

Summary
Fusobacterium nucleatum is an invasive anaerobic bacterium that is associated with periodontal disease. Previous studies have focused on virulence factors produced by F. nucleatum, but early recognition of the pathogen by the immune system remains poorly understood. Although an inflammasome in gingival epithelial cells (GECs) can be stimulated by danger-associated molecular patterns (DAMPs) (also known as danger signals) such as ATP, inflammasome activation by this periodontal pathogen has yet to be described in these cells. This study therefore examines the effects of F. nucleatum infection on pro-inflammatory cytokine expression and inflammasome activation in GECs. Our results indicate that infection induces translocation of NF-κB into the nucleus, resulting in cytokine gene expression. In addition, infection activates the NLRP3 inflammasome, which in turn activates caspase-1 and stimulates secretion of mature IL-1β. Unlike other pathogens studied until now, F. nucleatum activates the inflammasome in GECs in the absence of exogenous DAMPs such as ATP. Finally, infection promotes release of other DAMPs that mediate inflammation, such as high-mobility group box 1 protein and apoptosis-associated speck-like protein, with a similar time-course as caspase-1 activation. Thus, F. nucleatum expresses the pathogen-associated molecular patterns necessary to activate NF-κB and also provides an endogenous DAMP to stimulate the inflammasome and further amplify inflammation through secretion of secondary DAMPs.
Introduction
Oral inflammatory diseases represent the most common chronic diseases in the world, affecting over half of the adult population. In 2010, oral conditions affected approximately 3.9 billion people worldwide, with the prevalence of periodontitis being 35% (Richards, 2013) and severe periodontitis, 11 % (Oliver et al., 1998). An estimated 47.2% of American adults have periodontitis, ranging from mild to severe forms (Richards, 2014). Furthermore, periodontitis is more prevalent and severe in developing countries because of poorer oral hygiene and greater plaque retention (Pilot, 1998; Pihlstrom et al., 2005). Untreated periodontitis can lead to damage of the gum tissue surrounding the tooth and alveolar bone damage, resulting in tooth loss (Han et al., 2000; Saini et al., 2009; Atanasova and Yilmaz, 2015).
One of the first barriers to oral disease is represented by the gingival epithelium, which not only forms attachments to the surface of teeth and but also acts as a physical and chemical shield against infection (Ji et al., 2009; Signat et al., 2011). Gingival epithelial cells (GECs) release antimicrobial peptides such as human β-defensins (hBDs) as a mechanism of defence to prevent biofilm formation (Dommisch et al., 2007; Yin and Chung, 2011; Signat et al., 2011). hBDs have a broad spectrum of activity against Gram-negative and Gram-positive bacteria, and they also stimulate adaptive and innate immune responses, resulting in a coordinated defence of the epithelia against invading bacteria. The production of hBDs in GECs is pathogen-specific and is regulated by epigenetic changes in various genes (Yin and Chung, 2011).
The innate immune system consists of cells of hematopoietic origin such as macrophages and neutrophils, which can become activated at early times of infection. While epithelial cells are not typically considered to be specialized innate immune cells, they can also recognize infection and respond by secreting chemokines and cytokines such as IL-8 and IL-1β, as in the case of GECs (Yilmaz et al., 2010; Hung et al., 2013; Johnson et al., 2015) and other epithelial cells (Santana et al., 2015). Receptors on both immune cells and epithelial cells recognize microbial products such as lipopolysaccharide and peptidoglycan, also known as pathogen-associated molecular patterns (PAMPs), through pattern recognition receptors such as toll-like receptors, which stimulate expression of antimicrobial genes, and inflammatory cytokines and chemokines (Janeway and Medzhitov, 2002; Takeda and Akira, 2005; Cassel et al., 2009; Said-Sadier and Ojcius, 2012). At the same time, cells that are dying or stressed because of infection can release host-derived molecules, such as ATP, that are called alarmins, danger signals or danger-associated molecular patterns (DAMPs) (Said-Sadier and Ojcius, 2012; Santana et al., 2015). For the cytokines IL-1β and IL-18, ligation of pattern recognition receptors by PAMPs leads to intracellular production of pro-IL-1β and pro-IL-18, but not their secretion. Simultaneous ligation of receptors for DAMPs, such as the ATP receptor P2×7, leads to assembly of the NLRP3 inflammasome and auto-cleavage of pro-caspase-1 to its activated form, caspase-1 (Cassel et al., 2009; Tschopp and Schroder, 2010; Said-Sadier and Ojcius, 2012; Kim and Jo, 2013; Harijith et al., 2014). Activated caspase-1 can then cleave pro-IL-1β and pro-IL-18 to their mature and secreted form, IL-1β and IL-18 (Srinivasula et al., 2002).
Fusobacterium nucleatum, a Gram-negative anaerobic bacterium, stimulates inflammation in the human oral cavity and gut mucosa (Strauss et al., 2011) and has been identified in high frequencies in adults afflicted with mild to acute periodontitis (Signat et al., 2011). F. nucleatum can also invade host tissues and obstruct healing of damaged oral tissues (Bartold et al., 1991; Jeng et al., 1999). Infection of murine macrophages with F. nucleatum has been shown to result in NLPR3 inflammasome activation and IL-1β secretion (Taxman et al., 2012); however, the mechanism was not characterized, and infection of the preferred host cells of the bacteria, GECs, has not been previously examined.
Studies have shown a significantly high level of IL-1β expression in gingival crevicular fluid at sites of recent bone and attachment loss in periodontitis patients (Lee et al., 1995). Furthermore, IL-1β deficient mice demonstrated less Porphyromonas gingivalis lipopolysaccharide-induced periodontium destruction compared with wild-type mice undergoing the same treatment (Chiang et al., 1999), suggesting a strong association between severity of periodontitis and elevated levels of IL-1β (Hodge and Michalowicz, 2001), which may directly or indirectly stimulate osteoclast formation (Chiang et al., 1999). IL-1β also plays a prominent role in promoting tissue pathology and inflammatory responses in periodontal lesions and stimulating the loss of connective tissue and bone (Chiang et al., 1999).
High-mobility group box 1 (HMGB1) is normally associated with chromatin but mediates the response to injury, infection and inflammation when secreted into the cytosol (Lotze and Tracey, 2005; Lu et al., 2012). In the extracellular space, it has been proposed that HMGB1 behaves as a DAMP (Said-Sadier and Ojcius, 2012). Moreover, the secretion of HMGB1 from the nucleus to the extracellular space during inflammation and infection is thought to require the inflammasome and caspase-1 activation (Qin et al., 2006; Willingham et al., 2009; Lamkanfi et al., 2010; Andersson and Tracey, 2011). Similarly to HMGB1, it has recently been shown that the protein ASC (apoptosis-associated speck-like protein containing a carboxy-terminal CARD), which functions as an adaptor in assembly of the NLPR3 inflammasome, can be secreted from macrophages during inflammasome activation and can function as a DAMP (Baroja-Mazo et al., 2014; Franklin et al., 2014).
This study aims to determine whether F. nucleatum infection can activate the NLRP3 inflammasome in GECs, which represents the first barrier to infection in the oral cavity. Additionally, we examined potential mechanisms that could lead to NLRP3 inflammasome and evaluated whether inflammasome activation in GECs could be amplified further through secretion of additional danger signals, which has not been previously examined in GECs.
Results
F. nucleatum infection induces caspase-1 activation and NF-κB translocation
F. nucleatum infection of GECs was confirmed by immunofluorescence imaging, which showed intracellular F. nucleatum aggregation at 1 h after the start of infection (Fig. 1). Despite the F. nucleatum infection at an MOI of 100, GECs remained healthy and showed no significant cell death at 8 h post-infection. Cell viability assays via TB staining also showed normal cell morphology (data not shown) and no significant cell death after 0, 2, 4, 6 and 8 h in Opti-MEM media (Fig. 1).
Fig. 1.
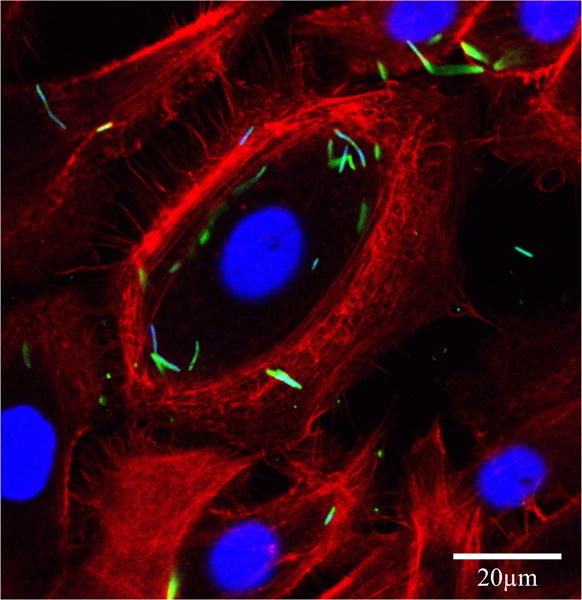
Intracellular localization of Fusobacterium nucleatum in GECs. Immunofluorescence confocal micrograph GECs infected with F. nucleatum (MOI of 100) for 1 h. The single optical section through the middle of the host cell confirms the intracellular localization of the bacteria. Fixed GECs were stained with phalloidin–tetramethylrhodamine B isothiocyanate (red) to show actin filaments, and anti-F. nucleatum antibody (green). Bar represents 20 μm.
To assess host cell responses to F. nucleatum infection, supernatants from F. nucleatum-infected cells were collected and caspase-1 activation was measured by Western blot at different infection time points and MOIs. The level of caspase-1 secretion (a reflection of caspase-1 activation) measured by Western blot showed a significant increase as a function of the MOI, being highest at an MOI of 100 (Fig. 2A). Furthermore, caspase-1 activation was observed within 2 h of infection and was significant after 6 h of infection, compared with control samples (Fig. 2B).
Fig. 2.
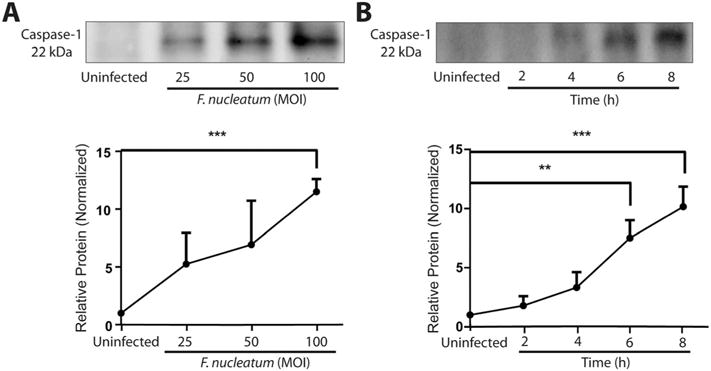
F. nucleatum infection activates caspase-1 in GECs Western blot analysis of supernatants from GECs infected with F. nucleatum A. at MOI of 25, 50 and 100 for 8 h or B. MOI of 100 for 2, 4, 6 or 8 h. Caspase-1 (22 kDa) activation was detected by Western blot, showing the small (22 kDa) fragment of activated caspase-1 in the supernatant. Uninfected samples were collected and processed after 8 h incubation. Results represent an average of three independent experiments, quantified by densitometry. Error bars represent mean ± standard deviations (SD). Asterisks designate statistically significant differences compared with control (**p < 0.01 and ***p < 0.001, Student’s t-test).
NF-κB confocal microscopy was used to visualize NF-κB translocation to the nucleus in uninfected and F. nucleatum-infected cells, which reflects NF-κB activation in the cells. A high level of NF-κB translocation was in fact observed when cells were infected at an MOI of 100 for 1 h (Fig. 3A). The time-course of nuclear translocation was followed by acquiring immunofluorescence images after 30 min, 1 and 2 h of infection (Fig. 2). Analysis of the images showed that was a significant increase in NF-κB staining in the nucleus in GECs infected with F. nucleatum at an MOI of 100 for 30 min, with highest levels of NF-κB activation observed after 1 h of infection and decreasing afterwards (Fig. 3B). NF-κB activation after 6 h of infection remained above basal levels (Fig. 3B).
Fig. 3.
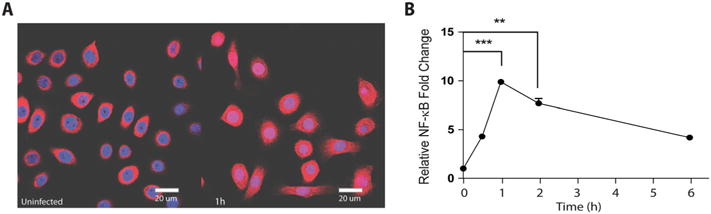
NF-κB translocation to the nucleus during F. nucleatum infection. Cells infected with F. nucleatum (MOI of 100) were analysed by confocal microscopy at multiple time points.
A. Image shows cells at 1 h post-infection stained with anti-NF-κB antibodies (red) and DAPI (blue) to visualize the nucleus.
B. Quantification of NF-κB because of fluorescence intensity of staining relative to control at 30 min, 1, 2 and 6 h. Uninfected controls were collected at after 6 h of incubation. Data are representative of two independent experiments ran in duplicates with at least 100 cells counted per time point. Bar represents 20 μm. Asterisks designate statistically significant difference compared with control (**p < 0.01 and ***p < 0.001, Student’s t-test).
F. nucleatum infection leads to IL-1β gene expression and IL-1β protein secretion, in the absence of exogenous danger signal
We examined whether inflammasomes may play a role during F. nucleatum infection by measuring IL-1β processing and secretion in collected supernatants from F. nucleatum-infected GECs with different MOI and times of infection by Western blot. As expected, F. nucleatum infection induced IL-1β secretion as a function of the MOI, with the highest levels observed at an MOI of 100 (Fig. 4A). Furthermore, IL-1β secretion was measurable at 4 h and was most intense at 8 h post-infection (Fig. 4B). As a confirmation of the Western blot analysis, ELISA was used to measure the time-course of IL-1β secretion after 2, 4 and 8 h of infection (Fig. 4C). A significantly high level of IL-1β secretion was detected by 4 h post-infection and became dramatically higher by 8 h post-infection (Fig. 4C).
Fig. 4.
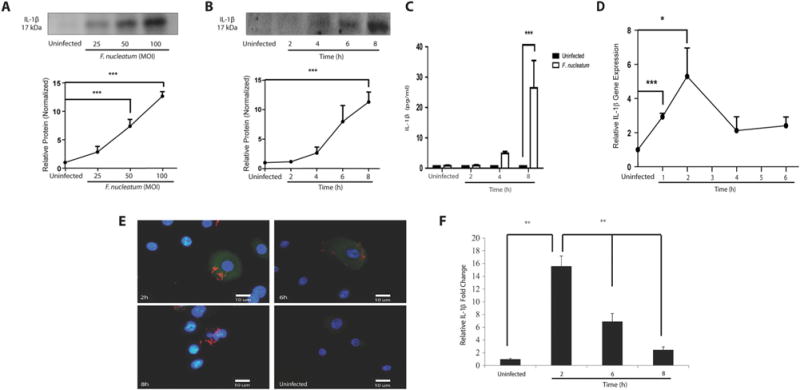
F. nucleatum infection stimulates IL1B gene transcription and processing of IL-1β in GECs. Secretion of IL-1β (17 kDa) was measured in cell supernatants by SDS-PAGE Western blot assay. GECs were infected with F. nucleatum
A. at an MOI of 25, 50 and 100 for 8 h, or
B. at an MOI of 100 for 2, 4, 6 and 8 h. Western blots were quantified by densitometry.
C. IL-1β protein secretion in GECs was confirmed by ELISA after 2, 4 and 8 h of F. nucleatum infection.
D. Relative IL-1β mRNA expression compared with control was evaluated by real-time PCR of GECs infected with F. nucleatum (MOI of 100) for 1, 2, 4 and 6 h.
E. Immunofluorescence staining images to evaluate IL-1β protein expression during F. nucleatum infection (MOI of 100) for 2, 6 and 8 h. Immunofluorescence intensity levels were measured using NIH-ImageJ and the values of at least 2 standard deviations above the average intensity values of the control group were considered significant. GECs were stained with FITC-conjugated antibody against IL-1β and the nuclear stain DAPI, as indicated in the Materials and methods section. Bar represents 10 μm.
F. Quantification of immunofluorescence intensity measured relative to controls at 2, 6 and 8 h. Results represent an average of three independent experiments performed in duplicates. Error bars represent the mean ± SD. Asterisks designate statistically significant difference compared with control (*p < 0.05, **p < 0.01 and ***p < 0.001, Student’s t-test).
Gene expression of IL1B was measured by qPCR, RT-PCR, showing a high level of IL1B gene expression at 1 h and 2 h post-infection (Fig. 4D), which, as expected, preceded the secretion of IL-1β protein (Fig. 4B). The IL1B gene expression level decreased after 2 h of infection and remained at a plateau above basal levels after 4 h of infection (Fig. 4D). The results are therefore consistent with the interpretation that NF-κB activation, which is maximal at 1 h post-infection (Fig. 3B), precedes IL1B gene expression, which in turn results in pro-IL-1β protein synthesis.
IL-1β protein secretion requires cleavage of pro-IL-1β by caspase-1, which is activated within 2 h of infection (Fig. 2B). We therefore used immunofluorescence staining to examine the time course for pro-IL-1β accumulation in F. nucleatum-infected primary GECs. The results showed a high level of pro-IL-1β accumulation within F. nucleatum infected GECs at 2 h post-infection, which subsequently dropped by approximately half at 6 h post-infection and continued decreasing at 8 h post-infection (Fig. 4E and 4F). These results are consistent with the appearance of mature, processed IL-1β in the supernatant after 4 h of infection (Fig. 4B). Pro-IL-1β levels in cell lysates were measured using Western blot analysis, showing that pro-IL-1β accumulation starts at 4 h post-infection and reaches a plateau at 6 h and 8 h, which is presumably because of IL-1β secretion (Fig. 3).
Together, the different assays confirmed that NF-κB is activated by F. nucleatum infection of GECs, which causes expression of IL1B and subsequent production of pro-IL-1β protein. Activation of caspase-1 within 2 h of infection is consistent with secretion of IL-1β protein after 4 h of infection. Finally, these results demonstrate that F. nucleatum infection, on its own, is sufficient to provide the PAMPs needed for NF-κB activation and the danger or stress signal required for caspase-1 activation.
F. nucleatum infection stimulates secretion of the danger signals ASC and HMGB1
The adapter protein ASC is involved in most cases of NLRP3 inflammasome activation in response to different stimuli (Tschopp and Schroder, 2010; Gomes et al., 2013; Lamkanfi and Dixit, 2014). Furthermore, recent studies showed that ASC can be secreted by cells into the extracellular medium, where ASC can behave as a danger signal (Baroja-Mazo et al., 2014; Franklin et al., 2014). Western blot analysis of supernatant was therefore used to measure the time-course of ASC secretion from F. nucleatum-infected GECs. The highest level of ASC secretion was observed at an MOI of 100, with the secretion reaching a plateau by an MOI of 50 (Fig. 5A). Little ASC secretion was observed after 2 h of infection, but high levels were measured after 4 h of infection, reaching a plateau by 6 h of infection with an MOI of 100 (Fig. 5B), suggesting that inflammasome and caspase-1 activation take place before or at the same time as ASC secretion.
Fig. 5.
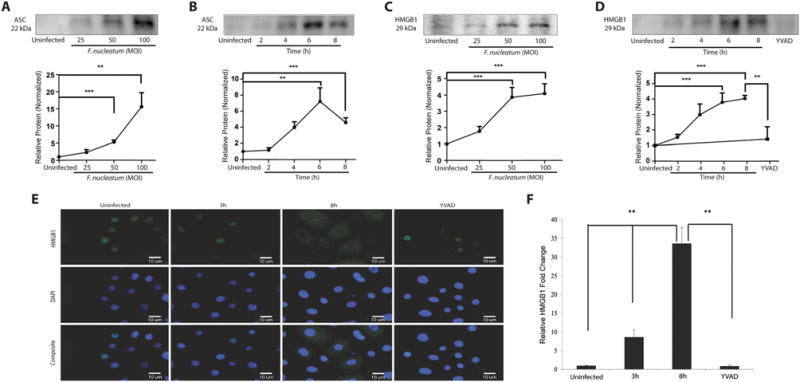
ASC and HMGB1 release following F. nucleatum infection. Release of ASC protein (22 kDa) in cell supernatants was detected using Western blot assay. F. nucleatum infected GECs were infected
A. at MOI of 25, 50 and 100 for 8 h, and
B. at MOI of 100 for 2, 4, 6, and 8 h. HMGB1 protein (29 kDa) was measured using Western blot of supernatant from cells infected with F. nucleatum
C. at an MOI of 25, 50 and 100 for 8 h an
D. at MOI 100 for 2, 4, 6 and 8 h, or for 8 h in GECs pretreated with 50 mM z-YVAD-fmk for 30 min prior to infection. Western blots were quantified by densitometry.
E. Immunofluorescence images of HMGB1 were captured at 3 and 8 h for infected cells (MOI of 100). Bar represents 10 μm.
F. Quantification of immunofluorescence measured relative to control at 3 and 8 h with or without z-YVAD-fmk pretreatment. Results represent an average of three independent experiments performed in duplicates. Error bars represent the mean ± SD. Asterisks designate statistically significant difference compared with control (**p < 0.01, ***p < 0.001, Student’s t-test).
Inflammasome activation has previously been shown to stimulate release of HMGB1 by some cells (Lu et al., 2012). Furthermore, the enzymatic activity of caspase-1 is required for the secretion of HMGB1 and other proteins (Vande Walle et al., 2011). In the extracellular medium, HMGB1 can act as a danger signal and promote innate immune responses (Qin et al., 2006; Vande Walle et al., 2011). We therefore measured HMGB1 localization in infected GECs by immunofluorescence and the HMGB1 secretion from GECs by Western blot analysis of supernatants of GECs, with and without pre-treatment of GECs with the caspase-1 inhibitor, z-YVAD-fmk. Western blot analysis revealed significant levels of HMGB1 release at an MOI of 25, with highest levels and a plateau at an MOI of 50 (Fig. 5C). The time-course of HMGB1 release was also similar to the time course of ASC release, with high levels of HMGB1 release observed after 4 h of infection and a plateau being reached by 6 h of infection (Fig. 5D). Little HMGB1 was found in the supernatant after 8 h of infection if the GECs were pretreated with the caspase-1 inhibitor, z-YVAD-fmk (Fig. 5D), implying that caspase-1 activation is required for HMGB1 secretion.
Redistribution of HMGB1 within GECs was also visualized by immunofluorescence staining, which showed that almost all of the HMGB1 was localized within the nucleus after 3 h of infection, but a high level of cytosolic staining was observed after 8 h of infection (Fig. 5E and 5F), suggesting that F. nucleatum infection leads to release of HMGB1 from the nucleus to the cytosol. In addition, treatment with the caspase-1 inhibitor, z-YVAD-fmk, decreased almost completely the redistribution of HMGB1 from the nucleus to the cytosol (Fig. 5E and 5F), confirming that caspase-1 is required for release of HMGB1 from the nucleus to the cytosol. The time-course of HMGB1 distribution within the cell was similar to the time-course of release of HMGB1 to the extracellular space (Fig. 5D), consistent with redistribution to the cytosol taking place before or at the same time as secretion from the cell.
The NLRP3 inflammasome is required for caspase-1 activation and IL-1β secretion during F. nucleatum infection
Four main types of inflammasomes have been characterized to date, relying on NLRP1, NLRP3, NLRC4 and AIM2 (Tschopp and Schroder, 2010; Said-Sadier and Ojcius, 2012; Lamkanfi and Dixit, 2014). To characterize the importance of NLRP3 in inflammasome activation, we prepared NLRP3-deficient GECs using an RNAi approach with lentiviral particles and evaluated caspase-1 activation and IL-1β secretion after 8 h of infection with F. nucleatum. The efficiency of NLRP3 (117 kDa) depletion was first verified by Western blot, which confirmed that NLRP3 protein levels were decreased with an efficiency of 49% in these cells (Fig. 4). As expected, F. nucleatum-infected NLRP3-deficient cells showed significantly lower caspase-1 activation in comparison with infected control cells (Fig. 6A). Similarly, there was a significant reduction of IL-1β secretion from NLRP3-depleted cells compared with control cells after 8 h of infection (Fig. 6B). These results imply that the NLRP3 inflammasome is involved in caspase-1 activation and IL-1β secretion during F. nucleatum infection of GECs.
Fig. 6.
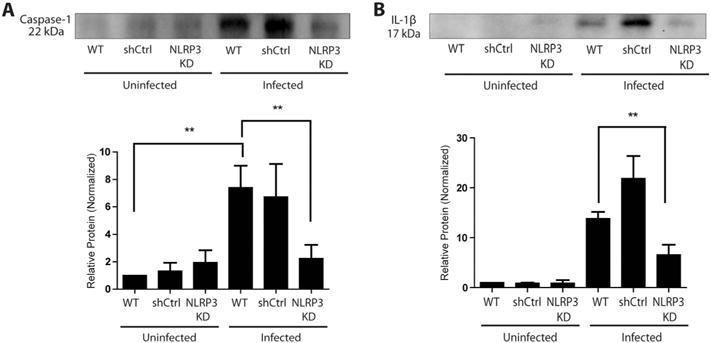
Depletion of NLRP3 inhibits caspase-1 activation and IL-1β secretion.
A, B. Wildtype and NLRP3-depleted (knockdown, KD) GECs were infected with F. nucleatum (MOI of 100) for 8 h. Supernatants were collected and analyzed by Western blot assay for A. caspase-1 activation and B. IL-1β secretion. Results represent three independent experiments. Error bars represent the mean + SD. Asterisks designate statistically significant difference compared with control (**p < 0.01).
Discussion
Periodontal diseases are induced by a complex interaction of periodontal bacteria, composed mainly of gram-negative anaerobes (Han et al., 2000). F. nucleatum is one of the first species to become established in plaque biofilms and is distinguished from other oral pathogens because of its highly invasive ability (Han et al., 2000). A recent in vivo study found that chronic oral infection is tightly linked with F. nucleatum colonization, inducing significant antibody responses along with alveolar bone resorption and intrabony defects (Saini et al., 2009; Velsko et al., 2015). Furthermore, F. nucleatum has recently been shown to directly contribute to inflammation of colonic epithelium cells and increase the risk of inflammatory bowel disease, resulting in colorectal tumorigenesis (Strauss et al., 2011; Bashir et al., 2015) and to colonize the mouse placenta and induce placental inflammatory responses (Liu et al., 2007). Given the ability of F. nucleatum to induce inflammation, we hypothesized that F. nucleatum might also have the ability to activate an NLRP3 inflammasome, which can detect different types of cellular stress or damage.
Infection by many types of intracellular bacteria stimulates synthesis of pro-IL-1β, but not its secretion (Tschopp and Schroder, 2010; Said-Sadier and Ojcius, 2012; Lamkanfi and Dixit, 2014). As described for GECs or macrophages infected by P. gingivalis (Yilmaz et al., 2010; Taxman et al., 2012), a second signal, due often to a danger signal such as extracellular ATP, activates an inflammasome and caspase-1, which results in processing and secretion of the mature IL-1β protein (Tschopp and Schroder, 2010; Lamkanfi and Dixit, 2014). In the case of F. nucleatum infection, we found, instead, that infection can both stimulate expression of the IL1B gene and secretion of mature IL-1β protein, implying that F. nucleatum infection provides both PAMPs and a danger signal. These results are consistent with a recent report that F. nucleatum infection of bone marrow derived macrophages from mice leads to caspase-1 activation and IL-1β secretion (Taxman et al., 2012); although the effect of infection in a physiologically relevant host cell has not been examined.
In our studies, we found that F. nucleatum infection in GECs led to early NF-κB activation and translocation into the nucleus, which stimulated expression of IL1B and the appearance of intracellular pro-IL-1β. Activation of caspase-1 then led to secretion of IL-1β from the infected cells, through a mechanism requiring the presence of the NLRP3 inflammasome.
Soon after caspase-1 was activated in the infected cells, the danger signal HMGB1 was redistributed from the nucleus to the cytosol, and was secreted to the extracellular space (Fig. 5). HMGB1 is the second most abundant protein in chromatin and is associated with DNA in healthy cells (Lotze and Tracey, 2005) and can be released from damaged cells and is secreted actively by some immune cells (Lamkanfi et al., 2010). In the extracellular medium, HMGB1 can interact with innate immune receptors such as RAGE and toll-like receptors (Sims et al., 2010; Yang et al., 2013; Tsung et al., 2014). It has recently been shown that the NLRP3 inflammasome can promote secretion of HMGB1, and we find that caspase-1 was required for HMGB1 release from the nucleus in F. nucleatum-infected GECs.
Similarly, the adaptor protein ASC is involved in NLRP3 inflammasome activation, but recent studies have demonstrated that it can also be secreted from cells during inflammasome activation (Baroja-Mazo et al., 2014; Franklin et al., 2014). In the extracellular space, ASC can amplify inflammatory responses by behaving as a danger signal. We find that infection of cells with F. nucleatum also leads to secretion of ASC. We therefore propose that F. nucleatum infection by itself is detected by GECs as a danger signal and that caspase-1 dependent secretion of HMGB1 and ASC participate in an auto-amplification loop in order to further enhance inflammation (Fig. 7).
Fig. 7.
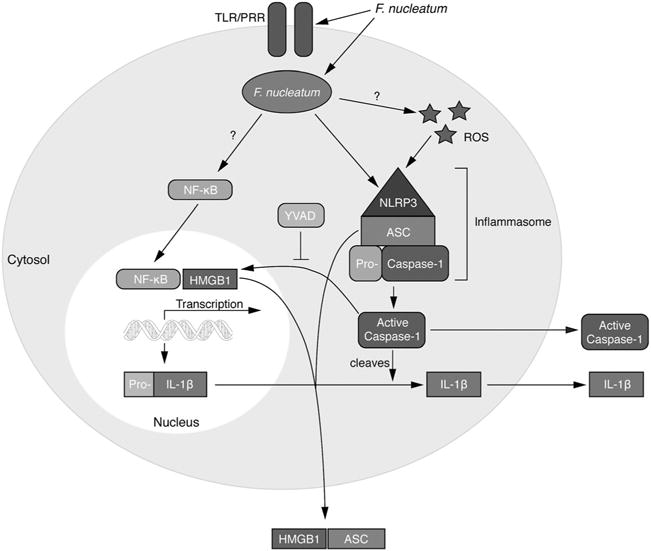
Model of pro-inflammatory cytokine expression and the inflammasome activation cascade during F. nucleatum infection F. nucleatum infection activates NF-κB, which translocates to the nucleus, where it stimulates expression of pro-inflammatory genes, including genes encoding pro-IL-1β. In addition, infection activates the NLRP3 inflammasome, which in turns activates caspase-1, resulting in processing and release of IL-1β. Inflammation is magnified further by release of the danger signal ASC, and caspase-1 secretion of HMGB1.
Experimental procedures
Bacterial culture
Fusobacterium nucleatum (ATCC 25586) was cultured under anaerobic condition at 37°C for 24 h in brain–heart infusion broth supplemented with yeast extract (5 mg ml−1), hemin (5 μg ml−1) and menadione (1 mg ml−1). Erythromycin (5 mg ml−1) was added to the media as a selective agent for growth, which was previously described (Zaborina et al., 1999). Bacteria were harvested by centrifugation at 6000 × g for 10 min at 4°C. The bacterial pellets were washed twice and resuspended with Dulbecco’s phosphate-buffered saline (PBS) containing calcium and magnesium (Life Technologies). Bacteria were quantified using a Klett-Summerson photoelectric colorimeter.
Primary and immortalized GECs
Healthy gingival tissue was obtained from healthy human subjects by oral surgery to produce primary GEC cultures as previously described (Yilmaz et al., 2004). With the consent of patients, gingival epithelial tissues were collected under the approval of the Institution Review Board of the University of Florida. GECs were cultured as monolayers in serum-free keratinocyte growth medium (Lonza) at 37°C in a 5% CO2 incubator. The human immortalized GEC cell line was obtained as previously described (Oda et al., 1996) and grown in defined keratinocyte serum-free media (Life Technologies) at 37°C in a 5% CO2 incubator. Prior to F. nucleatum infection, media was changed to Opti-MEM media (Life Technologies).
Cell viability assay
To measure cell viability of GECs after incubation in Opti-MEM media for 0, 2, 4, 6 and 8 h, we used trypan blue (TB) exclusion staining. GECs were collected at the indicated time points and resuspended with 1× PBS and 0.4% TB solution (Thermo Fisher Scientific) with a 1:1 ratio and mixed thoroughly. The resuspended mixture was then loaded onto a hemocytometer and counted under a light microscope after 5 min of incubation. Approximately 250 cells were counted using cell counter for each time point in duplicate.
Production of NLRP3-deficient GECs
The plasmids pMD.G, pCMVdelR8.91 and pLKO-shNL RP3 (Clone ID: TRCN0000431574) were purchased from National RNAi Core Facility at Academia Sinica of Taiwan. The plasmids pMD.G and pCMVdelR8.91 were used for the packaging of lentiviral particles as described (Huang et al., 2015). The shRNA sequence for NLRP3 was 5′-GAGACTCAGGAGTCGCAATTT-3′. The shRNA sequence 5′-TTGGCAACCGCTTTTTG-3′, which targets firefly luciferase mRNA, was used as the control.
Lentiviral particles were generated as described (Huang et al., 2015). HEK293T cells were cotransfected with lentiviral plasmid (pLKO-shNLRP3) and packaging plasmids (pMD.G and pCMVdelR8.91) by Arrest-In (Open Biosystems) according to manufacturer’s instructions. After 16 h of transfection, media containing the transfection reagents were removed and replenished with fresh media. After incubating for another 48 h, the media containing viral particles were centrifuged a 6000 × g for 5 min at 4°C, and the supernatants were collected for the infection studies. Infection of cells with lentiviral particles was carried out at 37°C in the presence of polybrene at 8 μg ml−1. Cells that stably express shRNA were obtained by culturing the infected cells in the presence of puromycin at 0.6 μg ml−1 for 2 weeks. Prior to F. nucleatum infection, media was changed to Opti-MEM media.
Infection of GECs with F. nucleatum
Primary and immortalized GECs were seeded on 6-well plates and grown until 75–80% confluence prior to bacterial infection or the indicated treatments. Cells were infected at a multiplicity of infection (MOI) of 25, 50, or 100. Prior to F. nucleatum infection, cells were treated with the inhibitors and incubated in a 5% CO2 incubator at 37°C. Fifty micromolar of caspase-1 inhibitor, YVAD (R&D Systems), was added for at least 15 min before infection (Cruz et al., 2007; Chen et al., 2012; Hung et al., 2013). Supernatant was collected at various time points for experiments described in the succeeding texts.
Western blot
Supernatant sample preparation
An equal number of cells were seeded per sample in order to ensure equal sample loading for all supernatants analysed by Western blot. Thus, 750 000 cells per well were seeded when using 6-well plates, or they were seeded to 75–80% confluence in cell-culture flasks. Proteins from cell-free culture supernatants were concentrated using trichloroacetic acid (TCA) (Koontz, 2014). One hundred percent TCA was added to supernatants for a final concentration of 20%. Samples were vortexed and incubated on ice for 30 min. After incubation, samples were centrifuged at 800 × g and supernatants were discarded. The pellets were washed twice with 100% cold acetone and allowed to air-dry. Pellets were resuspended with Pierce Lane Marker Reducing Sample Buffer (Thermo Scientific) and boiled at 95°C for 5 min. Samples were loaded and run on SDS-PAGE gels and transferred onto PVDF membranes. Membranes were blocked with 5% BSA for 1 h at RT, and then incubated with anti-TMS1 (Abcam), caspase-1 (Nathan), IL-1β (Abcam), anti-NLRP3 (Cell Signaling) or HMGB1 (Nathan) primary antibodies overnight at 4°C. After primary incubation, blots were washed and incubated with secondary goat anti-rabbit HRP (Millipore) for another hour. Finally, membranes were washed and exposed to Luminata Forte (Millipore) substrate. Images were acquired using ChemiDoc XRS+ system and analysed using NIH-ImageJ.
Cell lysate sample preparation
After removing supernatant, cells were washed once with 1X PBS (Dulbecco’s). M-PER Mammalian Protein Extract Reagent (Thermo Scientific) was added for 15 min to lyse cells and extract proteins from each sample. The protein concentration of the lysates was determined using Quick Start Bradford Assay (Bio-Rad) according to manufacturer’s instructions. Samples were resuspended with water and Pierce Lane Marker Reducing Sample Buffer and boiled at 95°C for 5 min. Samples were run and analysed as described in the preceding texts. After revealing the Western blot for experimental protein, the membranes were stripped using stripping buffer (Thermo Scientific) for 5 min and reprobed with GAPDH antibodies (Cell Signaling), which also confirmed equal loading of protein for the different lanes of the blot.
Measurement of IL-1β secretion by ELISA
GECs were seeded in 12-well plates and cultured until 75–80% confluence. The cells were then infected with F. nucleatum at an MOI of 100. Supernatants were collected at various time points and centrifuged at 800 × g for 5 min to remove cellular debris. Cell-free supernatants were used to test for IL-1β by ELISA assay. Human IL-1β was tested by ELISA (eBioscience) according to the manufacturer’s instructions.
RNA isolation, reverse transcription and quantitative PCR
Total RNA was isolated from GECs using TRIzol according to the manufacturer’s protocol (Invitrogen, Life Technologies). Taqman Reverse Transcription Reagents kit (Applied Biosystem) was used to amplify cDNA from 2 μg RNA. Real-time PCR RT-PCR and quantitative PCR (qPCR) were performed using 1/50 of the prepared cDNA with Mx3000P (Stratagene) and 25 μl of Briliant QPCR Master Mix (Stratagene). Amplification of cDNA was performed with the addition of sense and antisense primers at 200 nM concentration. Quantitative PCR was conducted at 95°C for 10 min, followed by 40 cycles at 95°C for 30 s, 55°C for 1 min and 72°C for 30 s. The IL-1β gene expression was normalized to GAPDH as instructed by the manufacturer (Stratagene) through comparative cycle threshold method as loading control. The following primers were used for IL-1β: 5′- AAATACCTGTGGCCTTGGGC -3′ and 5′- TTTGGGATCTACACTCTCCAGCT-3′. The primers used for GAPDH are 5′-TTAAAAGCAGCCCTGGTGAC-3′ and 5′-CTCTGCTCCTCCTGTTCGAC-3′.
Immunofluorescence microscopy
F. nucleatum-infected GECs were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton-X for 40 min. The cells were blocked with 2% BSA for 30 min before applying the primary antibody. Rabbit anti-human NF-κB antibody (Cell Signaling) was then added and incubated for 1 h followed by three washes with PBS containing 0.1% Triton-X. Cells were then incubated with goat anti-rabbit IgG antibody conjugated with red fluorescent Alexa Fluor 546 dye (Invitrogen) for 40 min and stained with DAPI (Millipore) for another 10 min. Finally, the cells were observed by wide-field fluorescence microscope (Leica) or laser scanning confocal microscope (Nikon TE-2000).
Immunofluorescence detection of F. nucleatum infection in GECs
Primary GECs were cultured in 4-well culture plates (Nunc) containing round glass inserts (Warner Instruments). Cells at approximately 80% confluence were infected with F. nucleatum at an MOI of 100 for 1 h. The cells were fixed in 10% neutral buffered formalin and permeabilized with 0.01% Triton X-100 in PBS. F. nucleatum infection was detected using a rabbit anti-F. nucleatum polyclonal antibody (Pacific Immunology) at 1:100 dilution and secondary Alexa Fluor 488-conjugated goat anti-rabbit antibody (Invitrogen) at 1:1000 dilution. Actin was labelled using phalloidin–tetramethylrhodamine B isothiocyanate (Sigma). The immunostained cells were mounted onto glass slides using Vectashield mounting medium (Fisher) containing DAPI stain and examined using a Leica confocal microscope and LAS AF (Leica) software.
Immunofluorescence examination of intracellular IL-1β and HMGB1
Primary GECs were cultured in 4-well culture plates containing round glass inserts. Cells were infected at a confluence of 70% with F. nucleatum at an MOI of 100 for 2, 6 and 8 h (for IL-1β) or for 3 and 8 h (for HMGB1). The effect of caspase-1 on HMGB1 translocation after 8 h F. nucleatum-infection was assessed using the caspase-1 inhibitor z-YVAD-fmk at a 50 μM final concentration for 30 min before infection. All cells were washed three times with PBS, fixed with 4% paraformaldehyde for 15 min and permeabilized using 0.01% Triton X-100 solution in PBS. IL-1β was detected using a FITC-conjugated mouse monoclonal antibody (R&D Systems). HMGB1 was detected using a rabbit polyclonal anti-human HMGB1 antibody and secondary Alexa Fluor® 488-conjugated goat anti-rabbit antibody (Invitrogen). F. nucleatum infection was detected using a rabbit anti-F. nucleatum polyclonal antibody (Pacific Immunology) and secondary Alexa Fluor® 594-conjugated goat anti-rabbit antibody (Invitrogen). All inserts were mounted onto glass slides using Vectashield mounting medium (Fisher) containing DAPI stain and examined using a Zeiss AxioImager fluorescence microscope connected to a cooled charge-coupled camera controlled by QCAPTURE software (Surrey). The intensity densities of cytoplasmic IL-1β and HMGB1 staining were measured using NIH-ImageJ. Intensity values at least 2 standard deviations above average were considered significant. Results indicate the average of measurements of at least 25 cells, originating from at least three separate replicate experiments.
Statistical analysis
Data are presented as mean ± standard deviation (SD). Standard t-tests were used to determine the statistical significance between groups, with P values < 0.05 considered as statistically significant.
Supplementary Material
Fig. S1. Time course of gingival epithelial cell (GEC) viability. GECs were stained with Trypan blue to assess viability with a hemocytometer after incubation in Opti-MEM media for 0, 2, 4, 6 and 8 h. Cell viability data shown represents three independent experiments counted in duplicates, giving similar results. Error bars represent the mean + SD.
Fig. S2. Immunofluorescence of NF-κB translocation time course. Localization of NF-κB in GECs induced by Fusobacterium nucleatum (MOI of 100) at 30 min, 1 and 2 h of infection. The images are representative of at least 100 cells from at three separate experiments. Bar represents 100 μm.
Fig. S3. Pro-IL-1β Western blot and loading control Pro-IL-1β protein (31 kDa) was extracted from cell lysates of F. nucleatum infected GECs. A time course of 2, 4, 6 and 8 h post-infection was analysed by Western blot with IL-1β antibodies. Blots were then stripped and probed with GAPDH antibodies as a loading control.
Fig. S4. Western blot confirmation of NLRP3 knockdown efficiency in GECs. Knockdown of NLRP3 expression was confirmed at the level of NLRP3 protein. Western blot analysis showed knockdown efficiency in comparison with controls. Actin was used as a loading control. The data shown represent three independent tests giving similar results.
Acknowledgments
This study was supported by intramural funds from the University of the Pacific, NIH/NIDCR grants R01DE019444, R01DE016593 and T90DE021990, and the Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro. We also would like to thank Dr. Nestor Oviedo (UC Merced) for technical support.
Footnotes
Supporting information
Additional Supporting Information may found in the online version of this article at the publisher’s web-site.
References
- Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasova KR, Yilmaz O. Prelude to oral microbes and chronic diseases: past, present and future. Microbes Infect. 2015;17:369–377. doi: 10.1016/j.micinf.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15:738–748. doi: 10.1038/ni.2919. [DOI] [PubMed] [Google Scholar]
- Bartold PM, Gully NJ, Zilm PS, Rogers AH. Identification of components in Fusobacterium nucleatum chemostat-culture supernatants that are potent inhibitors of human gingival fibroblast proliferation. J Periodontal Res. 1991;26:314–322. doi: 10.1111/j.1600-0765.1991.tb02069.x. [DOI] [PubMed] [Google Scholar]
- Bashir A, Miskeen AY, Bhat A, Fazili KM, Ganai BA. Fusobacterium nucleatum: an emerging bug in colorectal tumorigenesis. Eur J Cancer Prev. 2015;24:373–385. doi: 10.1097/CEJ.0000000000000116. [DOI] [PubMed] [Google Scholar]
- Cassel SL, Joly S, Sutterwala FS. The NLRP3 inflammasome: a sensor of immune danger signals. Semin Immunol. 2009;21:194–198. doi: 10.1016/j.smim.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Tsai SH, Lu CC, Hu ST, Wu TS, Huang TT, et al. Activation of an NLRP3 inflammasome restricts Mycobacterium kansasii infection. PLoS One. 2012;7:e36292. doi: 10.1371/journal.pone.0036292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CY, Kyritsis G, Graves DT, Amar S. Interleukin-1 and tumor necrosis factor activities partially account for calvarial bone resorption induced by local injection of lipopolysaccharide. Infect Immun. 1999;67:4231–4236. doi: 10.1128/iai.67.8.4231-4236.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282:2871–2879. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dommisch H, Chung WO, Rohani MG, Williams D, Rangarajan M, Curtis MA, Dale BA. Protease-activated receptor 2 mediates human beta-defensin 2 and CC chemokine ligand 20 mRNA expression in response to proteases secreted by Porphyromonas gingivalis. Infect Immun. 2007;75:4326–4333. doi: 10.1128/IAI.00455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol. 2014;15:727–737. doi: 10.1038/ni.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes MT, Campos PC, Oliveira FS, Corsetti PP, Bortoluci KR, Cunha LD, et al. Critical role of ASC inflammasomes and bacterial type IV secretion system in caspase-1 activation and host innate resistance to Brucella abortus infection. J Immunol. 2013;190:3629–3638. doi: 10.4049/jimmunol.1202817. [DOI] [PubMed] [Google Scholar]
- Han YW, Shi W, Huang GT, Kinder Haake S, Park NH, Kuramitsu H, Genco RJ. Interactions between periodontal bacteria and human oral epithelial cells: Fusobacterium nucleatum adheres to and invades epithelial cells. Infect Immun. 2000;68:3140–3146. doi: 10.1128/iai.68.6.3140-3146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harijith A, Ebenezer DL, Natarajan V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Frontiers in physiology. 2014;5:352. doi: 10.3389/fphys.2014.00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge P, Michalowicz B. Genetic predisposition to periodontitis in children and young adults. Periodontology. 2001;2000(26):113–134. doi: 10.1034/j.1600-0757.2001.2260106.x. [DOI] [PubMed] [Google Scholar]
- Huang PR, Hung SC, Pao CC, Wang TC. N-(1-pyrenyl) maleimide induces bak oligomerization and mitochondrial dysfunction in jurkat cells. Biomed Res Int. 2015;2015:798489. doi: 10.1155/2015/798489. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hung SC, Choi CH, Said-Sadier N, Johnson L, Atanasova KR, Sellami H, et al. P2X4 assembles with P2X7 and pannexin-1 in gingival epithelial cells and modulates ATP-induced reactive oxygen species production and inflammasome activation. PLoS One. 2013;8:e70210. doi: 10.1371/journal.pone.0070210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Jeng JH, Chan CP, Ho YS, Lan WH, Hsieh CC, Chang MC. Effects of butyrate and propionate on the adhesion, growth, cell cycle kinetics, and protein synthesis of cultured human gingival fibroblasts. J Periodontol. 1999;70:1435–1442. doi: 10.1902/jop.1999.70.12.1435. [DOI] [PubMed] [Google Scholar]
- Ji S, Shin JE, Kim YS, Oh JE, Min BM, Choi Y. Toll-like receptor 2 and NALP2 mediate induction of human beta-defensins by fusobacterium nucleatum in gingival epithelial cells. Infect Immun. 2009;77:1044–1052. doi: 10.1128/IAI.00449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L, Atanasova KR, Bui PQ, Lee J, Hung SC, Yilmaz O, Ojcius DM. Porphyromonas gingivalis attenuates ATP-mediated inflammasome activation and HMGB1 release through expression of a nucleoside-diphosphate kinase. Microbes Infect. 2015;17:369–377. doi: 10.1016/j.micinf.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Jo EK. NLRP3 inflammasome and host protection against bacterial infection. J Korean Med Sci. 2013;28:1415–1423. doi: 10.3346/jkms.2013.28.10.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koontz L. TCA precipitation. Methods Enzymol. 2014;541:3–10. doi: 10.1016/B978-0-12-420119-4.00001-X. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185:4385–4392. doi: 10.4049/jimmunol.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Kang IK, Chung CP, Choi SM. The subgingival microflora and gingival crevicular fluid cytokines in refractory periodontitis. J Clin Periodontol. 1995;22:885–890. doi: 10.1111/j.1600-051x.1995.tb01788.x. [DOI] [PubMed] [Google Scholar]
- Liu H, Redline RW, Han YW. Fusobacterium nucleatum induces fetal death in mice via stimulation of TLR4-mediated placental inflammatory response. J Immunol. 2007;179:2501–2508. doi: 10.4049/jimmunol.179.4.2501. [DOI] [PubMed] [Google Scholar]
- Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488:670–674. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda D, Bigler L, Lee P, Blanton R. HPV immortalization of human oral epithelial cells: a model for carcinogenesis. Exp Cell Res. 1996;226:164–169. doi: 10.1006/excr.1996.0215. [DOI] [PubMed] [Google Scholar]
- Oliver RC, Brown LJ, Loe H. Periodontal diseases in the United States population. J Periodontol. 1998;69:269–278. doi: 10.1902/jop.1998.69.2.269. [DOI] [PubMed] [Google Scholar]
- Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–1820. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- Pilot T. The periodontal disease problem. A comparison between industrialised and developing countries. Int Dent J. 1998;48:221–232. doi: 10.1111/j.1875-595x.1998.tb00710.x. [DOI] [PubMed] [Google Scholar]
- Qin S, Wang H, Yuan R, Li H, Ochani M, Ochani K, et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med. 2006;203:1637–1642. doi: 10.1084/jem.20052203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards D. Oral diseases affect some 3.9 billion people. Evid Based Dent. 2013;14:35. doi: 10.1038/sj.ebd.6400925. [DOI] [PubMed] [Google Scholar]
- Richards D. Review finds that severe periodontitis affects 11% of the world population. Evid Based Dent. 2014;15:70–71. doi: 10.1038/sj.ebd.6401037. [DOI] [PubMed] [Google Scholar]
- Said-Sadier N, Ojcius DM. Alarmins, inflammasomes and immunity. Biomed J. 2012;35:437–449. doi: 10.4103/2319-4170.104408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini R, Marawar PP, Shete S, Saini S. Periodontitis, a true infection. J Glob Infect Dis. 2009;1:149–150. doi: 10.4103/0974-777X.56251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana P, Martel J, Lai HC, Perfettini JL, Kanellopoulos J, Young JD, et al. Is the inflammasome relevant for epithelial cell function? Microbes Infect. 2015 doi: 10.1016/j.micinf.2015.10.007. [DOI] [PubMed] [Google Scholar]
- Signat B, Roques C, Poulet P, Duffaut D. Fusobacterium nucleatum in periodontal health and disease. Curr Issues Mol Biol. 2011;13:25–36. [PubMed] [Google Scholar]
- Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem. 2002;277:21119–21122. doi: 10.1074/jbc.C200179200. [DOI] [PubMed] [Google Scholar]
- Strauss J, Kaplan GG, Beck PL, Rioux K, Panaccione R, Devinney R, et al. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm Bowel Dis. 2011;17:1971–1978. doi: 10.1002/ibd.21606. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- Taxman DJ, Swanson KV, Broglie PM, Wen H, Holley-Guthrie E, Huang MT, et al. Porphyromonas gingivalis mediates inflammasome repression in polymicrobial cultures through a novel mechanism involving reduced endocytosis. J Biol Chem. 2012;287:32791–32799. doi: 10.1074/jbc.M112.401737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nature reviews. Immunology. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- Tsung A, Tohme S, Billiar TR. High-mobility group box-1 in sterile inflammation. J Intern Med. 2014;276:425–443. doi: 10.1111/joim.12276. [DOI] [PubMed] [Google Scholar]
- Vande Walle L, Kanneganti TD, Lamkanfi M. HMGB1 release by inflammasomes. Virulence. 2011;2:162–165. doi: 10.4161/viru.2.2.15480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velsko IM, Chukkapalli SS, Rivera-Kweh MF, Chen H, Zheng DH, Bhattacharyya I, et al. Fusobacterium nucleatum alters atherosclerosis risk factors and enhances inflammatory markers with an atheroprotective immune response in ApoE(null) mice. Plos One. 2015;10:e0129795. doi: 10.1371/journal.pone.0129795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ, et al. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol. 2009;183:2008–2015. doi: 10.4049/jimmunol.0900138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. 2013;93:865–873. doi: 10.1189/jlb.1212662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O, Jungas T, Verbeke P, Ojcius DM. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect Immun. 2004;72:3743–3751. doi: 10.1128/IAI.72.7.3743-3751.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O, Sater AA, Yao L, Koutouzis T, Pettengill M, Ojcius DM. ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cell Microbiol. 2010;12:188–198. doi: 10.1111/j.1462-5822.2009.01390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Chung WO. Epigenetic regulation of human beta-defensin 2 and CC chemokine ligand 20 expression in gingival epithelial cells in response to oral bacteria. Mucosal Immunol. 2011;4:409–419. doi: 10.1038/mi.2010.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborina O, Li X, Cheng G, Kapatral V, Chakrabarty AM. Secretion of ATP-utilizing enzymes, nucleoside diphosphate kinase and ATPase, by Mycobacterium bovis BCG: sequestration of ATP from macrophage P2Z receptors? Mol Microbiol. 1999;31:1333–1343. doi: 10.1046/j.1365-2958.1999.01240.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Time course of gingival epithelial cell (GEC) viability. GECs were stained with Trypan blue to assess viability with a hemocytometer after incubation in Opti-MEM media for 0, 2, 4, 6 and 8 h. Cell viability data shown represents three independent experiments counted in duplicates, giving similar results. Error bars represent the mean + SD.
Fig. S2. Immunofluorescence of NF-κB translocation time course. Localization of NF-κB in GECs induced by Fusobacterium nucleatum (MOI of 100) at 30 min, 1 and 2 h of infection. The images are representative of at least 100 cells from at three separate experiments. Bar represents 100 μm.
Fig. S3. Pro-IL-1β Western blot and loading control Pro-IL-1β protein (31 kDa) was extracted from cell lysates of F. nucleatum infected GECs. A time course of 2, 4, 6 and 8 h post-infection was analysed by Western blot with IL-1β antibodies. Blots were then stripped and probed with GAPDH antibodies as a loading control.
Fig. S4. Western blot confirmation of NLRP3 knockdown efficiency in GECs. Knockdown of NLRP3 expression was confirmed at the level of NLRP3 protein. Western blot analysis showed knockdown efficiency in comparison with controls. Actin was used as a loading control. The data shown represent three independent tests giving similar results.