

Abstract
Although Chronic Graft-versus-Host Disease (CGVHD) is the primary non-relapse complication of allogeneic transplantation, understanding of its pathogenesis is limited. To identify the main operant pathways across the spectrum of CGVHD, we analyzed gene expression in circulating monocytes, chosen as in situ systemic reporter cells. Microarrays identified two interrelated pathways: (1) Interferon-inducible genes and (2) innate receptors for cellular damage. Corroborating these with multiplex RNA quantitation, we found that multiple IFN-inducible genes (affecting lymphocyte trafficking, differentiation and antigen presentation) were concurrently upregulated in CGVHD monocytes compared to normal and nonCGVHD controls. IFN-inducible chemokines were elevated in both lichenoid and sclerotic CGHVD plasma and linked to CXCR3+ lymphocyte trafficking. Furthermore, the IFN-inducible genes CXCL10 and TNFSF13B (BAFF) levels were correlated at both the gene and plasma levels, implicating IFN-induction as a factor in elevated BAFF levels in CGVHD. In the second pathway, DAMP/PAMP receptor genes capable of inducing Type I IFN were upregulated. Type I IFN-inducible MxA was expressed in proportion to CGVHD activity in skin, mucosa and glands, and expression of TLR and RIG-1 receptor genes correlated with upregulation of Type I IFN-inducible genes in monocytes. Finally, in serial analyses following transplant, IFN-inducible and damage-response genes were upregulated in monocytes at CGVHD onset and declined upon therapy and resolution in both lichenoid and sclerotic CGVHD patients. This interlocking analysis of IFN-inducible genes, plasma analytes and tissue immunohistochemistry strongly supports a unifying hypothesis of induction of IFN by innate response to cellular damage as a mechanism for initiation and persistence of CGVHD.
Keywords: Graft versus host disease, transplantation, human, cytokines
Introduction
Chronic Graft Versus Host Disease (CGVHD) remains the principal source of non-relapse mortality and morbidity, affecting 40 – 70% of long-term survivors of allogeneic hematopoietic stem cell transplantation (allo-HSCT) (1). CGVHD encompasses a broad spectrum of inflammatory and fibrotic pathologies in multiple organs (2), and no common pathway has been implicated across the range of CGVHD manifestations. A deeper understanding of the pathogenesis of CGVHD is needed to support improved diagnosis, to design and evaluate targeted immunomodulatory therapies and to improve clinical response.
Based upon our prior immunohistochemical studies on lichenoid infiltrates in oral CGVHD, we proposed a role for Type I IFN production by pDC in the induction of chemokines that recruited cytotoxic T-bet+ Th1/Tc1 effector cells into oral tissues (3). These studies have been substantiated by immunohistochemistry showing infiltration by T-bet+ Th1/Tc1 cells and expression of IFN-inducible chemokines (CXCL9, CXCL10) in cutaneous CGVHD, and by the presence of elevated plasma levels of these chemokines at onset of CGVHD (4–11)}. Yet these studies do not identify mechanisms initiating IFN production, nor has this model been examined across the broad spectrum of CGVHD, ranging from newly developed inflammatory disease to severe, established CGVHD with sclerotic features.
We chose to use peripheral blood monocytes as ‘reporter’ cells for the dominant cytokine pathways within the multiple target tissues of CGVHD. Monocytes are relatively undifferentiated cells with high plasticity, that have been found to rapidly upregulate discrete profiles of genes in response to different cytokines such as IFN, IL-4/IL-13, TGFβ or glucocorticosteroids(12). Monocytes circulate throughout CGVHD patients, being exposed to the operant cytokine conditions in multiple locations. Monocyte-derived lineages are highly relevant to CGVHD, because the circulating monocytes emigrate into tissues, differentiating into scavenger macrophages, antigen presenting cells and regulators of fibrosis in CGVHD (4, 13). Since these recruited monocytes are constantly replenished from the marrow, their transcriptome provides a snapshot of prevailing cytokine conditions over a narrow window of time, yet this information can be tracked serially through multiple time points in CGVHD development.
We focused upon identifying pathways rather than individual highly upregulated genes, as pathways rely upon altered expression of multiple genes, and are less subject to individual variation in a diverse patient population. Blood and tissues were collected from the well-characterized patients in the NCI natural history study of CGVHD, because this study includes a high proportion of patients with severe sclerotic CGVHD as well as a broad range of early and late CGVHD manifestations. Pathways identified in CGVHD monocytes by microarray were verified using multiplex RNA assays in normal controls, nonCGVHD and CGHVD patients and were tracked at serial timepoints after during CGVHD onset and therapy. We corroborated these findings through plasma ELISA, FACS analysis of peripheral blood and immunohistochemistry of affected tissues. The results from these comprehensive analyses support a single common pathway model of CGVHD pathogenesis in which Type I and Type II Interferon (IFN), induced by innate immune processes, plays a central role in the initiation and persistence of CGVHD across the diverse range of inflammatory and fibrotic presentations.
Materials and Methods
Patient population
CGVHD patients were evaluated between 2004 and 2013 at the NIH Clinical Center, as part of a single-visit, cross-sectional NCI protocol “Natural History Study of Clinical and Biological Factors Determining Outcomes in Chronic Graft-Versus-Host Disease” (clinicaltrials.gov identifier: NCT00092235). A comprehensive evaluation was performed, including detailed history and clinical assessment according to the NIH Consensus Criteria (Table I); patients with late AGVHD were excluded. (2). Cutaneous CGVHD was further characterized by the percentage of the total body surface area (BSA) with erythema, dermal sclerosis, or deep sclerosis. Additionally, patients who never developed CGVHD (nonCGVHD) following reduced intensity allo-HSCT regimens utilizing mobilized T-replete PBSC (clinicaltrials.gov; NCT00520130 and NCT00074490) served as controls. Patients who developed CGVHD in these protocols were assessed at serial time points. Patients and normal healthy volunteers were all enrolled on IRB-approved NCI/CCR studies permitting sample acquisition and storage for research studies on transplantation; all human subjects provided written informed consent.
Table I.
CGVHD patient demographics
| Patient | Microarray N = 26 | Nanostring N = 69* | ||
|---|---|---|---|---|
| Median (Range) | Number of Patients | Median (Range) | Number of Patients | |
| Age | 42 (21–63) | 46 (21–68) | ||
| Sex (Male/Female) | 17/9 | 38/31 | ||
| HSCT Myeloablative/Nonmyeloablative | 16/10 | 39/30 | ||
| Donor: Related/ Unrelated/Haplo | 14/12/0 | 43/26/0 | ||
| Donor Source: BM/PBSC/CB | 3/23/0 | 13/55/1 | ||
| TBI | 13/13 | 27/42 | ||
| Months from Transplant to Evaluation | 29 (6–74) | 38 (5–258) | ||
| Months from CGVHD Diagnosis | 20 (2–67) | 27 (0–240) | ||
| Onset: (Progressive/Quiescent/De novo) | 7/10/9 | 26/20/24 | ||
| Lines of Prior Systemic Therapy | 4 (2–8) | 4 (0–9) | ||
| Current Systemic Therapy | ||||
| Prednisone | 17 | 46 | ||
| Tacrolimus | 15 | 24 | ||
| MMF | 8 | 20 | ||
| Sirolimus | 4 | 14 | ||
| ECP | 4 | 14 | ||
| Imatinib | 4 | 5 | ||
| Cyclosporine | 1 | 6 | ||
| None | 2 | 9 | ||
| Other† | 1 | 4 | ||
| Number of Organs Involved | 5 (2–7) | 5 (2–8) | ||
| CGVHD Organ Involvement (0/1/2/3) | ||||
| Vulvo/Vaginal (female only) | 1/4/2/2 | 13/10/4/4 | ||
| Oral | 9/13/3/1 | 20/37/9/3 | ||
| Eye | 2/11/9/3 | 12/26/26/5 | ||
| Liver | 12/11/3/0 | 35/23/10/1 | ||
| GI | 16/6/3/1 | 44/18/5/2 | ||
| Lung | 6/14/3/3 | 17/28/17/7 | ||
| Joints/Fascia | 6/5/13/2 | 18/16/28/7 | ||
| Skin | 0/2/6/18 | 9/6/15/39 | ||
| %Body Surface Area‡ (0, <25%, <50%, >50%) | ||||
| Erythematous | 2/19/4/1 | 21/38/8/2 | ||
| Dermal Sclerotic | 8/14/2/2 | 25/32/9/3 | ||
| Deep Sclerotic | 7/7/8/4 | 27/17/15/10 | ||
| Severity§ (Mild/Moderate/Severe) | 0/7/19 | 0/19/50 | ||
All 26 CGVHD patients assessed by microarray, as well as 43 additional CGVHD patients, were analyzed in the Nanostring cohort
Other systemic therapies: hydroxychloroquine, montelukast, methotrexate, rituximab
Some patients had combinations of erythematous, dermal and deep sclerotic areas of their skin and were included in more than one category
CGVHD Severity as assessed at time of NIH protocol enrollment
Assessment of patient blood and tissues
Concurrent with clinical assessment of CGVHD, peripheral blood and biopsies of skin, oral mucosa and labial minor salivary gland were obtained. Tissues were confirmed to have CGVHD by pathology and were assessed for MxA as previously described (3). Heparinized plasma, stored at −80°, was assessed for BAFF (R & D Systems) following manufacturer’s recommended procedures; levels of CXCL10, CXCL9, IFNα, IL-15 and IL-1β were determined in coded plasma samples by Aushon Technologies. Using cryopreserved PBMC, expression of CXCR3 (Becton Dickinson) on gated CD3+CD4+ and CD3+CD8+ T-cells were determined on a Gallios flow cytometer (Beckman Coulter, Inc) using FlowJo 9.6.4 (TreeStar, Inc) as previously described (6).
Gene expression
Monocytes from cryopreserved PBMC of normal and CGVHD patients were enriched by gating on FSChi SSChi CD3− CD4dim cells using an Influx Cell Sorter (Becton Dickinson Immunocytometry Systems); sorted monocytes were >95% CD14+. 300ng of total RNA per sample was used for Human Gene 1.0 ST Array analysis (Affymetrix) according manufacture’s instruction. We used both BRBArrayTools (http://linus.nci.nih.gov/BRB-ArrayTools.html) and Partek Genomic Suite 6.4 (Partek Inc.) for data visualization, identification of differentially expressed transcripts (p-value≤0.05). The Ingenuity Pathway Analysis tool (Ingenuity System Inc.) was used for analysis of functional pathways. The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus(14), and are accessible through GEO Series accession number GSE6074 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE60674). A custom nCounter® Gene Expression CodeSet (NanoString Technologies, Inc.), including 5 housekeeping genes (B2M, GAPDH, PPIB, SDHA, TBP) was generated consisting of two sequence-specific probes for each gene (a biotin-labeled capture probe and a fluorochrome-coded detection probe) (15), 200 ng of monocyte RNA, CodeSet and buffer were hybridized overnight on steptavidin-coated cartridges in an automated nCounter Prep Station, and then digitally counted in the nCounter Digital Analyzer. Data was analyzed on nSolver Analysis software, version 1.1. Heatmaps of Nanostring data were generated using GeneCluster 3.0 and Treeview 1.1.6r4.
Statistics
Nonparametric tests (Mann-Whitney U Test and Spearman Rank Correlation) were used on continuous variables. Multiple t test comparisons of log2 transformed gene copy data were performed in GraphPad Prism using the method of Benjamini and Hochberg with a False Discovery Rate was set at 5%. Values for B cells (cells/μl), BAFF (pg/ml), and IP-10 (pg/ml) were normalized for multiple regression analysis by log transformation (with values of 1 substituted for ≤1 B cell/μl) (Statview). All tests performed were 2-sided and considered significant at the .05 level.
Results
Microarray pathway analyses identify involvement of IFN-inducible and damage-response pathways
For the initial microarray, we compared monocytes collected from 10 normal healthy controls (ND) and 26 CGVHD patients chosen based upon severe lichen planus-like CGVHD of the skin or oral mucosa, or widespread sclerotic CGVHD affecting dermal or deep fascia. Overall, CGVHD patients were moderately to severely affected, according to NIH consensus criteria, with multiple organ involvement (see Table I) (2). Five were within 3 months of the initial CGVHD diagnosis; the remainder were patients with established, treatment-refractory CGVHD. All had received immunosuppressive regimens; only two were not currently on systemic therapy (Table I).
Supervised hierarchical cluster analysis of monocyte gene expression data (Human Gene Chip Exon 1.0ST. array) identified 1146 genes that were differentially expressed (t-test and FDR <0.05) in 26 CGVHD and 10 healthy subjects, including 532 that were up-regulated in CGVHD (Supplemental Figure S1A). Ingenuity Pathway Analysis identified several significantly upregulated canonical pathways in CGVHD (Supplemental Figure S1B); their common elements were the up-regulation of genes inducible by Type I and Type II IFN, or genes involved in the innate immune response to pathogen and damage associated molecular patterns (PAMP, DAMP), that in turn induce IFN or inflammasome activity. Overall, the highest probability upstream regulators of the gene expression changes were IFNα and IFNγ (p<.0001 and p<.001) (Figure S1B). GSEA analysis substantiated these findings, demonstrating that 50–85% of the genes involved in both Type I and Type II IFN responses, and in TLR and inflammasome pathways were upregulated in CGVHD monocytes (Supplemental Figure S1C). Because most of these genes are upregulated by both Type I and II Interferon (16, 17), we will refer to these genes as IFN-inducible.
Based upon identification of multiple significantly upregulated genes in IFN-signaling and DAMP/PAMP pathways (Figure 1A, Table II), we developed a custom probe set for multiplex RNA analysis (nCounter Nanostring) to assess RNA from the original CGVHD and ND microarray cohort and from sorted cryopreserved monocytes from 43 additional CGVHD patients assessed in the NIH natural history protocol (69 total), 9 additional normal controls (19 total), and 14 nonCGVHD patients evaluated in the natural history study (Table I). Expression of the panel of IFN-inducible genes was consistently up-regulated in CGVHD monocytes compared to those of normal donors, while expression in nonCGVHD patient monocytes was intermediate and not significantly different from that in normal controls (Figure 1B, Table II). Some highly inducible genes varied broadly over a multi-log range (for example, CXCL10, GBP1, STAT1), whereas constitutively expressed genes, such as TNFSF13B, had a much narrower range of expression. Nonetheless, those patients with high expression of one had high expression of the other genes, consistent with expression controlled by a common factor (Figure 1C). Importantly, expression of IFN-inducible genes was comparably upregulated both in patients with extensive lichenoid (erythematous) CGVHD and in those with widespread skin sclerosis, including both patients in the original microarray analysis and in the broader Nanostring cohort. (Figure 1D,E). The upregulated genes not only encompassed expected IFN-inducible anti-viral and antibacterial functions, but also included many mediating functions relevant to CGVHD including IFN signal transduction, antigen uptake and presentation, cellular cytotoxicity, chemokine regulation of lymphocyte trafficking, and cytokine regulation of lymphocyte differentiation (Table II). We therefore assessed systemic evidence of the IFN-inducible gene signature in established CGVHD.
Figure 1.

Microarray and nanostring analysis of monocytes from CGVHD patients and healthy normal controls. A. Unsupervised hierarchical-cluster heatmap of normal donor and CGVHD patients including 63 genes associated with response to DAMP or with induction by IFN (see Table II). B. Expression of IFN-inducible genes in monocytes sorted from 19 normal controls (blue), 14 nonCGVHD (green) and 69 CGVHD patients (red). Nanostring gene probe count data normalized by log2 transformation. Box and whisker plots indicate median values by central bar, 25th and 75th quartiles by box and full range from minimum to maximum copies by whiskers. Multiple t Test values shown for comparisons of normal and CGHVD and for nonCGVHD and CGVHD. The gray band indicates 3 S.E. above the negative control in the Nanostring analysis. C. Scatter plots of Nanostring-assessed expression of GBP1, STAT1 and TNFSF13B as compared with CXCL10, demonstrating correlated regulation in expression of these genes in monocytes from CGVHD (red), nonCGVHD (green) and normal controls (blue). D. Polar plot of median fold change in Microarray gene expression (as compared to the median in normal controls (dotted black line) in all CGVHD patients (purple line), in patients with either severe oral CGVHD or moderate to severe lichenoid cutaneous CGVHD ((n=13) fuschia line) or in patients with widespread (>30% body surface area) deep and dermal sclerosis ((n=13) orange line). Genes specifically induced by Type I IFN are noted in red. E. Box and whisker plot of Nanostring analyses comparing monocyte gene expression in 19 normal (blue), 7 patients with severe lichenoid cutaneous CGVHD on more than 20% of body surface area (BSA) and no sclerotic change (fuschia) and 29 patients with deep and dermal sclerotic involvement of 20–91% BSA (orange) and less than 6% erythema. Both CGVHD patient subsets were significantly different from ND but not from each other.
Table II.
Monocyte Gene Expression
| Genes | Microarray* | Nanostring† | |||||
|---|---|---|---|---|---|---|---|
| Gene Symbol | RefSeq | Gene Name | FDR | Permutated p- value | Fold- change | ND vs CGVHD | Non GVHD vs CGVHD |
| Interferon Induced Genes | |||||||
| Activation | |||||||
| CD38 | NM_001775 | CD38 molecule | 0.019 | 0.0008 | 1.65 | ||
| Antigen processing and presentation | |||||||
| LAP3 | NM_015907 | leucine aminopeptidase 3 | 0.003 | < 1e-07 | 1.58 | ||
| MSR1 | NM_002445 | macrophage scavenger receptor 1 | 0.004 | 0.0001 | 2.06 | 0.0006 | ns |
| PSMB9 | NM_002800 | proteasome (prosome, macropain) subunit, β type, 9 | 0.032 | 0.0019 | 1.45 | ||
| PSME2 | NM_002818 | proteasome (prosome, macropain) activator subunit 2 | 0.013 | 0.0001 | 1.42 | ||
| TAP1 | NM_000593 | transporter 1, ATP-binding cassette, sub-family B) | 0.009 | 0.0002 | 1.51 | <.0001 | ns |
| Anti viral and antibacterial function | |||||||
| APOL1 | NM_145343 | apolipoprotein L, 1 | 0.003 | < 1e-07 | 1.57 | ||
| GBP1 | NM_002053 | guanylate binding protein 1, interferon-inducible | 0.001 | < 1e-07 | 2.28 | <.0001 | 0.008 |
| GBP2 | NM_004120 | guanylate binding protein 2, interferon-inducible | 0.015 | 0.0006 | 1.54 | ||
| GBP3 | NM_018284 | guanylate binding protein 3 | 0.006 | < 1e-07 | 1.73 | ||
| GBP4 | NM_052941 | guanylate binding protein 4 | 0.006 | 0.0001 | 1.88 | ||
| GBP5 | NM_052942 | guanylate binding protein 5 | 0.007 | 0.0002 | 2.18 | ||
| IFI35 | NM_005533 | interferon-induced protein 35 | 0.012 | 0.0003 | 1.59 | 0.0002 | 0.0003 |
| PARP9 | NM_031458 | poly (ADP-ribose) polymerase family, member 9 | 0.004 | < 1e-07 | 1.87 | <.0001 | <.0001 |
| Chemokines and Chemokine Receptors | |||||||
| CCR2 | NM_001123396 | chemokine (C-C motif) receptor 2 | 0.004 | 0.0002 | 2.65 | ||
| CXCL10 | NM_001565 | chemokine (C-X-C motif) ligand 10 | 0.002 | < 1e-07 | 3.58 | <.0001 | 0.0008 |
| CXCL11 | NM_005409 | chemokine (C-X-C motif) ligand 11 | 0.005 | 0.0001 | 1.93 | 0.0007 | <.0001 |
| CXCL9 | NM_002416 | chemokine (C-X-C motif) ligand 9 | 0.017 | 0.001 | 1.74 | 0.0002 | 0.0008 |
| Complement | |||||||
| C3AR1 | NM_004054 | complement component 3a receptor 1 | 0.008 | 0.0007 | 2.49 | <.0001 | 0.0002 |
| SERPING1 | NM_000062 | serpin peptidase inhibitor, clade G (C1 inhibitor) | 0.023 | 0.0014 | 2.12 | 0.0002 | ns |
| Cytokines | |||||||
| TNFSF13B | NM_006573 | tumor necrosis factor superfamily, member 13b | 0.009 | < 1e-07 | 1.28 | <.0001 | 0.0002 |
| Cytotoxicity | |||||||
| TNFSF10 | NM_003810 | tumor necrosis factor superfamily, member 10 | 0.003 | < 1e-07 | 1.88 | <.0001 | 0.0004 |
| IFN receptor and signaling | |||||||
| IFNAR1 | NM_000629 | interferon (alpha, beta and omega) receptor 1 | 0.029 | 0.0008 | 1.27 | <.0001 | ns |
| JAK2 | NM_004972 | Janus kinase 2 | 0.039 | 0.0025 | 1.46 | ||
| STAT1 | NM_007315 | signal transducer and activator of transcription 1 | 0.004 | 0.0001 | 1.72 | <.0001 | ns |
| STAT2 | NM_005419 | signal transducer and activator of transcription 2 | 0.025 | 0.0009 | 1.46 | ns | ns |
| Negative Regulators of Inflammation | |||||||
| SLAMF8 | NM_020125 | SLAM family member 8 | 0.008 | 0.0001 | 1.74 | ||
| TRIM21 | NM_003141 | tripartite motif-containing 21 (Ro52) | 0.050 | 0.0051 | 1.38 | 0.0002 | 0.02 |
| Other | |||||||
| ANKRD22 | NM_144590 | ankyrin repeat domain 22 | 0.001 | < 1e-07 | 3.24 | 0.0002 | 0.0015 |
| LGALS3BP | NM_005567 | lectin, galactoside-binding, soluble, 3 binding protein | 0.014 | 0.0001 | 1.77 | 0.0015 | ns |
| Type I Interferon Induced Genes | |||||||
| IRF7 | NM_004031 | IFN regulatory factor 7 | ns | ns | |||
| IFI44 | NM_006417 | IFN-induced protein 44 | ns | ns | |||
| IFIT1 | NM_001548 | IFN-induced protein with tetratricopeptide repeats 1 | 0.207 | 0.0502 | 1.86 | ns | 0.007 |
| IFIT2 | NM_001547 | IFN-induced protein with tetratricopeptide repeats 2 | 0.024 | 0.0008 | 2.09 | ||
| IFIT3 | NM_001031683 | IFN-induced protein with tetratricopeptide repeats 3 | 0.083 | 0.0093 | 1.81 | ||
| IFITM3 | NM_021034 | IFN-induced transmembrane protein 3 | 0.101 | 0.0145 | 1.55 | 0.016 | ns |
| MX1 | NM_002462 | myxovirus (influenza virus) resistance 1 | ns | 0.03 | |||
| OAS1 | NM_016816 | 2',5'-oligoadenylate synthetase 1 | 0.018 | 0.0004 | 1.69 | <.0001 | <.0001 |
| RSAD2 | NM_080657 | radical S-adenosyl methionine domain containing 2 | 0.215 | 0.0473 | 1.68 | ns | 0.04 |
| XAF1 | NM_017523 | XIAP associated factor 1 | 0.054 | 0.0042 | 1.55 | 0.007 | 0.001 |
| DAMP and PAMP Pathway Genes | |||||||
| Cytosolic Nucleic Acid Receptors | |||||||
| AIM2 | NM_004833 | absent in melanoma 2 | 0.0131 | 0.0001 | 1.98 | <.0001 | 0.007 |
| DDX58 | NM_014314 | DEAD box polypeptide 58 (RIG-I) | 0.0244 | 0.0009 | 1.85 | <.0001 | <.0001 |
| IFI16 | NM_005531 | interferon, gamma-inducible protein 16 | 0.0321 | 0.0026 | 1.40 | <.0001 | .0008 |
| IFIH1 | NM_022168 | IFN-induced with helicase C domain 1 (MDA5) | 0.0262 | 0.0003 | 1.59 | ns | ns |
| Plasma membrane receptors for dead cell products | |||||||
| CLEC4E | NM_014358 | C-type lectin domain family 4, member E | 0.002 | < 1e-07 | 2.36 | <.0001 | <.0001 |
| FCGR1A | NM_000566 | Fc fragment of IgG, high affinity Ia, receptor (CD64) | 0.006 | 0.0003 | 1.97 | <.0001 | <.0001 |
| FCGR2A | NM_001136219 | Fc fragment of IgG, low affinity IIa, receptor (CD32) | 0.015 | 0.0001 | 1.27 | ||
| FPR2 | NM_001462 | formyl peptide receptor 2 / Annexin A1 receptor | 0.0003 | < 1e-07 | 2.93 | <.0001 | <.0001 |
| ITGB3 | NM_000212 | Integrin beta 3 | 0.040 | 0.0018 | 2.37 | .0007 | <.0001 |
| P2RY12 | NM_022788 | purinergic receptor P2Y, G-protein coupled, 12 | 0.0009 | < 1e-07 | 2.55 | ||
| P2RX7 | NM_002562 | purinergic receptor P2X, ligand-gated ion channel, 7 | 0.195 | 0.0434 | 1.26 | ||
| PANX1 | NM_015368 | pannexin 1 | 0.015 | 0.0003 | 1.36 | ||
| TLR pathway | |||||||
| IRAK4 | NM_001114182 | IL-1 receptor-associated kinase 4 | 0.080 | 0.0075 | 1.24 | 0.022 | 0.009 |
| TLR2 | NM_003264 | toll-like receptor 2 | 0.007 | 0.0001 | 1.36 | <.0001 | 0.004 |
| TLR4 | NR_024168 | toll-like receptor 4 | 0.004 | 0.0002 | 1.47 | <.0001 | 0.018 |
| TLR7 | NM_016562 | toll-like receptor 7 | 0.106 | 0.0158 | 1.54 | 0.03 | 0.016 |
| Glucocorticoid-Induced Genes | |||||||
| CD163 | NM_004244 | CD163 | 0.028 | 0.0014 | 2.56 | <.0001 | 0.011 |
| IL10 | NM_000572 | interleukin 10 | 0.014 | 0.0003 | 2.32 | <.0004 | ns |
| IL1R2 | NM_004633 | interleukin 1 receptor, type II | 0.028 | 0.0018 | 4.62 | <.0002 | 0.011 |
In the microarray analysis, the FDR, Permutated P value and fold change in expression of genes in CGVHD patients vs normal donors were determined using Partek software.
In the Nanostring analysis, log2 normalized values of gene expression were compared by t tests in Graphpad Prism v6 using unpaired t tests with no assumption of constant standard deviation and adjusted for multiple comparisons using the method of Benjamini and Hochberg, with a False Discovery Rate of 5%.
Correlative studies of CGVHD plasma link IFN-inducible genes to lymphocyte trafficking and B cell activation
A key element in CGVHD pathogenesis is the trafficking of T cells into target tissues. Corroborating the upregulation of IFN-inducible chemokine genes in CGVHD monocytes, we found elevated levels of CXCL9 and CXCL10 levels in CGVHD plasma, as compared to levels in normal control or nonCGVHD plasma, including significant increases in both severe sclerotic and lichenoid CGVHD patient subsets of the study cohort (Figure 2A). Expression of CXCL9 and CXCL10 in lichenoid oral and cutaneous CGVHD tissues, as detected by immunohistochemistry, has implicated these chemokines in recruitment of CXCR3+ Th1/Tc1 cells into the infiltrate (3, 5, 6, 8). Chemokine-mediated recruitment of CXCR3+ Th1 cells into extensive areas of skin should be accompanied by corresponding depletion of these cells in the blood. We determined that expression of CXCR3 on circulating T-cells was significantly reduced in patients with extensive body surface area (BSAhi) of either lichenoid or sclerotic cutaneous CGVHD, as compared with those with less skin involvement or normal controls (Figure 2B). These results support a role for IFN-induced chemokines in regulating lymphocyte trafficking in cutaneous CGVHD.
Figure 2.

IFN-inducible chemokines and cytokines in CGVHD plasma. A. Box and whisker plots comparing levels of the chemokines CXCL9 and CXCL10 in 10 normal controls (blue), 20 nonCGVHD (green) and 146 CGVHD (red) patients (including 36 CGVHD patients assessed by Nanostring), as well as the subsets of these CGVHD patients with lichenoid (n=21) or sclerotic (n=46) CGVHD. Kruskal-Wallis non-parametric ANOVA demonstrated significant differences in the groups. Multiple paired comparisons corrected by Dunn’s test found that All CGVHD and Lich and Scl CGVHD patient subsets differed significantly from ND. B. Box and whisker plots comparing the level of CXCR3 expression in CD4 and CD8 T cells in 9 normal controls (blue), 15 patients with lichenoid or sclerotic cutaneous CGVHD involving <25% Body Surface Area (BSAlo) (orange) and 53 patients with >25% (BSAhi). These patients included 23 examined by Nanostring. Expression gates were determined using full minus one controls. C-G. Plasma levels of BAFF (TNFSF13B) in CGVHD patients. C. Plasma levels of BAFF in 20 normal controls (blue), 21 nonCGVHD (green) and 125 CGVHD (red) patients (including 36 in the Nanostring assays). D. Graph of plasma levels of CXCL10 and BAFF in 125 CGVHD (red) patients and 13 nonCGVHD patients (green). E. Same graph identifying patients with >20% lichenoid BSA (fuschia) or >20% BSA with sclerotic CGVHD (orange). F. Graph of B cell number and plasma level of BAFF in 125 CGVHD (red) patients and 13 nonCGVHD patients (green). G. Graph of plasma BAFF levels of BAFF in 72 CGVHD (red) patients with B-cell numbers greater than 100/μl. Spearman correlates reflect BAFF levels in only the CGVHD patients.
BAFF (TNFSF13B) has been recognized not only as a useful plasma biomarker for CGVHD (Figure 2C), but also one that may contribute to the activated state of CGVHD B-cells (18–21). Expression of TNFSF13B, the gene for B cell activation factor (BAFF), was elevated above normal constitutive levels in CGVHD monocytes, in parallel with other IFN-inducible genes (Figure 1B,C) (22). When plasma levels of BAFF and CXCL10 were compared in 125 CGVHD patients, BAFF levels were positively correlated with CXCL10 (Figure 2D); again this was evident in both sclerotic as well as lichenoid CGVHD patients (Figure 2E). Plasma levels of BAFF vary dependent on levels of peripheral B-cells expressing the BAFF receptor (23, 24), hence, rising at transplant (due to lymphodepletion) and falling upon B cell repopulation (25). Yet many CGVHD patients have high levels of BAFF despite recovery of normal or elevated B-cell numbers (Figure 2F, G). Tested in a multiple regression model incorporating B cells, CXCL10 levels, time from transplant and degree of immune suppression, only CXCL10 levels (p<.0001) and B-cell numbers (p<.0001) were found to have a significant effect on BAFF plasma levels, contributing to a strong final adjusted R value (.656) and adjusted R2 (.430) (Supplemental Figure S2). This analysis implicates IFN-induction in the elevated plasma BAFF levels in CGVHD, and supports an indirect role of IFN-inducible factors in B cell dysregulation in CGVHD.
Expression of innate immune receptor genes inducing Type I Interferon production is upregulated in CGVHD
CGVHD monocyte transcriptome profiling also identified upregulation of many genes involved in the innate immune response to cellular damage (DAMP) and pathogens (PAMP (26–28)). Irrespective of lichenoid infiltrates or sclerotic manifestations of CGVHD, increases were found in receptors for nucleic acids (AIM2, DDX58, TLR7, TLR4, CLEC4E), annexin (FPRL2/ALYX) and phagocytosis (FCRG1A, MSR) (Table II, Figure 1A, 3A, B, C). Uptake of material from damaged cells can trigger formation of inflammasomes, resulting in Caspase 1-mediated release of IL-1β (29, 30). AIM2, IFI16, and NLRC4 (the first two inducible by IFN), were all upregulated in CGVHD monocytes (Figures 3A, C, Table II). Triggering of TLR (TLR7), CLR (CLEC4E) and RIG-1 (DDX58) pathways can increase inflammation through production of IFNα (via IRF7) and IL-6 (via NFκb). Whereas IRF7 levels were not upregulated overall in CGVHD natural history patient monocytes in the microarray, in those with levels of IRF7 above median, IFN-inducible genes were more strongly expressed, including those specifically induced by Type I IFN, again in both sclerotic and lichenoid patients (red genes, Figure 1D, 3D) (16). Moreover, higher expression of RIG-I helicase (DDX58) or TLR7, both in DAMP pathways inducing IRF7 (26), correlated with higher expression of Type I IFN-inducible genes (Figure 3E).
Figure 3.

Monocyte expression of innate immune receptors for DAMP and of Type I IFN induced genes. A. Polar plot of median fold change in microarray-assessed gene expression (as compared to the median in normal controls (dotted black line)) of innate immune receptors for DAMP in all CGVHD patients (purple line), in patients with either severe oral CGVHD or moderate to severe lichenoid cutaneous CGVHD (fuschia line) or in patients with widespread (>30% body surface area) deep and dermal sclerosis (orange line). B–C. Box and whisker plot comparing Nanostring-analyzed expression of several DAMP responsive genes in monocytes sorted from 19 normal controls (blue), 14 nonCGVHD (green) and 69 CGVHD patients (red). C. Box and whisker plots from a 12 severe CGVHD patients from the main cohort, retested with additional DAMP receptors. D. Polar plot of median fold change in gene expression of IFN-inducible genes in CGVHD microarray patients whose IRF7 expression was greater than the median (brown line) versus those with less than the median (green line). Type I IFN-specific genes marked in red. E. Scatter plots of Nanostring-assessed expression of OAS1, IFIT1, and XAF1 as compared with DDX58 and TLR7, demonstrating correlated expression of DAMP receptors and Type I IFN-inducible genes in CGVHD monocytes (red). (F–I) Fluorescence immunohistochemistry of the Type I-IFN induced protein MxA, showing expression in biopsies collected from affected (F) lichenoid skin, (G) sclerotic skin, (H) oral mucosa and (I) minor salivary gland. Tissues were formalin fixed, paraffin embedded, and following antigen retrieval and staining with MxA and AlexaFluor 555 secondary antibodies, were imaged on a Leica SP2 confocal microscope. Dotted lines mark the interface between epidermis and dermis or outline secretory acini. J–K. Immunofluorescent expression (mean fluorescent intensity) of MxA in epidermal keratinocytes in CGVHD tissues. J. Comparison of biopsies of skin from CGVHD patients with no skin involvement, with lichenoid (lich) involvement, or sclerotic (scl) involvement. Paired biopsies of affected and unaffected skin were collected from each lichenoid or sclerotic patient. K. Comparison of biopsies of oral mucosa from patients with no oral CGVHD or only mild symptoms, versus those with severe oral CGVHD. L. Scatter plots showing correlated increases in IFNα and IL-15 in 49 CGVHD natural history patients, of which 24 had more than 20% of body surface area affected with Lich-CGVHD or Scl-CGVH. M. Scatter plots showing correlated increases in IFNα inducible CXCL10 and IL-6 in 49 CGVHD natural history patients, including 23 of the microarray patients, and 12 normal controls. This assay was repeated 3 times with plasma from different cohorts of 40–50 CGVHD patients.
Furthermore, expression of proteins specifically induced by Type I IFN was elevated in commonly affected CGVHD tissues, with expression increasing in proportion to the degree of clinical activity. In lichenoid and sclerotic skin, oral mucosa and salivary glands, the Type I IFN-inducible protein MxA was expressed in both infiltrating myeloid cells and epidermal keratinocytes (Figures 3F–I) (3). MxA levels in the keratinocytes in both lichenoid and sclerotic skin biopsies were significantly increased compared to levels in skin from patients lacking skin involvement (Figure 3J). Similarly, MxA expression was higher in severely affected oral mucosa than in mildly affected or unaffected tissue (Figure 3K). In plasma, increases of IFNα correlated with increases in IL-15, an IFN-inducible cytokine supporting differentiation of CD8 Tc1 effectors (Figure 3L), which is also present with the same distribution as MxA in CGVHD affected oral mucosa (3). These data demonstrate that Type I IFN is widely produced in CGVHD tissues, concurrent with increases in Type I-IFN specific gene products. Finally, in addition to inducing IFNα, DAMP stimulation of TLR7 and RIG-1 will also induce production of TNFα and IL-6 via NFκB. Plasma levels of the IFN-inducible chemokine CXCL10 and NFκB-inducible IL-6 were correlated, consistent with concurrent activation of both pathways in individual CGVHD patients (Figure 3M). These results support the presence of innate immune activation that could support production of inflammatory mediators such as IFNα and IL-6.
Upregulation of IFN-inducible genes occurs at onset of CGVHD
Because the NIH CGVHD natural history protocol is a single-visit cross-sectional study, we then examined sorted monocytes from 18 NCI allo-HSCT patients at serial time points in the development and treatment of CGVHD using the Nanostring panel; results on six of these are shown, with gene upregulation expressed as fold changes from the mean levels found in normal controls (Figure 4A, patients a – f). Assessed IFN-inducible genes included those rapidly up-regulated by IFN (first row), those supporting antigen processing and presentation (second row), those specifically up-regulated by Type I IFN (third row) and those encoding endosomal and cytosolic DAMP receptors for nucleic acids (fourth row). (16, 17). All were at low levels prior to CGVHD onset, but were upregulated in parallel on the day of CGVHD diagnosis. Expression declined upon subsequent therapy, concurrent with declines in CGVHD symptoms, and remained low upon CGVHD resolution and discontinuation of therapy. In patient E, localized edema and lichenoid infiltrates in limbs at 12 months were succeeded by sclerotic changes at 18 months, with elevated levels of IFN-inducible and damage response genes present at both lichenoid and sclerotic phases of cutaneous CGVHD. Gene expression at diagnosis was comparable to that in established severe CGVHD and significantly higher than in nonCGVHD (Figure 4B). Thus serial data from patients followed at clinically relevant time points is consistent with elevation in IFN-inducible and DAMP receptor genes observed in CGVHD natural history patients, rising with onset of lichenoid or sclerotic disease and declining with effective therapy.
Figure 4.

Time courses of IFN-inducible gene expression by Nanostring analysis in monocytes from 7 patients developing CGVHD. A. Patients (a – f) had been transplanted at the NCI using a reduced intensity regimen followed by T-replete PBSC infusions from matched sibling (a, b, e) or matched unrelated (c, d, f) donors. In each patient the graphs in the top row represents the time course (in months from transplant) of expression of 6 genes strongly induced by both Type I and Type II IFN. The second row shows 3 IFN-inducible genes associated with antigen processing and presentation. The third row shows 6 genes specifically modulated by Type I IFN, including the transcription factor IRF7. The bottom graph shows the expression receptors responding to nucleic acids released from damaged cells, including of cytosolic (AIM2, DDX58) and endosomal (TLR7) nucleic acid receptors capable of inducing IRF7 and triggering inflammasome formation. The vertical axes are fold changes in gene expression, normalized relative to expression in normal controls (dotted lines). GC = period of treatment with high dose glucocorticoids. B. A series of 13 severe CGVHD and 14 nonCGVHD patient monocytes were compared for the same genes as in A, expressed as fold change above normal controls.
Patient F demonstrates the confounding effects of high dose corticosteroids. At 6 months, IFN-inducible genes were first upregulated, concurrent with diagnosis of moderate oral CGVHD. High dose systemic prednisone therapy (80mg/day – marked as GC) was initiated at the onset of a severe erythematous rash, 10 days prior to collection of the 9 month blood sample. All IFN-inducible genes were very low during corticosteroid treatment at 9 months. Upon rapid improvement in skin erythema, systemic prednisone was tapered off. Following taper of steroids, at the 12 month time point, the patient was diagnosed with oral, ocular and gastrointestinal CGVHD, accompanied once again by high levels of IFN-inducible genes; gene expression declined, once again, upon therapy.
Although high dose glucocorticoid therapy can rapidly resolve inflammatory symptoms and reduce expression of IFN-inducible genes, many in the CGVHD natural history cohort are refractory to steroid therapies (Table I). To track the effects of steroids on gene expression, we assessed expression of three potent anti-inflammatory genes (IL1R2, CD163 and IL10) that are induced by treatment with glucocorticoids (31, 32). These genes were found to be elevated only in patients receiving glucocorticoids; levels were comparable to normal controls in patients on other immune-suppressants, but increased in a dose-dependent manner from low maintenance glucocorticoid levels up to high therapeutic doses (Figure 5A,B). We therefore ascribe elevation in IL1R2, CD163 and IL10 to the effect of systemic glucocorticoid therapies. In patient F, treated with Prednisone 80mg/day for 10 days, the levels of IL1R2, IL-10 and CD163 were elevated 121-, 27- and 4-fold above normal levels respectively. Reciprocal expression of IFN- and glucocorticoid-induced genes in Patient F demonstrates that CGVHD gene pathways can be suppressed under first line therapy with high prednisone doses.
Figure 5.

Monocyte expression of genes inducible by glucocorticoids. A. Box and whisker plot comparing expression of IL10, IL1R2 and CD163 in monocytes from 19 normal controls (white), 23 CGVHD patients receiving no glucocorticoids (yellow), 18 CGVHD patients receiving <20mg/day (orange), and 26 CGVHD patients receiving 20–80mg/day (red). B. Graphic combining glucocorticoid dose, expression of glucocorticoid and IFN-inducible and DAMP pathway genes, and CGVHD patient organ involvement. Heatmap of Nanostring data generated from all normal, nonCGVHD and CGVHD patients by unsupervised hierarchical clustering of 3 glucocorticoid-induced genes and 18 IFN-induced genes, including 4 involved in response to DAMP (genes listed on right edge). Superimposed on the top is a bar graph of prednisone dose/day for each CGVHD (reddish brown) and nonCGVHD (green) patients (assumed to be zero for normal controls (blue)) and a line of markers (purple) indicating systemic treatments with non-steroidal immunosuppressants. At the bottom is a graphic showing involvement of the 8 organ systems assessed by the CGVHD consensus criteria, graded as not present (white), mild (yellow), moderate (medium orange) and severe (dark orange). The last row shows the extent of sclerotic involvement in skin (as in Table I). Gray boxes represent not applicable (vulvar vaginal CGVHD in males).
Continued expression of these IFN and DAMP response genes despite high doses of steroids (and high expression of glucocorticoid-inducible genes) may reflect the steroid-refractory nature of severe CGVHD in many patients. Inclusion of glucocorticoid-induced genes in the Nanostring panel can discriminate the effects of glucocorticoid therapy from those of CGVHD (Figure 5B). Many established CGVHD patients, at the right in Figure 5B, particularly those with sclerotic skin (bottom row organ score), are little different in gene expression from normal controls or nonCGVHD patients (blue or green bands). Those patients not receiving glucocorticoids (in the center) have significantly elevated expression of multiple IFN-induced and DAMP-responsive genes. On the left, patients on steroids are clearly identifiable by upregulation of the three steroid-inducible genes: IL1R2, CD163 and IL10. Some of these patients had reduced expression of the IFN signature, as was seen in newly treated patient F (Figure 5A), but others were refractory to steroid treatment. This disparity was independent of the wide range of organ involvement noted at the bottom of the graphic.
Discussion
In this study we determined that monocytes from CGVHD patients expressed many well-characterized Type I and Type II IFN-inducible genes at levels significantly elevated over those in healthy controls and nonCGVHD patients. These genes were expressed in a correlated manner, consistent with a common pathway, and were upregulated to comparable degrees in CGVHD patients with either lichenoid or sclerotic manifestations. In patients followed longitudinally, these genes were upregulated upon onset of CGVHD and declined upon treatment and resolution. Gene expression data was corroborated by plasma ELISA and flow cytometry that implicated IFN-inducible chemokines in T cell trafficking into tissues. Elevation of CXCL10 correlated with elevated plasma levels of BAFF, a cytokine associated with B-cell dysregulation in CGVHD. Indeed, the pathways defined in the monocyte transcriptome provide a mechanistic basis for the elevated expression of CXCL9, CXCL10 and BAFF, all currently considered as promising plasma biomarkers for CGVHD onset (5, 7, 9, 11, 20). Indeed, ROC analysis suggests that several of the IFN-inducible genes may similarly provide a useful gene expression index (Supplemental Figure S3).
Furthermore, we have demonstrated the concurrent upregulation of multiple receptors for pathogen and cell damage-derived molecules, both upon onset of CGVHD and in established CGVHD. Through their capacity to induce IFN and other inflammatory mediators, these receptors may both initiate and sustain CGVHD. TLR7 receptors triggered by endosomal uptake of complexes of nuclear debris and anti-nuclear autoantibodies (which develop in many CGVHD patients (33, 34)) can initiate Type I IFN production through IRF7. Uptake of debris into phagolysosomes can result in leakage of nucleic acids that trigger cytosolic RIG-1 receptors (DDX58). We have demonstrated correlations between expression levels of damage receptors (DDX58, TLR7) and expression of Type I IFN-inducible genes, as well as demonstrating IFNα-inducible MxA production in the tissues most commonly affected by CGVHD (Figure 3E–K).
The evidence of IFN-inducible pathways presented here supports a new perspective on human CGVHD initiation and persistence (Figure 6). We propose that molecules released by damaged cells initiate the inflammatory process by activating tissue-resident macrophages and DC and triggering local production of Type I IFN and IL-6 (Figure 3E–L). Type I IFN released in tissues induces local chemokine production by myeloid and epidermal cells; these in turn mediate recruitment of CXCR3+ Th1/Tc1 cells. IFNγ from Th1/Tc1 cells, in synergy with Type I IFN (16), upregulates expression of antigen processing and presentation components, receptors for cellular damage, chemokines supporting T cell recruitment, and cytokines increasing B cell activation (Table II). Meanwhile, the local release of Type I and II IFN in tissues would trigger specific gene upregulation in circulating monocytes, creating the changes in ‘reporter’ populations that we assessed.
Figure 6.
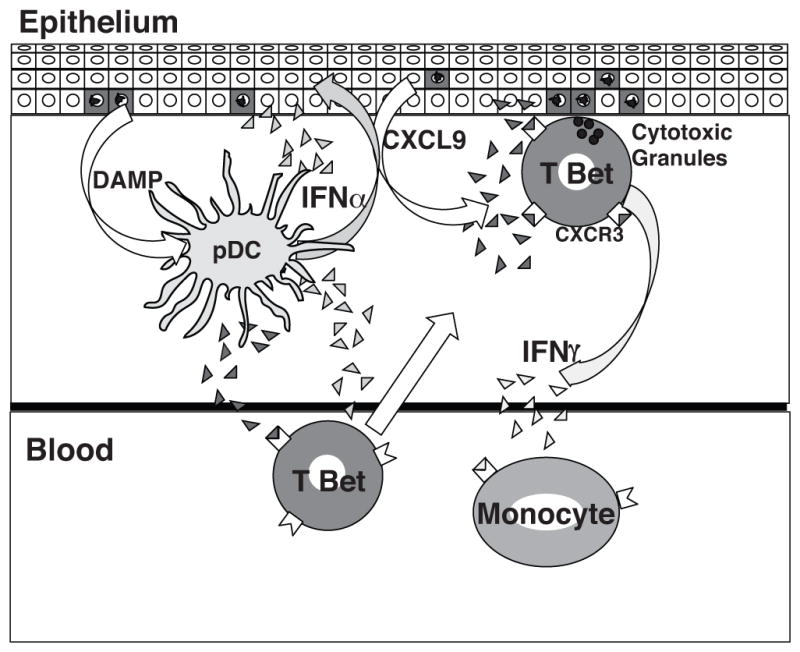
Model of initiation of CGVHD in tissues. Receptors for DAMP molecules (NLR, TLR and CLR) in pDC are triggered by nucleic acids and other alarmins released by epithelial cells damaged by conditioning, AGVHD, viral reactivation or local physical stress, stimulating IFNα production (light gray triangles). IFNα then induces production of chemokines CXCL9 (dark gray triangles) in myeloid cells and in the epithelial keratinocytes that attract T-bet+ Th1/Tc1 cells. These cells in turn both add IFNγ to local IFN levels and differentiate into cytotoxic effectors that continue the cycle of damage. Both IFNα and IFNγ generated in the tissues contribute to upregulation of IFN inducible genes in circulating monocytes.
Our evidence of increases in the levels of inflammasome-associated genes (AIM2, IFI16, CASP1, NLRC4) supports speculation of broader T lineage involvement (Figure 3A–C). Activation of inflammasomes can increase availability of IL-1β. The combination of IL-6 and IL-1β, both induced by response to damaged cells, have been found to be the critical cytokine combination supporting differentiation of pathogenic human Th17 cells (35–37). The potency of inflammasome activation has been demonstrated in AGVHD, wherein blockade of NLRP3 generation of IL-1β reduced Th17 differentiation and AGVHD (29, 30, 38). We would speculate that inflammasome-mediated induction of increased IL-1β in CGVHD may similarly contribute to the differentiation of the Th17 cells that have now been identified in CGVHD tissues, just as Type I IFN can skew lineage commitment toward Th1/Tc1 (4, 39, 40). The combined presence of Th1/Tc1 and Th17 cells in turn would exacerbate tissue damage, sustaining innate immune activation.
Murine studies of AGVHD have provided convincing evidence that transplant regimen-induced tissue damage and exposure to gut microbial antigens can initiate T cell activation and tissue infiltration, through involvement of inflammasomes and IFN-inducible pathways (29, 30, 38, 41). Recent work on well-established murine models of sclerotic CGVHD (B10.D2->BALB/c) using IFNγ−/− and anti-IFNAR blockade have similarly demonstrated a requirement an early requirement for Type I and Type II IFN signaling (42, 43). Tissue infiltrates of predominately Th1/Tc1 effectors were followed by an increased presence of cells with mixed IFN and Th17 production (43–45). Indeed the persistence of Teffectors and full development of sclerotic CGVHD in a comparable model (DBA/2->BALB/c) was dependent not only upon initial Th1 and Th17 cells, but upon B cells, as well, for APC function and autoantibody deposition in skin(45). Analysis of autoantibodies in plasma from the NIH CGVHD natural history cohort demonstrated more than half the patients developed one or more autoantibodies against molecules released by damaged cells (antibodies against nuclei, DNA, RNA, mitochondria, neutrophil cytoplasm (ANCA))(34). These studies support a model of a serial recruitment of multiple T and B effector lineages to produce the full manifestations of CGVHD.
Clinical analyses of CGVHD risk factors suggest that activation of the innate immune system by cellular damage after allo-HSCT may be a continuing factor that contributes to the initiation and persistence of GVHD. Total body irradiation is a risk factor in the later development of sclerotic CGVHD (46). AGVHD and CMV infection, both sources of cell destruction and induction of Type I and Type II IFN, are established cofactors in the development of CGVHD (47, 48). Indeed, localized areas of minor tissue damage after allo-HSCT – mild pressure from waistbands and bra straps, repeated blood draws, targeted irradiation and localized skin lesions from varicella – have all been associated with localized sclerotic CGVHD (49). Effects of earlier immune activation by these factors may not be apparent clinically. Two thirds of the established CGVHD patients included in this study had quiescent or de novo onset (Table I); only one third had progressive CGVHD, developing out of AGVHD seguing into CGVHD.
The limitation of the reporter-monocyte approach is that the gene expression profiles primarily reflect the early steps of monocyte differentiation. These relatively undifferentiated monocytes do not represent the complex populations of myeloid-derived cells in CGVHD affected tissue. Plasmacytoid and myeloid dendritic cells, and specialized scavenger and CX3CR1+ inflammatory macrophages are key elements of CGVHD infiltrates in tissues (4, 13). The precursors of these cells are often depleted in the circulation of CGVHD patients (see dendritic cells in Supplemental Figure S4 A,B) (13). Alterations in gene patterns that occur during terminal differentiation of myeloid infiltrates in tissues may not be present in circulating monocytes. TGFβ, PDGF and IL-4/IL-13 have all been proposed to play a role in the fibrosis affecting skin and other organs in CGVHD (50, 51). These pathways were not upregulated in microarray or Nanostring assays on monocytes, even within severely sclerotic patients, nor were Th2-recruiting chemokines increased in plasma (Supplemental Figures S4 C,D,E). These results cannot, however, exclude later involvement of these pathways in tissues. Further examination of changes in gene expression in the tissues is needed.
The objective of this study was to identify the common pathways that contribute to the full spectrum of manifestations of CGVHD. We have demonstrated a pattern of overexpression of IFN-inducible factors and DAMP receptors, in both erythematous and sclerotic patients, that aligns this model with current transcriptome analyses of systemic autoimmune disorders, including both systemic lupus erythematosus and systemic sclerosis (52, 53). IFNα treatment exacerbates disease in lupus-prone murine models (54, 55), and produces lupus-like rashes and anti-nuclear antibodies in man (56). Similarly, use of IFNα after transplant to treat patients relapsing with chronic myelogenous leukemia has been reported to produce severe CGVHD (57, 58). The commonalities between CGVHD and systemic autoimmune disorders are particularly relevant to the development of new approaches to CGVHD therapy. Recent clinical trials of SLE patients utilizing monoclonal antibodies against Type I IFN have demonstrated a marked decline in leukocyte transcripts of IFN-inducible genes, as well as plasma levels of CXCL10 and BAFF levels, concurrent with improvements in SLE symptoms (SLEDAI scale) (59, 60). Use of an anti Type I IFN receptor (IFNAR) has reduced Th1 activation and collagen production in preliminary trials in Systemic Sclerosis (SSc), and similar use of anti-IFNAR blockade has prevented skin and vascular changes in a sclerotic CGVHD murine model of SSc (42, 61). Furthermore, blockade of STAT1 signaling through JAK1/2 inhibitors (which block both IFN and IL-6 signaling) has shown much promise in both murine GVHD and clinical trials in AGVHD and CGVHD (62–64). Demonstration of the involvement of IFN-dependent pathways in the initiation and persistence of CGVHD may therefore lead both to new treatment modalities and preventive strategies.
Supplementary Material
Acknowledgments
Research Support: This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH. The Nanostring assays were supported by a National Cancer Institute Director’s Intramural Innovation Award to FTH.
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH; the Nanostring assays were supported by a National Cancer Institute Director’s Intramural Innovation Award to FTH. We would like to thank the clinical staff of Experimental Transplantation Branch and the multi-institute CGVHD clinical team for their continuing support of CGVHD patient evaluation and care, and the students and staff of the Preclinical Development and Clinical Monitoring Core for supporting the collection and archiving of patient materials. Finally we are grateful to all the patients who have participated in this study.
Footnotes
Presented in abstract form at the 57th annual meeting of the American Society of Hematology, Orlando, FL on December 7, 2015.
Authorship:
Contributions: SM, PJ, HW, DS and SD performed molecular assays, analyzed gene expression and identified pathways. XYY, VK and WT prepared sorted monocytes for transcriptome analyses. JR, SD, FTH performed flow cytometric analyses. NR and JD performed cytokine assays. MMI and JWM performed immunohistochemistry. MMI, JWM, EWC and HN clinically evaluated oral and cutaneous CGVHD and collected biopsy materials used in this study. DF, DH and SZP provided clinical care and evaluated CGVHD in transplant patients studied at serial time points. KB provided pediatric CGVHD evaluations. SZP established and has overseen the continuing cGVHD natural history study. REG and SZP provided critical advice on study design and data interpretation. FTH designed and supervised the research, performed statistical analyses, prepared figures and tables and wrote the manuscript. All authors reviewed and commented on the manuscript.
COI disclosure: The authors declare that they have no conflicts of interest.
The online version of this article contains a data supplement.
The publisher or recipient acknowledges the right of the US government to retain a nonexclusive, royalty-free license in and to any copyright covering the article.
References
- 1.Arai S, Arora M, Wang T, Spellman SR, He W, Couriel DR, Urbano-Ispizua A, Cutler CS, Bacigalupo AA, Battiwalla M, Flowers ME, Juckett MB, Lee SJ, Loren AW, Klumpp TR, Prockup SE, Ringden OT, Savani BN, Socie G, Schultz KR, Spitzer T, Teshima T, Bredeson CN, Jacobsohn DA, Hayashi RJ, Drobyski WR, Frangoul HA, Akpek G, Ho VT, Lewis VA, Gale RP, Koreth J, Chao NJ, Aljurf MD, Cooper BW, Laughlin MJ, Hsu JW, Hematti P, Verdonck LF, Solh MM, Norkin M, Reddy V, Martino R, Gadalla S, Goldberg JD, McCarthy PL, Perez-Simon JA, Khera N, Lewis ID, Atsuta Y, Olsson RF, Saber W, Waller EK, Blaise D, Pidala JA, Martin PJ, Satwani P, Bornhauser M, Inamoto Y, Weisdorf DJ, Horowitz MM, Pavletic SZ and C. Graft-vs-Host Disease Working Committee of the. Increasing incidence of chronic graft-versus-host disease in allogeneic transplantation: a report from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant. 2015;21:266–274. doi: 10.1016/j.bbmt.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Filipovich AH, Weisdorf D, Pavletic S, Socie G, Wingard JR, Lee SJ, Martin P, Chien J, Przepiorka D, Couriel D, Cowen EW, Dinndorf P, Farrell A, Hartzman R, Henslee-Downey J, Jacobsohn D, McDonald G, Mittleman B, Rizzo JD, Robinson M, Schubert M, Schultz K, Shulman H, Turner M, Vogelsang G, Flowers ME. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant. 2005;11:945–956. doi: 10.1016/j.bbmt.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Imanguli MM, Swaim WD, League SC, Gress RE, Pavletic SZ, Hakim FT. Increased T-bet+ cytotoxic effectors and type I interferon-mediated processes in chronic graft-versus-host disease of the oral mucosa. Blood. 2009;113:3620–3630. doi: 10.1182/blood-2008-07-168351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruggen MC, Klein I, Greinix H, Bauer W, Kuzmina Z, Rabitsch W, Kalhs P, Petzelbauer P, Knobler R, Stingl G, Stary G. Diverse T-cell responses characterize the different manifestations of cutaneous graft-versus-host disease. Blood. 2014;123:290–299. doi: 10.1182/blood-2013-07-514372. [DOI] [PubMed] [Google Scholar]
- 5.Croudace JE, Inman CF, Abbotts BE, Nagra S, Nunnick J, Mahendra P, Craddock C, Malladi R, Moss PA. Chemokine-mediated tissue recruitment of CXCR3+ CD4+ T cells plays a major role in the pathogenesis of chronic GVHD. Blood. 2012;120:4246–4255. doi: 10.1182/blood-2012-02-413260. [DOI] [PubMed] [Google Scholar]
- 6.Imanguli MM, Cowen EW, Rose J, Dhamala S, Swaim W, Lafond S, Yagi B, Gress RE, Pavletic SZ, Hakim FT. Comparative analysis of FoxP3 regulatory T cells in the target tissues and blood in chronic graft versus host disease. Leukemia. 2014 doi: 10.1038/leu.2014.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitko CL, Levine JE, Storer BE, Chai X, Fox DA, Braun TM, Couriel DR, Martin PJ, Flowers ME, Hansen JA, Chang L, Conlon M, Fiema BJ, Morgan R, Pongtornpipat P, Lamiman K, Ferrara JL, Lee SJ, Paczesny S. Plasma CXCL9 elevations correlate with chronic GVHD diagnosis. Blood. 2014;123:786–793. doi: 10.1182/blood-2013-08-520072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wenzel J, Lucas S, Zahn S, Mikus S, Metze D, Stander S, von Stebut E, Hillen U, Bieber T, Tuting T. CXCR3 <-> ligand-mediated skin inflammation in cutaneous lichenoid graft-versus-host disease. J Am Acad Dermatol. 2008;58:437–442. doi: 10.1016/j.jaad.2007.10.647. [DOI] [PubMed] [Google Scholar]
- 9.Kariminia A, Holtan SG, Ivison S, Rozmus J, Hebert MJ, Martin PJ, Lee SJ, Wolff D, Subrt P, Abdossamadi S, Sung S, Storek J, Levings M, Aljurf M, Arora M, Cutler C, Gallagher G, Kuruvilla J, Lipton J, Nevill TJ, Newell LF, Panzarella T, Pidala J, Popradi G, Szwajcer D, Tay J, Toze CL, Walker I, Couban S, Storer BE, Schultz KR. Heterogeneity of chronic graft-versus-host disease biomarkers: association with CXCL10 and CXCR3+ NK cells. Blood. 2016;127:3082–3091. doi: 10.1182/blood-2015-09-668251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paczesny S, Abu Zaid M. CXCL10: most consistent cGVHD biomarker? Blood. 2016;127:2950–2951. doi: 10.1182/blood-2016-04-709543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu J, Storer BE, Kushekhar K, Abu Zaid M, Zhang Q, Gafken PR, Ogata Y, Martin PJ, Flowers ME, Hansen JA, Arora M, Cutler C, Jagasia M, Pidala J, Hamilton BK, Chen GL, Pusic I, Lee SJ, Paczesny S. Biomarker Panel for Chronic Graft-Versus-Host Disease. J Clin Oncol. 2016;34:2583–2590. doi: 10.1200/JCO.2015.65.9615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177:7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 13.Namba N, Shinagawa K, Fujii N, Maeda Y, Ishimaru F, Ikeda K, Matsui T, Tanimoto M, Katayama Y. Predominant infiltration of monocytes in chronic graft-versus-host disease. Transplantation. 2007;83:220–224. doi: 10.1097/01.tp.0000245080.71722.87. [DOI] [PubMed] [Google Scholar]
- 14.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic acids research. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, James JJ, Maysuria M, Mitton JD, Oliveri P, Osborn JL, Peng T, Ratcliffe AL, Webster PJ, Davidson EH, Hood L, Dimitrov K. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 16.Sanda C, Weitzel P, Tsukahara T, Schaley J, Edenberg HJ, Stephens MA, McClintick JN, Blatt LM, Li L, Brodsky L, Taylor MW. Differential gene induction by type I and type II interferons and their combination. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. 2006;26:462–472. doi: 10.1089/jir.2006.26.462. [DOI] [PubMed] [Google Scholar]
- 17.Waddell SJ, Popper SJ, Rubins KH, Griffiths MJ, Brown PO, Levin M, Relman DA. Dissecting interferon-induced transcriptional programs in human peripheral blood cells. PloS one. 2010;5:e9753. doi: 10.1371/journal.pone.0009753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flynn R, Allen JL, Luznik L, MacDonald KP, Paz K, Alexander KA, Vulic A, Du J, Panoskaltsis-Mortari A, Taylor PA, Poe JC, Serody JS, Murphy WJ, Hill GR, Maillard I, Koreth J, Cutler CS, Soiffer RJ, Antin JH, Ritz J, Chao NJ, Clynes RA, Sarantopoulos S, Blazar BR. Targeting Syk-activated B cells in murine and human chronic graft-versus-host disease. Blood. 2015;125:4085–4094. doi: 10.1182/blood-2014-08-595470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujii H, Cuvelier G, She K, Aslanian S, Shimizu H, Kariminia A, Krailo M, Chen Z, McMaster R, Bergman A, Goldman F, Grupp SA, Wall DA, Gilman AL, Schultz KR. Biomarkers in newly diagnosed pediatric-extensive chronic graft-versus-host disease: a report from the Children's Oncology Group. Blood. 2008;111:3276–3285. doi: 10.1182/blood-2007-08-106286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarantopoulos S, Stevenson KE, Kim HT, Bhuiya NS, Cutler CS, Soiffer RJ, Antin JH, Ritz J. High levels of B-cell activating factor in patients with active chronic graft-versus-host disease. Clin Cancer Res. 2007;13:6107–6114. doi: 10.1158/1078-0432.CCR-07-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarantopoulos S, Stevenson KE, Kim HT, Cutler CS, Bhuiya NS, Schowalter M, Ho VT, Alyea EP, Koreth J, Blazar BR, Soiffer RJ, Antin JH, Ritz J. Altered B cell homeostasis and excess BAFF in human chronic graft versus host disease. Blood. 2009;113:3865–3874. doi: 10.1182/blood-2008-09-177840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harigai M, Kawamoto M, Hara M, Kubota T, Kamatani N, Miyasaka N. Excessive production of IFN-gamma in patients with systemic lupus erythematosus and its contribution to induction of B lymphocyte stimulator/B cell-activating factor/TNF ligand superfamily-13B. J Immunol. 2008;181:2211–2219. doi: 10.4049/jimmunol.181.3.2211. [DOI] [PubMed] [Google Scholar]
- 23.Gorelik L, Gilbride K, Dobles M, Kalled SL, Zandman D, Scott ML. Normal B cell homeostasis requires B cell activation factor production by radiation-resistant cells. J Exp Med. 2003;198:937–945. doi: 10.1084/jem.20030789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kreuzaler M, Rauch M, Salzer U, Birmelin J, Rizzi M, Grimbacher B, Plebani A, Lougaris V, Quinti I, Thon V, Litzman J, Schlesier M, Warnatz K, Thiel J, Rolink AG, Eibel H. Soluble BAFF levels inversely correlate with peripheral B cell numbers and the expression of BAFF receptors. J Immunol. 2012;188:497–503. doi: 10.4049/jimmunol.1102321. [DOI] [PubMed] [Google Scholar]
- 25.Jacobson CA, Sun L, Kim HT, McDonough SM, Reynolds CG, Schowalter M, Koreth J, Cutler CS, Ho VT, Alyea EP, Armand P, Blazar BR, Soiffer RJ, Antin JH, Ritz J, Sarantopoulos S. Post-transplantation B cell activating factor and B cell recovery before onset of chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2014;20:668–675. doi: 10.1016/j.bbmt.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol. 2008;9:1179–1188. doi: 10.1038/ni.1651. [DOI] [PubMed] [Google Scholar]
- 28.Maderna P, Cottell DC, Toivonen T, Dufton N, Dalli J, Perretti M, Godson C. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24:4240–4249. doi: 10.1096/fj.10-159913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jankovic D, Ganesan J, Bscheider M, Stickel N, Weber FC, Guarda G, Follo M, Pfeifer D, Tardivel A, Ludigs K, Bouazzaoui A, Kerl K, Fischer JC, Haas T, Schmitt-Graff A, Manoharan A, Muller L, Finke J, Martin SF, Gorka O, Peschel C, Ruland J, Idzko M, Duyster J, Holler E, French LE, Poeck H, Contassot E, Zeiser R. The Nlrp3 inflammasome regulates acute graft-versus-host disease. J Exp Med. 2013;210:1899–1910. doi: 10.1084/jem.20130084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilhelm K, Ganesan J, Muller T, Durr C, Grimm M, Beilhack A, Krempl CD, Sorichter S, Gerlach UV, Juttner E, Zerweck A, Gartner F, Pellegatti P, Di Virgilio F, Ferrari D, Kambham N, Fisch P, Finke J, Idzko M, Zeiser R. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med. 2010;16:1434–1438. doi: 10.1038/nm.2242. [DOI] [PubMed] [Google Scholar]
- 31.van de Garde MD, Martinez FO, Melgert BN, Hylkema MN, Jonkers RE, Hamann J. Chronic exposure to glucocorticoids shapes gene expression and modulates innate and adaptive activation pathways in macrophages with distinct changes in leukocyte attraction. J Immunol. 2014;192:1196–1208. doi: 10.4049/jimmunol.1302138. [DOI] [PubMed] [Google Scholar]
- 32.Ehrchen J, Steinmuller L, Barczyk K, Tenbrock K, Nacken W, Eisenacher M, Nordhues U, Sorg C, Sunderkotter C, Roth J. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood. 2007;109:1265–1274. doi: 10.1182/blood-2006-02-001115. [DOI] [PubMed] [Google Scholar]
- 33.Patriarca F, Skert C, Sperotto A, Zaja F, Falleti E, Mestroni R, Kikic F, Calistri E, Fili C, Geromin A, Cerno M, Fanin R. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389–396. doi: 10.1016/j.exphem.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 34.Kuzmina Z, Gounden V, Curtis L, Avila D, Taylor T, Baruffaldi J, Cowen EW, Naik HB, Hasni SA, Mays JW, Mitchell S, Baird K, Steinberg SM, Pavletic SZ. Clinical significance of autoantibodies in a large cohort of patients with chronic graft-versus-host disease defined by NIH criteria. American journal of hematology. 2015;90:114–119. doi: 10.1002/ajh.23885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cosmi L, De Palma R, Santarlasci V, Maggi L, Capone M, Frosali F, Rodolico G, Querci V, Abbate G, Angeli R, Berrino L, Fambrini M, Caproni M, Tonelli F, Lazzeri E, Parronchi P, Liotta F, Maggi E, Romagnani S, Annunziato F. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J Exp Med. 2008;205:1903–1916. doi: 10.1084/jem.20080397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O'Shea JJ. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeiser R, Penack O, Holler E, Idzko M. Danger signals activating innate immunity in graft-versus-host disease. J Mol Med (Berl) 2011;89:833–845. doi: 10.1007/s00109-011-0767-x. [DOI] [PubMed] [Google Scholar]
- 39.Amarnath S, Flomerfelt FA, Costanzo CM, Foley JE, Mariotti J, Konecki DM, Gangopadhyay A, Eckhaus M, Wong S, Levine BL, June CH, Fowler DH. Rapamycin generates anti-apoptotic human Th1/Tc1 cells via autophagy for induction of xenogeneic GVHD. Autophagy. 2010;6:523–541. doi: 10.4161/auto.6.4.11811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malard F, Bossard C, Brissot E, Chevallier P, Guillaume T, Delaunay J, Mosnier JF, Moreau P, Gregoire M, Gaugler B, Mohty M. Increased Th17/Treg ratio in chronic liver GVHD. Bone Marrow Transplant. 2014;49:539–544. doi: 10.1038/bmt.2013.215. [DOI] [PubMed] [Google Scholar]
- 41.Choi J, Ziga ED, Ritchey J, Collins L, Prior JL, Cooper ML, Piwnica-Worms D, DiPersio JF. IFNgammaR signaling mediates alloreactive T-cell trafficking and GVHD. Blood. 2012;120:4093–4103. doi: 10.1182/blood-2012-01-403196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Delaney TA, Morehouse C, Brohawn PZ, Groves C, Colonna M, Yao Y, Sanjuan M, Coyle AJ. Type I IFNs Regulate Inflammation, Vasculopathy, and Fibrosis in Chronic Cutaneous Graft-versus-Host Disease. J Immunol. 2016 doi: 10.4049/jimmunol.1502190. [DOI] [PubMed] [Google Scholar]
- 43.Nishimori H, Maeda Y, Teshima T, Sugiyama H, Kobayashi K, Yamasuji Y, Kadohisa S, Uryu H, Takeuchi K, Tanaka T, Yoshino T, Iwakura Y, Tanimoto M. Synthetic retinoid Am80 ameliorates chronic graft-versus-host disease by down-regulating Th1 and Th17. Blood. 2012;119:285–295. doi: 10.1182/blood-2011-01-332478. [DOI] [PubMed] [Google Scholar]
- 44.Okamoto S, Fujiwara H, Nishimori H, Matsuoka K, Fujii N, Kondo E, Tanaka T, Yoshimura A, Tanimoto M, Maeda Y. Anti-IL-12/23 p40 antibody attenuates experimental chronic graft-versus-host disease via suppression of IFN-gamma/IL-17-producing cells. J Immunol. 2015;194:1357–1363. doi: 10.4049/jimmunol.1400973. [DOI] [PubMed] [Google Scholar]
- 45.Jin H, Ni X, Deng R, Song Q, Young J, Cassady K, Zhang M, Forman S, Martin PJ, Liu Q, Zeng D. Antibodies from donor B cells perpetuate cutaneous chronic graft-versus-host disease in mice. Blood. 2016;127:2249–2260. doi: 10.1182/blood-2015-09-668145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martires KJ, Baird K, Steinberg SM, Grkovic L, Joe GO, Williams KM, Mitchell SA, Datiles M, Hakim FT, Pavletic SZ, Cowen EW. Sclerotic-type chronic GVHD of the skin: clinical risk factors, laboratory markers, and burden of disease. Blood. 2011;118:4250–4257. doi: 10.1182/blood-2011-04-350249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wojnar J, Giebel S, Holowiecka-Goral A, Krawczyk-Kulis M, Markiewicz M, Wozniczka K, Holowiecki J. The incidence and risk factors for chronic graft-versus-host-disease. Annals of transplantation : quarterly of the Polish Transplantation Society. 2006;11:14–20. discussion 32–43. [PubMed] [Google Scholar]
- 48.Remberger M, Kumlien G, Aschan J, Barkholt L, Hentschke P, Ljungman P, Mattsson J, Svennilson J, Ringden O. Risk factors for moderate-to-severe chronic graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2002;8:674–682. doi: 10.1053/bbmt.2002.v8.abbmt080674. [DOI] [PubMed] [Google Scholar]
- 49.Martires KJ, Baird K, Citrin DE, Hakim FT, Pavletic SZ, Cowen EW. Localization of Sclerotic-type Chronic Graft-vs-Host Disease to Sites of Skin Injury: Potential Insight Into the Mechanism of Isomorphic and Isotopic Responses. Archives of dermatology. 2011;147:1081–1086. doi: 10.1001/archdermatol.2011.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol. 2012;12:443–458. doi: 10.1038/nri3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yi T, Chen Y, Wang L, Du G, Huang D, Zhao D, Johnston H, Young J, Todorov I, Umetsu DT, Chen L, Iwakura Y, Kandeel F, Forman S, Zeng D. Reciprocal differentiation and tissue-specific pathogenesis of Th1, Th2, and Th17 cells in graft-versus-host disease. Blood. 2009;114:3101–3112. doi: 10.1182/blood-2009-05-219402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Assassi S, Mayes MD, Arnett FC, Gourh P, Agarwal SK, McNearney TA, Chaussabel D, Oommen N, Fischbach M, Shah KR, Charles J, Pascual V, Reveille JD, Tan FK. Systemic sclerosis and lupus: points in an interferon-mediated continuum. Arthritis and rheumatism. 2010;62:589–598. doi: 10.1002/art.27224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, Brohawn P, Kiener PA, Richman L, Fiorentino D, Greenberg SA, Jallal B, Yao Y. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Annals of the rheumatic diseases. 2011;70:2029–2036. doi: 10.1136/ard.2011.150326. [DOI] [PubMed] [Google Scholar]
- 54.Jacob N, Guo S, Mathian A, Koss MN, Gindea S, Putterman C, Jacob CO, Stohl W. B Cell and BAFF dependence of IFN-alpha-exaggerated disease in systemic lupus erythematosus-prone NZM 2328 mice. J Immunol. 2011;186:4984–4993. doi: 10.4049/jimmunol.1000466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, Theofilopoulos AN. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med. 2003;197:777–788. doi: 10.1084/jem.20021996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho V, McLean A, Terry S. Severe systemic lupus erythematosus induced by antiviral treatment for hepatitis C. Journal of clinical rheumatology : practical reports on rheumatic & musculoskeletal diseases. 2008;14:166–168. doi: 10.1097/RHU.0b013e3181775e80. [DOI] [PubMed] [Google Scholar]
- 57.Hamaki T, Kami M, Momomura S, Mineishi S, Kusumi E, Kanda Y, Ueyama J, Miyakoshi S, Morinaga S, Takaue Y, Mutou Y. Sustained molecular remission in a patient with CML in blastic crisis receiving dose-reduced hematopoietic stem-cell transplantation followed by early withdrawal of cyclosporine and prophylactic use of interferon-alpha. American journal of hematology. 2002;71:196–199. doi: 10.1002/ajh.10203. [DOI] [PubMed] [Google Scholar]
- 58.Serrano J, Prieto E, Mazarbeitia F, Roman A, Llamas P, Tomas JF. Atypical chronic graft-versus-host disease following interferon therapy for chronic myeloid leukaemia relapsing after allogeneic BMT. Bone Marrow Transplant. 2001;27:85–87. doi: 10.1038/sj.bmt.1702746. [DOI] [PubMed] [Google Scholar]
- 59.Merrill JT, Wallace DJ, Petri M, Kirou KA, Yao Y, White WI, Robbie G, Levin R, Berney SM, Chindalore V, Olsen N, Richman L, Le C, Jallal B, White B. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon alpha monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised study. Annals of the rheumatic diseases. 2011;70:1905–1913. doi: 10.1136/ard.2010.144485. [DOI] [PubMed] [Google Scholar]
- 60.Petri M, Wallace DJ, Spindler A, Chindalore V, Kalunian K, Mysler E, Neuwelt CM, Robbie G, White WI, Higgs BW, Yao Y, Wang L, Ethgen D, Greth W. Sifalimumab, a human anti-interferon-alpha monoclonal antibody, in systemic lupus erythematosus: a phase I randomized, controlled, dose-escalation study. Arthritis and rheumatism. 2013;65:1011–1021. doi: 10.1002/art.37824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo X, Higgs BW, Bay-Jensen AC, Karsdal MA, Yao Y, Roskos LK, White WI. Suppression of T Cell Activation and Collagen Accumulation by an Anti-IFNAR1 mAb, Anifrolumab, in Adult Patients with Systemic Sclerosis. J Invest Dermatol. 2015;135:2402–2409. doi: 10.1038/jid.2015.188. [DOI] [PubMed] [Google Scholar]
- 62.Kontzias A, Kotlyar A, Laurence A, Changelian P, O'Shea JJ. Jakinibs: a new class of kinase inhibitors in cancer and autoimmune disease. Current opinion in pharmacology. 2012;12:464–470. doi: 10.1016/j.coph.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, Verbeek M, Fischer J, Otten V, Schmickl M, Maas-Bauer K, Finke J, Peschel C, Duyster J, Poeck H, Zeiser R, von Bubnoff N. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood. 2014;123:3832–3842. doi: 10.1182/blood-2013-12-543736. [DOI] [PubMed] [Google Scholar]
- 64.Zeiser R, Burchert A, Lengerke C, Verbeek M, Maas-Bauer K, Metzelder SK, Spoerl S, Ditschkowski M, Ecsedi M, Sockel K, Ayuk F, Ajib S, de Fontbrune FS, Na IK, Penter L, Holtick U, Wolf D, Schuler E, Meyer E, Apostolova P, Bertz H, Marks R, Lubbert M, Wasch R, Scheid C, Stolzel F, Ordemann R, Bug G, Kobbe G, Negrin R, Brune M, Spyridonidis A, Schmitt-Graff A, van der Velden W, Huls G, Mielke S, Grigoleit GU, Kuball J, Flynn R, Ihorst G, Du J, Blazar BR, Arnold R, Kroger N, Passweg J, Halter J, Socie G, Beelen D, Peschel C, Neubauer A, Finke J, Duyster J, von Bubnoff N. Ruxolitinib in corticosteroid-refractory graft-versus-host disease after allogeneic stem cell transplantation: a multicenter survey. Leukemia. 2015;29:2062–2068. doi: 10.1038/leu.2015.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.