

Abstract
Receptor-interacting protein 1(RIPK1) has an essential role in the signaling pathways triggered by death receptors through activation of NF-κB and regulation of caspase-dependent apoptosis and RIPK3/mixed lineage kinase domain-like protein(MLKL)-mediated necroptosis. We examined the effect of RIPK1 antisense knockdown on the immune-mediated liver injury in C57BL/6 mice caused by α-galactosylceramide (αGalCer), a specific activator for invariant NKT cells. We found that knockdown of RIPK1 markedly exacerbated αGalCer-mediated liver injury and induced lethality. This was associated with increased hepatic inflammation and massive apoptotic death of hepatocytes as indicated by TUNEL staining and caspase-3 activation. Pretreatment with either zVAD.fmk, a pan-caspase inhibitor, or neutralizing Abs against TNF, almost completely protected against the exacerbated liver injury and lethality. Primary hepatocytes isolated from RIPK1 knockdown mice were sensitized to TNF-induced cell death that was completely inhibited by adding zVAD.fmk. Unexpectedly, the exacerbated liver injury was not due to impaired hepatic NF-κB activation in terms of IκBα phosphorylation and degradation in both in vivo and in vitro studies. Lack of RIPK1 kinase activity by either pretreatment with necrostatin-1, a RIPK1 kinase inhibitor, or in the RIPK1 kinase-dead knock-in (RIPK1D138N) mice, did not exacerbate αGalCer-mediated liver injury. Furthermore, both RIPK3 and MLKL knockout mice behaved similarly as wild type control mice in response to αGalCer with or without knockdown of RIPK1, excluding a switch to RIPK3/MLKL-mediated necroptosis. Our findings for the first time revealed a critical kinase-independent platform role of RIPK1 in protecting against TNF/caspase-dependent apoptosis of hepatocytes in immune-mediated liver injury.
Keywords: Natural killer T cell, TNF, RIPK3, MLKL, animal model
Introduction
Receptor-interacting protein kinase 1(RIPK1) is the founding member of serine/threonine kinase family that has an essential role in regulating caspase-dependent apoptosis and RIPK3/MLKL-dependent necroptosis as well as in promoting cell survival and inflammation by serving as a platform for activation of the canonical NF-κB pathway (1–4). Complete RIPK1 deficiency (RIPK1−/−) in mice results in perinatal death characterized by multi-organ cell death and systemic inflammation (5, 6). Cell death in multiple organs including the liver is the most striking feature of histological findings in RIPK1−/− mice and apoptosis (cleaved caspase-3) is increased in the liver (5, 6). Although the mechanisms by which RIPK1 prevent perinatal lethality is complex, the recent studies of genetic rescue of the perinatal death in RIPK1−/− mice have demonstrated that combined loss of caspase-8 and RIPK3 protects against the perinatal lethality and systemic inflammation, indicating that RIPK1 can function as an inhibitor to suppress both caspase-mediated apoptosis and RIPK3-mediated necroptosis (6–8). In support of this, the conditional deletion of RIPK1 in intestinal epithelial cells (IECs) was reported to result in apoptosis of IECs and intestinal inflammation (9, 10), suggesting that RIPK1 maintains epithelial homeostasis by inhibiting apoptosis in the intestine (9).
Hepatocyte death, either apoptosis or necrosis, has been critically implicated in liver pathology. Necroptosis, a recently defined necrosis regulated by RIPK1/RIPK3/MLKL signaling pathway (1–3, 11), has also been suggested to account for hepatocyte death in liver diseases including alcoholic liver disease (12), drug-induced liver injury (13, 14) and immune-mediated liver injury (15, 16). In the studies of the perinatal lethality in RIPK1−/− mice, the liver is amongst the multi-organs exhibiting cell death and inflammation (5, 6), suggesting a possible role of RIPK1 in regulating apoptosis and/or necroptosis of hepatocytes. However, in more recent studies using inducible conditional knockout of RIPK1 in adult mice (17), the liver appears to be spared from cell death and inflammation in spite of the fact that acute ablation of RIPK1 in adult mice results in intestinal epithelial and hematopoietic cell death. Thusfar little is known about the physiological role of RIPK1 in regulating hepatocyte death, either apoptosis or necroptosis, triggered by death receptors such as TNF and Fas in immune-mediated liver injury.
CD1d-restricted invariant natural killer T (iNKT) cells are a specialized subset of T cells that recognize lipid antigens presented by CD1d molecules on antigen-presenting cells (18). iNKT cells are enriched in the liver, accounting for up to 40% of hepatic lymphocytes in mice, and have been implicated in the pathophysiology of a variety of liver diseases including viral and autoimmune hepatitis, alcoholic liver disease and non-alcoholic fatty liver disease (19–22). Direct activation of hepatic iNKT cells by α-galactosylceramide (αGalCer), a specific high-affinity ligand for iNKT cells, results in rapid release of large quantities of various cytokines including TNF and IFNγ from iNKT cells, promoting subsequent activation and infiltration of NK cells, macrophages, and neutrophils, all of which lead to a mild immune-mediated liver injury (18, 19, 23, 24). In this study, we employed this well-established animal model of hepatitis to explore the function of RIPK1 in regulating the immune-mediated liver injury. After RIPK1 antisense oligonucleotide (ASO) treatment, we found that knockdown of RIPK1 markedly exacerbates αGalCer-mediated liver injury and induces lethality associated with increased hepatic inflammation. We provide evidence that the exacerbation of liver injury is due to TNF-and caspase-dependent massive apoptosis of hepatocytes as neutralization TNF or inhibition of caspases by pan-caspase inhibitor zVAD.fmk confers almost complete protection. We further show that RIPK3/MLKL-mediated necroptosis does not contribute to the exacerbation of liver injury because either RIPK3−/− or MLKL−/− mice exhibit exacerbated liver injury and lethality and respond similarly as wild type (WT) control mice. Unexpectedly, the exacerbation of liver injury is not due to impairment of pro-survival NF-κB signaling pathway. Moreover, lack of RIPK1 kinase activity in RIPK1D168N mice does not exacerbate αGalCer-mediated liver injury, indicating that it is the platform function of RIPK1 protein and not the kinase activity that regulates TNF/caspase-dependent apoptosis of hepatocytes in response to iNKT cell activation.
Materials and Methods
Reagents
Abs to RIPK1, Caspase-3, IκBα, p-IκBα (Ser32/36), β-Actin were from Cell Signaling (Danvers, MA). Anti-c-FLIP mAb (Dave-2) from Adipogen Corporation (San Diego, CA), cIAP1 mAb (1E1-1-10) from Enzo Life Sciences (Farmingdale, NY). Anti-RIPK3 was provided by Genentech. Purified TNFα mAb from BioLegend (San Diego, CA). Anti-Fas Ab (Jo2) from BD Biosciences (San Jose, CA). Necrostatin-1 (Nec-1) and inactive Nec-1 from Calbiochem (San Diego, CA). α-galactosylceramide (αGalCer) from Funakoshi Co. (Tokyo, Japan). Pan-caspase inhibitor zVAD.fmk (zVAD) from R&D Systems (Minneapolis, MN). Lipopolysaccharide (LPS) and Concanavalin A (ConA) from Sigma-Aldrich. RIPK1 and scrambled antisense oligonucleotide (ASO) was provided by Ionis Pharmaceuticals (Carlsbad, CA).
Animals, Treatments, and Assays for Serum ALT
Pathogen-free male C57BL/6J mice, 6 to 8 weeks of age, were obtained from the Jackson Laboratory (Bar Harbor, ME). MLKL−/− mice (on a C57BL/6J background) were provided by Professor Warren Alexander at Walter and Eliza Hall Institute of Medical Research (Parkville, Australia). The RIPK1 kinase-dead knock-in (RIPK1D138N) mice and RIPK3−/− mice on a C57BL/6N background were provided by Dr. Vishva Dixit at Genentech, and male C57/BL6N mice (6–8 weeks old) were obtained from Harlan Bioproducts for Science Inc. (Indianapolis, IN) as controls. RIPK1 and scrambled ASO was dissolved in PBS, and the animals were injected i.p. with ASO at the dose of 50 mg/kg every other day for a total of 5 injections as previously described (25). The additional treatments were started the next day after the last ASO injection. The highly efficiency of hepatic RIPK1 knockdown was confirmed by Western blot analysis and mRNA levels of liver tissues. We also confirmed that either scrambled or RIPK1 ASO treatment alone are not hepatotoxic evidenced by normal serum ALT and liver histology. αGalCer was stored as a stock solution of 200μg/ml in vehicle (0.5% w/v polysorbate-20), and diluted in PBS before i.p. injection of 4μg per mouse in a total volume of 200 μl. For LPS-induced liver injury, non-lethal dose of LPS (3mg/kg) was injected i.p. For Fas-mediated liver injury, 4μg of anti-Fas (jo2) Ab was injected i.p. For ConA-induced liver injury, low-toxic dose of ConA (10mg/kg) was injected via tail vein. For inhibition of caspases, zVAD (10mg/kg) was injected i.p. 15min prior to αGalCer or ConA treatment. For neutralization of TNF, 0.6 mg per mouse of anti-TNFα mAb was administered i.p. 15 min prior to αGalCer. Serum ALT levels were measured with ALT reagents from Teco Diagnostics (Anaheim, CA). Animal experiments were carried out in accordance with guidelines from the USC Institutional Animal Care and Use Committee.
Preparation of Liver Extracts and Western Blot Analyses
Total liver lysates or whole cell lysates of cultured primary mouse hepatocytes were prepared in RIPA buffer with complete protease and phosphatase inhibitor cocktail (Roche) as previously described (26). Aliquots of protein extracts were subjected to 12% SDS-PAGE. After electrophoresis, proteins were transferred to a PVDF membrane (Amersham Biosciences, UK) and then incubated with the desired primary and secondary Abs. Finally, the proteins were detected by Luminol ECL reagent (Thermo Scientific). Densitometry was done using Image J software. All gels and densitometry shown are representative of at least three experiments otherwise indicated.
Hepatocytes Isolation, Culture and cell death assay
Primary mouse hepatocytes were isolated and cultured as described previously (27). Three hours after plating of isolated hepatocytes, TNF (20 ng/ml) with or without pan-caspase inhibitor zVAD (10 μM) was added. After 16hr, cells were double-stained with Hoechst 33258 (8 μg/ml; Invitrogen) and Sytox Green (1 μmol/L; Invitrogen). Quantitation of cell death was performed by counting a minimum of 300 cells in different fields.
Histology and TUNEL staining
For histology, liver tissue was fixed in 10% neutral buffered formalin and sections were stained with H&E to determine morphologic changes. TUNEL staining was performed by using fluorescent detection kit (Roche Diagnostics) following the manufacturer’s instruction.
RNA isolation and real-time PCR (qPCR)
Liver tissues were homogenized in TRI reagent (Sigma Aldrich) and RNA was extracted using RNeasy Plus Mini Kit (Qiagen). Total RNA (2 μg) was reverse-transcribed in a 20μL reaction mixture containing 4U Omniscript Reverse Transcriptase (Qiagen) and oligo dT(12–18mer) primer (Life Technologies). The following sense and antisense primers, respectively, were used in the PCR reaction: cIAP1 sense 5′-GGCCACCTAGTGTTCCTGTT-3′ and antisense 5′-CAACATCTCAAGCCACCATC-3′, and c-FLIP sense 5′-TCAGGGTTGTCTCGTCAGAG-3′ and antisense 5′-CAGAGACCACCAGCTACGAA-3′. qPCR for cIAP1 and c-FLIP and qPCR array analyses of NF-κB target genes (RT2 Profiler PCR Arrays, Qiagen) were performed with 7900HT Sequence Detection System (Life Technologies). GAPDH was analyzed as an internal control. Data are expressed as fold changes.
Bead-based multiplex immunoassays for cytokine analysis
Concentrations of a panel of cytokines and chemokines in mouse serum samples were determined by the AimPlex® Mouse 18-Plex Assay Kit (YSL Bioprocess Development Co., Pomona, CA) according to the manufacturer’s instructions. In brief, 15 μL of serum sample was diluted with 30 μL of assay buffer in each well of a 96-well filter plate and incubated with capture antibody conjugated AimPlex beads for 1 hr. After washing the beads 3 times using the AimPlex EZPrep Filter Plate Washer, a cocktail of biotinylated detection antibodies was applied and followed by an addition of PE-conjugated streptavidin. Data were acquired on a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and analyzed with FCAP Array v3.0 (SoftFlow Hungary Ltd., Hungary).
Preparation of hepatic leukocytes and flow cytometric Analysis
Hepatic leukocytes were isolated and prepared as described previously (20, 26). For flow cytometric analysis, 106 cells were first incubated for 10 min with Fcγ receptor blocker (anti-mouse CD16/32, Clone 2.4G2; Tonbo Biosciences, San Diego, CA), then stained with mAbs at a concentration of 0.5–1μg per 100μl PBS containing 0.2% BSA (Boehringer Mannheim, Indianapolis, IN) and 0.02% sodium azide for 30 min on ice. The following antibodies were used: FITC-conjugated anti-CD3 (145-2C11), anti-Ly6C (HK1.4); PE-conjugated anti NK1.1 (PK136), anti-F4/80 (BM8.1); PerCP-conjugated anti-Ly6G (1A8), anti-CD19 (1D3); APC-Cy7-conjugated anti CD45 (Biolegend); PE-Cy7-conjugated anti CD11b (M1-70) (BD Biosciences). Flow cytometric analysis was performed on a FACSVerse (BD Biosciences).
Statistical Analysis
Data are expressed as mean±SEM. Group comparisons were performed using Student’s t test or analysis of variance. The log-rank test was used for mouse survival. Differences were considered statistically significant at p<0.05.
Results
Knockdown of RIPK1 markedly exacerbates aGalCer-mediated liver injury and induces lethality
First, we examined the effect of RIPK1 knockdown on the liver injury induced by i.p. injection of αGalCer. In control mice pretreated with scrambled control ASO (Cont ASO), αGalCer caused a mild to moderate liver injury evidenced by increased serum ALT values starting at 6hr, which peaked around 12~24hr (Fig. 1A), without any mortality (Fig. 1B). In striking contrast, when hepatic RIPK1 was knocked down by RIPK1 ASO pretreatment, αGalCer markedly increased serum ALT at 6hr and induced ~80% mortality by 8hr (Fig. 1A, B). For the remaining RIPK1 ASO mice surviving to 12 and 24hr, the serum ALT levels remained much higher compared to control mice (Fig. 1A). RIPK1 knockdown caused massive hepatocyte death and hemorrhage in the liver at 6 and 24hr compared to small patchy necrotic foci with inflammatory cell infiltration in the H&E staining of liver sections from control ASO treated mice at 6 and 24hr (Fig. 1C). Accompanying the exacerbated liver injury, RIPK1 knockdown also significantly increased serum levels of pro-inflammatory cytokines and chemokines including TNF and IFNγ at 6hr after αGalCer and caused highly increased inflammatory cell infiltration into the liver compared to control mice (Supplemental Fig. 1). However, no differences of the serum levels of cytokines/chemokines were observed at 3hr after αGalCer (Supplemental Fig. 1A), suggesting that RIPK1 knockdown has no direct effect on the rapid activation of iNKT cells by αGalCer. We confirmed that RIPK1 ASO efficiently knocked down hepatic RIPK1 by Western blotting (Fig. 1D) and qPCR (not shown). Together, these results indicate that the lack of RIPK1 proteins in the liver markedly exacerbates αGalCer-mediated liver injury.
FIGURE 1.

Knockdown of RIPK1 markedly exacerbates α-galactosylceramide (αGalCer)-induced liver injury and causes mortality. Groups of mice were pretreated with scrambled (Cont ASO) or RIPK1 ASO every other day for 5 times to silence RIPK1 prior to αGalCer (4μg/mouse i.p.). (A) Serum ALT values at indicated times after αGalCer. *p<0.05 vs. scrambled controls at 8hr; representative of three separate experiments. (B) Mouse survival by 24 hr after αGalCer. *p<0.05 vs. scrambled controls. (C) H&E staining of liver sections at indicated times after αGalCer (x40). (D) Western blotting of whole liver lysates for RIPK1 at indicated times after αGalCer.
Knockdown of RIPK1 causes massive caspase-dependent apoptosis of hepatocytes in response to aGalCer
Apoptotic death of hepatocytes is often associated with immune-mediated hepatitis. To determine whether apoptosis contributes to the exacerbated liver injury in RIPK1 ASO-treated mice in response to αGalCer, we performed TUNEL staining of liver sections at 6hr after αGalCer. As shown in Fig. 2A, αGalCer treatment drastically increased TUNEL positive hepatocytes in RIPK1 ASO mice compared with that in control mice. Furthermore, liver extracts from RIPK1 ASO treated mice displayed increased protein levels of cleaved caspase-3 at 3hr and 6hr after αGalCer while no cleaved caspase-3 was detected in the liver of control mice (Fig. 2B). These data suggest an involvement of caspase-mediated apoptosis. To further validate the role of apoptosis, pretreatment with zVAD afforded almost complete protection against the exacerbated αGalCer-mediated liver injury in RIPK1 ASO mice in terms of serum ALT levels and histologic liver injury (Fig. 2C, D). By 8hr after αGalCer, only ~20% of the RIPK1 ASO mice without zVAD pretreatment survived, whereas all of the RIPK1 ASO mice with pretreatment of zVAD were alive (Fig. 2E). Thus these results indicate that the exacerbated αGalCer-mediated liver injury and the lethality in RIPK1 ASO mice are due to caspase-dependent massive apoptotic death of hepatocytes.
FIGURE 2.

Knockdown of RIPK1 causes massive hepatic apoptosis in response to αGalCer. Animals were pretreated with Cont ASO or RIPK1 ASO to silence RIPK1 prior to αGalCer (4μg/mouse i.p.). (A) TUNEL staining of liver sections at 6hr after αGalCer. DAPI was used for nucleus staining, representative of images with similar results. (B) Western blotting for hepatic caspase-3 and cleaved caspase-3 in whole liver lysates at 6hr after αGalCer. Representative of three separate experiments.
(C–E) Pre-treatment by pan-caspase inhibitor zVAD almost completely protects against the exacerbated liver injury and lethality in RIPK1 ASO+αGalCer-treated mice. RIPK1 ASO were either pre-treated with vehicle or zVAD (10mg/kg i.p.) 15min prior to αGalCer treatment (4μg/mouse i.p.). (C) Serum ALT measured at 6hr after αGalCer (n=5 per group, p<0.05). (D) Representative images of H&E staining of liver sections at 6hr after αGalCer. (E) Mouse survival by 8 hr after αGalCer (n=6).
Knockdown of RIPK1 sensitizes to TNF-dependent apoptosis
Activation of iNKT cells by αGalCer rapidly releases the pro-inflammatory cytokine TNF, which plays a critical role in αGalCer-mediated liver injury (20, 24). To determine the role of TNF in the exacerbation of liver injury in RIPK1 ASO mice, animals were pre-treated with neutralizing antibodies against TNF. As indicated in Fig. 3, neutralization of TNF conferred almost complete protection against the exacerbated liver injury caused by αGalCer in RIPK1 ASO mice evidenced by dramatic reduction of serum ALT and histologic liver injury (Fig. 3A, B), and complete protection against lethality (Fig. 3C). Next we performed in vitro studies to examine the direct role of TNF in primary mouse hepatocytes. As shown in Fig. 3D, hepatocytes isolated from RIPK1 ASO mice were sensitized to TNF-mediated cell death, displaying a significantly increased cell death indicated by Sytox green staining. Co-treatment with zVAD almost completely abrogated the cell death. Thus, RIPK1 knockdown sensitized to TNF induced caspase-dependent apoptotic death of hepatocytes in vitro or after αGalCer in vivo. Although increased serum TNF levels due to RIPK1 knockdown in iNKT or other inflammatory cells may be contributing to the enhanced apoptosis of hepatocytes, our in vitro findings indicate that the hepatocytes in the absence of RIPK1 become intrinsically more susceptible to TNF. In addition to TNF, Fas/FasL pathway has been implicated in αGalCer-mediated liver injury (20). To examine whether knockdown of RIPK1 sensitizes to Fas-mediated apoptotic death of hepatocytes, animals were injected with a sub-lethal dose of anti-Fas (Jo2) to induce acute liver injury (28). The liver injury (ALT and histology) was comparable between scrambled ASO control mice and RIPK1 ASO mice in response to Jo2 treatment (Supplemental Fig. 2), thus excluding the role of Fas-mediated apoptosis in the exacerbated cell death in response to αGalCer.
FIGURE 3.

Pre-treatment by neutralizing Abs against TNFα almost completely protects against the exacerbated liver injury and lethality in RIPK1 ASO+αGalCer-treated mice. RIPK1 ASO treated mice were either pre-treated with vehicle or anti-TNFα Ab (600μg/mouse, i.p.) 15min prior to αGalCer treatment (4μg/mouse i.p.). (A) Serum ALT values at 6hr after αGalCer (n=4–6, p<0.05). (B) Representative H&E stained liver sections at 6hr after αGalCer. (C) Mouse survival by 8 hours after αGalCer (n=6). (D–E) RIPK1 knockdown sensitizes hepatocytes to TNF-induced apoptotic cell death. Primary mouse hepatocytes isolated from Cont ASO or RIPK1 ASO-treated mice were treated with TNFα (20 ng/ml) for 16hr in the absence or presence of pan-caspase inhibitor zVAD (10 μM). (D) Representative of microscopic images of primary hepatocytes stained by Hoechst and Sytox green. (E) The percentages of cell death at 16hr. *p<0.05 vs hepatocytes from Cont ASO-treated mouse.
Knockdown of RIPK1 does not impair NF-κB activation in the liver and hepatocytes
Ubiquitination of RIPK1 is believed to be required for TNF-mediated pro-survival signaling (3, 5, 29). Therefore, it is conceivable that the exacerbated apoptotic hepatocyte death in RIPK1 ASO treated mice may simply be attributed to impairment of NF-κB activation. To this end, we examined the activity of NF-κB by Western blot analysis of phosphorylated IκBα and total IκBα proteins in the liver and primary mouse hepatocytes. Surprisingly, RIPK1 ASO mice displayed comparable levels of hepatic phosphorylation and degradation of IκBα proteins to that observed in control mice at 1, 3 and 6hr in response to αGalCer (Fig. 4A). Furthermore, primary mouse hepatocytes from RIPK1 ASO treated mice also exhibited a similar time course of phosphorylation and degradation of IκBα proteins compared to that from control mice in response to TNF stimulation in vitro (Fig. 4C). Moreover, we examined mRNA expressions of a group of downstream genes regulated by NF-κB activation in both the liver tissues and isolated primary hepatocytes at 3hr after αGalCer treatment (Supplemental Fig. 3). The results showed that these genes, including IκBα, c-FLIP, cIAP1, TNF, IFNγ, iNOS and SOD2, were significantly induced by αGalCer treatment not only in the liver tissues, but also in the primary hepatocytes from both scrambled control and RIPK1 ASO treated mice. Together, these results indicate that NF-κB activation is intact when RIPK1 is knocked down in the liver and hepatocytes, suggesting that the exacerbated liver injury and hepatocyte death in RIPK1 ASO treated mice in response to TNF are not due to the impairment of NF-κB signaling.
FIGURE 4.

Knockdown of RIPK1 has no effect on hepatic NF-κB activation and causes degradation of c-FLIP and cIAP1. (A) Western blot for detection of phosphorylated IκBa (p-IκBα), total IκBα, RIPK1, c-FLIP and cIAP1 in the liver tissues from Cont ASO and RIPK1 ASO mice at indicated times after αGalCer. (B) Densitometry of Western blotting bands for hepatic c-FLIP and cIAP1 from three separate experiments. *P<0.05. (C) Western blot for detection of phosphorylated IκBa (p-IκBα), total IκBα and RIPK1 in primary hepatocytes isolated from Cont ASO and RIPK1 ASO mice. Primary hepatocytes were cultured with TNFα (20 ng/ml) for indicated times before subjected to whole cell lysates protein extracts. Representative of three separate experiments. (D) Western blot for detection of c-FLIP and cIAP1 in primary hepatocytes isolated from Cont ASO and RIPK1 ASO mice and cultured with TNF (20ng/ml) for indicated times. Representative of two separate experiments with similar results.
RIPK1 has been shown to promote cell survival in TNF-stimulated cells by stabilizing anti-apoptotic proteins such as cIAP1 and c-FLIP (9, 29, 30). c-FLIP-deficient hepatocytes have been demonstrated to be highly susceptible to death receptor (TNFR1, Fas, and TRAIL)-induced apoptosis (31, 32). To this end, we examined the protein levels of hepatic c-FLIP and cIAP1 by Western blot analysis. As shown in Fig. 4A, B, we found that the induction of hepatic protein levels of c-FLIP and cIAP1 was decreased after αGalCer treatment in RIPK1 ASO treated mice compared to control mice. We further examined the protein levels of c-FLIP and cIAP1 in primary hepatocytes in response to TNF stimulation in vitro. As shown in Fig. 4D, the induction of cIAP1 and c-FLIP at 3 and 6hr after TNF was decreased in primary hepatocytes from RIPK1 ASO treated mice compared to that of control mice. Coupled with no impairment of NF-κB induced increase of mRNA expressions of c-FLIP and cIAP1 in both liver tissues and primary hepatocytes, these results suggest that degradation of c-FLIP and cIAP1 in the hepatocytes of RIPK1 ASO treated mice in the absence of RIPK1 may contribute to the massive apoptosis of hepatocytes and the exacerbated liver injury.
RIPK1 kinase activity is dispensable for the exacerbation of aGalcer-mediated liver injury
Next we used transgenic RIPK1 kinase-dead knock-in mice (RIPK1D138N) (33) to examine whether the exacerbation of αGalCer-mediated liver injury in RIPK1 ASO mice is due to lack of the platform function or the kinase activity of RIPK1. As shown in Fig. 5, RIPK1D138N mice lacking RIPK1 kinase activity exhibited similar response to αGalCer as WT control mice (Fig. 5A, B). The lack of exacerbation of αGalCer-mediated liver injury in RIPK1D138N mice suggests that the scaffolding function of RIPK1 plays a critical role in regulating hepatocyte death. In support of this hypothesis, pretreatment with Nec-1, a specific RIPK1 kinase inhibitor, showed no exacerbation of αGalCer-mediated liver injury in WT mice (Supplemental Fig. 4).
FIGURE 5.

Loss of RIPK1 kinase activity does not exacerbate αGalCer-mediated liver injury in the RIPK1 kinase-dead knock-in (RIPK1D138N) mice. (A) Serum ALT values at 0 and 8hr after αGalCer treatment (4μg/mouse, i.p.) (n=5, p>0.05 RIPK1D138N vs. WT ). (B) representative of H & E staining of liver sections (x40) at 0 and 8hr after αGalCer.
RIPK3/MLKL dependent necroptosis is not involved in the exacerbated liver injury and lethality in RIPK1 ASO mice in response to aGalCer
RIPK1 is known to regulate both caspase-dependent apoptosis and RIPK3/MLKL-mediated necroptosis (1–8). To exclude the possibility that necroptosis is implicated in the exacerbation of αGalCer-mediated liver injury in RIPK1 ASO treated mice, we performed a series of experiments by using RIPK3−/− and MLKL−/− mice. In Fig. 6, RIPK3−/− mice behaved similarly as WT control mice, exhibiting a mild to moderate liver injury in response to αGalCer alone. However, when RIPK1 was knocked down in RIPK3−/− mice, αGalCer still induced markedly exacerbated liver injury and high mortality by 8hr (Fig. 6). Furthermore, MLKL−/− mice also behaved similarly to WT mice and RIPK3−/−mice, exhibiting markedly exacerbated αGalCer-mediated liver injury and lethality after RIPK1 ASO (Fig. 7). Together these results showed that loss of either RIPK3 or the necroptosis executioner MLKL did not protect against the markedly exacerbated αGalCer-mediated liver injury and lethality after RIPK1 ASO treatment, excluding the involvement of RIPK3/MLKL-mediated necroptosis of hepatocytes as the mechanism for increased injury. Furthermore, as shown above, inhibition of caspases with zVAD treatment did not cause a switch to necroptosis (Fig. 2 and Fig. 3).
FIGURE 6.
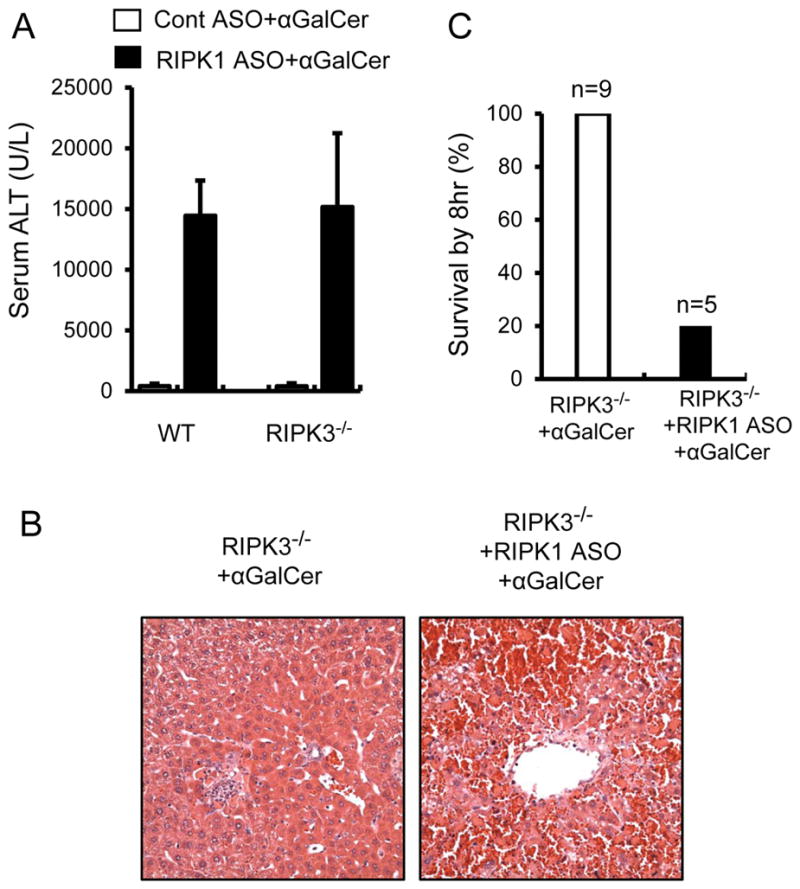
Similar to WT control mice, knockdown of RIPK1 markedly increases αGalCer-mediated liver injury and induces lethality in RIPK3−/− mice. RIPK3−/− mice were pretreated with Cont ASO or RIPK1 ASO prior to αGalCer treatment (4μg/mouse, i.p.). (A) Serum ALT values at 6hr after αGalCer (n=5–8, *p<0.05 vs. αGalCer alone). (B) Representative H&E stained liver sections at 6hr after αGalCer. (C) Mouse survival by 8hr after αGalCer.
FIGURE 7.
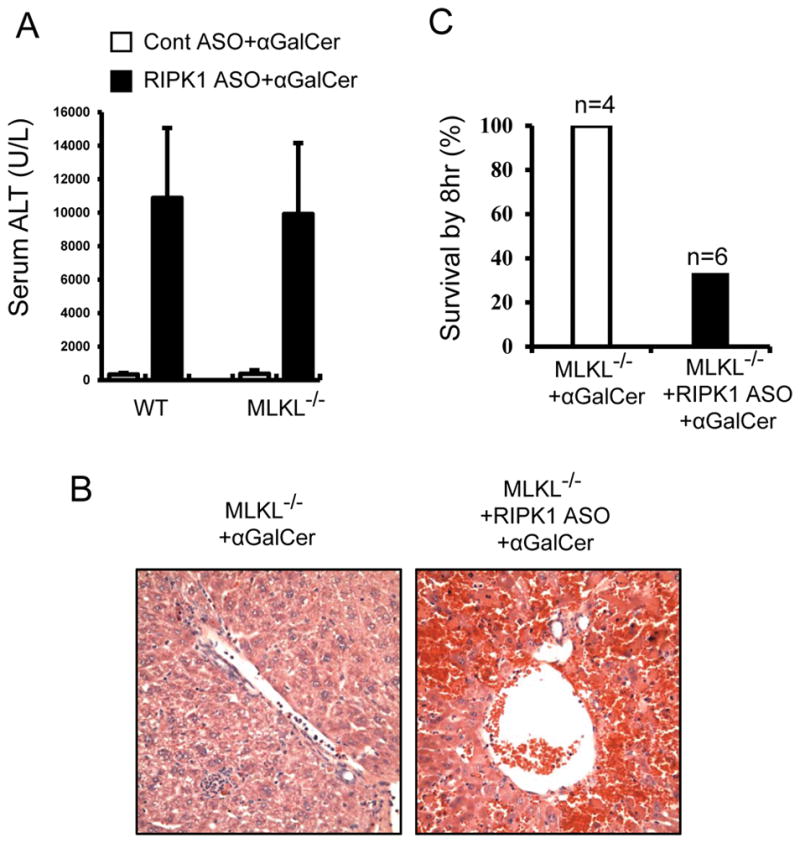
Similar to WT and RIPK3−/− mice, knockdown of RIPK1 markedly increases αGalCer-mediated liver injury and induces lethality in MLKL−/− mice. MLKL−/− mice were pretreated with Cont ASO or RIPK1 ASO prior to αGalCer treatment (4μg/mouse, i.p.). (A) Serum ALT values at 6hr after αGalCer (n=4–6, *p<0.05 vs. αGalCer alone). (B) Representative H&E stained liver sections at 6hr after αGalCer. (C) Mouse survival by 8hr after αGalCer.
Knockdown of RIPK1 markedly sensitizes to LPS- and Concanavalin A (ConA)-mediated liver injury and causes lethality
Finally, we examined whether the effect of knockdown of RIPK1 in αGalCer toxicity extends to other TNF related models, namely LPS- and ConA-mediated liver injury. LPS toxicity is associated with activation of macrophages/Kupffer cells in the liver and release of inflammatory cytokines, particularly TNF. On the other hand, ConA causes T cell-mediated hepatitis in which TNF also plays a critical role in hepatocyte death, although necrosis rather than apoptosis appears to be the predominant mode of cell death. We performed studies on LPS-induced liver injury using a dose that is not lethal to normal mice. As shown in Fig. 8A & B, a low non-lethal dose of LPS (3mg/kg, i.p.) caused very mild liver injury (serum ALT and liver histology) in scrambled ASO treated control mice. However, as in the αGalCer model, knockdown of RIPK1 markedly enhanced LPS-induced liver injury as evidenced by dramatically increased serum ALT and massive hepatocyte death and hemorrhage in the liver accompanied by increased positive TUNEL staining of hepatocytes (Fig. 8C). Furthermore, all of the RIPK1 ASO treated mice (n=5) succumbed to LPS treatment by 8hr whereas all of the control ASO treated mice (n=5) survived. Similarly, a low toxic dose of ConA (10mg/kg) markedly increased liver injury (ALT and histology) in RIPK1 ASO treated mice compared with control ASO treated mice (Fig. 8 E, F). ConA also induced lethality in RIPK1 ASO treated mice with a ~60% mortality by 6hr (n=11), while all of the control ASO treated mice survived (n=10). Furthermore, the exacerbated ConA-mediated liver injury in RIPK1 ASO treated mice was almost completely prevented by pretreatment of zVAD (Fig. 8E, F), indicating that a massive apoptotic death of hepatocytes contributes to the exacerbation of liver injury in RIPK1 ASO treated mice.
FIGURE 8.

(A–D) RIPK1 ASO markedly exacerbated LPS-mediated liver injury characterized by massive hepatic apoptosis. (A) Serum ALT at 6hr after LPS (3mg/kg i.p.) (n=5 per group). (B)Histology of liver sections at 6hr after LPS, showing massive cell death and hemorrhage in the liver of RIPK1 ASO treated mice. (C) TUNEL staining of liver sections at 6hr after LPS, displaying massive TUNEL positive cells in the liver of RIPK1 ASO mice. (D) Western blotting for RIPK1 in the liver lysates, confirming the efficient knockdown of RIPK1 proteins in RIPK1 ASO mice. (E–F) Knockdown of RIPK1 markedly exacerbated ConA-mediated liver injury and zVAD pretreatment prevented the the exaerbation of liver injury. (E) Serum ALT at 6hr after ConA (10mg/kg, i.v.) (n=6–10, *p<0.05). For caspase inhibition, zVAD (10mg/kg) was administered i.p. 15min prior to ConA treatemnt. (F)Representive H&E staining of liver sections at 6hr after ConA treatment, showing massive hepatocyte death and hemorrhage in the liver of RIPK1 ASO treated mice, which is prevented by pretreatment of zVAD.
Discussion
In this study, we found that knockdown of hepatic RIPK1 markedly exacerbates αGalCer-mediated liver injury and induces lethality, and clearly demonstrated that exacerbation of liver injury and lethality in RIPK1 ASO treated mice is due to TNF/caspase-dependent massive apoptotic death of hepatocytes. Our conclusions are based upon the following findings: 1) increased TUNEL positive staining of hepatocytes associated with increased caspase-3 activity in the liver; 2) almost complete protection by neutralization of TNF or inhibition of caspases by zVAD; 3) Sensitization of primary mouse hepatocytes from RIPK1 ASO treated mice to TNF-mediated cell death in the absence of transcription inhibitor e.g. actinomycin D, which could be blocked by caspase inhibition. Together, these data indicate that RIPK1 in hepatocytes acts as an inhibitor of apoptosis mediated through TNFR signaling and has a pro-survival role in hepatocytes in immune-mediated liver injury. Interestingly, our findings are consistent with a recent report on the conditional deletion of RIPK1 in IECs (9). IEC-specific RIPK1−/− mice spontaneously develop intestinal inflammation associated with IEC apoptosis leading to early death (9). The lethality could be rescued by antibiotic treatment, and deficiency of TNFR1 or caspase-8, indicating that gut commensal bacteria-mediated TNF/caspase-dependent apoptosis contributes to the cell death and the lethality (9). In agreement with recent genetic studies showing that the platform function of RIPK1 can suppress both caspase 8-dependent apoptosis and RIPK3/MLKL-mediated necroptosis (6–8), we found that it is the platform function of RIPK1, not its kinase activity, that contributes to the exacerbation of αGalCer-mediated liver injury in RIPK1 ASO mice. The lack of RIPK1 kinase activity either by Nec-1 treatment in WT mice or use of knock-in of dominant negative RIPK1(RIPK1D138N mice) does not exacerbate αGalCer-mediated liver injury. This is also supported by the findings in IEC-specific RIPK1−/− mice showing that the platform function of RIPK1 instead of its kinase activity maintains homeostasis of IECs by suppressing TNF-mediated apoptosis (9). Collectively, the platform function of RIPK1 in both hepatocytes and IECs appears to protect these epithelial cells against apoptotic death in response to TNFR signaling pathway.
After TNFR stimulation, it is generally believed that RIPK1 protein, not its kinase activity, in TNFR1 complex serve as a platform for recruitment of kinases such as NEMO(IKKγ) and TAK1 that are required to activate NF-κB, inducing pro-survival and pro-inflammatory gene expression (3, 5, 29). Previous studies have shown that NF-κB plays a critical role in maintaining the homeostasis of the liver by preventing death receptor induced apoptosis (34). Interestingly, our results showed that RIPK1 knockdown did not impair NF-κB activation in the hepatocytes in response to TNF stimulation. Increased sensitivity to TNF-mediated apoptosis of MEF cells from RIPK1−/− mice has been thought to be due to defective NF-κB activation. Thus the perinatal lethality of RIPK1−/− mice was originally believed to be caused by defective NF-κB-induced survival genes (5). However, recent studies argue against the requirement of RIPK1 in the activation of NF-κB (35). IEC-specific RIPK1−/− mice display no impairment of NF-κB activation in IECs, and the IEC apoptosis and intestinal inflammation are not due to defective NF-κB activation (9). Consistent with these findings, we found that TNF-mediated NF-κB activation is not impaired in the liver from RIPK1 ASO+αGalCer mice in vivo or in cultured primary hepatocytes from RIPK1 ASO treated mice with TNF in vitro. Therefore, the exacerbation of αGalCer-mediated liver injury and lethality in RIPK1 ASO treated mice and the sensitization of hepatocytes by RIPK1 knockdown to TNF-mediated apoptosis are not simply due to impaired NF-κB pro-survival pathway, suggesting that RIPK1-independent pathways may exist in hepatocytes to activate NF-κB as in IECs. Although RIPK1 knockdown did not impair gene expression of anti-apoptotic protein c-FLIP and cIAP1 in the liver tissues and primary mouse hepatocytes, we found that the induction of hepatic protein levels of c-FLIP and cIAP1 was decreased after αGalCer treatment in both liver tissue lysates and whole cell lysates of hepatocytes from RIPK1 ASO treated mice. Therefore, degradation of anti-apoptotic proteins in the absence of RIPK1 may render the hepatocytes more susceptible to caspase-dependent apoptosis, thus contributing to the exacerbation of TNF/caspase-dependent apoptotic death of hepatocytes (29–32).
Our data showed that both RIPK3−/− and MLKL−/− mice behaved similarly as WT control mice and were not sensitized to αGalCer treatment alone. Furthermore, they were not protected from the markedly exacerbated liver injury and lethality in response to αGalCer treatment when RIPK1 was knocked down. These results clearly indicate that a possible RIPK3/MLKL-mediated necroptosis in the absence of RIPK1 is not involved in the exacerbation of αGalCer-mediated liver injury and lethality in RIPK1 ASO treated mice. In certain cell types including macrophages, T cells and skin keratinocytes, death receptor stimulation can induce necroptosis under the conditions of inhibition of caspase activity or lack of caspase-8 (1–4, 10, 29, 36). However, our results showed that the pan-caspase inhibitor, zVAD, almost completely prevented the enhanced liver injury and lethality in vivo and blocked TNF-mediated cell death of hepatocytes from RIPK1 ASO treated mice in vitro. The abrogation of injury by caspase inhibition and lack of “necroptotic switch” in hepatocytes with caspase inhibition is not surprising as hepatocytes do not express RIPK3 under basal conditions (25). Conditional deletion of caspase-8 or caspase inhibition has been demonstrated to confer protection against acute liver failure due to massive apoptotic death of hepatocytes in D (+)-galactosamine/LPS model (37). This is also supported by in vitro studies showing no switch from apoptosis to necroptosis when primary mouse hepatocytes were treated with actinomycin D/TNF and zVAD (38). Together, these results argue against the role of TNF-mediated necroptosis in hepatocytes. Our recent studies have extensively examined the protein expression of RIPK3 in the liver (25), a key molecule for MLKL-mediated necroptosis. We found that RIPK3 is exclusively expressed on hepatic non-parenchymal cells (Kupffer cells, sinusoidal endothelial cells and leukocytes), with little expression of RIPK3 protein in hepatocytes under basal conditions and in acute acetaminophen (APAP) hepatotoxicity (25). The low expression of RIPK3 at both mRNA and protein levels are also supported by other studies (38, 39). In support of previous findings, we did not detect RIPK3 protein in primary mouse hepatocytes from control and RIPK1 ASO treated mice with or without TNF stimulation (data not shown). RIPK3 can be activated independently of RIPK1 to cause necroptosis, particularly when the cells express relatively high levels of RIPK3 (40–42). The levels of RIPK3 expression has been demonstrated to correlate with the sensitivity of cells to necroptosis (2, 29, 33, 43, 44). Although the role of necroptosis in acute injury models is unlikely, it is conceivable that RIPK3 expression can be induced in chronic injury and result in necroptotic death of hepatocytes. This may play a role in certain chronic inflammatory liver diseases such as alcoholic and non-alcoholic steatohepatitis(11, 12, 45). There is still much controversy on the role of necroptosis in the liver (12, 13, 15, 16, 25, 38, 46); however, our data indicate that in this TNF-dependent immune mediated acute liver injury model, the mode of cell death is caspase-dependent apoptosis and there is no contribution of necroptosis.
In contrast to the protective role of RIPK1 in the current study, in APAP induced necrosis, we have recently shown that knockdown of RIPK1 protects against necrotic cell death of hepatocytes both in vivo and in vitro (25), indicating that RIPK1 participates in the pathways leading to necrosis of hepatocytes caused by APAP toxicity. However mechanistically APAP hepatotoxicity is different from αGalCer-mediated liver injury triggered by death receptor signaling. The mode of cell death in APAP hepatotoxicity is cell autonomous and APAP is directly toxic to hepatocytes (25). Cell death in the APAP model is predominantly regulated necrosis and involves the sustained activation of JNK, leading to impairment of mitochondria and resultant oxidative stress (27, 47), and is not TNFα and caspase dependent (48). Interestingly we have demonstrated that in both models, RIPK3/MLKL-mediated necroptosis does not contribute to hepatocyte death presumably due to little expression of RIPK3 proteins in hepatocytes. The apparent opposite roles of RIPK1 in APAP and αGalCer-induced liver injury are very interesting, which confirms the critical role of RIPK1 as a signaling node and a central regulator of cell death and survival pathways (1–4). It is clear that the function of RIPK1 in many cell types including hepatocytes is complex. Detailed studies are needed to help understand the various roles of RIPK1 in different types of liver diseases and experimental conditions.
In conclusion, knockdown of RIPK1 markedly exaerbates immune-mediated liver injury and induces lethality through massive TNFα/caspase-dependent apoptosis of hepatocytes, not explained by defective NF-kB activation or a switch to RIPK3/MLKL-dependent necroptosis. Therefore, RIPK1 has a unique kinase-independent platform role in protecting against TNF-induced apoptosis of hepatocytes in immune-mediated liver injury.
Supplementary Material
Acknowledgments
This work was supported by NIH grant RO1DK067215 (NK), P30DK48522 (NK), RO1DK078586 (ZL), and U01AA021857 (ZL).
We acknowledge the Analytical/Instrumentation Core, Histology/Imaging Core, and Cell Isolation Core of the University of Southern California Research Center for Liver Diseases (P30DK48522) for the use of various instruments, for providing services for histology/imaging, isolation of primary mouse hepatocytes, and flow cytometric analysis.
List of abbreviations
- αGalCer
alpha-galactosylceramide
- ALT
alanine aminotransferase
- ASO
antisense oligonucleotide
- c-FLIP
cellular FLICE inhibitory protein
- cIAP
cellular inhibitor of apoptosis protein
- FADD
Fas-associated protein with death domain
- IEC
intestinal epithelial cell
- LSEC
liver sinusoidal endothelial cell
- MEF
mouse embryonic fibroblast
- MLKL
mixed lineage kinase domain-like protein
- NPCs
non-parenchymal cells
- RIPK1
receptor interacting protein kinase 1
- RIPK3
receptor interacting protein kinase 3
- TAK1
transforming growth factor β-activated kinase 1
- zVAD.fmk
benzyloxycarbonyl Val-Ala-Asp (OMe) fluoromethylketone
References
- 1.Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16:689–697. doi: 10.1038/ni.3206. [DOI] [PubMed] [Google Scholar]
- 2.Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 2015;25:347–353. doi: 10.1016/j.tcb.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 3.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 4.Weinlich R, Green DR. The two faces of receptor interacting protein kinase-1. Mol Cell. 2014;56:469–480. doi: 10.1016/j.molcel.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- 6.Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, Vince JE, Lawlor KE, Ninnis RL, Anderton H, Hall C, Spall SK, Phesse TJ, Abud HE, Cengia LH, Corbin J, Mifsud S, Di Rago L, Metcalf D, Ernst M, Dewson G, Roberts AW, Alexander WS, Murphy JM, Ekert PG, Masters SL, Vaux DL, Croker BA, Gerlic M, Silke J. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157:1175–1188. doi: 10.1016/j.cell.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 7.Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, Janke LJ, Kelliher MA, Kanneganti TD, Green DR. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157:1189–1202. doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, Sundararajan A, Guo H, Roback L, Speck SH, Bertin J, Gough PJ, Balachandran S, Mocarski ES. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci U S A. 2014;111:7753–7758. doi: 10.1073/pnas.1401857111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takahashi N, Vereecke L, Bertrand MJ, Duprez L, Berger SB, Divert T, Goncalves A, Sze M, Gilbert B, Kourula S, Goossens V, Lefebvre S, Gunther C, Becker C, Bertin J, Gough PJ, Declercq W, van Loo G, Vandenabeele P. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature. 2014;513:95–99. doi: 10.1038/nature13706. [DOI] [PubMed] [Google Scholar]
- 10.Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, Eftychi C, Lin J, Corona T, Hermance N, Zelic M, Kirsch P, Basic M, Bleich A, Kelliher M, Pasparakis M. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513:90–94. doi: 10.1038/nature13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology. 2014;147:765–783e764. doi: 10.1053/j.gastro.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57:1773–1783. doi: 10.1002/hep.26200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramachandran A, McGill MR, Xie Y, Ni HM, Ding WX, Jaeschke H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology. 2013;58:2099–2108. doi: 10.1002/hep.26547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Deutsch M, Graffeo CS, Rokosh R, Pansari M, Ochi A, Levie EM, Van Heerden E, Tippens DM, Greco S, Barilla R, Tomkotter L, Zambirinis CP, Avanzi N, Gulati R, Pachter HL, Torres-Hernandez A, Eisenthal A, Daley D, Miller G. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis. 2015;6:e1759. doi: 10.1038/cddis.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F, Takahashi N, Sergent O, Lagadic-Gossmann D, Vandenabeele P, Samson M, Dimanche-Boitrel MT. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012;19:2003–2014. doi: 10.1038/cdd.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roderick JE, Hermance N, Zelic M, Simmons MJ, Polykratis A, Pasparakis M, Kelliher MA. Hematopoietic RIPK1 deficiency results in bone marrow failure caused by apoptosis and RIPK3-mediated necroptosis. Proc Natl Acad Sci U S A. 2014;111:14436–14441. doi: 10.1073/pnas.1409389111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 19.Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol. 2009;86:513–528. doi: 10.1189/jlb.0309135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minagawa M, Deng Q, Liu ZX, Tsukamoto H, Dennert G. Activated natural killer T cells induce liver injury by Fas and tumor necrosis factor-alpha during alcohol consumption. Gastroenterology. 2004;126:1387–1399. doi: 10.1053/j.gastro.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 21.Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, Witek RP, Choi SS, Guy CD, Fearing CM, Teaberry V, Pereira FE, Adams DH, Diehl AM. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology. 2010;51:1998–2007. doi: 10.1002/hep.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takeda K, Hayakawa Y, Van Kaer L, Matsuda H, Yagita H, Okumura K. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc Natl Acad Sci U S A. 2000;97:5498–5503. doi: 10.1073/pnas.040566697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Osman Y, Kawamura T, Naito T, Takeda K, Van Kaer L, Okumura K, Abo T. Activation of hepatic NKT cells and subsequent liver injury following administration of alpha-galactosylceramide. Eur J Immunol. 2000;30:1919–1928. doi: 10.1002/1521-4141(200007)30:7<1919::AID-IMMU1919>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 24.Biburger M, Tiegs G. Alpha-galactosylceramide-induced liver injury in mice is mediated by TNF-alpha but independent of Kupffer cells. J Immunol. 2005;175:1540–1550. doi: 10.4049/jimmunol.175.3.1540. [DOI] [PubMed] [Google Scholar]
- 25.Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D, Kaplowitz N. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology. 2015;62:1847–1857. doi: 10.1002/hep.27939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology. 2006;43:1220–1230. doi: 10.1002/hep.21175. [DOI] [PubMed] [Google Scholar]
- 27.Win S, Than TA, Han D, Petrovic LM, Kaplowitz N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J Biol Chem. 2011;286:35071–35078. doi: 10.1074/jbc.M111.276089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, Itoh N, Suda T, Nagata S. Lethal effect of the anti-Fas antibody in mice. Nature. 1993;364:806–809. doi: 10.1038/364806a0. [DOI] [PubMed] [Google Scholar]
- 29.Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol. 2015;33:79–106. doi: 10.1146/annurev-immunol-032414-112248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gentle IE, Wong WW, Evans JM, Bankovacki A, Cook WD, Khan NR, Nachbur U, Rickard J, Anderton H, Moulin M, Lluis JM, Moujalled DM, Silke J, Vaux DL. In TNF-stimulated cells, RIPK1 promotes cell survival by stabilizing TRAF2 and cIAP1, which limits induction of non-canonical NF-kappaB and activation of caspase-8. J Biol Chem. 2011;286:13282–13291. doi: 10.1074/jbc.M110.216226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schattenberg JM, Zimmermann T, Worns M, Sprinzl MF, Kreft A, Kohl T, Nagel M, Siebler J, Schulze Bergkamen H, He YW, Galle PR, Schuchmann M. Ablation of c-FLIP in hepatocytes enhances death-receptor mediated apoptosis and toxic liver injury in vivo. J Hepatol. 2011;55:1272–1280. doi: 10.1016/j.jhep.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Piao X, Komazawa-Sakon S, Nishina T, Koike M, Piao JH, Ehlken H, Kurihara H, Hara M, Van Rooijen N, Schutz G, Ohmuraya M, Uchiyama Y, Yagita H, Okumura K, He YW, Nakano H. c-FLIP maintains tissue homeostasis by preventing apoptosis and programmed necrosis. Sci Signal. 2012;5:ra93. doi: 10.1126/scisignal.2003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose-Girma M, Warming S, Dixit VM. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 34.Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, Roskams T, Trautwein C, Pasparakis M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 35.Wong WW, I, Gentle E, Nachbur U, Anderton H, Vaux DL, Silke J. RIPK1 is not essential for TNFR1-induced activation of NF-kappaB. Cell Death Differ. 2010;17:482–487. doi: 10.1038/cdd.2009.178. [DOI] [PubMed] [Google Scholar]
- 36.He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011;108:20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaufmann T, Jost PJ, Pellegrini M, Puthalakath H, Gugasyan R, Gerondakis S, Cretney E, Smyth MJ, Silke J, Hakem R, Bouillet P, Mak TW, Dixit VM, Strasser A. Fatal hepatitis mediated by tumor necrosis factor TNFalpha requires caspase-8 and involves the BH3-only proteins Bid and Bim. Immunity. 2009;30:56–66. doi: 10.1016/j.immuni.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang YJ, Bang BR, Han KH, Hong L, Shim EJ, Ma J, Lerner RA, Otsuka M. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat Commun. 2015;6:8371. doi: 10.1038/ncomms9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun X, Lee J, Navas T, Baldwin DT, Stewart TA, Dixit VM. RIP3, a novel apoptosis-inducing kinase. J Biol Chem. 1999;274:16871–16875. doi: 10.1074/jbc.274.24.16871. [DOI] [PubMed] [Google Scholar]
- 40.Moujalled DM, Cook WD, Okamoto T, Murphy J, Lawlor KE, Vince JE, Vaux DL. TNF can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis. 2013;4:e465. doi: 10.1038/cddis.2012.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–31279. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kearney CJ, Cullen SP, Clancy D, Martin SJ. RIPK1 can function as an inhibitor rather than an initiator of RIPK3-dependent necroptosis. FEBS J. 2014;281:4921–4934. doi: 10.1111/febs.13034. [DOI] [PubMed] [Google Scholar]
- 43.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 45.Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, Kreggenwinkel K, Schneider AT, Bartneck M, Neumann UP, Canbay A, Reeves HL, Luedde M, Tacke F, Trautwein C, Heikenwalder M, Luedde T. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med. 2014;6:1062–1074. doi: 10.15252/emmm.201403856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang X, Chao X, Wang ZT, Ding WX. The End of RIPK1-RIPK3-MLKL-Mediated Necroptosis in Acetaminophen-Induced Hepatotoxicity? Hepatology. 2015 doi: 10.1002/hep.28263. [DOI] [PubMed] [Google Scholar]
- 47.Han D, Dara L, Win S, Than TA, Yuan L, Abbasi SQ, Liu ZX, Kaplowitz N. Regulation of drug-induced liver injury by signal transduction pathways: critical role of mitochondria. Trends Pharmacol Sci. 2013;34:243–253. doi: 10.1016/j.tips.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaeschke H, Williams CD, Farhood A. No evidence for caspase-dependent apoptosis in acetaminophen hepatotoxicity. Hepatology. 2011;53:718–719. doi: 10.1002/hep.23940. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.