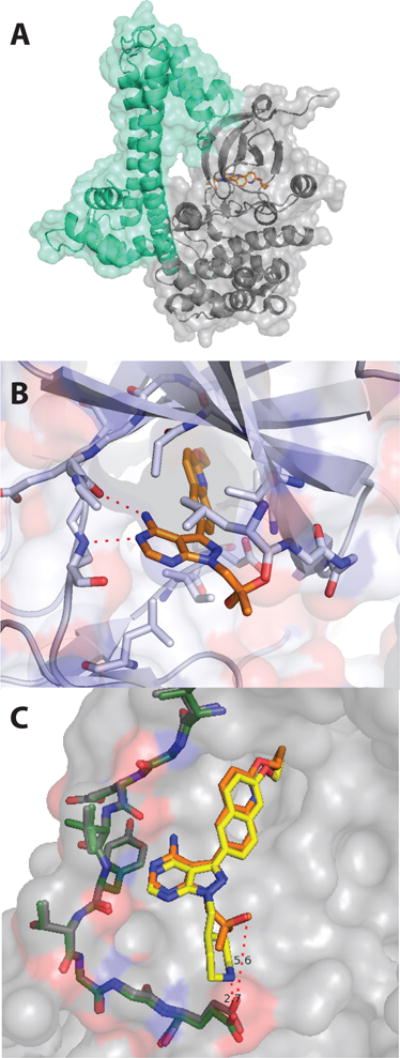
Structure of the TgCDPK1·33 complex (PDB accession code: 4TZR). (A) Entire view of the 33·TgCDPK1 co-crystal structure. Inhibitor 33 is shown as orange sticks. The CDPK activation domain (CAD) of TgCDPK1 is in mint and the kinase domain is shown in grey. (B) Expanded view of the ATP-binding site from the TgCDPK1·33 complex. The backbone amides of residues Glu129 and Tyr131 in the hinge region (shown as sticks) form two hydrogen bonds (dashed red lines) with the pyrazolopyrimidine scaffold of 33. The 2-cyclopropyloxyquinolin-6-yl R2 group of 33 is directed towards the Gly128 gatekeeper residue. (C) An overlay of an analog of 1 (yellow) bound to TgCDPK1 (PDB accession code: 3SX9) and the TgCDPK1·33 (orange) complex (PDB accession code: 3SX9)
Large Animal Pharmacokinetics
The lack of toxicity in the initial mouse toxicity screens allowed us to move forward into large animal pharmacokinetic and tolerability profiling. Male calves (n=2 for each compound) were dosed orally at 10 mg/kg for compound 32 and 9.3 mg/kg for compound 33 and blood sampled up to 12 days after dosing (Figure 7). Similar to rats, the absorption of both compounds was slow with maximum concentrations reached at 24 hours for compound 33 and 12 hours for compound 32. The maximum plasma concentrations were similar for the two compounds, 7.9 μM and 9.8 μM for compound 32 and 33, respectively. However, the overall exposure to 33 was greater than that of 32 due to its lower oral clearance and longer half-life (Table 6). Yet, both compounds had very low oral clearances and long systemic half-lives. Similar to rats, compound 33 had a higher apparent volume of distribution than compound 32 and the overall distribution characteristics were similar to those observed in rats.
Figure 7.
Calf plasma levels. Comparison of oral PK for compounds 32 and 33. Each compound was tested in two calves. Compounds 32 was dosed at 9.3 mg/kg and compound 33 was dosed at 10 mg/kg, PO. Each point shows the mean and range of measurements in two calves.
Table 6.
In vivo pharmacokinetic parameters of 32 and 33 in calves following PO administration of 10 mg/kg of 32 and 9.3 mg/kg of 33. Data are shown as the mean and range between animals.
| AUC (h*μ mol/L) | CL/F (mL/hr/kg) | V/F (L/kg) | t1/2 (hr) | |
|---|---|---|---|---|
| 32 | 588 (353–825) | 52 (31–72) | 1.5 (1.3–1.8) | 23 (17–28) |
| 33 | 849 (607–1091) | 30 (22–39) | 2.3 (1.4–3.2) | 51 (46–56) |
There were some notable calf health concerns following the single oral dose of compound 33. For compound 33, both calves developed neurologic signs of toxicity on day 4–5 post-dose. Both calves were observed to have a plantigrade stance and ataxia. Despite abnormalities, both calves were ambulatory, alert, and responsive with normal appetites. The signs became progressively worse, with notable ataxia, followed by marked improvement by day 10 but were never completely resolved by the end of the experiment on day 14. These signs were not observed for compound 32. One possibility for the increased CNS toxicity was the greater brain exposure for compound 33 compared with compound 32. The observed neurological signs of toxicity in compound 33 gave us reasons to no longer pursue further studies of this compound. Moving forward we chose to further characterize compound 32 before performing small animal efficacy studies and large animal PK studies.
Further Toxicity Profiling of 32
Potential further toxicity of compound 32 was examined in mice by testing two doses (30 mg/kg and 100 mg/kg PO) daily for five days, while observing mice for signs of toxicity and collecting blood samples. Both groups of mice remained active, well-groomed, and appeared normal throughout the study. Upon necropsy, there were no gross abnormalities. Histology revealed mild focal inflammation in the spleen in two of three mice in the 30 mg/kg group. The only abnormality seen in the 100 mg/kg group was inflammatory infiltrate in the hepatic lobules in one of three mice. The concentrations following 30 mg/kg doses were slightly higher than those predicted from single dose PK data while the exposures following the 100 mg/kg doses were similar to those predicted from single 10 mg/kg dose data (Figure 8). Similar to the early multiple dosing experiments in mice, after the last 100 mg/kg dose the elimination of compound 32 was slower than predicted with considerable plasma concentrations persisting at 72 hours after the final dose.
Figure 8.
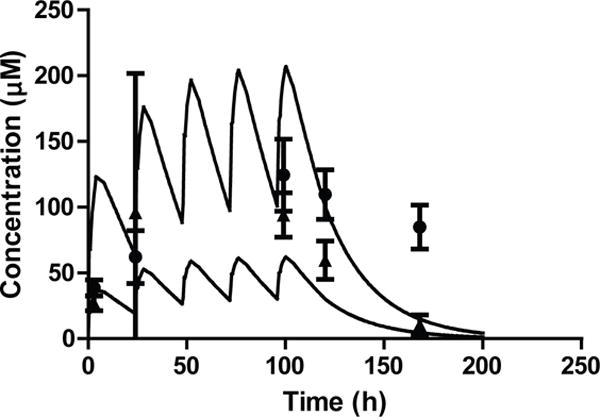
Exposure to compound 32 in multiple dose toxicity studies. The concentration profiles of 32 following 30 mg/kg and 100 mg/kg daily doses were simulated based on the single dose 10 mg/kg data. The mean observed concentrations (n=3) with standard deviation are plotted as triangles for 30 mg/kg dosing and circles for the 100 mg/kg dosing) with the simulation.
Next, we completed a maximally tolerated dose study to determine the lowest observable adverse effect level (LOAEL) and the no observable adverse effect level (NOAEL) of compound 32. Mice were dosed with single PO doses of 32 ranging from 200 mg/kg to 1000 mg/kg. The LOAEL was observed at 500 mg/kg and the NOAEL was observed at 400 mg/kg. Mice had slightly ruffled fur and were less active than the control mice at 3 hours following the 500 mg/kg dose. The lowered activity persisted at 24 hours but was resolved by 30 hours. The lack of toxicity up to 500 mg/kg confirmed the safety of the compound for small animal efficacy studies.
Large Animal Pharmacokinetics of 32
The pharmacokinetics of 32 was further explored in dogs and monkeys following IV and PO administration. Similar to rats, 32 had a very low clearance in both species and a relatively long half-life (Table 7). The plasma concentrations time-profile following IV dosing was biphasic in both species with a similar extent of distribution in dogs and monkeys as observed in rats and calves (Figure 9). The bioavailability of compound 32 was 66 ±17% in monkeys and 88 ±12% in dogs demonstrating good bioavailability in both species as predicted from rats.
Table 7.
In vivo pharmacokinetic parameters of 32 following IV and PO administration of 1 mg/kg to dogs and monkeys (n=3 for each). Data is show as mean and standard deviation.
| AUC (h*μ mol/L) | CL (mL/hr/kg) | Vss (L/kg) | t1/2 (hr) | |
|---|---|---|---|---|
| Dogs | ||||
| IV | 11200 ±1100 | 90 ±10 | 1.7 ±0.1 | 13.2 |
| PO | 9620 ±1350 | |||
| Monkeys | ||||
| IV | 8720 ±3400 | 130 ±50 | 1.8 ±0.3 | 9.6 |
| PO | 5730 ±1500 | |||
Figure 9.
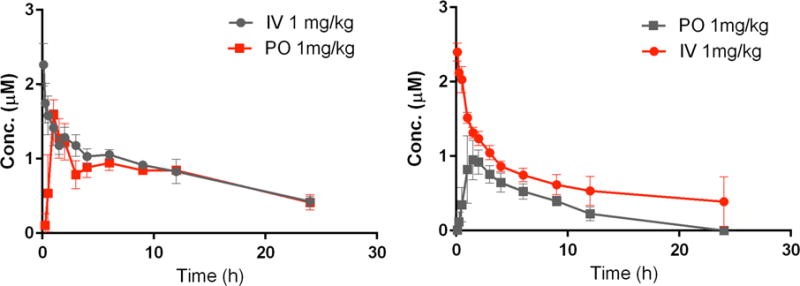
Plasma concentration time curves for compound 32 following IV and PO dosing to dogs (left) and monkeys (right).
In Vivo Efficacy of 32 Against Acute and Latent T. gondii Infection in Mice
We tested 32 against a high inoculum of type I RH strain T. gondii to determine efficacy against fulminate toxoplasmosis in two experiments (Figure 10). Unlike type II T. gondii strains, type I strains do not typically form tissue cysts but rather cause death within 10 days. Compound 32 was administered via oral gavage two days after infection and the burden of infection was measured 5 days after the initiation of treatment. Treatment with 32 at 20 mg/kg for 5 days reduced the number of T. gondii tachyzoites in the peritoneal fluid below the limits of detection (less than 100 tachyzoites/mL). Treatment with 32 was highly effective at 20 mg/kg daily for 5 days in reducing infection in the spleen (more than 99%), and infection in the brain (95%). The efficacy of 32 against an acute T. gondii infection that is not adequately controlled by the mouse immune system suggests that 32 would be active against toxoplasmosis in an immunocompromised human.
Figure 10.
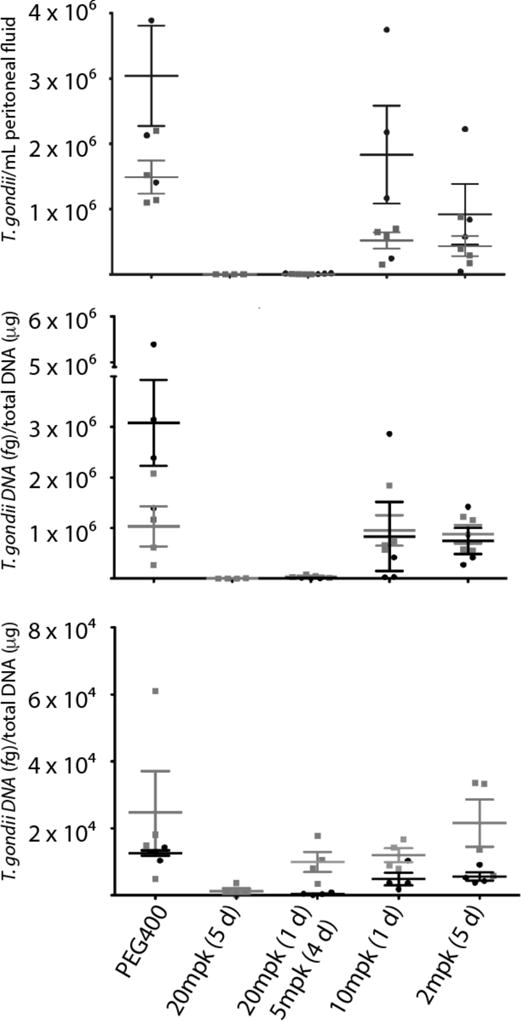
Inhibitor 32 is efficacious against acute toxoplasmosis. Efficacy was evaluated by measurement of T. gondii in peritoneal fluid (top), spleen (middle), and brain (bottom) in two experiments. Mice were treated daily for 5 days, beginning 2 days after IP infection with T. gondii. Mice were analyzed one day after the last dose. Peritoneal fluid was analyzed by fluorescent microscopy and spleen and brain tissue were analyzed by quantitative real-time PCR. Groups consisted of 4 mice. Bars represent the mean and the standard error of the mean. PEG = polyethylene glycol; mpk = mg/kg.
We next tested 32 for its efficacy against latent murine toxoplasmosis by determining its ability to reduce T. gondii brain cysts (Fig. 11).34 Mice were infected IP with the type II ME49 strain of T. gondii, which recapitulates the non-lethal course of infection seen in humans, culminating in formation of cysts in the brain. Five weeks after infection, mice were treated once daily for 14 days via oral gavage with either 30 mg/kg of 32 or a vehicle control. An additional group of mice was dosed twice–days 1 and 7–with 105 mg/kg of 32. Mice were sacrificed and cyst numbers were determined from brain homogenate by microscopy two weeks after the last drug dose. The mean number of brain cysts for vehicle-treated control mice was 1537 ±623. Treatment with 30 mg/kg of 32 led to a reduction of brain cyst number of 88.7%. The percent reduction in brain cyst number for the 105 mg/kg-treated group was essentially the same (87.6%). Importantly, no signs of toxicity or increased morbidity were observed in either drug-treated group. The favorable safety profile and brain permeability of 32 make it an attractive candidate to treat toxoplasmosis in pregnancy, as well as CNS toxoplasmosis, although we have not fully investigated it for effects on fetal development.
Fig. 11.
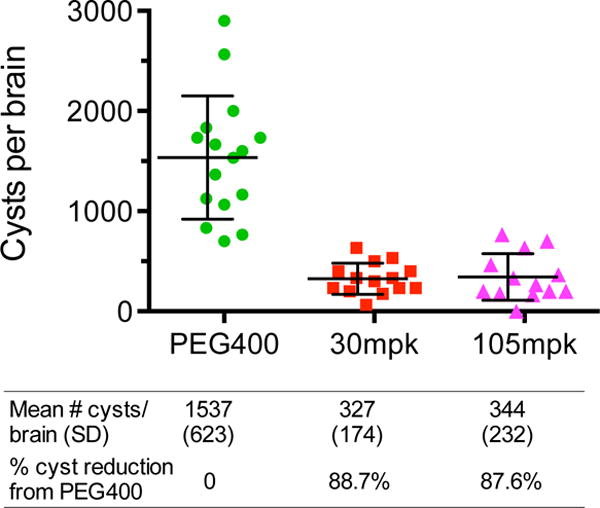
Inhibitor 32 is efficacious against latent T. gondii infection. 32 reduced the number of T. gondii cysts per brain compared to a vehicle control. Five weeks after being inoculated by the ME49 T. gondii strain, mice were treated once daily for 14 days via oral gavage with 30 mg/kg of 32 or PEG400 vehicle control. A group of mice was also treated twice–days 1 and 7–via oral gavage with 105 mg/kg of 32. Two weeks after the final drug treatment, mice were sacrificed and the number of brain cysts was determined. The number of cysts per brain was >85% lower for both dosing regimens of 32 relative to a vehicle control. Bars represent the mean and the standard error of the mean. PEG = polyethylene glycol; mpk = mg/kg.
Conclusions
In the present study, we sought to identify PP-based inhibitors that possess all of the favorable properties of lead compound 1 but lack its hERG activity. To do this, analogs of 1, which contain variable R1 and R2 groups displayed from the PP-scaffold, were first screened for potent inhibition of the TgCDPK1 enzyme and T. gondii invasion/proliferation. Inhibitors of sufficient potency were then screened for the lack of growth inhibition of mammalian cell lines and minimal hERG activity. In this series of inhibitors, the key to eliminating hERG activity was replacing the N-methyl-4-piperidinemethylene R2 position group of 1 with a non-basic substituent that is able to confer a similar level of TgCDPK1 selectivity. While a number of non-basic R2 substituents provided inhibitors with potent activity against T. gondii and minimal mammalian cell cytotoxicity and hERG activity, inhibitors 32 and 33, which contain a 2, 2-dimethylpropan-1-ol group at the R2 position, stood out due to their superior PK properties and adequate exposure to the CNS. However, the superior safety profile of inhibitor 32 coupled with its favorable pharmacokinetic characteristics in small and large animals made this compound the focus of anti-toxoplasmosis efficacy studies. The efficacy of 32 in mouse models of acute and latent toxoplasmosis points to the promise of this drug for further development.
EXPERIMENTAL PROCEDURES
General Synthetic Methods
All chemicals were purchased from commercial suppliers and used without further purification unless otherwise stated. Reactions were monitored with thin-layer chromatography using silica gel 60 F254 coated glass plates (EM Sciences). Compound purification was performed with an IntelliFlash 280 automated flash chromatography system/Combi flash Rf+ using pre-packed Varian Super Flash/Redi sep Rf silica gel columns (hexanes/EtOAc or CH2Cl2/MeOH gradient solvent systems). A Varian Dynamax Microsorb 100-5 C18 column (250 mm × 21.4 mm), eluting with H2O/CH3CN and H2O/ MeOH gradient solvent systems (+0.05% TFA) was used for preparatory HPLC purification. Products were detected by UV at λ=254 & 220 nm. The purity of all final compounds was determined by two analytical RP-HPLC methods, using an Agilent ZORBAX SB-C18 (2.1 mm × 150 mm) or Varian Microsorb-MV 100-5 C18 column (4.6 mm × 150 mm), and eluting with either H2O/CH3CN or H2O/MeOH gradient solvent systems (+0.05% TFA) run over 30 min. Products were detected by UV at λ=254 nm, with all final compounds displaying >95% purity. NMR spectra were recorded on Bruker 300 or 500 MHz spectrometers at ambient temperature. Chemical shifts are reported in parts per million (δ) and coupling constants in Hz. 1H-NMR spectra were referenced to the residual solvent peaks as internal standards (7.26 ppm for CDCl3, 2.50 ppm for d6-DMSO, and 3.34 ppm for CD3OD). Mass spectra were recorded with a Bruker Esquire Liquid Chromatograph - Ion Trap Mass Spectrometer. The synthetic routes used to generate inhibitors are shown in Schemes 1–3.
Synthesis and purification methods for compounds 1–9 in Table 1 are described in previous publications.19,26 Protocols for synthesis of intermediates are provided in the Supporting Information.
General R2 alkylation procedure
Pyrazolopyrimidine (1 equiv.), K2CO3 or Cs2CO3 or K2CO3:NaH2PO4 (1.5–2 equiv.), and an alkylhalide (1.1 equiv.) or alkylmesylate (1.1 equiv.) or oxirane (1.1 equiv.) were stirred in dry DMF at room temperature or 80 °C. The reaction was monitored by thin layer chromatography. After completion, ethyl acetate and water were added and the organic phase was separated. The water phase was extracted with ethyl acetate. The combined organic phases were washed with brine, dried over Na2SO4 and evaporated under reduced pressure. The crude product was then purified via flash chromatography over silica, eluting with either a hexanes/EtOAc or CH2Cl2/MeOH gradient. If necessary, further purification was performed with preparatory RP-HPLC.
General Suzuki coupling procedure
3-Iodopyrazolopyrimidines or 3-Bromopyrazolopyrimidines (1 equiv.), Na2CO3 or K3PO4 (2–4 equiv.), Pd(PPh3)4 or Pd(II)Cl2dppf.DCM, (0.05 equiv.) and boronic acids or boronate pinacol esters (1–2 equiv.) were dissolved in a mixture of dimethoxyethane (1.5 mL) and water (0.5 mL) and then heated in a microwave at 80 °C for one hour. The reaction was monitored by thin layer chromatography. After cooling, ethyl acetate and water were added and the organic phase was separated. The water phase was extracted with ethyl acetate. The combined organic phases were washed with brine, dried over Na2SO4 and evaporated under reduced pressure. The crude product was then purified via flash chromatography over silica, eluting with either a hexanes/EtOAc or CH2Cl2/MeOH gradient. If necessary, further purification was performed with preparatory RP-HPLC.
General naphthol alkylation procedure
6-Hydroxy-2-naphthalene pyrazolopyrimidines (1 equiv.), K2CO3 or Cs2CO3 (1.5–2 equiv.), and alkyl halides/epoxides (1.1 equiv.), NaH2PO4:K2CO3 (1:1 equiv.), were stirred in dry DMF at room temperature or 60–80 °C and monitored by thin layer chromatography. After completion, ethyl acetate and water were added and the organic phase was separated. The water phase was extracted with ethyl acetate. The combined organic phases were washed with brine, dried over Na2SO4, and evaporated under reduced pressure. The crude product was then purified via flash chromatography over silica, eluting with either a hexanes/EtOAc or CH2Cl2/MeOH gradient. If necessary, further purification was performed with preparatory RP-HPLC.
General boc-deprotection procedure
Boc-amine-containing pyrazolopyrimidine was stirred in a TFA/CH2Cl2 (1:1) mixture for ~3 h. The reaction was then concentrated and purified via preparatory RP-HPLC. After HPLC purification, the product was then re-concentrated from 1.25 M HCl in EtOH to afford the final, purified product as a bis-HCl salt.
General reductive amination procedure
Deprotected pyrazolopyrimidines (1 equiv.) were dissolved in methanol and neutralized with sodium methoxide. A solution containing 2% acetic acid and an aldehyde or ketone (5–10 equiv.) was stirred at room temperature for 10 min. Sodium cyanoborohydride (5 equiv.) was then added and the reaction was stirred until reaching completion, as determined by thin layer chromatography (typically ~2 h). The crude reaction was then purified via preparatory RP-HPLC. After HPLC purification, the residue was dissolved in a small amount of 2 M HCl in methanol and, after concentration in vacuo, the final product was obtained as an HCl salt.
3-(6-(Cyclopropylmethoxy)naphthalen-2-yl)-1-((1-methylpiperidin-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (10)
Tert-butyl-4-((4-amino-3-(6-hydroxynaphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)piperidine-1-carboxylate26 and (bromomethyl)cyclopropane were subjected to the General naphthol alkylation procedure followed by the General boc-deprotection procedure and General reductive amination procedure in order to afford 10. 1H NMR (300 MHz, CD3OD) δ 8.51 (s, 1H), 8.17 (s, 1H), 8.01 (d, J = 8.5 Hz, 1H), 7.93 (d, J = 9.1 Hz, 1H), 7.81 (d, J = 9.3 Hz, 1H), 7.36 (s, 1H), 7.30 (d, J = 9.3 Hz, 1H), 4.56 (d, J = 6.4 Hz, 2H), 4.03 (d, J = 6.6 Hz, 2H), 3.57 (m, 2H), 3.06 (m, 2H), 2.88 (s, 3H), 2.45 (m, 1H), 2.02 (m, 2H), 1.77 (m, 2H), 1.44 (m, 1H), 0.71 (m, 2H), 0.46 (m, 2H); MS (ESI) 443.5 m/z [MH+], C26H31N6O requires 443.5.
3-(6-Cyclobutoxynaphthalen-2-yl)-1-((1-methylpiperidin-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (11)
2-(6-Cyclobutoxynaphthalen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (38) and tert-butyl-4-((4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)piperidine-1-carboxylate19 were subjected to the General Suzuki coupling procedure followed by the General boc-deprotection procedure and General reductive amination procedure in order to afford 11. 1H NMR (300 MHz, CD3OD) δ 8.47 (s, 1H), 8.15 (s, 1H), 7.99 (d, J = 8.3 Hz, 1H), 7.92 (d, J = 9.5 Hz, 1H), 7.80 (d, J = 8.9 Hz, 1H), 7.24 (m, 2H), 4.53 (d, J = 6.6 Hz, 2H), 3.56 (m, 2H), 3.03 (m, 2H), 2.86 (s, 3H), 2.62 (m, 2H), 2.44 (m, 1H), 2.22 (m, 2H), 2.04–1.60 (m, 6H); MS (ESI) 443.4 m/z [MH+], C26H31N6O requires 443.2.
3-(6-Cyclopropoxynaphthalen-2-yl)-1-((1-methylpiperidin-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (12)
2-(6-Cyclopropoxynaphthalen-2-yl)-4,4,5,5-tetramethyl-1,3,2- dioxaborolane (41) and tert-butyl-4-((4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)piperidine-1-carboxylate were subjected to the General Suzuki coupling procedure followed by the General boc-deprotection procedure and General reductive amination procedure in order to afford 12. 1H NMR (300 MHz, CD3OD) δ 8.49 (s, 1H), 8.17 (s, 1H), 8.03 (d, J = 8.5 Hz, 1H), 7.92 (d, J = 9.1 Hz, 1H), 7.80 (d, J = 8.5 Hz, 1H), 7.64 (d, J = 2.0 Hz, 1H), 7.27 (dd, J = 8.9, 1.8 Hz, 1H), 4.52 (d, J = 6.0 Hz, 2H), 3.98 (m, 1H), 3.56 (m, 2H), 3.05 (m, 2H), 2.87 (s, 3H), 2.44 (m, 1H), 2.00 (m, 2H), 1.76 (m, 2H), 0.92 (m, 2H), 0.80 (m, 2H); MS (ESI) 429.5 m/z [MH+], C25H29N6O requires 429.6.
3-(6-(2-Methoxyethoxy)naphthalen-2-yl)-1-((1-methylpiperidin-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (13)
Tert-butyl-4-((4-amino-3-(6-hydroxynaphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)piperidine-1-carboxylate26 and bromomethoxyethane were subjected to the General naphthol alkylation procedure followed by the General boc-deprotection procedure and General reductive amination procedure in order to afford 13. 1H NMR (300 MHz, CD3OD) δ 8.50 (s, 1H), 8.16 (s, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.93 (d, J = 8.5 Hz, 1H), 7.80 (d, J = 8.3 Hz, 1H), 7.40 (s, 1H), 7.30 (d, J = 7.8 Hz, 1H), 4.52 (d, J = 4.2 Hz, 2H), 4.31 (m, 2H), 3.86 (m, 2H), 3.62 (m, 2H), 3.54 (s, 3H), 3.04 (m, 2H), 2.87 (s, 3H), 2.44 (m, 1H), 1.99 (m, 2H), 1.77(m, 2H); MS (ESI) 447.5 m/z [MH+], C25H31N6O2 requires 447.5.
1-((1-Methylpiperidin-4-yl)methyl)-3-(6-(oxetan-3-yloxy)naphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (14)
Tert-butyl-4-((4-amino-3-(6-hydroxynaphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)piperidine-1-carboxylate19 and 3-bromooxetane were subjected to the General naphthol alkylation procedure followed by the General boc-deprotection procedure and General reductive amination procedure in order to afford 14. 1H NMR (300 MHz, CD3OD) δ 8.49 (s, 1H), 8.18 (s, 1H), 8.06–7.92 (m, 2H), 7.82 (d, J = 8.5 Hz, 1H), 7.51 (s, 1H), 7.36 (d, J = 9.1 Hz, 1H), 4.81 (m, 1H), 4.53 (d, J = 5.6 Hz, 2H), 3.99–3.84 (m, 4H), 3.57 (m, 2H), 3.05 (m, 2H), 2.86 (s, 3H), 2.44 (m, 1H), 2.00 (m, 2H), 1.74 (m, 2H); MS (ESI) 445.2 m/z [MH+], C25H29N6O2 requires 445.2.
2-(6-(4-Amino-1-((1-methylpiperidin-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)naphthalen-2-yloxy)ethanol (15)
Tert-butyl-4-((4-amino-3-(6-hydroxynaphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)piperidine-1-carboxylate19 and oxirane were subjected to the General naphthol alkylation procedure followed by the General boc-deprotection procedure and General reductive amination procedure in order to afford 15. 1H NMR (300 MHz, CD3OD) δ 8.50 (s, 1H), 8.17 (s, 1H), 8.02 (d, J = 8.3 Hz, 1H), 7.94 (d, J = 8.9 Hz, 1H), 7.80 (d, J = 8.9 Hz, 1H), 7.41 (d, J = 2.2 Hz, 1H), 7.34 (dd, J = 8.7, 2.0 Hz, 1H), 4.53 (d, J = 6.4 Hz, 2H), 4.25 (t, J = 4.5 Hz, 2H), 3.99 (t, J = 4.5 Hz, 2H), 3.56 (m, 2H), 3.04 (m, 2H), 2.86 (s, 3H), 2.44 (m, 1H), 2.00 (m, 2H), 1.72 (m, 2H); MS (ESI) 433.3 m/z [MH+], C24H29N6O2 requires 433.5.
1-(6-(4-Amino-1-((1-methylpiperidin-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)naphthalen-2-yloxy)-2-methylpropan-2-ol (16)
Tert-butyl-4-((4-amino-3-(6-hydroxynaphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)piperidine-1-carboxylate19 and 2,2-dimethyloxirane were subjected to the General naphthol alkylation procedure followed by the General boc-deprotection procedure and General reductive amination procedure in order to afford 16. 1H NMR (300 MHz, CD3OD) δ 8.48 (s, 1H), 8.17 (s, 1H), 8.02 (d, J = 8.9 Hz, 1H), 7.94 (d, J = 8.7 Hz, 1H), 7.79 (d, J = 8.5 Hz, 1H), 7.43–7.32 (m, 2H), 4.53 (d, J = 6.8 Hz, 2H), 3.98 (s, 2H), 3.56 (m, 2H), 3.04 (m, 2H), 2.87 (s, 3H), 2.44 (m, 1H), 2.00 (m, 2H), 1.77 (m, 2H), 1.41 (s, 6H); MS (ESI) 461.5 m/z [MH+], C26H33N6O2 requires 461.5.
3-(2-Ethoxyquinolin-6-yl)-1-((1-methylpiperidin-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (17)
Tert-butyl-4-((4-amino-3-(2-ethoxyquinolin-6-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)piperidine-1-carboxylate26 was subjected to the General boc-deprotection procedure and General reductive amination procedure in order to afford 17. 1H NMR (300 MHz, CD3OD) δ 8.29 (s, 1H), 8.22 (d, J = 8.2 Hz, 1H), 8.12 (s, 1H), 7.98 (m, 2H), 7.04 (d, J = 8.5 Hz, 1H), 4.55 (q, J = 6.8 Hz, 2H), 4.38 (d, J = 4.3 Hz, 2H), 2.95 (m, 2H), 2.30 (s, 3H), 2.99 (m, 2H),1.67 (m, 2H), 1.48(m, 4H); MS (ESI) 418.3 m/z [MH+], C23H28N7O requires 418.5.
1-((1-Methylpiperidin-4-yl)methyl)-3-(2-(2,2,2-trifluoroethoxy)quinolin-6-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (18)
2-(2,2,2-Trifluoroethoxy)quinolin-6-ylboronic acid (39) and tert-butyl-4-((4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)piperidine-1-carboxylate19 were subjected to the General Suzuki coupling procedure followed by the General boc-deprotection procedure and General reductive amination procedure in order to afford 18. 1H NMR (300 MHz, CD3OD) δ 8.49 (s, 1H), 8.39 (d, J = 8.5 Hz, 1H), 8.24 (s, 1H), 8.04 (s, 2H), 7.18 (d, J = 7.0 Hz, 1H), 5.09 (q, J = 8.7 Hz, 2H), 4.52 (s, 2H), 3.54 (m, 2H), 3.04 (m, 2H), 2.84 (s, 3H), 2.45 (m, 1H), 1.98 (m, 2H), 1.75 (m, 2H); MS (ESI) 472.2 m/z [MH+], C23H25F3N7O requires 472.5.
3-(2-Cyclopropoxyquinolin-6-yl)-1-((1-methylpiperidin-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (19)
2-Cyclopropoxyquinolin-6-ylboronic acid (40) and tert-butyl-4-((4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)piperidine-1-carboxylate19 were subjected to the General Suzuki coupling procedure followed by the General boc-deprotection procedure and General reductive amination procedure in order to afford 19. 1H NMR (300 MHz, CD3OD) δ 9.17 (s, 1H), 8.75–8.33 (m, 3H), 8.28–7.98 (m, 2H),4.72 (m, 1H), 4.54 (d, J = 6.0 Hz, 2H), 3.56 (m, 2H), 3.10 (m, 2H), 3.00 (s, 3H), 2.48 (m, 1H), 2.00 (m, 2H), 1.60 (m, 2H), 1.20–1.10 (m, 4H); MS (ESI) 430.5 m/z [MH+], C24H28N7O requires 430.6.
3-(2-Cyclopropoxyquinolin-6-yl)-1-(3-(dimethylamino)-2,2-dimethylpropyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (20)
2-Cyclopropoxyquinolin-6-ylboronic acid (40) and 1-(3-(dimethylamino)-2,2-dimethylpropyl)-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine (42) were subjected to the General Suzuki coupling procedure to afford 20. 1H NMR (300 MHz, CD3OD) δ 8.29 (s, 1H), 8.22 (d, J = 8.9 Hz, 1H,), 8.11 (d, J = 1.6 Hz, 1H) 8.01 (s, 1H), 8.00 (dd, J = 8.5, 1.5 Hz, 1H,), 7.07 (d, J = 8.9 Hz, 1H), 4.46 (m, 1H), 4.36 (s, 2H), 2.46 (s, 2H), 2.44 (s, 6H), 1.03 (s, 6H), 0.93–0.82 (m, 4H). MS (ESI) 432.6 m/z [MH+], C24H30N7O requires 432.5.
3-(6-Ethoxynaphthalen-2-yl)-1-isobutyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (21)
6-Ethoxynaphthalen-2-ylboronic acid and 3-iodo-1-isobutyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine26 were subjected to the General Suzuki coupling procedure in order to afford 21. 1H NMR (300 MHz, CD3OD) δ 8.27 (s, 1H), 8.09 (s, 1H), 7.96 (d, J = 8.1 Hz, 1H), 7.89 (d, J = 9.1 Hz, 1H), 7.75 (dd, J = 8.2, 2.0 Hz, 1H), 7.32 (s, 1H), 7.22 (dd, J = 9.1, 2.4 Hz, 1H), 4.28–4.17 (m, 4H), 2.39 (m, 1H), 1.48 (t, J = 6.8 Hz, 3H), 0.96 (d, J = 6.6 Hz, 6H); MS (ESI) 362.4 m/z [MH+], C21H24N5O requires 362.2.
3-(6-Cyclopropoxynaphthalen-2-yl)-1-isobutyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (22)
6-(4-Amino-1-isobutyl-1H-pyrazolo[3,4-d]pyrimidin-3-yl)naphthalen-2-ol (46) and bromocyclopropane were subjected to the General naphthol alkylation procedure in order to afford 22. 1H NMR (300 MHz, CD3OD) δ 8.27 (s, 1H), 8.10 (s, 1H), 7.98 (d, J = 8.7 Hz, 1H), 7.89 (d, J = 8.9 Hz, 1H), 7.76 (dd, J = 8.5, 1.6 Hz, 1H), 7.61 (d, J = 2.2 Hz, 1H), 7.25–7.18 (dd, J = 8.9, 2.2 Hz, 1H), 4.24 (d, J = 7.25 Hz, 2H), 3.95 (m, 1H), 2.38 (m, 1H), 0.97 (d, J = 6.6 Hz, 6H), 0.90 (m, 2H), 0.79 (m, 2H); MS (ESI) 374.2 m/z [MH+], C22H24N5O requires 374.4.
3-(2-Cyclopropoxyquinolin-6-yl)-1-isobutyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (23)
2-Cyclopropoxyquinolin-6-ylboronic acid (40) and 3-iodo-1-isobutyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine26 were subjected to the General Suzuki coupling procedure in order to afford 23. 1H NMR (300 MHz, CD3OD) δ 8.25 (s, 1H), 8.23 (d, J = 8.9 Hz, 1H), 8.10 (s, 1H), 8.00–7.91 (m, 2H), 7.02 (d, J = 8.7 Hz, 1H), 4.46 (s, 1H), 4.22 (d, J = 7.4 Hz, 2H), 2.36 (m, 1H), 0.96 (d, J = 6.6 Hz, 6H), 0.91–0.72 (m, 4H); MS (ESI) 375.4 m/z [MH+], C21H23N6O requires 375.4.
3-(6-Cyclopropoxynaphthalen-2-yl)-1-((3-methyloxetan-3-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (24)
2-(6-Cyclopropoxynaphthalen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (41) and 3-iodo-1-((3-methyloxetan-3-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (44) were subjected to the General Suzuki coupling procedure in order to afford 24. 1H NMR (300 MHz, CDCl3) δ 8.39 (s, 1H), 8.08 (s, 1H), 7.93 (d, J = 8.7 Hz, 1H), 7.83 (d, J = 8.9 Hz, 1H), 7.77 (d, J = 9.4 Hz, 1H), 7.52 (s, 1H), 7.24 (dd, J = 10.0, 3.0 Hz, 1H), 5.68 (s, 2H), 4.90 (d, J = 6.1 Hz, 2H), 4.64 (s, 2H), 4.45 (d, J = 6.1 Hz, 2H), 3.91 (m, 1H), 1.36 (3, 3H), 0.96–0.83 (m, 4H); MS (ESI) 402.2 m/z [MH+], C23H24N5O2 requires 402.4.
3-(2-Cyclopropoxyquinolin-6-yl)-1-((3-methyloxetan-3-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (25)
2-Cyclopropoxyquinolin-6-ylboronic acid (40) and 3-iodo-1-((3-methyloxetan-3-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (44) were subjected to the General Suzuki coupling procedure in order to afford 25. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 8.12–8.01 (m, 3H), 7.96 (d, J = 8.3 Hz, 1H), 6.97 (d, J = 8.0 Hz, 1H), 5.54 (s, 2H), 4.91 (d, J = 6.1 Hz, 2H), 4.64 (s, 2H), 4.56 (m, 1H), 4.45 (d, J = 6.1 Hz, 2H), 1.36 (s, 3H), 0.95–0.82 (m, 4H); MS (ESI) 403.2 m/z [MH+], C22H23N6O2 requires 403.4.
3-(6-Cyclopropoxynaphthalen-2-yl)-1-((tetrahydro-2H-pyran-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (28)
2-(6-Cyclopropoxynaphthalen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (41) and 3-iodo-1-((tetrahydro-2H-pyran-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine26 were subjected to the General Suzuki coupling procedure in order to afford 28. 1H NMR (300 MHz, CD3OD) δ 8.47 (s, 1H), 8.16 (s, 1H), 8.03 (d, J = 8.2 Hz, 1H), 7.92 (d, J = 9.1 Hz, 1H), 7.78 (d, J = 8.2 Hz, 1H), 7.64 (s, 1H), 7.27 (dd, J = 9.1, 2.2 Hz, 1H) 4.45 (d, J = 6.8 Hz, 2H,), 3.99 (m, 1H), 3.76 (m, 2H), 3.05 (m, 2H), 2.40 (m, 1H), 1.61 (m, 2H), 1.46 (m, 2H), 0.93–0.80 (m, 4H); MS (ESI) 416.4 m/z [MH+], C24H25N5O2 requires 416.4.
3-(2-Cyclopropoxyquinolin-6-yl)-1-((tetrahydro-2H-pyran-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (29)
2-Cyclopropoxyquinolin-6-ylboronic acid (40) and 3-iodo-1-((tetrahydro-2H-pyran-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine26 were subjected to the General Suzuki coupling procedure in order to afford 29. 1H NMR (300 MHz, CD3OD) δ 8.31 (s, 1H), 8.22 (d, J = 8.9 Hz, 1H), 8.12 (d, J = 1.8 Hz, 1H), 8.06 (d, J = 8.7 Hz, 1H), 8.00 (dd, J = 8.5, 1.2 Hz, 1H), 7.07 (d, J = 8.9 Hz, 1H), 4.47 (m, 1H) 4.36 (d, J = 7.2 Hz, 2H,), 3.98 (m, 2H), 3.42 (m, 2H), 2.34 (m, 1H), 1.59 (m, 2H), 1.49 (m, 2H), 0.93–0.83 (m, 4H); MS (ESI) 416.4 m/z [MH+], C23H24N6O2 requires 416.4.
1-(4-Amino-3-(6-ethoxynaphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2-methylpropan-2-ol (30)
6-Ethoxynaphthalen-2-ylboronic acid (41) and 1-(4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2-methylpropan-2-ol (43) were subjected to the General Suzuki coupling procedure to afford compound 30; 1H NMR (300 MHz, CD3OD) δ 8.26 (s, 1H), 8.09 (s, 1H), 7.97–7.83 (m, 2H), 7.76 (dd, J = 8.2, 1.4 Hz, 1H), 7.30 (d, J = 2.0 Hz, 1H), 7.20 (dd, J = 8.9, 1.4 Hz, 1H), 4.41 (s, 2H), 4.19 (q, J = 7.0 Hz, 2H), 1.47 (t, J = 6.8 Hz, 3H), 1.28 (s, 6H). MS (ESI) 378.2 m/z [MH+], C21H24N5O2 requires 378.1.
1-(4-Amino-3-(2-ethoxyquinolin-6-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2-methylpropan-2-ol (31)
2-Ethoxy-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)quinoline19 and 1-(4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2-methylpropan-2-ol (43) were subjected to the General Suzuki coupling procedure to afford compound 31; 1H NMR (300 MHz, CD3OD) δ 8.69 (d, J = 9.1 Hz, 1H), 8.45 (s, 1H), 8.34 (s, 1H), 8.17 (d, J = 8.0 Hz, 1H), 8.04 (d, J = 7.4 Hz, 1H), 7.45 (d, J = 7.4 Hz, 1H), 4.67 (q, J = 6.0 Hz, 2H), 4.50 (s, 2H), 1.54 (t, J = 6.5 Hz, 3H), 1.31 (s, 6H). MS (ESI) 379.2 m/z [MH+], C20H23N6O2 requires 379.1.
1-(4-Amino-3-(6-cyclopropoxynaphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2-methylpropan-2-ol (32)
2-(6-Cyclopropoxynaphthalen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (41) and 1-(4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2-methylpropan-2-ol (43) were subjected to the General Suzuki coupling procedure to afford title compound 32; 1H NMR (500 MHz, CD3OD) δ 8.26 (s, 1H), 8.08 (s, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 8.81 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.57 (s, 1H), 7.20 (dd, J = 8.8, 1.8 Hz, 1H), 4.40 (s, 2H), 3.91 (m, 1H), 1.27 (s, 6H), 0.88 (m, 2H), 0.78 (m, 2H). MS (ESI) 390.2 m/z [MH+], C22H24N5O2 requires 390.1.
1-(4-Amino-3-(2-cyclopropoxyquinolin-6-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2-methylpropan-2-ol (33)
2-Cyclopropoxyquinolin-6-ylboronic acid (40) and 1-(4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2-methylpropan-2-ol (43) were subjected to the General Suzuki coupling procedure in order to afford 33. NMR (300 MHz, CD3OD) δ 8.28 (s, 1H), 8.28–8.24 (d, J = 8.9 Hz, 1H), 8.15 (t, J = 1.4 Hz, 1H), 8.01 (d, J = 1.2 Hz, 2H), 7.06 (d, J = 8.7 Hz, 1H), 4.53–4.45 (m, 1H), 4.42 (s, 2H), 1.28 (s, 6H), 0.92–0.85 (m, 2H), 0.85–0.77 (m, 2H); MS (ESI) 391.1 m/z [MH+], C21H23N6O2 requires 391.2.
3-(4-Amino-3-(6-cyclopropoxynaphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2,2-dimethylpropan-1-ol (34)
2-(6-Cyclopropoxynaphthalen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (41) and previously reported 3-(4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2,2-dimethylpropan-1-ol26 were subjected to the General Suzuki coupling procedure in order to afford 34. 1H NMR (300 MHz, CD3OD) δ 8.30 (s, 1H), 8.12 (s, 1H), 8.00 (d, J = 8.7 Hz, 1H), 7.90 (d, J = 8.9 Hz, 1H), 7.78 (dd, J = 8.2, 1.5 Hz, 1H), 7.62 (d, J = 2.4, 1H), 7.24 (dd, J = 9.1, 2.4 Hz, 1H), 4.33 (s, 2H), 3.97 (m, 1H), 3.32 (s, 2H), 1.22 (s, 6H), 0.94–0.81 (m, 4H); MS (ESI) 404.5 m/z [MH+], C23H26N5O2 requires 404.4.
3-(4-Amino-3-(2-cyclopropoxyquinolin-6-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2,2-dimethylpropan-1-ol (35)
2-Cyclopropoxyquinolin-6-ylboronic acid (40) and previously reported 3-(4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2,2-dimethylpropan-1-ol26 were subjected to the General Suzuki coupling procedure in order to afford 35. 1H NMR (300 MHz, CD3OD) δ 8.65 (s, 1H), 8.50 (s, 1H), 8.36 (m, 1H), 8.21–8.05 (m, 2H), 7.51 (d, J = 8.3 Hz, 1H), 4.59 (s, 2H), 4.47 (s, 2H), 4.46 (m, 1H), 1.01 (s, 6H), 0.70 (m, 4H); MS (ESI) 405.2 m/z [MH+], C22H25N6O2 requires 405.4.
3-(4-Amino-3-(2-(2,2,2-trifluoroethoxy)quinolin-6-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2,2-dimethylpropan-1-ol (36)
2-(2,2,2-Trifluoroethoxy)quinolin-6-ylboronic acid (39) and previously reported 3-(4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2,2-dimethylpropan-1-ol26 were subjected to the General Suzuki coupling procedure in order to afford 36. 1H NMR (300 MHz, CD3OD) δ 8.40 (s, 1H), 8.17 (d, J = 8.7 Hz, 1H), 8.07 (d, J = 1.6 Hz, 1H), 8.01–7.95 (m, 2H), 7.12 (d, J = 8.9 Hz, 1H), 4.98 (q, J = 8.5 Hz, 2H), 4.35 (s, 2H), 3.15 (s, 2H), 1.05 (s, 6H); MS (ESI) 447.5 m/z [MH+], C21H21F3N6O2 requires 447.4.
2-((4-Amino-3-(6-cyclopropoxynaphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)-1,1,1,3,3,3-hexafluoropropan-2-ol (37)
2-(6-Cyclopropoxynaphthalen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (41) and 2-((4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)-1,1,1,3,3,3-hexafluoropropan-2-ol (45) were subjected to the General Suzuki coupling procedure in order to afford 37. 1H NMR (300 MHz, CD3OD) δ 8.32 (s, 1H), 8.11 (s, 1H), 7.97 (d, J = 8.5 Hz, 1H), 7.87 (d, J = 9.1 Hz, 1H), 7.76 (dd, J = 8.5, 1.6 Hz, 1H), 7.59 (d, J = 2.0, 1H), 7.23 (dd, J = 8.9, 2.2 Hz, 1H), 5.03 (s, 2H), 3.94 (m, 1H), 0.94–0.76 (m, 4H); MS (ESI) 498.2 m/z [MH+], C22H18F6N5O2 requires 498.2.
Physiochemical Procedures
Pharmacokinetic analysis in mice
For mouse oral PK studies, three female BALB/c mice (10 to 12 weeks old) were used in each group (performed under IACUC protocol number 2145-01 (UW, Seattle)). Each group received a test compound at a dose of 10 mg/kg body weight dissolved in 3% ethanol/7% Tween 80/90% normal saline by oral gavage. Blood samples were taken at the designated time points by tail bleeding and centrifuged to obtain plasma. The samples were frozen at −20 °C. The test compounds were extracted from the plasma samples using acetonitrile/0.1% formic acid with an internal standard. A standard mix of all test compounds was prepared for comparison and quantification. The compounds were quantified by LC/MS analysis. PK calculations were performed using Phoenix WinNonlin software (Pharsight).
Pharmacokinetic analysis in rats
Test compound was administered to Sprague-Dawley jugular canulated rats (Charles River) by either oral gavage or IV injection followed by blood sampling from the jugular vein at designated time points (performed under IACUC protocol number 2145-01 (UW, Seattle)). The oral dose was administered to each rat at 20 mg/kg for compound 32 and 5 mg/kg for compound 33 at time = 0 in a 1 mL volume of dosing solution (7% Tween 80, 3% EtOH, 5% DMSO, 0.9% saline.) IV injections were administered at 5 mg/kg from time = 0 to 3 minutes in a 1 mL volume of dosing solution, and blood was sampled at the same time points via the jugular vein. Experiments were performed with groups of 2 rats each for the oral and IV dosing. Plasma was separated and extracted with acetonitrile and quantified by LC/MS analysis. PK calculations were performed using Phoenix WinNonlin software (Pharsight).
Pharmacokinetic analysis in calves
Previously described procedures were used for calf enrollment, housing, management, and for test compound administration (performed under WSU IACUC # ASAF 04477 “Novel Therapeutics for Cryptosporidiosis and other Parasites: Bumped Kinase Inhibitors”).36
Pharmacokinetic analysis in dogs and monkeys
In each study, groups of three animals received a 1 mg/kg intravenous or oral dose of compound 32. The dose was administered as a solution in PEG-400 containing 10% DMSO (0.5 ml/kg dose volume); the oral dose solution was placed in a capsule just prior to dosing. Blood samples for plasma concentration analysis were obtained from each animal for 24 hours after dosing. Plasma concentrations of parent drug were determined by LC-MS/MS. All studies involving vertebrate animals at AbbVie were approved by an animal ethics approval committee (IACUC).
Distribution of compounds between mouse plasma and brain
Mice were injected with 32 or 33 (5 mg/kg IP) and sacrificed at the indicated times for collection of plasma and brain (performed under IACUC protocol number 2145-01 (UW, Seattle)). Compound was dissolved in 0.4 mL of dosing solution (7% Tween 80, 3% ethanol, 5% DMSO, 0.9% saline) for IP injections. The brains were weighed and immediately frozen, then later homogenized in acetonitrile. Prior to animal studies, recovery of test compound was carried out by adding a known amount to a mouse brain in the test extraction solvent and performing the homogenization. Compound recovery was determined by liquid chromatography/tandem mass spectrometry analysis relative to a standard compound amount. Blood was taken from the same mice in heparinized capillary tubes for determination of compound concentration in plasma. The concentration of compound in the brain was obtained by dividing the moles of compound in the brain by the brain volume (obtained from the brain weight assuming 1 g is 1 mL) and correcting for the brain vasculature volume of 3% by weight.
Supplementary Material
Table 5.
In vivo pharmacokinetic parameters of 32 and 33 in rats following IV administration of 5 mg/kg. Data is shown as mean and range between animals.
| AUC (h*μ mol/L) | CL (mL/hr/kg) | Vss (L/kg) | T1/2 (hr) | |
|---|---|---|---|---|
| 32 | 167 (160–175) | 77 (73–80) | 0.91(0.89–0.93) | 9.6 (9.5–9.7) |
| 33 | 45 (31–59) | 310 (216–412) | 5.8 (4.1–7.5) | 13 (12.6–13.4) |
Acknowledgments
Funding Sources
We thank DeAnne Stolarik (Abbvie) and Lance Heinle (Abbvie) for assistance with the dog and monkey PK studies. Research reported in this publication was supported by the National Institute of General Medical Sciences, National Institute of Allergy and Infectious Diseases and National Institute of Child Health and Human Development of the National Institutes of Health under award numbers R01GM086858, R01AI089441, R01AI111341, and R01HD080670. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was also supported in part by Award # 2014-06183 from the United States Department of Agriculture, by Career Development Award # BX002440 from the United States Department of Veterans Affairs Biomedical Laboratory Research and Development, and by Award 07R1857 from the Stanley Medical Research Institute. Crystallography performed in support of the work benefitted from remote access to resources at the Stanford Synchrotron Radiation Lightsource supported by the U.S. Department of Energy Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515 and by the National Institutes of Health (P41GM103393).
ABBREVIATIONS USED
- AUC
area under plasma concentration time curve
- ATP
adenosine triphosphate
- Boc
tert-butyloxycarbonyl
- CDPK1
calcium-dependent protein kinase 1
- CL
clearance
- Cmax
maximum plasma concentration
- CNS
central nervous system
- DIAD
diisopropyl azodicarboxylate
- DMF
dimethylformamide
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- EC50
half maximal effective concentration
- GI50
half maximal growth inhibition
- hERG
human Ether-à-go-go-Related Gene
- IC50
half maximal inhibitory concentration
- IV
intravenous
- Km
Michaelis constant
- PO
per os
- tmax
time at which maximum concentration is reached
- TBDMS
tert-butyldimethylsilyl
- TgCDPK1
Toxoplasma gondii CDPK1
Footnotes
Supporting Information Available: IC50 values of compounds 32 and 33 for human kinases; crystallographic data; synthesis and characterization data for all intermediate compounds; biological assay procedures; aqueous solubility assays; NMR spectra for final compounds; This material is available free of charge via the internet at http://pubs.acs.org.
Accession Codes
The PDB code for the X-ray crystallographic structure of compound 33 bound to wild type T.gondii CDPK1 is 4TZR and compound 1 bound to wild type T.gondii CDPK1 is 3SX9.
Notes
The authors declare the following competing financial interest: Gail Freiberg, Kennan Marsh, and Dale Kempf are employees of Abbvie.
References
- 1.Tenter AM, Heckeroth AR, Weiss LM. Toxoplasma gondii: from animals to humans. Int J Parasitol. 2000;30:1217–1258. doi: 10.1016/s0020-7519(00)00124-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pappas G, Roussos N, Falagas ME. Toxoplasmosis snapshots: global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. Int J Parasitol. 2009;39:1385–1394. doi: 10.1016/j.ijpara.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Schwartzman JD, Maguire JH. In Tropical Infectious Diseases: Principles, Pathogens and Practice. 3rd. W. B. Saunders; Edinburgh, U.K.: 2011. Toxoplasmosis; pp. 722–728. Chapter 103. [Google Scholar]
- 4.Dupont CD, Christian DA, Hunter CA. Immune response and immunopathology during toxoplasmosis. Semin Immunopathol. 2012;34:793–813. doi: 10.1007/s00281-012-0339-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanchard N, Dunay IR, Schluter D. Persistence of Toxoplasma gondii in the central nervous system: a fine-tuned balance between the parasite, the brain and the immune system. Parasite Immunol. 2015;37:150–158. doi: 10.1111/pim.12173. [DOI] [PubMed] [Google Scholar]
- 6.Flegr J, Prandota J, Sovickova M, Israili ZH. Toxoplasmosis-a global threat. Correlation of latent toxoplasmosis with specific disease burden in a set of 88 countries. PLoS One. 2014;9:e90203. doi: 10.1371/journal.pone.0090203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones JL, Muccioli C, Belfort R, Jr, Holland GN, Roberts JM, Silveira C. Recently acquired Toxoplasma gondii infection, Brazil. Emerging Infect Dis. 2006;12:582–587. doi: 10.3201/eid1204.051081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlier Y, Truyens C, Deloron P, Peyron F. Congenital parasitic infections: a review. Acta Trop. 2012;121:55–70. doi: 10.1016/j.actatropica.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 9.Dubey JP, Jones JL. Toxoplasma gondii infection in humans and animals in the United States. Int J Parasitol. 2008;38:1257–1278. doi: 10.1016/j.ijpara.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 10.Wei HX, Wei SS, Lindsay DS, Peng HJ. A Systematic Review and Meta-Analysis of the Efficacy of Anti-Toxoplasma gondii Medicines in Humans. PLoS One. 2015;10:e0138204. doi: 10.1371/journal.pone.0138204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaplan JE, Benson C, Holmes KK, Brooks JT, Pau A, Masur H, Centers for Disease Control and Prevention (CDC); NIH Publ HIV Medicine Asso. of the Infect. Dis. Soc. of America Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR. 2009;58:1–207. [PubMed] [Google Scholar]
- 12.Kieschnick H, Wakefield T, Narducci CA, Beckers C. Toxoplasma gondii attachment to host cells is regulated by a calmodulin-like domain protein kinase. J Biol Chem. 2001;276:12369–12377. doi: 10.1074/jbc.M011045200. [DOI] [PubMed] [Google Scholar]
- 13.Lourido S, Shuman J, Zhang C, Shokat KM, Hui R, Sibley LD. Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma. Nature. 2010;465:359–362. doi: 10.1038/nature09022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lourido S, Zhang C, Lopez MS, Tang K, Barks J, Wang Q, Wildman SA, Shokat KM, Sibley LD. Optimizing small molecule inhibitors of calcium-dependent protein kinase 1 to prevent infection by Toxoplasma gondii. J Med Chem. 2013;56:3068–3077. doi: 10.1021/jm4001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doggett JS, Ojo KK, Fan E, Maly DJ, Van Voorhis WC. Bumped kinase inhibitor 1294 treats established Toxoplasma gondii infection. Antimicrob Agents Chemother. 2014;58:3547–3549. doi: 10.1128/AAC.01823-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Z, Ojo KK, Johnson SM, Larson ET, He P, Geiger JA, Castellanos-Gonzalez A, White AC, Jr, Parsons M, Merritt EA, Maly DJ, Verlinde CL, Van Voorhis WC, Fan E. Benzoylbenzimidazole-based selective inhibitors targeting Cryptosporidium parvum and Toxoplasma gondii calcium-dependent protein kinase-1. Bioorg Med Chem Lett. 2012;22:5264–5267. doi: 10.1016/j.bmcl.2012.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Z, Ojo KK, Vidadala R, Huang W, Geiger JA, Scheele S, Choi R, Reid MC, Keyloun KR, Rivas K, Siddaramaiah LK, Comess KM, Robinson KP, Merta PJ, Kifle L, Hol WG, Parsons M, Merritt EA, Maly DJ, Verlinde CL, Van Voorhis WC, Fan E. Potent and selective inhibitors of CDPK1 from T. gondii and C. parvum Based on a 5-aminopyrazole-4-carboxamide scaffold. ACS Med Chem Lett. 2014;5:40–44. doi: 10.1021/ml400315s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murphy RC, Ojo KK, Larson ET, Castellanos-Gonzalez A, Perera BG, Keyloun KR, Kim JE, Bhandari JG, Muller NR, Verlinde CL, White AC, Jr, Merritt EA, Van Voorhis WC, Maly DJ. Discovery of Potent and Selective Inhibitors of Calcium-Dependent Protein Kinase 1 (CDPK1) from C. parvum and T. gondii. ACS Med Chem Lett. 2010;1:331–335. doi: 10.1021/ml100096t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson SM, Murphy RC, Geiger JA, DeRocher AE, Zhang Z, Ojo KK, Larson ET, Perera BG, Dale EJ, He P, Reid MC, Fox AM, Mueller NR, Merritt EA, Fan E, Parsons M, Van Voorhis WC, Maly DJ. Development of Toxoplasma gondii calcium-dependent protein kinase 1 (TgCDPK1) inhibitors with potent anti-toxoplasma activity. J Med Chem. 2012;55:2416–2426. doi: 10.1021/jm201713h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ojo KK, Larson ET, Keyloun KR, Castaneda LJ, Derocher AE, Inampudi KK, Kim JE, Arakaki TL, Murphy RC, Zhang L, Napuli AJ, Maly DJ, Verlinde CL, Buckner FS, Parsons M, Hol WG, Merritt EA, Van Voorhis WC. Toxoplasma gondii calcium-dependent protein kinase 1 is a target for selective kinase inhibitors. Nat Struct Mol Biol. 2010;17:602–607. doi: 10.1038/nsmb.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ojo KK, Reid MC, Kallur Siddaramaiah L, Muller J, Winzer P, Zhang Z, Keyloun KR, Vidadala RS, Merritt EA, Hol WG, Maly DJ, Fan E, Van Voorhis WC, Hemphill A. Neospora caninum calcium-dependent protein kinase 1 is an effective drug target for neosporosis therapy. PLoS One. 2014;9:e92929. doi: 10.1371/journal.pone.0092929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winzer P, Muller J, Aguado-Martinez A, Rahman M, Balmer V, Manser V, Ortega-Mora LM, Ojo KK, Fan E, Maly DJ, Van Voorhis WC, Hemphill A. In Vitro and In Vivo Effects of the Bumped Kinase Inhibitor 1294 in the Related Cyst-Forming Apicomplexans Toxoplasma gondii and Neospora caninum. Antimicrob Agents Chemother. 2015;59:6361–6374. doi: 10.1128/AAC.01236-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ojo KK, Eastman RT, Vidadala R, Zhang Z, Rivas KL, Choi R, Lutz JD, Reid MC, Fox AM, Hulverson MA, Kennedy M, Isoherranen N, Kim LM, Comess KM, Kempf DJ, Verlinde CL, Su XZ, Kappe SH, Maly DJ, Fan E, Van Voorhis WC. A specific inhibitor of PfCDPK4 blocks malaria transmission: chemical-genetic validation. J Infect Dis. 2014;209:275–284. doi: 10.1093/infdis/jit522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Noord C, Eijgelsheim M, Stricker BH. Drug- and non-drug-associated QT interval prolongation. Br J Clin Pharmacol. 2010;70:16–23. doi: 10.1111/j.1365-2125.2010.03660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larson ET, Ojo KK, Murphy RC, Johnson SM, Zhang Z, Kim JE, Leibly DJ, Fox AM, Reid MC, Dale EJ, Perera BG, Kim J, Hewitt SN, Hol WG, Verlinde CL, Fan E, Van Voorhis WC, Maly DJ, Merritt EA. Multiple determinants for selective inhibition of apicomplexan calcium-dependent protein kinase CDPK1. J Med Chem. 2012;55:2803–2810. doi: 10.1021/jm201725v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vidadala RS, Ojo KK, Johnson SM, Zhang Z, Leonard SE, Mitra A, Choi R, Reid MC, Keyloun KR, Fox AM, Kennedy M, Silver-Brace T, Hume JC, Kappe S, Verlinde CL, Fan E, Merritt EA, Van Voorhis WC, Maly DJ. Development of potent and selective Plasmodium falciparum calcium-dependent protein kinase 4 (PfCDPK4) inhibitors that block the transmission of malaria to mosquitoes. Eur J Med Chem. 2014;74:562–573. doi: 10.1016/j.ejmech.2013.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J biol chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 28.Bridal TR, Margulis M, Wang X, Donio M, Sorota S. Comparison of human Ether-a-go-go related gene screening assays based on IonWorks Quattro and thallium flux. Assay Drug Dev Technol. 2010;8:755–765. doi: 10.1089/adt.2010.0267. [DOI] [PubMed] [Google Scholar]
- 29.Bowes J, Brown AJ, Hamon J, Jarolimek W, Sridhar A, Waldron G, Whitebread S. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat Rev Drug Discov. 2012;11:909–22. doi: 10.1038/nrd3845. [DOI] [PubMed] [Google Scholar]
- 30.Hitchcock SA, Pennington LD. Structure-brain exposure relationships. J Med Chem. 2006;49:7559–7583. doi: 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]
- 31.Cheng Y, Brown J, Judd TC, Lopez P, Qian W, Powers TS, Chen JJ, Bartberger MD, Chen K, Dunn RT, Epstein O, Fremeau RT, Jr, Harried S, Hickman D, Hitchcock SA, Luo Y, Minatti AE, Patel VF, Vargas HM, Wahl RC, Weiss MM, Wen PH, White RD, Whittington DA, Zheng XM, Wood S. An Orally Available BACE1 Inhibitor That Affords Robust CNS Aβ Reduction without Cardiovascular Liabilities. ACS Med Chem Lett. 2015;6:210–215. doi: 10.1021/ml500458t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Epstein O, Bryan MC, Cheng AC, Derakhchan K, Dineen TA, Hickman D, Hua Z, Human JB, Kreiman C, Marx IE, Weiss MM, Wahl RC, Wen PH, Whittington DA, Wood S, Zheng XM, Fremeau RT, Jr, White RD, Patel VF. Lead optimization and modulation of hERG activity in a series of aminooxazoline xanthene β-site amyloid precursor protein cleaving enzyme (BACE1) inhibitors. J Med Chem. 2014;57:9796–9810. doi: 10.1021/jm501266w. [DOI] [PubMed] [Google Scholar]
- 33.Keyloun KR, Reid MC, Choi R, Song Y, Fox AM, Hillesland HK, Zhang Z, Vidadala R, Merritt EA, Lau AO, Maly DJ, Fan E, Barrett LK, WC VANV, Ojo KK. The gatekeeper residue and beyond: homologous calcium-dependent protein kinases as drug development targets for veterinarian Apicomplexa parasites. Parasitology. 2014;141:1499–1509. doi: 10.1017/S0031182014000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doggett JS, Nilsen A, Forquer I, Wegmann KW, Jones-Brando L, Yolken RH, Bordón C, Charman SA, Katneni K, Schultz T, Burrows JN, Hinrichs DJ, Meunier B, Carruthers VB, Riscoe MK. Endochin-like quinolones are highly efficacious against acute and latent experimental toxoplasmosis. Proc Natl Acad Sci USA. 2012;109:15936–15941. doi: 10.1073/pnas.1208069109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edvinsson B, Lappalainen M, Evengård B, ESCMID Study Group for Toxoplasmosis Real-time PCR targeting a 529-bp repeat element for diagnosis of toxoplasmosis. Clin Microbiol Infect. 2006;12:131–136. doi: 10.1111/j.1469-0691.2005.01332.x. [DOI] [PubMed] [Google Scholar]
- 36.Ollivett TL, Nydam DV, Bowman DD, Zambriski JA, Bellosa ML, Linden TC, Divers TJ. Effect of nitazoxanide on cryptosporidiosis in experimentally infected neonatal dairy calves. J Dairy Sci. 2009;92:1643–1648. doi: 10.3168/jds.2008-1474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.