

ABSTRACT
Hepatitis C virus (HCV) species tropism is incompletely understood. We have previously shown that at the level of entry, human CD81 and occludin (OCLN) comprise the minimal set of human factors needed for viral uptake into murine cells. As an alternative approach to genetic humanization, species barriers can be overcome by adapting HCV to use the murine orthologues of these entry factors. We previously generated a murine tropic HCV (mtHCV or Jc1/mCD81) strain harboring three mutations within the viral envelope proteins that allowed productive entry into mouse cell lines. In this study, we aimed to characterize the ability of mtHCV to enter and infect mouse hepatocytes in vivo and in vitro. Using a highly sensitive, Cre-activatable reporter, we demonstrate that mtHCV can enter mouse hepatocytes in vivo in the absence of any human cofactors. Viral entry still relied on expression of mouse CD81 and SCARB1 and was more efficient when mouse CD81 and OCLN were overexpressed. HCV entry could be significantly reduced in the presence of anti-HCV E2 specific antibodies, suggesting that uptake of mtHCV is dependent on viral glycoproteins. Despite mtHCV’s ability to enter murine hepatocytes in vivo, we did not observe persistent infection, even in animals with severely blunted type I and III interferon signaling and impaired adaptive immune responses. Altogether, these results establish proof of concept that the barriers limiting HCV species tropism can be overcome by viral adaptation. However, additional viral adaptations will likely be needed to increase the robustness of a murine model system for hepatitis C.
IMPORTANCE
At least 150 million individuals are chronically infected with HCV and are at risk of developing serious liver disease. Despite the advent of effective antiviral therapy, the frequency of chronic carriers has only marginally decreased. A major roadblock in developing a vaccine that would prevent transmission is the scarcity of animal models that are susceptible to HCV infection. It is poorly understood why HCV infects only humans and chimpanzees. To develop an animal model for hepatitis C, previous efforts focused on modifying the host environment of mice, for example, to render them more susceptible to HCV infection. Here, we attempted a complementary approach in which a laboratory-derived HCV variant was tested for its ability to infect mice. We demonstrate that this engineered HCV strain can enter mouse liver cells but does not replicate efficiently. Thus, additional adaptations are likely needed to construct a robust animal model for HCV.
INTRODUCTION
At least 150 million individuals worldwide are chronically infected with hepatitis C virus (HCV). Chronic HCV infection is associated with severe liver disease, including fibrosis, cirrhosis, and hepatocellular carcinoma. Treatment options have drastically improved in recent years, and it is now possible to effectively cure HCV infection in the majority of patients. However, effective ways to prevent infection, for example via vaccination, remain elusive. To systematically test and prioritize vaccine candidates, development of an immunocompetent animal model, optimally with inheritable susceptibility to HCV, remains a high priority.
HCV exhibits a narrow and mechanistically incompletely understood species tropism. While humans and chimpanzees are readily susceptible to HCV infection, most other species—with the exception of intermittent, sporadic viremia in tree shrews—appear to be resistant (reviewed in reference 1). Resistance of mice to HCV is multifactorial and determined at least by blocks in viral entry and replication (reviewed in reference 2). HCV utilizes a large number of host factors to enter its host cells, the human hepatocyte (reviewed in reference 3). These factors include glycosaminoglycans (GAGs) present on heparan sulfate proteoglycans (HSPGs), low-density-lipoprotein receptor (LDLR) (4, 5), CD81 (6), scavenger receptor class B member 1 (SCARB1) (7), the tight junction claudin proteins, including claudin-1 (CLDN1) (8) and CLDN6 and CLDN9 (9), occludin (OCLN) (10, 11), the receptor tyrosine kinases epidermal growth factor receptor (EGFR), and ephrin receptor A2 (EphA2) (12), the cholesterol transporter Niemann-Pick C1-like 1 (NPC1L1) (13), transferrin receptor 1 (TfR1) (14), and the cell death-inducing DFFA-like effector b (CIDEB) (15). It was previously shown that CD81 (6), SCARB1 (7) and the two tight junction proteins CLDN1 (8) and OCLN (10, 11) are all required for uptake into human and also rodent cells but that only CD81 and OCLN need to be of human origin to facilitate this process (11). Indeed, transient adenoviral (16) or stable transgenic (17) expression of human CD81 and OCLN in mouse hepatocytes facilitates HCV uptake. The differential abilities of mouse and human CD81 and OCLN to support viral entry have been mapped to amino acids in their second extracellular loops (11, 18). While infectious HCV particles can assemble in mouse liver cells in vitro (19) and in vivo (17), HCV RNA replication in mice is limited, presumably by a combination of innate antiviral immune responses (17, 20–23) and possibly by poor compatibility between murine orthologues of replication cofactors and the virally encoded components of the HCV replication machinery (1).
Complementary to the host genetic adaptation approach, adaptation of HCV to rodent hosts is an alternative strategy for establishing a mouse model for hepatitis C. We previously used an unbiased selection approach to adapt an HCV genotype 2a strain, Jc1, to use mouse CD81 (24). We identified three adaptive mutations in the HCV envelope proteins E1 and E2 that facilitated uptake into cell lines expressing human SCARB1, CLDN1, OCLN, and mouse, rat, or hamster CD81 (24). The mutations significantly increased the affinity of the virus for the large extracellular loops of human CD81, suggesting an indirect enhancement by exposing a CD81 binding site. The mutations in the mouse CD81 (mCD81)-adapted, i.e., murine tropic, virus (mtHCV or Jc1/mCD81) altered usage of human SCARB1 (hSCARB1) and human OCLN (hOCLN). Blocking antibodies against hSCARB1 and silencing of hOCLN had a less pronounced effect on the entry of the mutant virus compared to the parental strain, suggesting that the mCD81-adapted virus was less dependent on hSCARB1 and hOCLN. Finally, mouse fibroblasts expressing murine CD81, SCARB1, CLDN1, and OCLN supported the uptake of adapted virus. This entry could be blocked with anti-mCD81 antibodies, indicating that the species-specific restriction to human OCLN was altered while dependence on CD81 was maintained.
Here, we aimed to extend this work and directly test whether our mtHCV harboring mutations that facilitate efficient engagement of murine CD81 and OCLN could infect murine primary hepatocytes in vitro and in vivo. We demonstrate that mtHCV can enter mouse hepatocytes in cell culture and that this uptake is dependent on the adaptive mutations present in HCV E1 and E2. Using a sensitive reporter, we show that mtHCV enters mouse hepatocytes in vivo in an HCV glycoprotein-dependent manner. Although hepatic overexpression of mouse CD81 and OCLN enhances HCV entry, uptake is not dependent on ectopic expression, and mtHCV is capable of utilizing the endogenous proteins, albeit at low efficiency. Previous data suggest that innate immunity restricts HCV replication in mouse hepatocytes in vitro (20–22, 25) and in vivo (17). Mice with targeted disruptions of signal transducer and activator of transcription factor 1 (STAT1), which have severely impaired type I and III interferon (IFN) responses, did not develop persistent viremia following infection. Engraftment of STAT1-deficient mouse hepatocytes into immunodeficient mice with liver injuries allowed us to directly test whether murine adaptive immune responses further antagonize HCV replication. However, in the absence of functional B, T, and natural killer cells, mtHCV infection in murine hepatocytes was not further enhanced. Collectively, these data suggest that additional barriers limit propagation of mtHCV in mice. The low uptake efficiency of mtHCV into mouse hepatocytes expressing endogenous levels of viral entry factors may preclude efficient spread, which may be necessary to establish persistence. In addition, the efficiency of postentry steps of the viral life cycle could conceivably be improved with human host factors important for HCV replication, assembly, and/or egress. Thus, additional, currently unknown proviral factors and/or negative regulators antagonizing HCV will have to be identified to enhance HCV replication and assembly in mouse cells.
RESULTS
mtHCV-specific adaptations are maintained upon long-term replication in vivo.
It was previously demonstrated that cell culture adaptive mutations that increased the level of viral replication in vitro could potentially compromise viral fitness in vivo (26). Thus, we aimed to test whether the three adaptive mutations in Jc1/mCD81 affected its viral fitness in vivo and whether they were stably maintained during long-term in vivo replication.
We took advantage of a previously established human liver-uPA-SCID mouse model that stably supports HCV replication (27, 28). Stable engraftment with human hepatocytes was determined by monitoring serum levels of human albumin (data not shown). We infected the mice with 6 × 105 TCID50 (50% tissue culture infective doses) of Jc1 or Jc1/mCD81 and observed stable and comparable HCV RNA levels for both Jc1- and Jc1/mCD81-infected mice for about 56 days (Fig. 1a). Eight weeks after inoculation of Jc1/mCD81, we deep sequenced the genes of the envelope glycoproteins E1 and E2, the signal peptides of E1 and p7. All three adaptive mutations—L216F, V388G, and M405T—were conserved 100% between the viral inoculum (Fig. 1b) and the mouse-passaged virus (Fig. 1c). Taken together with the observation that infection with both Jc1 and Jc1/mCD81 leads to comparable HCV RNA serum levels, these data demonstrate that the three adaptive mutations do not diminish the replication capacity of Jc1/mCD81 in vivo. Furthermore, neither virus acquired any additional adaptive mutation(s) upon long-term in vivo replication.
FIG 1 .

Evolution of mtHCV during long-term replication in uPA-SCID mice repopulated with human hepatocytes. (a) Mice were inoculated with an equal dose of wild-type HCV and mtHCV. Virus plasma levels were determined at the indicated time points postinoculation. (b and c) Evolution of mtHCV sequence during chronic infection in mice. The E1-E2-p7 coding region of the virus inoculum (b) and of the virus population circulating 8 weeks postinoculation (c) was amplified and sequenced using the Roche 454 platform. The x axis represents nucleotide position relative to the reference sequence. The y axis represents nucleotide coverage across the E1-E2-p7 coding region with synonymous (dS) and nonsynonymous (dN) single nucleotide polymorphism (SNP) frequencies in the viral population relative to the reference sequence highlighted. The mtHCV-specific adaptations L216F, V388G, and M405T are marked with red asterisks.
Jc1/mCD81 is capable of entering murine hepatocytes in vitro and in vivo.
We previously demonstrated that ectopic expression of the murine orthologues of the four canonical HCV entry factors—SCARB1, CD81, CLDN1, and OCLN—supports cell entry of HCV pseudoparticles (HCVpp) packaged with envelopes of mtHCV (mtHCVpp) (24). However, initial attempts to quantify mtHCVpp uptake into primary murine hepatocytes in cell culture failed (24). Since HCV RNA replication is inefficient in murine hepatocytes, we adopted a Jc1 genome expressing a Gaussia luciferase reporter [Jc1(p7nsGluc2A)] (29) that allows highly sensitive detection of HCV entry by luciferase secretion following initial translation of the HCV genome. We engineered the three adaptive mutations of mtHCV into Jc1(p7nsGluc2A), yielding Jc1/mCD81(p7nsGluc2A). Infection of mouse primary hepatocytes isolated from C57BL/6 mice with Jc1/mCD81(p7nsGluc2A) resulted in a 2- to 4-fold-higher luciferase signal than infection with the parental virus (Fig. 2a), suggesting that the three adaptive mutations enabled viral uptake, albeit at low efficiency.
FIG 2 .
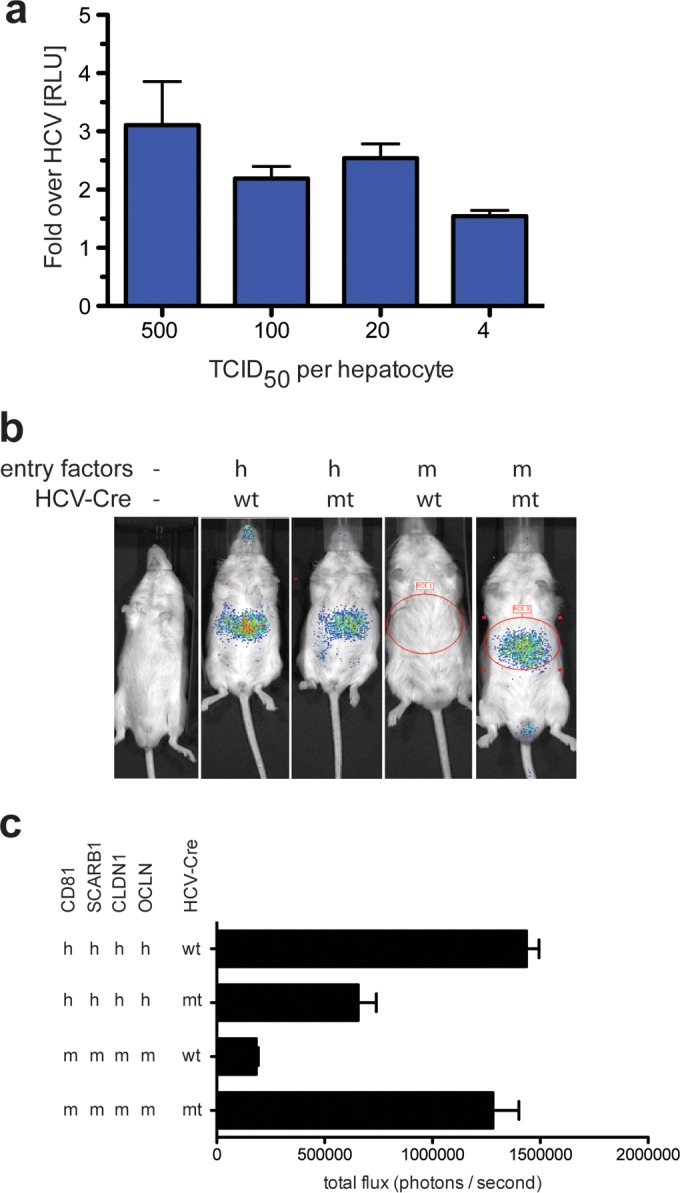
mtHCV can enter mouse hepatocytes in vitro and in vivo. R26-LSL-Fluc mice were injected with 1011 adenoviral particles encoding human (h) or mouse (m) CD81, SCARB1, CLDN, and OCLN. Bioluminescence was measured at 72 h postinjection of 2 × 107 50% tissue culture infectious doses (TCID50) of cell culture-produced murine tropic (mt) or wild-type (wt) HCV-Cre. (a) Primary hepatocytes isolated from C57BL/6 mice were plated on collagen-coated plates, and monolayers were infected with Jc1(p7nsGluc2A) or Jc1/mCD81Gluc2A with the indicated numbers of infectious particles (as titered on Huh-7.5 cells) per primary hepatocyte. Luciferase activity was measured 48 h postinfection. Data are represented as the signal of mtHCV over the parental strain. (b) Representative pseudo-colored images. (c) Quantitation of total photon flux/second (n ≥ 4). Values are means plus standard deviations (SD) (error bars).
To directly test whether mtHCV is capable of entering mouse hepatocytes in vivo, we utilized a highly sensitive reporter in which cellularly encoded firefly luciferase is activated by Cre recombinase expressed in a bicistronic HCV genome configuration (BiCre-Jc1) (16). Analogous to previous studies, we expressed human or mouse CD81, SCARB1, CLDN1, and OCLN via an adenoviral vector in the livers of Rosa26-Fluc mice (30) and infected animals with BiCre-Jc1 or BiCre-Jc1/mCD81. In accordance with previously published data, BiCre-Jc1 entered mouse hepatocytes overexpressing the human orthologues of the canonical entry factors but not mouse hepatocytes overexpressing murine orthologues of the canonical entry factors. In contrast, the bicistronic HCV genome harboring the adaptive mutations in E1 and E2 activated the reporter equally well in mice overexpressing entry factors from either humans or mice (Fig. 2b and c). Collectively, these data demonstrate that the three adaptive mutations in E1 and E2 enable uptake of HCV into mouse primary hepatocytes in vitro and in vivo.
Efficiency of mtHCV uptake in vivo positively correlates with the viral dose and expression levels of HCV entry factors.
To assess the efficiency of mtHCV uptake in vivo, we performed dose-response experiments titrating either the level of overexpression of mouse CD81, SCARB1, CLDN1, and OCLN while keeping the viral inoculum constant (Fig. 3a and b) or overexpressing mouse canonical entry factors and varying the inoculum dose (Fig. 3c and d). Rosa26-Fluc mice were injected intravenously with low (1 × 108 adenoviral [AdV] particles/mouse), intermediate (1 × 109 or 1 × 1010 AdV particles/mouse), or high (1 × 1011 AdV particles/mouse) doses of adenoviral particles or no adenovirus and subsequently infected with cell culture-produced BiCre-Jc1/mCD81 (2 × 107 TCID50/mouse). The highest expression of HCV entry factors enhanced the signal of the luminescent reporter 6- to 7-fold compared with mice that received the lowest dose of HCV entry factors (Fig. 3a and b). These data are in accordance with our previous data demonstrating that uptake of HCV with native E1/E2 glycoproteins is more efficient in mouse hepatocytes expressing high levels of human CD81, SCARB1, CLDN1, and OCLN (16).
FIG 3 .

Efficiency of mtHCV uptake positively correlates with HCV entry factor expression levels and challenge dose. (a and b) Rosa26-Fluc mice were injected with limiting dilutions of adenoviruses encoding mouse CD81 (mCD81), SCARB1, CLDN1, and OCLN prior to infection with 1 × 106 TCID50 mtHCV-Cre. (c and d) Rosa26-LSL-Fluc mice were injected with 1011 particles of adenoviruses encoding mouse CD81, SCARB1, CLDN1, and OCLN or mock injected, followed 24 h later by infection with the indicated doses of mtHCV-Cre or, as a control, PBS (n = 3). (a and c) Representative pseudocolored images; (b and d) quantitation of bioluminescent signal (n = 3). Data were acquired 72 h following HCV infection. Values are means ± SD (error bars).
Of note, even in the absence of ectopic expression of HCV entry factors, mtHCV was still capable of entering mouse hepatocytes, albeit at very low levels (Fig. 3a, left panel). This suggests that endogenous expression levels of HCV entry factors in the mouse liver may suffice for viral uptake.
We observed a similar correlation when mice overexpressing mouse CD81, SCARB1, CLDN1, and OCLN or just CD81 and OCLN were infected with various doses of cell culture-produced BiCre-Jc1/mCD81. The higher the inoculum, the more robustly the luminescent reporter was activated in mice overexpressing murine versions of all four HCV entry factors (ca. 4- to 5-fold over background) (Fig. 3c and d). Because viral uptake relied on endogenous expression of SCARB1 and CLDN1, in mice overexpressing only mouse CD81 and OCLN, the signal was about 3-fold over background. In mice not injected with HCV entry factor-expressing adenoviruses, luminescent reporter activity was even lower, only 2-fold over background levels (Fig. 3d).
Collectively, these data demonstrate that mtHCV entry into murine hepatocytes is more efficient when mouse CD81, SCARB1, CLDN1, and OCLN are not limiting. It should be noted that these constraints are due in part to the detection limit of the Cre-based reporter assay.
Quantification of the frequency of HCV-infected cells.
To estimate the number of HCV-infected liver cells, we used an indicator mouse strain in which Cre leads to activation of a nucleus-localized green fluorescent protein (GFP)/β-galactosidase (GNZ) reporter (Rosa26-GNZ) (31). In approximately 0.07 to 0.09% of mouse hepatocytes, GFP fluorescence was detected when uptake was solely mediated by the endogenous murine entry factors after a challenge with a dose of 2 × 107 TCID50 (Fig. 4). Upon adenoviral overexpression of mouse CD81, SCARB1, CLDN1, and OCLN, the frequency increased roughly 10-fold, reaching levels that were about one-half of those reported in mice expressing human orthologues of the canonical entry factors (1 to 1.5%) (16, 17). Following previously established protocols (32), we also included DNA intercalating dyes (Hoechst) to determine any differences in the ability of mono-, bi-, and multinucleated hepatocytes to support HCV uptake. Infection frequencies were largely equivalent among these subsets of hepatocyte populations, with slightly reduced numbers in multi- versus mono- or binucleated cells.
FIG 4 .
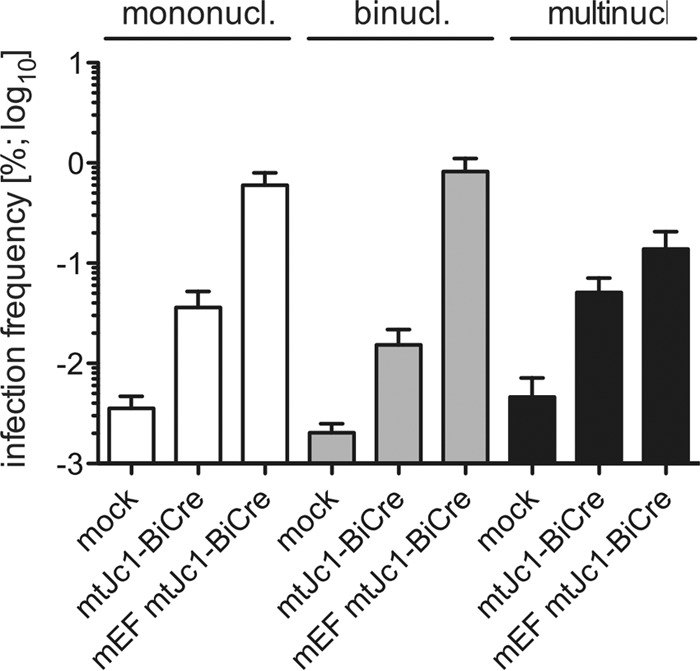
Quantitation of the frequency of mtHCV-infected hepatocytes in vivo. Rosa26-LSL-GNZ mice were injected with 1 × 1011 adenoviral particles encoding mouse entry factors (mEF) or not injected with AdV and subsequently infected with 1 × 106 TCID50 mtHCV-Cre. Seventy-two hours following infection, single-cell suspensions were prepared by liver perfusion with collagenase. Cells were counterstained with DRAQ5 to distinguish between mono-, bi-, and multinucleated (or multiploid) cells. The total frequency of infected hepatocytes within the subpopulations was determined by flow cytometry (n = 3). Values are means plus SD.
mtHCV enters hepatocytes in vivo in a glycoprotein-dependent manner.
To demonstrate that mtHCV entry into mouse hepatocytes is dependent on the interaction of the viral envelope with cellularly encoded receptors, we performed blocking and loss-of-function experiments (Fig. 5). First, we took advantage of the well-characterized broadly neutralizing antibody AR4A, which binds to an epitope in the HCV envelope protein E2 (33). mtHCV was preincubated with various concentrations of either AR4A or an isotype control antibody (B12) and injected into Rosa26-Fluc mice expressing mouse CD81, SCARB1, CLDN1, and OCLN. Blockade with AR4A led to a 30 to 40% decrease in the luciferase signal at the highest antibody concentrations (Fig. 5a). For comparison, preincubation of the parental BiCre-Jc1 strain resulted in comparable, although slightly more effective (60 to 70%), blockade in Rosa26-Fluc mice expressing human CD81, SCARB1, CLDN1, and OCLN (Fig. 5a). These data suggest that uptake of HCV and mtHCV engaging human or mouse HCV entry factors, respectively, is dependent on HCV glycoproteins.
FIG 5 .
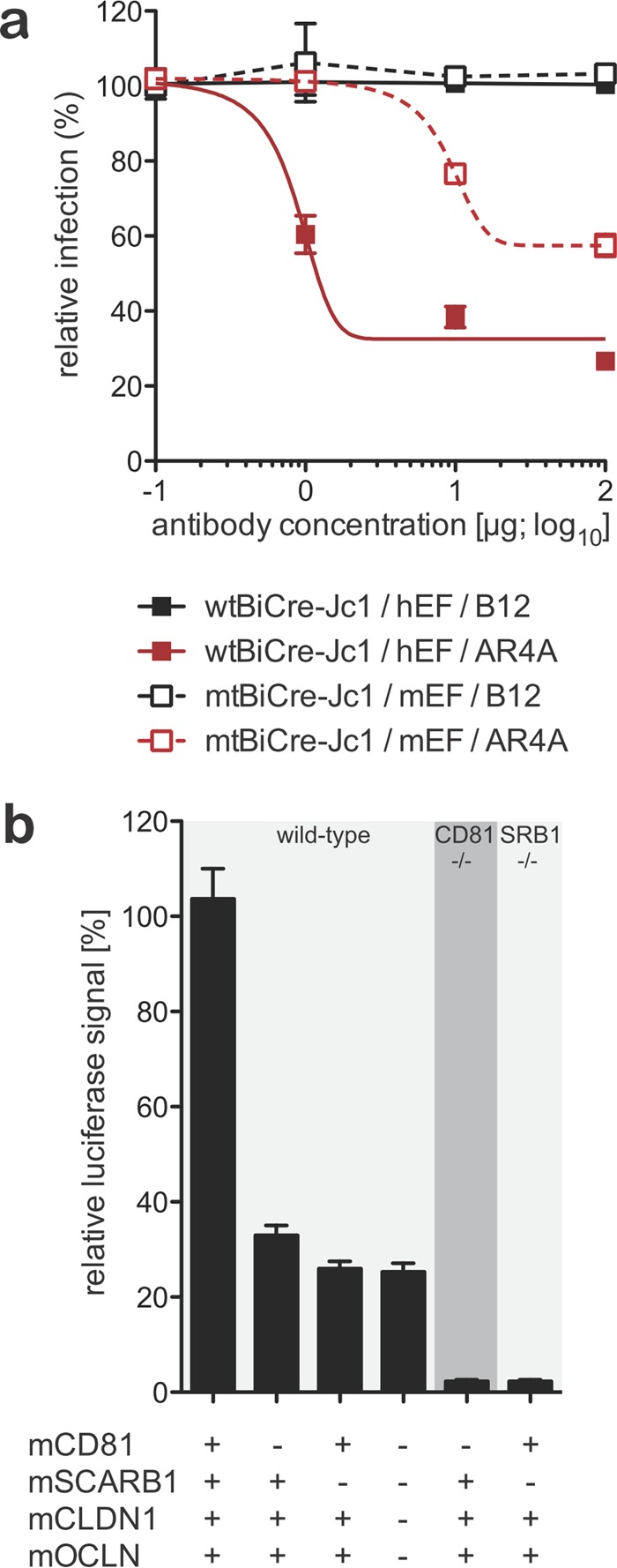
Uptake of mtHCV in vivo can be blocked with anti-HCV E2-specific antibodies and is dependent on endogenous expression of mouse CD81 and SCARB1. (a) R26-LSL-Fluc mice were injected with 1 × 1011 particles of adenoviruses expressing mouse CD81, SCARB1, CLDN1, and OCLN. mtHCV-Cre was incubated with the indicated doses of anti-HCV E2 (clone AR4A) or an anti-HIV isotype control antibody (clone B12) for 1 h prior to injection (1 × 106 TCID50) and 24 h after adenoviral delivery (n = 3). Data were acquired 72 h following HCV infection. Values are means ± SD. (b) Rosa26-Fluc mice were crossed with mCD81−/− or mSCARB1−/− mice, and offspring with the indicated zygosities for the respective mutant alleles were injected with the indicated combinations of AdVs encoding mouse CD81 (mCD81), SCARB1, CLDN1, and OCLN and 24 h later with 2 × 107 TCID50 BiCre-Jc1 (n ≥ 4). Luminescence was quantified 72 h following HCV infection. Values are means plus SD.
To further explore this process, we employed mice with targeted disruptions in the genes encoding CD81 (CD81−/−) or SCARB1 (SCARB1−/−) that we crossed to the Rosa26-Fluc background (Fig. 5b). We focused our analysis on these two molecules since there is strong evidence that both CD81 and SCARB1 interact directly with lipoviral HCV particles, unlike many of the other HCV entry factors. CD81−/− and SCARB1−/− mice and, as controls, mice with both alleles were injected with adenoviruses delivering the indicated combinations of the canonical entry factor mouse orthologues. In CD81−/− mice overexpressing mouse SCARB1, CLDN1, and OCLN (Fig. 5b, 5th column from the left), the uptake efficiency was reduced by 90% compared with mice with the endogenous CD81 and treated with the same combination of adenoviruses (Fig. 5b, 2nd column from the left). Likewise, in SCARB1−/− mice overexpressing mouse CD81, CLDN1, and OCLN (Fig. 5b, 6th column from the left), the uptake efficiency was reduced by ca. 85% compared to SCARB1+/+ mice treated with the same combination of adenoviruses (Fig. 5b, 3rd column from the left). While previous in vitro experiments using anti-SCARB1 blocking suggested that mtHCV entry may be less dependent on SCARB1, our in vivo experiment establishes that mtHCV requires both CD81 and SCARB1 to enter murine hepatocytes in vivo.
mtHCV does not replicate in STAT1-deficient mice.
To test whether mtHCV may not only enter but also go through later stages of the viral life cycle, we analyzed postentry steps in vivo. Previous studies demonstrated that antiviral defenses, in particular type I and III IFN-dependent pathways, interfere with HCV RNA replication in mouse cell lines and in mice (17, 20–23, 33, 34). Thus, we tested the replicative fitness of mtHCV in mice with a targeted disruption of STAT1 (Fig. 6). STAT1 knockout mice are severely impaired in type I and III IFN signaling. We previously showed that STAT-deficient mice transgenically expressing human CD81, SCARB1, CLDN1, and OCLN support low levels of HCV infection over several weeks (17). While we were also able to detect HCV RNA—presumably from the residual inoculum—4 h following infection of STAT1−/− mice with cell culture-produced mtHCV (5 × 106 TCID50/mouse, intravenously), none of the animals were consistently viremic at any of the later time points. Furthermore, we did not detect any differences between STAT1−/− and isogenic wild-type control animals.
FIG 6 .

Infection of mice with blunted innate immunity does not result in persistent infection. Mice with a fully intact innate immune system (wild-type [WT]) or lacking STAT1 were infected with 5 × 106 TCID50 of Jc1 or Jc1/mCD81. HCV RNA levels (number of copies [cop] per milliliter) were measured by qRT-PCR at the indicated time points. Each symbol represents the value f or an individual animal. The dashed line shows the limit of detection (l.o.d.).
Minimal evidence for mtHCV RNA replication in the absence of antiviral innate and adaptive immune responses.
It is conceivable that those cells in which mtHCV may start to replicate are rapidly cleared by the murine adaptive immune system. Thus, to test whether mtHCV may establish persistent infection in highly immunocompromised animals in which both innate and adaptive responses were compromised, we transplanted Alb-uPA SCID/beige mice (28) with hepatocytes isolated from STAT1−/− mice. Consistent with previously published data (35), transplantation of 2 × 105 or 4 × 105 mouse hepatocytes from STAT1−/− mice rescued Alb-uPA SCID/beige mice from acute liver failure and resulted in successive weight gain (data not shown). Stably engrafted animals were injected with cell culture-produced mtHCV (1.33 × 106 TCID50), and viremia was monitored for 8 weeks. In contrast to human liver chimeric mice in which mtHCV caused high-level, persistent viremia (Fig. 1), most murine liver chimeric mice remained below the limit of detection (Table 1). A single mouse (B620R) became viremic at a single time point 4 weeks following inoculation, but viremia was just slightly above the limit of detection of 300 IU/ml. These data suggest that mtHCV does not replicate in murine hepatocytes or replicates at levels below the detection limit of our assays.
TABLE 1 .
Infection of uPA-SCID mice engrafted with STAT1-deficient murine hepatocytes with mtHCV does not result in stable viremiaa
| Group | Mouse IDb | Hepatocyte donor | Hepatocyte dose | Viral RNA (IU/ml) at the following time postinfection (wk)c: |
||||
|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 8 | ||||
| RO | K1389L | STAT-1 KO | 2.10E+5 | <375 | <300 | <300 | <300 | <150 |
| K1389RL | STAT-1 KO | 2.10E+5 | <375 | <300 | <300 | <300 | <150 | |
| IP | B620 | STAT-1 KO | 4.10E+5 | <375 | <300 | <300 | <300 | <150 |
| B620L | STAT-1 KO | 4.10E+5 | <375 | <300 | <300 | <300 | <150 | |
| B620R | STAT-1 KO | 4.10E+5 | <375 | <300 | +, <300 | <300 | <150 | |
| Negative control (RO) | B623L | Wild-type | <375 | ND | ND | ND | ND | |
uPA-SCID mice were injected with 2 × 105 or 4 × 105 murine hepatocytes derived from STAT1-deficient (knockout [KO]) mice. Resultant cohorts (RO and IP) were infected intravenously with 1.3 × 106 TCID50 mtHCV-Cre.
ID, identifier.
HCV RNA levels in serum were measured for up to 8 weeks postinfection. Viral RNA was quantified for up to 8 weeks postinfection using the Roche COBAS AmpliPrep instrument and the COBAS TaqMan48 HCV test. The limit of detection was 300 IU/ml. ND, not determined.
DISCUSSION
The molecular basis for HCV’s narrow host range remains incompletely understood. This has posed challenges for developing small animal models for HCV infection. In rodent cells, the HCV life cycle is blocked at the level of entry and at postentry steps. HCV’s inability to enter rodent cells can be explained by differences in critical residues in the second extracellular loops of CD81 (36, 37) and OCLN (18). Consequently, expression of human CD81 and OCLN, along with human or mouse SCARB1 and CLDN1, facilitates HCV uptake in mouse cells in vitro (11) and in vivo (16, 17, 38).
As an alternative to genetic host adaptations, we have previously demonstrated that the block of HCV at the level of entry can also be overcome by viral adaptation (24). Using an in vitro selection approach, we identified mutations within HCV E1 and E2 that increased the affinity of the viral envelope for mouse CD81. These mutations appeared to more broadly affect the conformation of the viral envelope, as the resulting mCD81-adapted strain was also less dependent on human OCLN and could enter cell lines expressing only mouse CD81, SCARB1, CLDN1, and OCLN (24). Here, we show that Jc1/mCD81 can also infect murine primary hepatocytes in vitro and in vivo. Uptake of this mtHCV strain is dependent on interactions of the viral glycoproteins with murine orthologues of HCV entry factors, specifically CD81 and SCARB1. However, uptake of mtHCV into mouse hepatocytes appears to be rather inefficient, as viral uptake can be detected in only 0.05 to 0.1% of murine hepatocytes (Fig. 4). This is further corroborated by our observation that adenoviral overexpression of the murine orthologues increased uptake efficiency (Fig. 3). It is conceivable that additional adaptive mutations may be necessary to facilitate more efficient entry. At the same time, it cannot be ruled out that molecules present on the surfaces of mouse hepatocytes but presumably not human hepatocytes interfere with viral uptake.
We further explored whether Jc1/mCD81 could also proceed through other steps of the viral life cycle. Numerous in vitro studies have demonstrated that type I and III IFN-dependent antiviral defenses appear to act more efficiently to shut down HCV infection in rodent cells than in human cells (25, 39). For example, fibroblast and immortalized hepatic cell lines from mice deficient in interferon regulatory factor 3 (IRF3), IRF7, mitochondrial antiviral signaling protein (MAVS), and STAT1 support HCV RNA replication more efficiently than wild-type cells do (17, 20–23). Thus, we employed STAT1-deficient mice, which are severely impaired in type I and III IFN signaling and animals lacking both functional innate and adaptive immune responses. We previously showed that STAT−/− mice transgenically expressing human CD81, SCARB1, CLDN1, and OCLN became viremic, albeit at low levels (17). Other studies in HCV entry factor transgenic mice on an ICR background suggested that HCV infection could become chronic even in fully immunocompetent animals (38).
Blunting cell intrinsic immunity and even both innate and adaptive immunity was not sufficient to establish persistent HCV infection. This may be explained by a combination of the very low entry efficiency and replication of mtHCV in mouse hepatocytes in the absence of ectopic (over)expression of the human orthologues of HCV entry factors. Future efforts will have to focus on identifying putatively missing human-specific positive regulators of HCV replication in mouse hepatocytes or alternatively, identifying mutations that increase HCV replicative fitness in mouse cells. The latter approach has been attempted, but mutations that arose during replication of antibiotic-resistant HCV replicons in mouse cells seemed to be random rather than adaptive (40), as none substantially enhanced replication efficiency in the murine cell environment when reintroduced into the parental subgenome (41).
Conceivably, it may be simpler to adapt HCV to other species that are genetically more closely related to humans than mice. Results from prior studies suggested that several other nonhuman primate species are resistant to HCV infection (42, 43). However, more-recent work has shown that hepatocyte-like cells derived from pig-tailed macaques induced pluripotent stem cells (44) and primary adult hepatocytes from rhesus macaques (45) can support the entire HCV life cycle. When rhesus macaque hepatocytes were engrafted in immunodeficient liver injury recipients, the resultant simian liver chimeric mice supported persistent HCV infection, albeit with delayed kinetics and lower peak viremia levels than in control humanized mice. These data suggest that it may be possible to establish HCV infection in species more distantly related to humans. However, since mice and humans diverged phylogenetically approximately 65 million years ago, it may be rather challenging to bridge the genetic gap by viral adaptation despite HCV’s considerable genetic plasticity.
Over the last few years, other viruses related to HCV in either the Pegivirus genus, also a part of the Flaviviridae family, or the Hepacivirus genus have been identified in a variety of species, including dogs (46), horses (47, 48), wild mice (49), and rats (50). An in-depth understanding of how these viruses establish chronicity in species not known to be zoonotic reservoirs for HCV may guide further, more directed adaptations of HCV to robustly infect heterologous hosts.
MATERIALS AND METHODS
Cell lines.
Huh-7.5 (51), Huh-7.5.1 (52), and HEK293 cells were maintained in Dulbecco modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS).
Animals and cell lines.
Gt(ROSA)26Sortm1(Luc)Kaelin11 (Rosa26-Fluc), B6; 129-Gt(ROSA)26Sortm1Joe/J27 (Rosa26-GNZ), B6; 129S2-Scarb1tm1Kri/J17 (SCARB1−/−) mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and Cd81tm1Lvy (CD81−/−) mice were obtained from the European Mutant Mouse Archive (Munich, Germany). 129S6/SvEv-Stat1tm1Rds (STAT1−/−) and 129S6/SvEvTac (wild-type) mice were obtained from Taconic (Germantown, NY). Human liver-uPA-SCID mice were generated as previously described (28, 53). Rosa26-Fluc mice contain the firefly luciferase (luc) gene inserted into the Gt.(ROSA)26Sor locus. Expression of the luciferase gene is blocked by a loxP-flanked STOP fragment placed between the luc sequence and the Gt.(ROSA)26Sor promoter. Cre recombinase-mediated excision of the transcriptional stop cassette results in luciferase expression in Cre-expressing tissues. Rosa26-GNZ knock-in mice have widespread expression of a nucleus-localized green fluorescent protein/beta-galactosidase fusion protein (GFP-NLS-GNZ) once an upstream loxP-flanked STOP sequence is removed. When Cre recombinase is introduced into cells, the resulting GNZ fusion protein expression allows for enhanced (single-cell level) visualization. All infections of mice with the indicated strains were performed intravenously.
Mice were bred and maintained at the Laboratory Animal Resources of Princeton University, Comparative Bioscience Center of the Rockefeller University, and at the Animal Resource Center of the University of Ghent according to guidelines established by the respective Institutional Animal Care and Use Committees.
Hepatitis C virus.
The sequence of the genotype 2a/2a chimera J6/JFH1 has been deposited in a gene bank (accession number JF343782). Construction of Jc1 (54), and mCD81/Jc1 (24) (mtHCV) was described elsewhere. BiCre-Jc1/mCD81 was generated by introducing the L216F, V388G, and M405T point mutations into BiCre-Jc1 (16) via site-directed mutagenesis. Jc1(p7nsGluc2a) and Jc1/mCD81(p7nsGluc2a) were generated by ligating an EcoRI/BsaBI fragment containing core, E1, and parts of E2 into the EcoRI/BsaBI backbone of Jc1/Flag2(p7nsGluc2a) (29). To produce infectious virus, Huh-7.5.1 (52) or Huh-7.5 (55) cells were electroporated with in vitro-transcribed full-length HCV RNA (T7 RiboMAX express large-scale RNA production system; Promega, Madison, WI). Seventy-two hours postelectroporation, the medium was replaced with DMEM without FBS, and supernatants were harvested every 6 h starting from 72 h. Pooled supernatants were filtered through a 0.45-µm bottle top filter (Millipore, Billerica, MA) and concentrated using a stirred cell (Millipore). Viral titers (TCID50) were determined using Huh-7.5 cells as previously described (56).
Adenoviruses.
Construction of adenoviral constructs encoding human or mouse CD81, SCARB1, CLDN1, or OCLN was described previously (16) using the AdEasy adenoviral vector system (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s instructions. Briefly, cDNAs encoding HCV entry factors were PCR amplified and inserted into the pShuttle-CMV (CMV stands for cytomegalovirus) using KpnI/NotI sites. Recombinant pShuttle-CMV plasmids were linearized with PmeI and ligated to pAdEasy by homologous recombination followed by electroporation into BJ5183 cells (Agilent). Recombinant pShuttle-pAdEasy constructs were identified by PacI restriction analysis. All plasmid constructs were verified by DNA sequencing.
RT-PCR quantification of HCV RNA.
Total RNA was isolated from the indicated mouse tissues using the RNeasy kit (Qiagen, Valencia, CA) and from serum using the QIAamp viral RNA minikit. HCV genome copy number was quantified by one-step reverse transcription-PCR (RT-PCR) using MultiCode ISOlution (catalog no. 3691; Luminex, Madison, WI), reverse transcriptase (catalog no. 3693; Luminex, Madison, WI), Titanium Taq (catalog no. S1792; Clontech, Mountain View, CA), and a Step One Plus quantitative PCR (qPCR) machine (Life Technologies, Carlsbad, CA), according to manufacturers’ instructions. Data were analyzed using the MultiCode Analysis Software v1.6.5 (Luminex, Madison, WI). The following primers were used for the detection of HCV RNA: GCTCACGGACCTTTCA (sense) and GGCTCCATCTTAGCCC (antisense).
Quantification of HCV RNA in the chimeric Alb-uPA mice was performed with the Roche COBAS AmpliPrep instrument and the COBAS TaqMan 48 HCV test.
Antibodies.
Blocking antibodies against CD81 (JS81) and IgG1 control antibodies were obtained from BD Biosciences (Franklin Lakes, NJ). Antibodies against NS5A (56) and E2 (clone AR4A) (33) and the human IgG1 isotype control (B12) (57) have been described previously. As a secondary antibody for the detection of anti-NS5A, Alexa Fluor 647-labeled goat anti-mouse IgG (Life Technologies, Carlsbad, CA) was used.
Flow cytometry.
The frequency of infected hepatocytes and the frequency of infected Huh-7.5 cells after inoculation with sera from infected mice were confirmed by flow cytometry using an LSRII flow cytometer (BD Biosciences). For the determination of infection frequency, hepatocytes were isolated from Rosa26-GNZ mice, fixed in 4% paraformaldehyde, permeabilized in phosphate-buffered saline (PBS) plus 0.01% Triton X-100, stained with anti-NS5A, and counterstained with DRAQ5. Data were analyzed using FlowJo software (Treestar Software, Ashland, OR).
Isolation and infection of murine hepatocytes.
C57BL/6 mice were anesthetized by intraperitoneal injection of a mixture of 100 mg of ketamine/kg of body weight and 10 mg/kg xylazine. Livers were perfused through the portal vein with a chelating solution (0.01 M HEPES [pH 7.3] and 0.5 mM EGTA [pH 8.0] in Ca2+/Mg2+-free Earle’s balanced salt solution [EBSS]) at a flow rate of 2 ml/min until the liver bleached, followed by 40 ml of collagenase solution (0.01 M HEPES [pH 7.3] and 1 mg/ml collagenase type II in EBSS with Ca2+, Mg2+, and phenol red). The digested liver was cut into pieces, transferred into a washing solution (0.01 M HEPES [pH 7.3] and 10% FBS in DMEM), passed through a 100-µm cell strainer, washed, and passed through a 100-µm cell strainer. The resulting cell suspension was passed through a 70-µm cell strainer. The cell suspension was washed three more times with spinning steps at 140 × g for 5 min to remove unwanted cellular contaminants. The cells were resuspended in HBM basal medium after adding HCM Single Quots (Lonza, Basel, Switzerland), counted, and seeded on collagen-coated 24-well cell culture plates (Corning, Corning, NY) at a density of 1.25 × 105 cells/cm2. The medium was changed every other day. Confluent monolayers of primary hepatocytes were infected with different doses of Jc1(p7nsGluc2A) or Jc1/mCD81p7nsGluc2A for 12 to 18 h and washed 3 or 4 times with medium, and supernatants were collected at 48 h following infection. Gaussia luciferase activity was quantified in the cell culture supernatants with a luciferase assay system (Promega, Madison, WI) according to the manufacturer’s instructions using a Tristar 2 multimode reader LB942 (Berthold Technologies, Bad Wildbad, Germany).
Bioluminescence im aging.
Unless otherwise specified, mice were injected with 1011 adenoviral particles 24 h prior to intravenous injection with 2 × 107 TCID50 HCV-Cre. At 72 h postinfection, mice were anesthetized using ketamine/xylazine and injected intraperitoneally with 1.5 mg Luciferin (Caliper Life Sciences, Hopkinton, MA). Bioluminescence was measured using an IVIS Lumina II platform (Caliper Life Sciences).
HCV genome sequencing.
Serum samples from mice inoculated with Jc1/mCD81 were subjected to RNA purification (high pure viral RNA kit; Roche, Basel, Switzerland) and cDNA amplification (Transcriptor high-fidelity cDNA synthesis kit; Roche, Basel, Switzerland) with an antisense primer situated on the NS2 gene. cDNA content was determined by qRT-PCR. For amplicon library preparation, six contigs which covered the signal peptide genes of E1, E2, and p7 (amino acids 131 to 813, corresponding to the polyprotein sequence of the reference strain Jc1) were generated with the Expand Long Range dNTPack kit (Roche, Basel, Switzerland) by nested PCR utilizing 12 primers tagged at the 5′ end. Amplicons were prepared individually and quantified by using the Quant-iT PicoGreen dsDNA (double-stranded DNA) assay kit (Carlsbad, CA). Emulsion-PCR and bidirectional sequencing reaction was performed using the 454 platform (Roche, Basel, Switzerland) as described in the GS FLX emPCR Methods Manual (Roche, Basel, Switzerland). Sequencing data were analyzed using CLC genomics workbench v4.9 (Aarhus, Denmark). Contig assembly and viral population variant calling were performed as described previously (58). The average read depth was 11,000, ranging from 3,000 to 21,000 reads.
Statistical analysis.
Statistical analysis was performed with GraphPad Prism software (La Jolla, CA). Statistics were calculated using t test or Kruskal-Wallis one-way analysis of variance (ANOVA). P values less than 0.05 were considered statistically significant.
ACKNOWLEDGMENTS
We thank Jade Xiao, Rachael Labitt, Tamar Frilling, and Sabrina Woltemate for excellent technical help and Jenna Gaska, Qiang Ding, and Florian Douam for editing and critical discussions of the manuscript.
This study is supported by grants from the National Institutes of Health (R01 AI079031, R01 AI107301, and R21AI117213 to A.P.), a Research Scholar Award from the American Cancer Society (RSG-15-048-01-MPC to A.P.), a Burroughs Wellcome Fund Award for Investigators in Pathogenesis (to A.P.), and a grant from the German Research Foundation (SFB 900/Z1 to S.S.). T.P. was supported by a grant from the European Research Council ERC-2011-StG_281473 (VIRAFRONT) and by a grant from the Helmholtz Association SO-024. M.V.S. is a recipient of a postdoctoral fellowship from the German Research Foundation (Deutsche Forschungsgemeinschaft).
Footnotes
Citation von Schaewen M, Dorner M, Hueging K, Foquet L, Gerges S, Hrebikova G, Heller B, Bitzegeio J, Doerrbecker J, Horwitz JA, Gerold G, Suerbaum S, Rice CM, Meuleman P, Pietschmann T, Ploss A. 2016. Expanding the host range of hepatitis C virus through viral adaptation. mBio 7(6):e01915-16. doi:10.1128/mBio.01915-16.
REFERENCES
- 1.von Schaewen M, Ploss A. 2014. Murine models of hepatitis C: what can we look forward to? Antiviral Res 104:15–22. doi: 10.1016/j.antiviral.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sandmann L, Ploss A. 2013. Barriers of hepatitis C virus interspecies transmission. Virology 435:70–80. doi: 10.1016/j.virol.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ding Q, von Schaewen M, Ploss A. 2014. The impact of hepatitis C virus entry on viral tropism. Cell Host Microbe 16:562–568. doi: 10.1016/j.chom.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. 1999. Hepatitis C virus and other Flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci U S A 96:12766–12771. doi: 10.1073/pnas.96.22.12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Owen DM, Huang H, Ye J, Gale M Jr.. 2009. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology 394:99–108. doi: 10.1016/j.virol.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 7.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 9.Zheng A, Yuan F, Li Y, Zhu F, Hou P, Li J, Song X, Ding M, Deng H. 2007. Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J Virol 81:12465–12471. doi: 10.1128/JVI.01457-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu S, Yang W, Shen L, Turner JR, Coyne CB, Wang T. 2009. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J Virol 83:2011–2014. doi: 10.1128/JVI.01888-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert TF. 2011. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sainz B Jr, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL. 2012. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med 18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin DN, Uprichard SL. 2013. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc Natl Acad Sci U S A 110:10777–10782. doi: 10.1073/pnas.1301764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X, Lee EM, Hammack C, Robotham JM, Basu M, Lang J, Brinton MA, Tang H. 2014. Cell death-inducing DFFA-like effector b is required for hepatitis C virus entry into hepatocytes. J Virol 88:8433–8444. doi: 10.1128/JVI.00081-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorner M, Horwitz JA, Robbins JB, Barry WT, Feng Q, Mu K, Jones CT, Schoggins JW, Catanese MT, Burton DR, Law M, Rice CM, Ploss A. 2011. A genetically humanized mouse model for hepatitis C virus infection. Nature 474:208–211. doi: 10.1038/nature10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dorner M, Horwitz JA, Donovan BM, Labitt RN, Budell WC, Friling T, Vogt A, Catanese MT, Satoh T, Kawai T, Akira S, Law M, Rice CM, Ploss A. 2013. Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature 501:237–241. doi: 10.1038/nature12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michta ML, Hopcraft SE, Narbus CM, Kratovac Z, Israelow B, Sourisseau M, Evans MJ. 2010. Species-specific regions of occludin required by hepatitis C virus for cell entry. J Virol 84:11696–11708. doi: 10.1128/JVI.01555-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long G, Hiet MS, Windisch MP, Lee JY, Lohmann V, Bartenschlager R. 2011. Mouse hepatic cells support assembly of infectious hepatitis C virus particles. Gastroenterology 141:1057–1066. doi: 10.1053/j.gastro.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 20.Vogt A, Scull MA, Friling T, Horwitz JA, Donovan BM, Dorner M, Gerold G, Labitt RN, Rice CM, Ploss A. 2013. Recapitulation of the hepatitis C virus life-cycle in engineered murine cell lines. Virology 444:1–11. doi: 10.1016/j.virol.2013.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin LT, Noyce RS, Pham TN, Wilson JA, Sisson GR, Michalak TI, Mossman KL, Richardson CD. 2010. Replication of subgenomic hepatitis C virus replicons in mouse fibroblasts is facilitated by deletion of interferon regulatory factor 3 and expression of liver-specific microRNA 122. J Virol 84:9170–9180. doi: 10.1128/JVI.00559-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang KS, Cai Z, Zhang C, Sen GC, Williams BR, Luo G. 2006. Replication of hepatitis C virus (HCV) RNA in mouse embryonic fibroblasts: protein kinase R (PKR)-dependent and PKR-independent mechanisms for controlling HCV RNA replication and mediating interferon activities. J Virol 80:7364–7374. doi: 10.1128/JVI.00586-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nandakumar R, Finsterbusch K, Lipps C, Neumann B, Grashoff M, Nair S, Hochnadel I, Lienenklaus S, Wappler I, Steinmann E, Hauser H, Pietschmann T, Kröger A. 2013. Hepatitis C virus replication in mouse cells is restricted by IFN-dependent and -independent mechanisms. Gastroenterology 145:1414–1423.e1. doi: 10.1053/j.gastro.2013.08.037. [DOI] [PubMed] [Google Scholar]
- 24.Bitzegeio J, Bankwitz D, Hueging K, Haid S, Brohm C, Zeisel MB, Herrmann E, Iken M, Ott M, Baumert TF, Pietschmann T. 2010. Adaptation of hepatitis C virus to mouse CD81 permits infection of mouse cells in the absence of human entry factors. PLoS Pathog 6:e1000978. doi: 10.1371/journal.ppat.1000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frentzen A, Anggakusuma, Gürlevik E, Hueging K, Knocke S, Ginkel C, Brown RJ, Heim M, Dill MT, Kröger A, Kalinke U, Kaderali L, Kuehnel F, Pietschmann T. 2014. Cell entry, efficient RNA replication, and production of infectious hepatitis C virus progeny in mouse liver-derived cells. Hepatology 59:78–88. doi: 10.1002/hep.26626. [DOI] [PubMed] [Google Scholar]
- 26.Bukh J, Pietschmann T, Lohmann V, Krieger N, Faulk K, Engle RE, Govindarajan S, Shapiro M, St Claire M, Bartenschlager R. 2002. Mutations that permit efficient replication of hepatitis C virus RNA in Huh-7 cells prevent productive replication in chimpanzees. Proc Natl Acad Sci U S A 99:14416–14421. doi: 10.1073/pnas.212532699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mercer DF, Schiller DE, Elliott JF, Douglas DN, Hao C, Rinfret A, Addison WR, Fischer KP, Churchill TA, Lakey JR, Tyrrell DL, Kneteman NM. 2001. Hepatitis C virus replication in mice with chimeric human livers. Nat Med 7:927–933. doi: 10.1038/90968. [DOI] [PubMed] [Google Scholar]
- 28.Meuleman P, Libbrecht L, De Vos R, de Hemptinne B, Gevaert K, Vandekerckhove J, Roskams T, Leroux-Roels G. 2005. Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology 41:847–856. doi: 10.1002/hep.20657. [DOI] [PubMed] [Google Scholar]
- 29.Marukian S, Jones CT, Andrus L, Evans MJ, Ritola KD, Charles ED, Rice CM, Dustin LB. 2008. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 48:1843–1850. doi: 10.1002/hep.22550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Safran M, Kim WY, Kung AL, Horner JW, DePinho RA, Kaelin WG Jr.. 2003. Mouse reporter strain for noninvasive bioluminescent imaging of cells that have undergone Cre-mediated recombination. Mol Imaging 2:297–302. doi: 10.1162/153535003322750637. [DOI] [PubMed] [Google Scholar]
- 31.Awatramani R, Soriano P, Mai JJ, Dymecki S. 2001. An Flp indicator mouse expressing alkaline phosphatase from the ROSA26 locus. Nat Genet 29:257–259. doi: 10.1038/ng1101-257. [DOI] [PubMed] [Google Scholar]
- 32.Darzynkiewicz Z, Halicka HD, Zhao H. 2010. Analysis of cellular DNA content by flow and laser scanning cytometry. Adv Exp Med Biol 676:137–147. doi: 10.1007/978-1-4419-6199-0_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, Rice CM, Ploss A, Burton DR, Law M. 2012. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc Natl Acad Sci U S A 109:6205–6210. doi: 10.1073/pnas.1114927109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu D, Nishimura T, Nishimura S, Zhang H, Zheng M, Guo YY, Masek M, Michie SA, Glenn J, Peltz G. 2014. Fialuridine induces acute liver failure in chimeric TK-NOG mice: a model for detecting hepatic drug toxicity prior to human testing. PLoS Med 11:e1001628. doi: 10.1371/journal.pmed.1001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rhim JA, Sandgren EP, Palmiter RD, Brinster RL. 1995. Complete reconstitution of mouse liver with xenogeneic hepatocytes. Proc Natl Acad Sci U S A 92:4942–4946. doi: 10.1073/pnas.92.11.4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flint M, von Hahn T, Zhang J, Farquhar M, Jones CT, Balfe P, Rice CM, McKeating JA. 2006. Diverse CD81 proteins support hepatitis C virus infection. J Virol 80:11331–11342. doi: 10.1128/JVI.00104-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Higginbottom A, Quinn ER, Kuo CC, Flint M, Wilson LH, Bianchi E, Nicosia A, Monk PN, McKeating JA, Levy S. 2000. Identification of amino acid residues in CD81 critical for interaction with hepatitis C virus envelope glycoprotein E2. J Virol 74:3642–3649. doi: 10.1128/JVI.74.8.3642-3649.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen J, Zhao Y, Zhang C, Chen H, Feng J, Chi X, Pan Y, Du J, Guo M, Cao H, Chen H, Wang Z, Pei R, Wang Q, Pan L, Niu J, Chen X, Tang H. 2014. Persistent hepatitis C virus infections and hepatopathological manifestations in immune-competent humanized mice. Cell Res 24:1050–1066. doi: 10.1038/cr.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anggakusuma, Frentzen A, Gürlevik E, Yuan Q, Steinmann E, Ott M, Staeheli P, Schmid-Burgk J, Schmidt T, Hornung V, Kuehnel F, Pietschmann T. 2015. Control of hepatitis C virus replication in mouse liver-derived cells by MAVS-dependent production of type I and type III interferons. J Virol 89:3833–3845. doi: 10.1128/JVI.03129-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uprichard SL, Chung J, Chisari FV, Wakita T. 2006. Replication of a hepatitis C virus replicon clone in mouse cells. Virol J 3:89. doi: 10.1186/1743-422X-3-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu Q, Guo JT, Seeger C. 2003. Replication of hepatitis C virus subgenomes in nonhepatic epithelial and mouse hepatoma cells. J Virol 77:9204–9210. doi: 10.1128/JVI.77.17.9204-9210.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abe K, Kurata T, Teramoto Y, Shiga J, Shikata T. 1993. Lack of susceptibility of various primates and woodchucks to hepatitis C virus. J Med Primatol 22:433–434. [PubMed] [Google Scholar]
- 43.Bukh J, Apgar CL, Govindarajan S, Emerson SU, Purcell RH. 2001. Failure to infect rhesus monkeys with hepatitis C virus strains of genotypes 1a, 2a or 3a. J Viral Hepat 8:228–231. doi: 10.1046/j.1365-2893.2001.00284.x. [DOI] [PubMed] [Google Scholar]
- 44.Sourisseau M, Goldman O, He W, Gori JL, Kiem HP, Gouon-Evans V, Evans MJ. 2013. Hepatic cells derived from induced pluripotent stem cells of pigtail macaques support hepatitis C virus infection. Gastroenterology 145:966–969.e7. doi: 10.1053/j.gastro.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scull MA, Shi C, de Jong YP, Gerold G, Ries M, von Schaewen M, Donovan BM, Labitt RN, Horwitz JA, Gaska JM, Hrebikova G, Xiao JW, Flatley B, Fung C, Chiriboga L, Walker CM, Evans DT, Rice CM, Ploss A. 2015. Hepatitis C virus infects rhesus macaque hepatocytes and simianized mice. Hepatology 62:57–67. doi: 10.1002/hep.27773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kapoor A, Simmonds P, Gerold G, Qaisar N, Jain K, Henriquez JA, Firth C, Hirschberg DL, Rice CM, Shields S, Lipkin WI. 2011. Characterization of a canine homolog of hepatitis C virus. Proc Natl Acad Sci U S A 108:11608–11613. doi: 10.1073/pnas.1101794108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burbelo PD, Dubovi EJ, Simmonds P, Medina JL, Henriquez JA, Mishra N, Wagner J, Tokarz R, Cullen JM, Iadarola MJ, Rice CM, Lipkin WI, Kapoor A. 2012. Serology-enabled discovery of genetically diverse hepaciviruses in a new host. J Virol 86:6171–6178. doi: 10.1128/JVI.00250-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kapoor A, Simmonds P, Cullen JM, Scheel TK, Medina JL, Giannitti F, Nishiuchi E, Brock KV, Burbelo PD, Rice CM, Lipkin WI. 2013. Identification of a pegivirus (GB virus-like virus) that infects horses. J Virol 87:7185–7190. doi: 10.1128/JVI.00324-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kapoor A, Simmonds P, Scheel TK, Hjelle B, Cullen JM, Burbelo PD, Chauhan LV, Duraisamy R, Sanchez Leon M, Jain K, Vandegrift KJ, Calisher CH, Rice CM, Lipkin WI. 2013. Identification of rodent homologs of hepatitis C virus and pegiviruses. mBio 4:e00216-13. doi: 10.1128/mBio.00216-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Firth C, Bhat M, Firth MA, Williams SH, Frye MJ, Simmonds P, Conte JM, Ng J, Garcia J, Bhuva NP, Lee B, Che X, Quan PL, Lipkin WI. 2014. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. mBio 5:e01933-14. doi: 10.1128/mBio.01933-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meuleman P, Vanlandschoot P, Leroux-Roels G. 2003. A simple and rapid method to determine the zygosity of uPA-transgenic SCID mice. Biochem Biophys Res Commun 308:375–378. doi: 10.1016/S0006-291X(03)01388-3. [DOI] [PubMed] [Google Scholar]
- 54.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A 103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for hepatitis C virus genomic and subgenomic RNA replication. J Virol 76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 57.Burton DR, Pyati J, Koduri R, Sharp SJ, Thornton GB, Parren PW, Sawyer LS, Hendry RM, Dunlop N, Nara PL, Lamacchia M, Garratty E, Stiehm ER, Bryson YJ, Cao Y, Moore JP, Ho DD, Barbas CF III. 1994. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 266:1024–1027. doi: 10.1126/science.7973652. [DOI] [PubMed] [Google Scholar]
- 58.Irving WL, Rupp D, McClure CP, Than LM, Titman A, Ball JK, Steinmann E, Bartenschlager R, Pietschmann T, Brown RJ. 2014. Development of a high-throughput pyrosequencing assay for monitoring temporal evolution and resistance associated variant emergence in the hepatitis C virus protease coding-region. Antiviral Res 110:52–59. doi: 10.1016/j.antiviral.2014.07.009. [DOI] [PubMed] [Google Scholar]