

Abstract
Ehlers–Danlos syndrome (EDS)—hypermobility type (HT) is considered to be the most common subtype of EDS and the least severe one; EDS-HT is considered to be identical to the joint hypermobility syndrome and manifests with musculoskeletal complaints, joint instability, and soft tissue overuse injury. Musculoskeletal complaints manifest with joint pain of non-inflammatory origin and/or spinal pain. Joint instability leads to dislocation or subluxation and involves peripheral joints as well as central joints, including the temporomandibular joints, sacroiliac joints, and hip joints. Soft tissue overuse injury may lead to tendonitis and bursitis without joint inflammation in most cases. Ehlers–Danlos syndrome-HT carries a high potential for disability due to recurrent dislocations and subluxations and chronic pain. Throughout the years, extra-articular manifestations have been described, including cardiovascular, autonomic nervous system, gastrointestinal, hematologic, ocular, gynecologic, neurologic, and psychiatric manifestations, emphasizing the multisystemic nature of EDS-HT. Unfortunately, EDS-HT is under-recognized and inadequately managed, leading to neglect of these patients, which may lead to severe disability that almost certainly could have been avoided. In this review article we will describe the known manifestations of the extra-articular systems.
Keywords: Disability, Ehlers-Danlos syndrome, hypermobility syndrome, joint hypermobility, multisystemic, neglect
INTRODUCTION
The Ehlers–Danlos syndromes (EDSs) constitute a group of inherited disorders of connective tissue characterized by soft hyperextensible skin and joint hypermobility, distinguished by additional connective tissue manifestations.1 The Ehlers–Danlos syndrome was first described by Ehlers in Denmark in 1898 and Danlos in Paris in 1908. They published individual case studies with common features of ligamentous laxity and skin hyperextensibility.2 Ehlers–Danlos syndrome—hypermobility type (EDS-HT) is considered to be the most common subtype of EDS3,4 and the least severe one.3 It is characterized by joint laxity, soft, stretchy, and often semi-transparent skin, and musculoskeletal complications, without severe complications of arterial dissection or bowel rupture seen in EDS-vascular type,1,5 and without hemosiderotic scars and molluscoid pseudotumors seen in the EDS-classical type.1,6 Ehlers–Danlos syndrome-HT, now considered to be indistinguishable if not identical to the joint hypermobility syndrome (JHS), manifests with musculoskeletal complaints, joint instability, and soft tissue overuse injury.3,7–12 Musculoskeletal complaints manifest with joint pain of non-inflammatory origin and/or spinal pain. Joint instability leads to dislocation or subluxation and involves peripheral joints as well as central joints, including the temporomandibular joints (TMJ), sacroiliac joints, and hip joints.7–9 Soft tissue overuse injury may lead to tendonitis and bursitis10,11,12 without joint inflammation in most cases.3,11 Although an inflammatory component is rare, EDS-HT carries a high potential for disability13 due to recurrent dislocations and subluxations and chronic pain.8,11,12,14,15 Throughout the years, extra-articular manifestations have been described, including cardiovascular and autonomic nervous system,16–22 gastrointestinal,19,23 hematologic,24–26 ocular,27 gynecologic,19,28–31 neurologic,19,25,32,33 and psychiatric manifestations,7,8,11,19,34,35 emphasizing the multisystemic nature of EDS-HT. Unfortunately, EDS-HT is under-recognized and inadequately managed,36–38 leading to neglect of these patients which may lead to severe disability that almost certainly could have been avoided.39
GENERAL CHARACTERISTICS AND MANIFESTATIONS
Joint hypermobility (JH), defined as an excessive range of joint movement taking into consideration age, gender, and ethnic background, is inherited40,41 and may pose no problem. Acquired hypermobility may also result from changes in connective tissue in other diseases such as systemic lupus erythematosus.42 Joint hypermobility is recognized by the nine-point Beighton score43 (Figure 1) and includes passive dorsiflexion of each fifth finger greater than 90°, passive apposition of each thumb to the flexor surface of the forearm, hyperextension of each elbow greater than 10°, hyperextension of each knee greater than 10°, and ability to place the palms flat on the floor with the knees fully extended.
Figure 1.
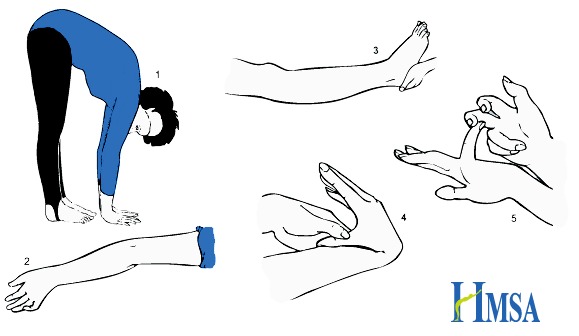
Calculation of the Beighton Score.
- One point if while standing forward bending you can place palms on the ground with legs straight
- One point for each elbow that bends backwards
- One point for each knee that bends backwards
- One point for each thumb that touches the forearm when bent backwards
- One point for each little finger that bends backwards beyond 90 degrees
Taken with permission from the Hypermobility Syndromes Association (HMSA) site (http://hypermobility.org/help-advice/hypermobility-syndromes/beighton-score/).
Ehlers–Danlos syndrome-HT, now considered to be indistinguishable if not identical to the joint hypermobility syndrome (JHS),44 is a clinical condition of JH with symptoms of joint instability, arthralgia, myalgia, soft tissue injuries, and arthritis.45,46 Diagnosis relies on the Brighton criteria (Table 1).47,48 The predominant presenting complaint is pain, which is often widespread and longstanding, with patients reporting pain ranging from 15 days to 45 years.39,49 Chronic pain may start in adolescence (with 75% of hypermobile adolescents reporting symptoms by the age of 15) or even as late as the fifth or sixth decade of life.3,39,45 Severity sometimes correlates with the degree of joint instability.3,15 Fatigue and sleep disturbance, most probably secondary to severe chronic pain, subluxations, and dislocations while changing posture during sleep, are frequently associated.3,11,12,15 Affected individuals are often misdiagnosed with chronic fatigue syndrome, fibromyalgia, depression, hypochondriasis, and/or malingering prior to recognition of joint laxity and establishment of the correct underlying diagnosis.3 Over the last three decades it has become apparent that EDS-HT has a widespread distribution and is not manifested solely in the joints (Table 2).
Table 1.
Revised Diagnostic Criteria for Ehlers-Danlos Hypermobility Type, a.k.a. Joint Hypermobility Syndrome (JHS).
| Revised Diagnostic Criteria for Ehlers–Danlos Hypermobility Type |
|---|
Major Criteria:
|
Minor Criteria:
|
| JHS is diagnosed in the presence two major criteria, or one major and two minor criteria, or four minor criteria. Two minor criteria will suffice where there is an unequivocally affected first-degree relative. |
Taken with permission from the Hypermobility Syndromes Association (HMSA) site (http://hypermobility.org/help-advice/hypermobility-syndromes/the-brighton-score/).50
Table 2.
Multisystemic Nature of EDS-HT.
| System | Manifestations |
|---|---|
| Cardiovascular | Aortic regurgitation, aortic root dilatation, mitral valve prolapse, mitral regurgitation, tricuspid regurgitation, Reynaud phenomenon |
| Autonomic Nervous System | Palpitations, dizziness, pre-syncope, syncope |
| Gastrointestinal | Gastroesophageal reflux, dyspepsia, gastritis, delayed gastric emptying, irritable bowel syndrome |
| Hematologic | Easy bruising, bleeding tendency, prolonged bleeding time, oral mucosal bruises, menometrorrhagia |
| Ocular | Myopia, strabismus |
| Gynecologic | Dysmenorrhea, menorrhagia, dyspareunia, uterine prolapse |
| Urologic | Constipation, fecal soiling, urinary tract infections, urinary incontinence, bladder prolapse, rectal prolapse, |
| Obstetric | Short labor and delivery, premature rupture of membranes, pelvic pain, varicose veins, worsening of dysautonomia during pregnancy, postpartum hemorrhage, complicated perineal wounds |
| Neurologic | Headache, local anesthesia failure, postural instability, increased frequency of falls, impaired proprioceptive acuity, Chiari 1 type 1 |
| Psychiatric | Kinesiophobia, anxiety, depression |
Cardiovascular and Autonomic Nervous System Manifestations
A mild degree of aortic root dilatation has been found in up to one-third of EDS-HT patients,20,21,22 necessitating echocardiographic evaluation and surveillance. Raynaud phenomenon was found in 38% of EDS-HT patients.19 Patients with EDS-HT may suffer from palpitations, chest pain, dizziness, pre-syncope, and syncope,17 which has been attributed in the past to mitral valve prolapse (MVP). Mitral valve prolapse was originally included in the earlier version of the Brighton criteria in 1986.47 With more modern evaluation techniques clinically significant MVP has not been found to be more prevalent among EDS-HT patients.21,22,50,51 For this reason MVP was removed from the Brighton criteria in 1998.48 The frequency of MVP among EDS-HT patients was found to be 28%–67% in more recent studies,52,53 but its clinical significance is not clear. Symptoms formerly attributed to MVP are now considered to be related to autonomic dysfunction, which was found to be highly prevalent among EDS-HT patients.16–18
Gastrointestinal Manifestations
Gastroesophageal reflux was found in 57% of EDS-HT patients.19,23 Chronic gastrointestinal discomfort was reported in 86% of patients with EDS-HT, attributed to dyspepsia, gastritis, or gastroesophageal reflux. Irritable bowel syndrome was found among 62% of patients. Early satiety and delayed gastric emptying are reported and exacerbated by opioids.3
Hematologic Manifestations
Easy bruising and bleeding tendency is common in all EDS types, including EDS-HT.25 It manifests with prolonged bleeding time,24,26 oral mucosa fragility with mucosal bruises,9 and menometrorrhagia.54 Since coagulation tests are normal,24–26 the underlying cause is presumed to be mechanically impaired collagen too weak to afford adequate protection to the capillaries. It is important to note that small and large arterial dissections have not been reported in EDS-HT.
Ocular Manifestations
Myopia has been found in up to 50% of EDS-HT patients,54 and high myopia of more than −6.0 diopters was found in 16% of patients compared with 0% in the control group.3,27 Strabismus was found in 7% of EDS-HT pediatric patients55 (as opposed to only 2%–4% of the general pediatric population), and it is often refractory to surgical correction.56 Meyer et al. found size variations and shape abnormalities of collagen fibrils in the extra-ocular muscles that control the movement of the eye.57
Gynecologic Manifestations
Dysmenorrhea and menorrhagia are common19,28,29,54,56 and thought to be due to muscle contractions occurring with greater force given the loose connective tissue. Dyspareunia was found among 30%–57% of EDS-HT women,28,29,58 thought to be caused by small tears in the vaginal surface and lack of appropriate vaginal secretions.56 Pelvic organ prolapse is common,19,28,29,56,59–62 including uterine prolapse which was found in almost 40% of women with EDS-HT.49
Urologic Manifestations
In children with hypermobility constipation and fecal soiling were found to be more common in boys, and urinary tract infection and urinary incontinence more common among girls.63 In another pediatric series 13% of girls and 6% of boys suffered from urinary tract infections.64 Stress urinary incontinence was found in 40%–70% of women with EDS-HT,28,58,65 often earlier in life, thought to be due to a weakened pelvic floor, which may be worsened to bladder prolapse.56 Fecal incontinence was found in up to almost 15% of EDS-HT patients, as compared to only 2.2% of the general population.65 Rectal prolapse may also be found among EDS-HT patients.66 Furthermore, Dordoni et al. reported on two EDS-HT family members who suffered from visceroptosis, including bilateral kidney prolapse, gastric ptosis, liver prolapse, and ovarian and heart prolapse.67
Obstetric Manifestations
While labor and delivery might be rapid (shorter than 4 hours),19,29 and premature rupture of membranes is common,54,68,69 pregnancy in women with EDS-HT is generally normal with good maternal and neonatal outcome.30,70 However, joint laxity and pain may increase during pregnancy.3,29,30,54,70 Pelvic pain and instability necessitate the use of pelvic belt, crutches, and/or bed rest in 26% of women with EDS, the majority being EDS-HT (compared to only 7% among non-affected women).56,70 Varicose veins in the legs and the vulva are more common among pregnant women with EDS-HT.56
Dysautonomia, characterized by lightheadedness, dizziness, fainting, etc., may worsen during pregnancy,56 and when postural orthostatic tachycardia syndrome (POTS) is present a blood pressure fall was reported.71 Women with EDS-HT are more prone to postpartum hemorrhage (19% versus 7%) and complicated perineal wounds (8% versus none).70 Premature delivery was found to be more related to EDS-HT of the infant (40%), and was less prevalent if the mother had EDS-HT (21%).70
Neurologic Manifestations
A total of 40% of children with EDS-HT72 and 50% of adults14 suffer from headaches, characterized as chronic recurrent headaches in the absence of structural, congenital, or acquired central nervous system lesions that correlate with their symptoms.73 Many complain of headaches related to the neck or facial pain that might be related to jaw or TMJ problems.56 Headaches may also be part of dysautonomia, which was found in 78% of EDS-HT patients versus 10% of controls,17 characterized by dizziness/ lightheadedness and pre-syncopal episodes, which were found in 88% and 83% of patients, respectively. Partial or complete failure of local anesthesia was described during biopsies and dental or obstetric procedures.74,75 Hakim and Grahame found local anesthesia resistance in 58% of EDS-HT patients versus 21% of controls.32 Proprioceptive acuity has been found to be impaired among EDS-HT adult patients76,77 and pediatric patients.78 Postural instability and balance and gait impairment, resulting in increased frequency of falls, were found among EDS-HT patients as compared to matched healthy controls.79 Impaired proprioceptive acuity is thought to influence muscle strength. Therefore, improving muscle strength on the basis of proprioceptive impairment may be more important for reducing activity limitations than just improving muscle strength.80 Chiari 1 malformation type 1 was found in 4.7% of EDS-HT patients19 and may be associated with cranio-cervical instability and/or the tethered cord syndrome.
Psychiatric Manifestations
Fear of joint pain and/or instability may lead to avoidance behavior (kinesiophobia) and exacerbate dysfunction and disability.3,7 Depression and anxiety are more common among EDS-HT patients7,19,34 and are exacerbated by fatigue and pain.11,15
GENERAL REMARKS
The multisystemic nature of EDS-HT results in patients having difficulty coping with the syndrome, as well as medical personnel failing to understand the true nature of the condition. This may adversely affect the therapeutic relationship, giving rise to skepticism, resentment, distrust, and hostility on the part of the patient.3,7
Although EDS-HT is the most common type and the least severe type of EDS, it tends to be underdiagnosed and mistreated, sometimes leading to severe disability that may have been preventable if diagnosed and treated properly.64,81,82 A survey among physiotherapists in the UK found that only 32% of respondents received formal training in EDS-HT management.83 Patients perceive a lack of awareness of the syndrome among health professionals and describe delays in diagnosis and access to appropriate health care services.84 Many patients reported lengthy diagnosis trajectories and treatment for individual symptoms rather than EDS-HT as a whole. Receiving a correct diagnosis is necessary in order to access appropriate care pathways, for example, referral for physiotherapy for EDS-HT rather than for an acute single joint problem.84 A study conducted among military personnel found misdiagnosis of EDS-HT has a disabling impact on military personnel with EDS-HT who are exposed to strenuous physical activities.85 Significant neuromuscular and motor development problems have been found among a pediatric population, and delay in diagnosis resulted in poor control of pain and disruption of normal home life, schooling, and physical activities.64 Furthermore, they conclude that knowledge of the diagnosis and appropriate interventions are likely to be highly effective in reducing the morbidity and cost to the health and social services.64
DIAGNOSIS
Diagnosis relies on the revised Brighton criteria, but it is important to rule out other connective tissue disorders, especially Marfan syndrome and other types of EDS. Unfortunately, no genetic defect has been found, and for such a prevalent and complex genetic disorder multiple genes might be involved.
MEDICAL MANAGEMENT
Treatment requires multidisciplinary co-operation and consulting with a cardiologist with echocardiogram monitoring every 2–5 years, orthopedic surgeon with a follow-up once a year, oral and maxillofacial surgeon for temporomandibular joint involvement, gastroenterologist when gastrointestinal manifestations are present, ophthalmologist to rule out other connective tissue diseases and when ocular manifestations are present, urologist and urogynecologist when urologic manifestations are suspected, neurologist and neurosurgeon when prolonged headache is present to rule out Chiari 1, and psychiatry when anxiety and/or depression are suspected. Allergologic consultation may also be needed when there are multiple drug reactions and/or food allergies. An autonomic nervous system specialist should be consulted when signs and symptoms of POTS or other autonomic nervous system manifestations are present. Management includes physiotherapy and hydrotherapy aimed at symmetric and generalized muscle strengthening and proprioception acuity improvement, including deep connective tissue manipulations after each session, occupational therapy when wrists and fingers are involved, and cognitive behavioral therapy for proper adjustment to the chronic nature of the condition. Nutrition has an important role in treating EDS-HT, and nutritional deficiencies should be sought out and treated.
CONCLUSION
Ehlers–Danlos syndrome-HT is a complex hereditary disorder which is multisystemic, probably due to the prevalence of connective tissue in all body systems. Its gene defect has yet to be found and might be of multigenetic nature, but until then we have to think about the possibility of EDS-HT in every chronic pain patient, and look for joint hypermobility as well as other multisystemic manifestations of this prevalent syndrome.
Abbreviations
- EDS
Ehlers–Danlos syndrome
- HT
hypermobility type
- JH
joint hypermobility
- JHS
joint hypermobility syndrome
- MVP
mitral valve prolapse
- TMJ
temporomandibular joints
Footnotes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK) Am J Med Genet. 1998;77:31–7. doi: 10.1002/(sici)1096-8628(19980428)77:1<31::aid-ajmg8>3.0.co;2-o. https://doi.org/10.1002/(SICI)1096-8628(19980428)77:1%3C31::AID-AJMG8%3E3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 2.Grahame R. Ehlers-Danlos syndrome. S Afr Med J. 2016;106:S45–6. doi: 10.7196/SAMJ.2016.v106i6.10991. https://doi.org/10.7196/SAMJ.2016.v106i6.10991. [DOI] [PubMed] [Google Scholar]
- 3.Levy HP. Ehlers Danlos Syndrome, Hypermobility Type. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet] Seattle, WA: University of Washington, Seattle; 1993–2016. [accessed October 7, 2016]. Updated March 31, 2016. Available at: http://bit.ly/2ePdTyk. [Google Scholar]
- 4.De Paepe A, Malfait F. The Ehler-Danlos syndrome, a disorder with many faces. Clin Genet. 2012;82:1–11. doi: 10.1111/j.1399-0004.2012.01858.x. https://doi.org/10.1111/j.1399-0004.2012.01858.x. [DOI] [PubMed] [Google Scholar]
- 5.Pepin MG, Murray ML, Byers PH. Vascular Ehlers-Danlos Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet] Seattle, WA: University of Washington, Seattle; 1993–2016. [accessed October 7, 2016]. Updated Nov 19, 2015. Available at: http://bit.ly/2fiszsr. [Google Scholar]
- 6.Malfait F, Wenstrup R, De Paepe A. Ehlers-Danlos Syndrome, Classic Type. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet] Seattle, WA: University of Washington, Seattle; 1993–2016. [accessed October 7, 2016]. Updated Nov 19, 2015. Available at: http://bit.ly/2dL9wGW. [Google Scholar]
- 7.Branson JA, Kozlowska K, Kaczynski KJ, Roesler TA. Managing chronic pain in a young adolescent girl with Ehlers-Danlos syndrome. Harv Rev Psychiatry. 2011;19:259–70. doi: 10.3109/10673229.2011.614484. https://doi.org/10.3109/10673229.2011.614484. [DOI] [PubMed] [Google Scholar]
- 8.Hagberg C, Berglund B, Korpe L, Andersson-Norinder J. Ehlers-Danlos syndrome (EDS) focusing on oral symptoms: a questionnaire study. Orthod Craniofac Res. 2004;7:178–85. doi: 10.1111/j.1601-6343.2004.00288.x. https://doi.org/10.1111/j.1601-6343.2004.00288.x. [DOI] [PubMed] [Google Scholar]
- 9.De Coster PJ, Martens LC, De Paepe A. Oral health in prevalent types of Ehlers-Danlos syndromes. J Oral Pathol Med. 2005;34:298–307. doi: 10.1111/j.1600-0714.2004.00300.x. https://doi.org/10.1111/j.1600-0714.2004.00300.x. [DOI] [PubMed] [Google Scholar]
- 10.Rombaut L, Malfait F, Cools A, De Paepe A, Calders P. Musculoskeletal complaints, physical activity and health-related quality of life among patients with the Ehlers-Danlos syndrome hypermobility type. Disabil Rehabil. 2010;32:1339–45. doi: 10.3109/09638280903514739. https://doi.org/10.3109/09638280903514739. [DOI] [PubMed] [Google Scholar]
- 11.Rombaut L, Malfait F, De Paepe A, et al. Impairment and impact of pain in female patients with Ehlers-Danlos syndrome: a comparative study with fibromyalgia and rheumatoid arthritis. Arthritis Rheum. 2011;63:1979–87. doi: 10.1002/art.30337. https://doi.org/10.1002/art.30337. [DOI] [PubMed] [Google Scholar]
- 12.Rombaut L, Malfait F, De Wandele I, et al. Medication, surgery, and physiotherapy among patients with the hypermobility type of Ehlers-Danlos syndrome. Arch Phys Med Rehabil. 2011;92:1106–12. doi: 10.1016/j.apmr.2011.01.016. https://doi.org/10.1016/j.apmr.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 13.Voermans NC, Knoop H. Both pain and fatigue are important possible determinants of disability in patients with the Ehlers-Danlos syndrome hypermobility type. Disab Rehabil. 2011;33:706–7. doi: 10.3109/09638288.2010.531373. https://doi.org/10.3109/09638288.2010.531373. [DOI] [PubMed] [Google Scholar]
- 14.Sacheti A, Szemere J, Bernstein B, Tafas T, Schechter N, Tsipouras P. Chronic pain is a manifestation of the Ehlers-Danlos syndrome. J Pain Symptom Manage. 1997;14:88–93. doi: 10.1016/s0885-3924(97)00007-9. https://doi.org/10.1016/S0885-3924(97)00007-9. [DOI] [PubMed] [Google Scholar]
- 15.Voermans NC, Knoop H, Bleijenberg G, van Engelen BG. Pain in ehlers-danlos syndrome is common, severe, and associated with functional impairment. J Pain Symptom Manage. 2010;40:370–8. doi: 10.1016/j.jpainsymman.2009.12.026. https://doi.org/10.1016/j.jpainsymman.2009.12.026. [DOI] [PubMed] [Google Scholar]
- 16.Rowe PC, Barron DF, Calkins H, Maumenee IH, Tong PY, Geraghty MT. Orthostatic intolerance and chronic fatigue syndrome associated with Ehlers-Danlos syndrome. J Pediatr. 1999;135:494–9. doi: 10.1016/s0022-3476(99)70173-3. https://doi.org/10.1016/S0022-3476(99)70173-3. [DOI] [PubMed] [Google Scholar]
- 17.Gazit Y, Nahir AM, Grahame R, Jacob G. Dysautonomia in the joint hypermobility syndrome. Am J Med. 2003;115:33–40. doi: 10.1016/s0002-9343(03)00235-3. https://doi.org/10.1016/S0002-9343(03)00235-3. [DOI] [PubMed] [Google Scholar]
- 18.Mathias CJ, Low DA, Iodice V, Owens AP, Kirbis M, Grahame R. Postural tachycardia syndrome--current experience and concepts. Nat Rev Neurol. 2011;8:22–34. doi: 10.1038/nrneurol.2011.187. https://doi.org/10.1038/nrneurol.2011.187. [DOI] [PubMed] [Google Scholar]
- 19.Castori M, Camerota F, Celletti C, et al. Natural history and manifestations of the hypermobility type Ehlers-Danlos syndrome: a pilot study on 21 patients. Am J Med Genet A. 2010;152A:556–64. doi: 10.1002/ajmg.a.33231. [DOI] [PubMed] [Google Scholar]
- 20.Wenstrup RJ, Meyer RA, Lyle JS, et al. Prevalence of aortic root dilation in the Ehlers-Danlos syndrome. Genet Med. 2002;4:112–17. doi: 10.1097/00125817-200205000-00003. https://doi.org/10.1097/00125817-200205000-00003. [DOI] [PubMed] [Google Scholar]
- 21.McDonnell NB, Gorman BL, Mandel KW, et al. Echocardiographic findings in classical and hypermobile Ehlers-Danlos syndromes. Am J Med Genet A. 2006;140:129–36. doi: 10.1002/ajmg.a.31035. https://doi.org/10.1002/ajmg.a.31035. [DOI] [PubMed] [Google Scholar]
- 22.Atzinger CL, Meyer RA, Khoury PR, Gao Z, Tinkle BT. Cross-sectional and longitudinal assessment of aortic root dilation and valvular anomalies in hypermobile and classic Ehlers-Danlos syndrome. J Pediatr. 2011;158:826–30.e1. doi: 10.1016/j.jpeds.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 23.Levy HP, Mayoral W, Collier K, Tio TL, Francomano CA. Gastroesophageal reflux and irritable bowel syndrome in classical and hypermobile Ehlers Danlos syndrome (EDS) Am J Hum Genet. 1999;65:A69. [Google Scholar]
- 24.Anstey A, Mayne K, Winter M, Van de Pette J, Pope FM. Platelet and coagulation studies in Ehlers-Danlos syndrome. Br J Dermatol. 1991;125:155–63. doi: 10.1111/j.1365-2133.1991.tb06063.x. https://doi.org/10.1111/j.1365-2133.1991.tb06063.x. [DOI] [PubMed] [Google Scholar]
- 25.De Paepe A, Malfait F. Bleeding and bruising in patients with Ehlers-Danlos syndrome and other collagen vascular disorders. Br J Haematol. 2004;127:491–500. doi: 10.1111/j.1365-2141.2004.05220.x. https://doi.org/10.1111/j.1365-2141.2004.05220.x. [DOI] [PubMed] [Google Scholar]
- 26.Mast KJ, Nunes ME, Ruymann FB, Kerlin BA. Desmopressin responsiveness in children with Ehlers-Danlos syndrome associated bleeding symptoms. Br J Haematol. 2009;144:230–3. doi: 10.1111/j.1365-2141.2008.07446.x. https://doi.org/10.1111/j.1365-2141.2008.07446.x. [DOI] [PubMed] [Google Scholar]
- 27.Gharbiya M, Moramarco A, Castori M, et al. Ocular features in joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type: a clinical and in vivo confocal microscopy study. Am J Ophthalmol. 2012;154:593–600.e1. doi: 10.1016/j.ajo.2012.03.023. https://doi.org/10.1016/j.ajo.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 28.McIntosh LJ, Mallett VT, Frahm JD, Richardson DA, Evans MI. Gynecologic disorders in women with Ehlers-Danlos syndrome. J Soc Gynecol Investig. 1995;2:559–64. doi: 10.1016/1071-5576(94)00050-b. [DOI] [PubMed] [Google Scholar]
- 29.Castori M, Morlino S, Dordoni C, et al. Gynecologic and obstetric implications of the joint hypermobility syndrome (a.k.a. Ehlers – Danlos syndrome hypermobility type) in 82 Italian patients. Am J Med Genet Part A. 2012;158A:2176–82. doi: 10.1002/ajmg.a.35506. https://doi.org/10.1002/ajmg.a.35506. [DOI] [PubMed] [Google Scholar]
- 30.Volkov N, Nisenblat V, Ohel G, Gonen R. Ehlers-Danlos syndrome: insights on obstetric aspects. Obstet Gynecol Surv. 2007;62:51–7. doi: 10.1097/01.ogx.0000251027.32142.63. https://doi.org/10.1097/01.ogx.0000251027.32142.63. [DOI] [PubMed] [Google Scholar]
- 31.Dutta I, Wilson H, Oteri O. Pregnancy and delivery in ehlers-danlos syndrome (hypermobility type): review of the literature. Obstet Gynecol Int. 2011;2011:306413. doi: 10.1155/2011/306413. https://doi.org/10.1155/2011/306413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hakim AJ, Grahame R, Norris P, Hopper C. Local anaesthetic failure in joint hypermobility syndrome. J R Soc Med. 2005;98:84–5. doi: 10.1258/jrsm.98.2.84. https://doi.org/10.1258/jrsm.98.2.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milhorat TH, Bolognese PA, Nishikawa M, McDonnell NB, Francomano CA. Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and chiari malformation type I in patients with hereditary disorders of connective tissue. J Neurosurg Spine. 2007;7:601–9. doi: 10.3171/SPI-07/12/601. https://doi.org/10.3171/SPI-07/12/601. [DOI] [PubMed] [Google Scholar]
- 34.Baeza-Velasco C, Gély-Nargeot MC, Bulbena Vilarrasa A, Bravo JF. Joint hypermobility syndrome: problems that require psychological intervention. Rheumatol Int. 2011;31:1131–6. doi: 10.1007/s00296-011-1839-5. https://doi.org/10.1007/s00296-011-1839-5. [DOI] [PubMed] [Google Scholar]
- 35.Bulbena A, Duro JC, Porta M, et al. Anxiety disorders in the joint hypermobility syndrome. Psychiatry Reserve. 1993;46:59–68. doi: 10.1016/0165-1781(93)90008-5. https://doi.org/10.1016/0165-1781(93)90008-5. [DOI] [PubMed] [Google Scholar]
- 36.Grahame R. Time to take hypermobility seriously (in adults and children) Rheumatology (Oxford) 2001;40:485–7. doi: 10.1093/rheumatology/40.5.485. https://doi.org/10.1093/rheumatology/40.5.485. [DOI] [PubMed] [Google Scholar]
- 37.Gurley-Green S. Living with the hypermobility syndrome. Rheumatology (Oxford) 2001;40:487–9. doi: 10.1093/rheumatology/40.5.487. https://doi.org/10.1093/rheumatology/40.5.487. [DOI] [PubMed] [Google Scholar]
- 38.Keer R, Grahame R. Hypermobility Syndrome—Recognition and Management for Physiotherapists. London: Butterworth-Heinemann; 2003. [Google Scholar]
- 39.Simmonds JV, Keer RJ. Hypermobility and the hypermobility syndrome. Man Ther. 2007;12:298–309. doi: 10.1016/j.math.2007.05.001. https://doi.org/10.1016/j.math.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 40.Child AH. Joint hypermobility syndrome: inherited disorder of collagen synthesis. J Rheumatol. 1986;13:239–43. [PubMed] [Google Scholar]
- 41.Beighton P, Grahame R, Bird HA. Genetic Aspects of the Hypermobility Syndrome. In: Beighton P, Grahame R, Bird HA, editors. Hypermobility of Joints. 2nd ed. Berlin, Germany: Springer; 1989. pp. 55–66. https://doi.org/10.1007/978-1-4471-3900-3_5. [Google Scholar]
- 42.Beighton P, Grahame R, Bird HA. Clinical Features of Hypermobility Syndrome. In: Beighton P, Grahame R, Bird HA, editors. Hypermobility of Joints. 2nd ed. Berlin, Germany: Springer; 1989. pp. 67–84. https://doi.org/10.1007/978-1-4471-3900-3_6. [Google Scholar]
- 43.Beighton PH, Solomon L, Soskolne CL. Articular mobility in an African population. Ann Rheum Dis. 1973;32:413–18. doi: 10.1136/ard.32.5.413. https://doi.org/10.1136/ard.32.5.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grahame R. Hypermobility and Hypermobility Syndrome. In: Keer R, Grahame R, editors. Hypermobility Syndrome—Recognition and Management For Physiotherapists. London, UK: Butterworth-Heinemann; 2003. pp. 1–14. https://doi.org/10.1016/B978-0-7506-5390-9.50005-8. [Google Scholar]
- 45.Kirk JA, Ansell BM, Bywaters EL. The hypermobility syndrome. Ann Rheum Dis. 1967;26:419–25. doi: 10.1136/ard.26.5.419. https://doi.org/10.1136/ard.26.5.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grahame R. Joint hypermobility: clinical aspects. Proc R Soc Med. 1971;64:32–4. [PMC free article] [PubMed] [Google Scholar]
- 47.Beighton P, De Paepe A, Danks D, et al. International Nosology of Heritable Disorders of Connective Tissue, Berlin, 1986. Am J Med Genet. 1988;29:581–94. doi: 10.1002/ajmg.1320290316. https://doi.org/10.1002/ajmg.1320290316. [DOI] [PubMed] [Google Scholar]
- 48.Grahame R, Bird HA, Child A. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS) J Rheumatol. 2000;27:1777–9. [PubMed] [Google Scholar]
- 49.El-Shahaly HA, el-Sherif AK. Is the benign joint hypermobility syndrome benign? Clin Rheumatol. 1991;10:302–7. doi: 10.1007/BF02208695. https://doi.org/10.1007/BF02208695. [DOI] [PubMed] [Google Scholar]
- 50.Dolan AL, Mishra MB, Chambers JB, Grahame R. Clinical and echocardiographic survey of the Ehlers-Danlos syndrome. Br J Rheumatol. 1997;36:459–62. doi: 10.1093/rheumatology/36.4.459. https://doi.org/10.1093/rheumatology/36.4.459. [DOI] [PubMed] [Google Scholar]
- 51.Mishra MB, Ryan P, Atkinson P, et al. Extra-articular features of benign joint hypermobility syndrome. Br J Rheumatol. 1996;35:861–6. doi: 10.1093/rheumatology/35.9.861. https://doi.org/10.1093/rheumatology/35.9.861. [DOI] [PubMed] [Google Scholar]
- 52.Camerota F, Castori M, Celletti C, et al. Heart rate, conduction and ultrasound abnormalities in adults with joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Clin Rheumatol. 2014;33:981–7. doi: 10.1007/s10067-014-2618-y. https://doi.org/10.1007/s10067-014-2618-y. [DOI] [PubMed] [Google Scholar]
- 53.Kozanoglu E, Coskun Benlidayi I, Eker Akilli R, Tasal A. Is there any link between joint hypermobility and mitral valve prolapse in patients with fibromyalgia syndrome? Clin Rheumatol. 2016;35:1041–4. doi: 10.1007/s10067-015-3024-9. https://doi.org/10.1007/s10067-015-3024-9. [DOI] [PubMed] [Google Scholar]
- 54.Ainsworth SR, Aulicino PL. A survey of patients with Ehlers-Danlos syndrome. Clin Orthop Rel Res. 1993;286:250–256. https://doi.org/10.1097/00003086-199301000-00037. [PubMed] [Google Scholar]
- 55.Pemberton JW, MacKenzie Freeman H, Schepens CL. Familial retinal detachment and the Ehlers-Danlos syndrome. Arch Ophthalmol. 1966;76:817–24. doi: 10.1001/archopht.1966.03850010819007. https://doi.org/10.1001/archopht.1966.03850010819007. [DOI] [PubMed] [Google Scholar]
- 56.Tinkle BT. A Guide for the Issues & Management of Ehlers-Danlos Syn-drome Hypermobility Type and The Hypermobility Syndrome. Niles, IL: Left Paw Press, LLC; 2010. Joint Hypermobility Handbook. [Google Scholar]
- 57.Meyer E, Ludatcher RM, Zonis S. Collagen fibril abnormalities in the extraocular muscles in Ehlers-Danlos syndrome. J Pediatr Opthalmol Strabismus. 1988;25:67–72. doi: 10.3928/0191-3913-19880301-05. [DOI] [PubMed] [Google Scholar]
- 58.Castori M, Camerota F, Celletti C, Grammatico P, Padua L. Quality of life in the classic and hypermobility types of Ehlers-Danlos syndrome. Ann Neurol. 2010;67:145–7. doi: 10.1002/ana.21934. https://doi.org/10.1002/ana.21934. [DOI] [PubMed] [Google Scholar]
- 59.Al-Rawi ZS, Al-Rawi ZT. Joint hypermobility in women with genital prolapse. Lancet. 1982;1:1439–41. doi: 10.1016/s0140-6736(82)92453-9. https://doi.org/10.1016/S0140-6736(82)92453-9. [DOI] [PubMed] [Google Scholar]
- 60.Norton PA, Baker JE, Sharp HC, Warenski JC. Genitourinary prolapsed and joint hypermobility in women. Obstet Gynecol. 1995;85:225–8. doi: 10.1016/0029-7844(94)00386-R. https://doi.org/10.1016/0029-7844(94)00386-R. [DOI] [PubMed] [Google Scholar]
- 61.Carley ME, Schaffer J. Urinary incontinence and pelvic organ prolapsed in women with Marfan or Ehlers Danlos syndrome. Am J Obstet Gynecol. 2000;182:1021–3. doi: 10.1067/mob.2000.105410. https://doi.org/10.1067/mob.2000.105410. [DOI] [PubMed] [Google Scholar]
- 62.Aydeniz A, Dikensoy E, Cebesoy B, Altinadaq O, Gursoy S, Balat O. The relation between genitourinary prolapsed and joint hypermobility in Turkish women. Arch Gynecol Obstet. 2010;281:301–4. doi: 10.1007/s00404-009-1103-3. https://doi.org/10.1007/s00404-009-1103-3. [DOI] [PubMed] [Google Scholar]
- 63.de Kort LM, Verhulst JA, Engelbert RH, Uiterwaal CS, de Jong TP. Lower urinary tract dysfunction in children with generalized hypermobility of joints. J Urol. 2003;170:1971–4. doi: 10.1097/01.ju.0000091643.35118.d3. https://doi.org/10.1097/01.ju.0000091643.35118.d3. [DOI] [PubMed] [Google Scholar]
- 64.Adib N, Davies K, Grahame R, Woo P, Murray KJ. Joint hypermobility syndrome in childhood A not so benign multisystem disorder. Rheumatology (Oxford) 2005;44:744–50. doi: 10.1093/rheumatology/keh557. https://doi.org/10.1093/rheumatology/keh557. [DOI] [PubMed] [Google Scholar]
- 65.Arunkalaivanan AS, Morrison A, Jha S, Blann A. Prevalence of urinary and faecal incontinence among female members of the Hypermobility Syndrome Association (HMSA) J Obstet Gyneacol. 2009;29:126–8. doi: 10.1080/01443610802664747. https://doi.org/10.1080/01443610802664747. [DOI] [PubMed] [Google Scholar]
- 66.Grahame R. Pain, distress and joint hyperlaxity. Joine Bone Spine. 2000;67:157–63. [PubMed] [Google Scholar]
- 67.Dordoni C, Ritelli M, Venturini M, et al. Recurring and generalized visceroptosis in Ehlers-Danlos syndrome hypermobility type. Am J Med Genet A. 2013;161A:1143–7. doi: 10.1002/ajmg.a.35825. https://doi.org/10.1002/ajmg.a.35825. [DOI] [PubMed] [Google Scholar]
- 68.Taylor DJ, Wilcox I, Russell JK. Ehlers-Danlos syndrome during pregnancy: a case report and review of the literature. Obstet Gynecol Surv. 1981;36:277–81. doi: 10.1097/00006254-198106000-00001. https://doi.org/10.1097/00006254-198106000-00001. [DOI] [PubMed] [Google Scholar]
- 69.De Vos M, Nuytinck L, Verellen C, De Paepe A. Preterm premature rupture of membranes in a patient with the hypermobility type of the Ehlers-Danlos syndrome. A case report. Fetal Diagn Ther. 1999;14:244–7. doi: 10.1159/000020930. https://doi.org/10.1159/000020930. [DOI] [PubMed] [Google Scholar]
- 70.Lind J, Wallenburg HC. Pregnancy and the Ehler-Danlos syndrome: a retrospective study in a Dutch population. Acta Obstet Gynecol Scand. 2002;81:293–300. doi: 10.1034/j.1600-0412.2002.810403.x. https://doi.org/10.1034/j.1600-0412.2002.810403.x. [DOI] [PubMed] [Google Scholar]
- 71.Jones TL, Ng C. Anaesthesia for caesarean section in a patient with Ehlers-Danlos syndrome associated with postural orthostatic tachycardia syndrome. Int J Obstet Anesth. 2008;17:365–9. doi: 10.1016/j.ijoa.2008.04.003. https://doi.org/10.1016/j.ijoa.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 72.Mato H, Berde T, Hasson N, Grahame R, Maillard S. A review of symptoms with benign joint hypermobility syndrome in children. Ped Rheumatol. 2008;6(Suppl 1):P155. https://doi.org/10.1186/1546-0096-6-S1-P155. [Google Scholar]
- 73.Jacome DE. Headache in Ehlers-Danlos syndrome. Cephalalgia. 1999;19:791–6. doi: 10.1046/j.1468-2982.1999.1909791.x. https://doi.org/10.1046/j.1468-2982.1999.1909791.x. [DOI] [PubMed] [Google Scholar]
- 74.Kaalund S, Hogsaa B, Grevy C, Oxlund H. Reduced strength of skin in Ehlers-Danlos syndrome type III. Scand J Rheumatol. 1990;19:67–70. doi: 10.3109/03009749009092623. https://doi.org/10.3109/03009749009092623. [DOI] [PubMed] [Google Scholar]
- 75.Arendt-Nielsen L, Kaalund S, Bjerring P, Hogsaa B. Insufficient effect of local analgesics in Ehlers-Danlos type III patients (connective tissue disorder) Acta Anaesth Scand. 1990;34:358–61. doi: 10.1111/j.1399-6576.1990.tb03103.x. https://doi.org/10.1111/j.1399-6576.1990.tb03103.x. [DOI] [PubMed] [Google Scholar]
- 76.Mallik AK, Ferrell WR, McDonals AG, Sturrock RD. Impaired proprioceptive acuity at the proximal interphalangeal joint in patients with the hypermobility syndrome. Br J Rheumatol. 1994;33:631–7. doi: 10.1093/rheumatology/33.7.631. https://doi.org/10.1093/rheumatology/33.7.631. [DOI] [PubMed] [Google Scholar]
- 77.Hall MG, Ferrell WR, Sturrock RD, Hamblen DL, Baxendale RH. The effect of the hypermobility syndrome on knee joint proprioception. Br J Rheumatol. 1995;34:121–5. doi: 10.1093/rheumatology/34.2.121. https://doi.org/10.1093/rheumatology/34.2.121. [DOI] [PubMed] [Google Scholar]
- 78.Fatoye F, Palmar S, Macmillan F, Rowe P, van der Linden M. Proprioception and muscle torque deficits in children with hypermobility syndrome. Rheumatology (Oxford) 2009;48:152–7. doi: 10.1093/rheumatology/ken435. https://doi.org/10.1093/rheumatology/ken435. [DOI] [PubMed] [Google Scholar]
- 79.Rombaut L, Malfait F, De Wandele I, et al. Balance, gait, falls, and fear of falling in women with the hypermobility type of Ehlers-Danlos syndrome. Arthritis Care Res (Hoboken) 2011;63:1432–9. doi: 10.1002/acr.20557. https://doi.org/10.1002/acr.20557. [DOI] [PubMed] [Google Scholar]
- 80.Scheper M, Rombaut L, de Vreis J, et al. The association between muscle strength and activity limitations in patients with the hypermobility type of Ehlers-Danlos syndrome: the impact of proprioception. Disabil Rehabil. 2016 Jun 24;:1–7. doi: 10.1080/09638288.2016.1196396. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 81.Grahame R. Joint hypermobility: emerging disease or illness behavior? Clin Med (Lond) 2013;13(Suppl 6):s50–2. doi: 10.7861/clinmedicine.13-6-s50. https://doi.org/10.7861/clinmedicine.13-6-s50. [DOI] [PubMed] [Google Scholar]
- 82.Wolf JM, Cameron KL, Owens BD. Impact of joint laxity and hypermobility on the musculoskeletal system. J Am Acad Orthop Surg. 2011;19:463–71. doi: 10.5435/00124635-201108000-00002. https://doi.org/10.5435/00124635-201108000-00002. [DOI] [PubMed] [Google Scholar]
- 83.Palmar S, Cramp F, Lewis R, Muhammad S, Clark E. Diagnosis, management and assessment if adults with joint hypermobility syndrome: a UK-wide survey of physiotherapy practice. Musculoskeletal Care. 2015;13:101–11. doi: 10.1002/msc.1091. https://doi.org/10.1002/msc.1091. [DOI] [PubMed] [Google Scholar]
- 84.Terry RH, Palmer ST, Rimes KA, Clark CJ, Simmonds JV, Horwood JP. Living with joint hypermobility syndrome: patient experience of diagnosis, referral and self-care. Fam Pract. 2015;32:354–8. doi: 10.1093/fampra/cmv026. https://doi.org/10.1093/fampra/cmv026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mullick G, Bhakuni DS, Shnmuganandan K, et al. Clinical profile of benign joint hypermobility syndrome from a tertiary care military hospital in India. Int J Rheum Dis. 2013;16:590–4. doi: 10.1111/1756-185x.12024. https://doi.org/10.1111/1756-185x.12024. [DOI] [PubMed] [Google Scholar]