

The crystal structure of a triazolylthioacetamide inhibitor in complex with Verona integron-encoded metallo-β-lactamase 2 (VIM-2) is reported at a resolution of 1.50 Å. The structure shows that the inhibitor binds to the active site of the enzyme and reveals detailed information on the inhibitor interactions.
Keywords: antibiotic resistance, DMSO-free co-crystallization, carbapenemases, VIM-2, metallo-β-lactamases
Abstract
The increasing number of pathogens expressing metallo-β-lactamases (MBLs), and in this way achieving resistance to β-lactam antibiotics, is a significant threat to global public health. A promising strategy to treat such resistant pathogens is the co-administration of MBL inhibitors together with β-lactam antibiotics. However, an MBL inhibitor suitable for clinical use has not yet been identified. Verona integron-encoded metallo-β-lactamase 2 (VIM-2) is a widespread MBL with a broad substrate spectrum and hence is an interesting drug target for the treatment of β-lactam-resistant infections. In this study, three triazolylthioacetamides were tested as inhibitors of VIM-2. One of the tested compounds showed clear inhibition of VIM-2, with an IC50 of 20 µM. The crystal structure of the inhibitor in complex with VIM-2 was obtained by DMSO-free co-crystallization and was solved at a resolution of 1.50 Å. To our knowledge, this is the first structure of a triazolylthioacetamide inhibitor in complex with an MBL. Analysis of the structure shows that the inhibitor binds to the two zinc ions in the active site of VIM-2 and revealed detailed information on the interactions involved. Furthermore, the crystal structure showed that binding of the inhibitor induced a conformational change of the conserved residue Trp87.
1. Introduction
The discovery of β-lactam antibiotics for the treatment of bacterial infections has revolutionized modern medicine and saved countless lives. Unfortunately, the overuse of these antibiotics has resulted in the rapid emergence of β-lactam-resistant bacteria, representing a major threat to global public health (World Health Organization, 2014 ▸). A main mechanism for bacteria to develop resistance to β-lactam antibiotics is the expression of β-lactamases, enzymes that are able to hydrolyse the β-lactam ring and render the antibiotics ineffective. The co-administration of β-lactamase inhibitors together with β-lactam antibiotics can prevent their hydrolysis and in this way overcome resistance. To date, two different groups of β-lactamases have been identified: metallo-β-lactamases (MBLs) and serine β-lactamases (SBLs). For SBLs, potent inhibitors co-administered with β-lactam antibiotics are already in clinical use (Drawz & Bonomo, 2010 ▸; Lucasti et al., 2016 ▸). However, no MBL inhibitors have yet reached clinical use, and hence there is a clear need to identify novel inhibitors (Fast & Sutton, 2013 ▸).
Verona integron-encoded metallo-β-lactamase 2 (VIM-2) is a widespread MBL with a broad substrate spectrum, including penicillins, cephalosporins, cephamycins and carbapenems (Poirel et al., 2000 ▸). In its mature form, VIM-2 is a monomeric enzyme consisting of 240 amino acids with a molecular mass of 25 kDa (Docquier et al., 2003 ▸; Garcia-Saez et al., 2008 ▸). Several crystal structures of VIM-2 have been reported (Garcia-Saez et al., 2008 ▸; Aitha et al., 2014 ▸). The structures show that the enzyme is folded in an αβ/βα sandwich and that two zinc ions bridged by a hydroxide are located in the active site. Furthermore, crystal structures of different inhibitors in complex with VIM-2 have been reported (Brem et al., 2014 ▸, 2016 ▸; Christopeit et al., 2015 ▸). VIM-2 has a high clinical relevance owing to its wide spread and broad substrate range, and therefore reflects an important drug target for the treatment of antibiotic-resistant infections.
Triazolylthioacetamides have been identified as inhibitors of different MBLs and hence are interesting scaffolds for the design of potent VIM-2 inhibitors (Zhang et al., 2014 ▸; Yang et al., 2015 ▸; Zhai et al., 2016 ▸). In this study, we tested three different substituted triazolylthioacetamides as inhibitors of VIM-2. Moreover, the complex structure of one of the inhibitors bound to VIM-2 is presented and protein–inhibitor interactions are explored.
2. Materials and methods
2.1. Expression and purification of VIM-2
The expression and purification of VIM-2 was carried out as described previously (Christopeit et al., 2015 ▸). In brief, the gene sequence coding for residues Val25–Glu300 of VIM-2 was isolated from Pseudomonas aeruginosa strain 301-5473. The sequence for a 6×His tag and a Tobacco etch virus (TEV) protease cleavage site was added and the construct was transferred into pDEST14 vector. Chemically competent Escherichia coli BL21(DE3)pLysS cells (Invitrogen) were transformed with the VIM-2 pDEST14 vector and cultured at 37°C in Terrific broth (TB) medium supplemented with 100 mg l−1 ampicillin and 34 mg l−1 chloramphenicol. Protein expression was induced overnight at 20°C with 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). The cells were harvested by centrifugation and stored at −80°C. Sonication was used to lyse the cells and insoluble material was removed by centrifugation. The supernatant was applied onto a HisTrap HP column (GE Healthcare) and bound VIM-2 was eluted with a linear imidazole gradient. The His tag was removed by adding in-house-produced His-tagged TEV protease. The cleaved His tag, uncleaved protein and the His-tagged TEV protease were removed by performing a second HisTrap purification. The flowthrough containing VIM-2 was collected and the protein was finally purified by anion-exchange chromatography. The purity of the enzyme was analysed by SDS–PAGE and the activity was confirmed by the hydrolysis of nitrocefin. The protein concentration was determined by measuring the absorption at 280 nm using a NanoDrop 2000c spectrophotometer (Thermo Scientific). The extinction coefficient of 29 910 M −1 cm−1 at 280 nm was estimated from the amino-acid sequence of VIM-2 using ProtParam (Gasteiger et al., 2005 ▸).
2.2. Synthesis of compounds 1, 2 and 3
Compound 1 was synthesized and characterized as described previously by Yang et al. (2015 ▸) and compounds 2 and 3 as described previously by Zhang et al. (2014 ▸). All compounds were dissolved in DMSO at a concentration of 100 mM and stored at ∼20°C.
2.3. Enzyme-inhibition assay and IC50 determination
The IC50 values were determined using a previously established enzyme-activity assay based on the hydrolysis of nitrocefin (Christopeit et al., 2015 ▸). Measurements were carried out at 25°C in 96-well plates using a SpectraMax M2e (Molecular Devices). Data were analysed using the SoftMax Pro 5.2 software (Molecular Devices) and GraphPad Prism 5 (GraphPad Software). A buffer consisting of 50 mM HEPES pH 7.2, 150 mM NaCl, 100 µM ZnCl2, 0.005% Tween 20, 2.5% DMSO and 20 µg ml−1 bovine serum albumin (BSA) was used in all measurements. The inhibitors were dissolved in buffer in a twofold dilution series and mixed with VIM-2. After 5 min of incubation, the enzyme reaction was started by adding nitrocefin (Calbiochem) to a final concentration of 3 µM. The final enzyme concentration was 150 pM. The hydrolysis of nitrocefin was followed by measurement of the absorption at 482 nm every 17 s for 20 min. The initial velocity was determined and the percentage of inhibition was calculated in relation to a positive control without inhibitor. The percentage of inhibition was plotted against the inhibitor concentration and the data were fitted to a dose–response curve with a constant top plateau (100%), a constant bottom plateau (0%) and a constant Hill slope of −1.
2.4. Crystallization, X-ray data collection and structure determination
VIM-2 was crystallized by the sitting-drop vapour-diffusion method (Table 1 ▸) using a new DMSO-free co-crystallization method. The reservoir wells of an MRC 96-well crystallization plate (Molecular Dimensions) were pre-coated with the inhibitor. 3 µl of compound 1 (100 mM) dissolved in DMSO was added to every reservoir well and the DMSO was evaporated by leaving the plate open in a fume hood for 24 h. After evaporation of the DMSO, 60 µl of reservoir solution was added consisting of 22–27% polyethylene glycol (PEG) 3350, 0.2 M magnesium formate. The 96-well plate was placed on a shaker for 24 h to dissolve the inhibitor in the reservoir solution. The reservoir solution was mixed with the protein solution (9.4 mg ml−1) in a 1:1 ratio and crystals appeared after 2–14 d. For cryoprotection, crystals were transferred to 25% PEG 3350, 0.2 M MgCl2, 15% ethylene glycol, 50 mM HEPES pH 7.2 and were flash-cooled in liquid nitrogen. X-ray data were collected on beamline BL14.1 operated by the Joint Berlin MX-Laboratory at the BESSY II electron-storage ring (Mueller et al., 2012 ▸). Statistics for data collection are shown in Table 2 ▸. XDS, POINTLESS and AIMLESS were used to integrate, scale and truncate the data set and to determine the space group (Kabsch, 2010 ▸; Evans, 2006 ▸, 2011 ▸; Evans & Murshudov, 2013 ▸). The phase problem was solved by molecular replacement using Phaser (McCoy et al., 2007 ▸) and a previously published VIM-2 structure (PDB entry 1ko3; Garcia-Saez et al., 2008 ▸). The final structure was obtained by manual model optimization in WinCoot according to the 2F o − F c and F o − F c electron-density maps and several refinement cycles using PHENIX (Emsley et al., 2010 ▸; Adams et al., 2010 ▸). Translation/libration/screw (TLS) parameters were used in the refinement process. Alternative rotamers were manually modelled in WinCoot and refined as implemented in PHENIX. Noncrystallographic symmetry (NCS) restraints were not used during the refinement. For all Zn2+ and Cl− ions, anisotropic B factors and occupancies were refined by PHENIX. 5% of the X-ray data were used for R free cross-validation. Refinement statistics are shown in Table 3 ▸. The PyMOL Molecular Graphics System v.1.4.1 (Schrödinger) and LIGPLOT were used to generate illustrations and to visualize interactions (Wallace et al., 1995 ▸). The class B β-lactamase numbering system is used for the numbering of all residues (Garau et al., 2004 ▸).
Table 1. Crystallization.
| Method | Sitting-drop vapour diffusion in a new DMSO-free co-crystallization method |
| Plate type | MRC 96-well crystallization plate (Molecular Dimensions) |
| Temperature (°C) | 22 |
| Protein concentration (mg ml−1) | 9.4 |
| Buffer composition of protein solution | 50 mM Tris–HCl pH 7.2, 100 µM ZnCl2 |
| Composition of reservoir solution | 22–27% PEG 3350, 0.2 M magnesium formate |
| Volume and ratio of drop | 2 µl, 1:1 |
| Volume of reservoir | 60 µl |
Table 2. Data collection.
Values in parentheses are for the outer shell.
| Diffraction source | MX beamline BL14.1, BESSY II |
| Wavelength (Å) | 0.918409 |
| Temperature (°C) | −173 |
| Detector | PILATUS 6M |
| Crystal-to-detector distance (mm) | 297 |
| Rotation range per image (°) | 0.1 |
| Total rotation range (°) | 200 |
| Exposure time per image (s) | 0.4 |
| Space group | C2 |
| a, b, c (Å) | 100.62, 79.25, 67.20 |
| α, β, γ (°) | 90.0, 90.0, 130.5 |
| Resolution range (Å) | 38.25–1.50 (1.52–1.50) |
| No. of unique reflections | 63893 |
| Multiplicity | 3.7 (3.6) |
| Completeness (%) | 99.1 (93.3) |
| R merge (%) | 4.8 (30.9) |
| Mean 〈I/σ(I)〉 | 18.8 (4.3) |
| Overall B factor from Wilson plot (Å2) | 12.12 |
Table 3. Refinement statistics.
| PDB code | 5lsc |
| Final R work (%) | 13.55 |
| Final R free (%) | 16.55 |
| No. of non-H atoms | |
| Protein | 3901 (chains A and B) |
| Ions | 6 Zn2+, 6 Cl− |
| Compound 1 | 56 (2 molecules) |
| Water | 644 |
| R.m.s. deviations | |
| Bonds (Å) | 0.008 |
| Angles (°) | 1.233 |
| Average B factors (Å2) | |
| Protein | 17.6 |
| Ion | 15.9 |
| Compound 1 (occupancy) | 21.3 (0.99/0.98) |
| Water | 32.1 |
| Ramachandran plot | |
| Most favoured (%) | 98.3 |
| Allowed (%) | 1.5 |
3. Results and discussion
3.1. Inhibition of VIM-2 by triazolylthioacetamides
In a previous study, we established a nitrocefin-based enzyme-activity assay suitable to study the influence of inhibitors on the activity of VIM-2 (Christopeit et al., 2015 ▸). This assay was used to determine the inhibition potency of three substituted triazolylthioacetamides (Fig. 1 ▸). All three compounds have previously been reported as inhibitors of different MBLs (Yang et al., 2015 ▸; Zhang et al., 2014 ▸). However, only compound 1 showed clear inhibition of VIM-2, whereas compounds 2 and 3 had no influence on the enzyme activity at concentrations of up to 100 µM (data not shown). To determine the IC50 for compound 1, the inhibition at different concentrations was measured and the data were fitted to a dose–response curve (Fig. 2 ▸). The IC50 was calculated to be 20 µM (pIC50 = 4.71 ± 0.04).
Figure 1.

Structures of the tested triazolylthioacetamide inhibitors: compound 1 (a), compound 2 (b) and compound 3 (c).
Figure 2.

Dose–response curve for the inhibition of VIM-2 by compound 1.
3.2. DMSO-free co-crystallization of VIM-2 with compound 1
We have previously reported crystal structures of VIM-2 in complex with small fragments (Christopeit et al., 2015 ▸). These complex structures were obtained by solving the fragments in DMSO and soaking them into native VIM-2 crystals. However, the soaking time was limited by the negative influence of DMSO on the crystal quality and soaking was not successful for compound 1. Furthermore, adding compound 1 directly to the reservoir or protein solution prevented crystal formation, most likely owing to the high DMSO concentration. Therefore, a DMSO-free co-crystallization strategy was explored in which the reservoir wells were pre-coated with compound 1 (Fig. 3 ▸). To achieve the pre-coating, the dissolved inhibitor was added to the reservoir wells and the DMSO was removed by evaporation. Afterwards, the inhibitor was redissolved in the reservoir solution, which was then used to set up the crystallization experiments. The structures obtained from crystals prepared by the DMSO-free co-crystallization showed additional electron density in the active site of VIM-2 corresponding to compound 1. Although the pre-coating of sitting-drop wells with ligands has been reported before (Gelin et al., 2015 ▸), to our knowledge the pre-coating of reservoir wells with ligands has not been used for co-crystallization.
Figure 3.
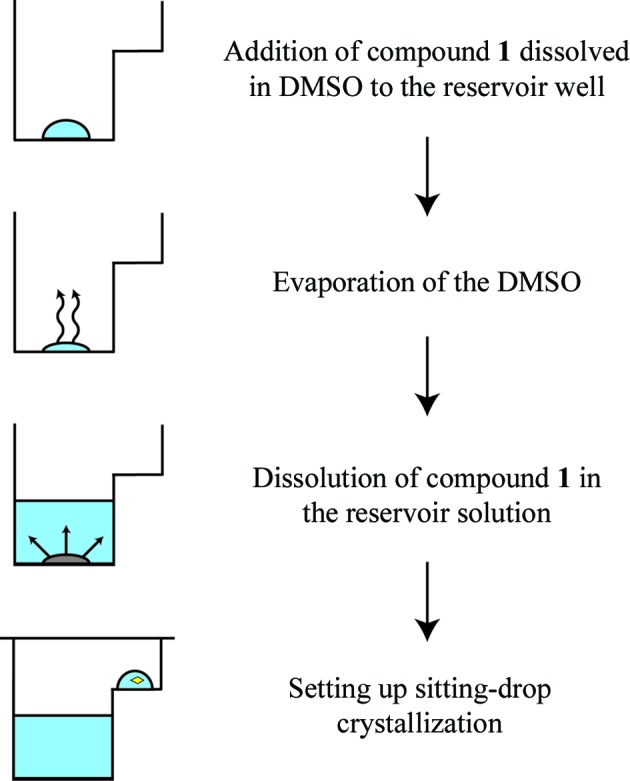
Strategy for DMSO-free co-crystallization of VIM-2 with compound 1. After adding compound 1 to the reservoir well, the DMSO was evaporated and compound 1 was redissolved in the reservoir solution. The reservoir solution was mixed with the protein solution to set up sitting-drop experiments.
3.3. Complex structure of VIM-2 with compound 1
The crystal structure of VIM-2 in complex with compound 1 was solved by molecular replacement to a resolution of 1.5 Å. The complex crystallized in space group C2, with two protein molecules in the asymmetric unit (Fig. 4 ▸). Details of the refinement statistics are shown in Table 3 ▸. The overall completeness of the data set was 99.1%, with a multiplicity of 3.7 and an R merge of 4.8%. The overall structures of the two VIM-2 molecules in the asymmetric unit were similar, with an r.m.s. deviation of 0.36 Å for the Cα atoms (WinCoot; Emsley et al., 2010 ▸).
Figure 4.

Ribbon diagram of the two VIM-2 molecules in the asymmetric unit. Helices are shown in red (chain A) or salmon (chain B) and β-strands are shown in yellow (chain A) or purple (chain B). The inhibitor bound to the active site is shown in stick representation with atom-type colouring and C atoms depicted in cyan. Zn2+ ions are shown as orange spheres and Cl− ions as dark blue spheres.
As is typical for MBLs, VIM-2 was folded in an αβ/βα sandwich. Two Zn2+ ions were observed in the active site of the enzyme, where Zn1 was coordinated by His116, His118 and His196, and Zn2 was coordinated by Asp120, Cys221 and His263. Between the two symmetry mates, a third Zn2+ ion was located, which has been observed before and is normally considered to be a crystallographic artefact. The overall protein structure and the coordination of the Zn2+ ions, as well as the Zn–ligand and Zn–Zn distances, were in agreement with previously published VIM-2 structures (Garcia-Saez et al., 2008 ▸; Aitha et al., 2014 ▸; Christopeit et al., 2015 ▸; Yamaguchi et al., 2007 ▸).
In the active site of both VIM-2 molecules, additional electron density corresponding to a chloride ion was observed. The ions showed high occupancy, and the B factors of 17.3 and 20.5 Å2 for chains A and B, respectively, were consistent with the average B factor of the protein (Table 3 ▸). Although the chloride ions were in proximity to the bound inhibitor, they showed no direct interaction. The ions were coordinated by the backbone N atom of Asp119, the side-chain N atom of Asn165 and one or three water molecules. To our knowledge, chloride ions at this position in the active site of VIM-2 have not been observed before. However, it is not clear whether the chloride ion has any biological relevance or whether it is a crystallographic artefact.
The experimental electron-density map clearly showed additional density corresponding to compound 1 in the active site of both VIM-2 molecules in the asymmetric unit (Fig. 5 ▸). The carboxyl group of the inhibitor interacts with Zn2 and forms hydrogen bonds to two water molecules and the backbone O atom of Asn233. Similar interactions have been observed before for fragments binding to the active site of VIM-2 (Christopeit et al., 2015 ▸). The importance of this interaction for inhibitor binding is reflected by the fact that replacing the carboxyl group by a hydroxide group, as in compound 2, or replacing the benzoic acid moiety by an 4-pyridyl moiety, as in compound 3, resulted in a strong reduction in the inhibitory potency. The triazole ring of compound 1 interacted with both zinc ions and replaced the hydroxide molecule that normally bridges the two zinc ions in the native structure of VIM-2 (Christopeit et al., 2015 ▸; Garcia-Saez et al., 2008 ▸). Furthermore, the benzothiazole ring formed parallel π–π stacking with Phe61 and T-shaped π–π stacking with Trp87. Compared with our previously published structures of native VIM-2 and of VIM-2 in complex with fragments (Christopeit et al., 2015 ▸), the binding of compound 1 induced a flip of the backbone and a 180° flip of the side chain of Trp87 (Fig. 6 ▸). Penicillins are well known substrates of VIM-2 and several of them have bulky hydrophobic groups, e.g. ampicillin or benzylpenicillin (Poirel et al., 2000 ▸). The interactions involved in the binding of different substrates to VIM-2 are not exactly known and no crystal structures in complex with these substrates have been reported. However, the structure of New Delhi metallo-β-lactamase 1 (NDM-1) in complex with hydrolysed benzylpenicillin has been reported (King et al., 2012 ▸). This structure shows that the bulky hydrophobic group of benzylpenicllin forms similar interactions with the conserved residue Trp87 in NDM-1 as compound 1 forms with Trp87 in VIM-2 (Fig. 7 ▸). Hence, it is likely that the binding of substrates with bulky hydrophobic groups to the active site of VIM-2 also induces the same conformational changes of Trp87 as compound 1.
Figure 5.

OMIT maps (a, b), final density maps (c, d) and crystal structures (e, f) of compound 1 bound to the active site of VIM-2. The complex crystallized with two copies in the asymmetric unit (chains A and B). C atoms of compound 1 are depicted in blue and VIM-2 C atoms in green. Water molecules are shown in red and Zn2+ ions in orange. The OMIT F o − F c maps are shown in dark green at 2.25σ. The final 2F o − F c electron-density maps around the ligand are shown in blue at 1σ. The final F o − F c difference density maps around the ligand are shown at 4σ (light green) and −4σ (red). The interactions were analysed using LIGPLOT (g, h). Hydrogen bonds are shown as red dashed lines and interatomic distances are given in Å. Hydrophobic interactions are indicated by red arcs.
Figure 6.
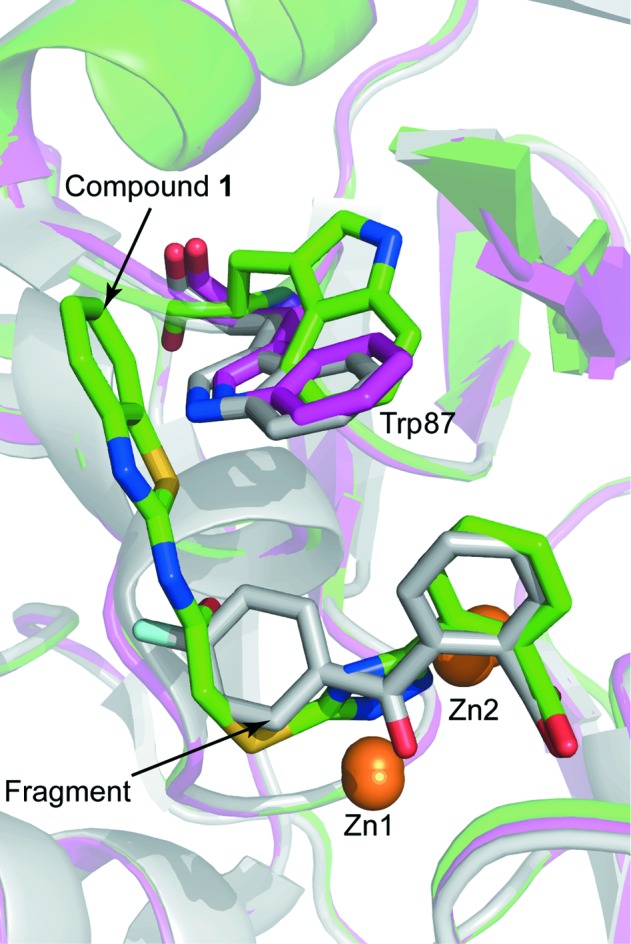
Structural alignment of a native VIM-2 structure (purple; PDB entry 5acu; Christopeit et al., 2015 ▸), the VIM-2 structure in complex with compound 1 (green) and the VIM-2 structure in complex with a fragment (grey; PDB entry 5acx; Christopeit et al., 2015 ▸). The protein backbones are shown as ribbon cartoons. Zinc ions are shown as orange spheres and Trp87, compound 1 and the fragment are shown as sticks.
Figure 7.
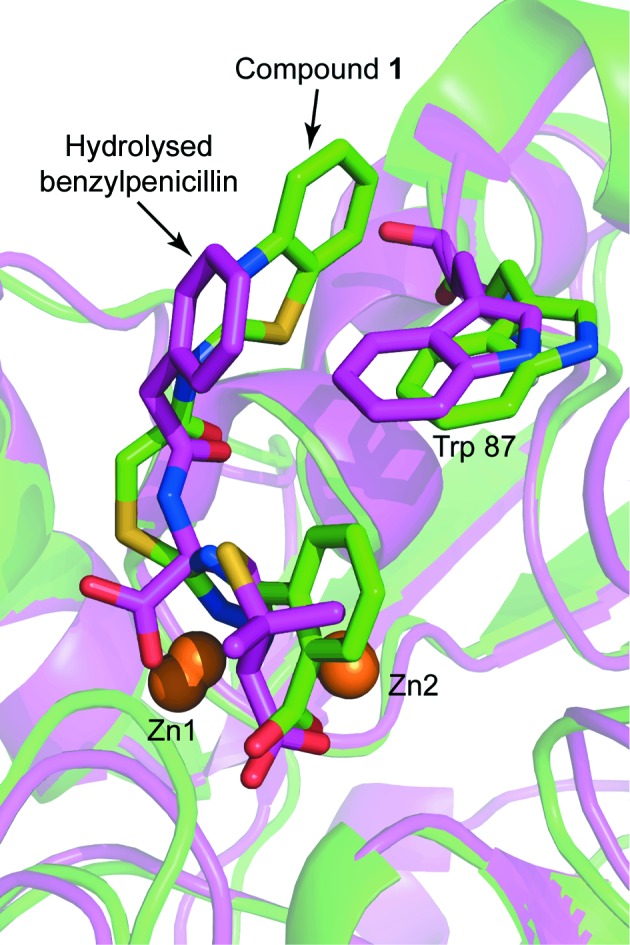
Structural alignment of the VIM-2 structure in complex with compound 1 (green) and a structure of NDM-1 bound to hydrolysed benzylpenicillin (purple; PDB entry 4eyf; King et al., 2012 ▸). The protein backbones are shown as ribbon cartoons. Zinc ions are shown as orange spheres and Trp87, compound 1 and the fragment are shown as sticks.
4. Conclusion
In this study, compound 1 was identified as an inhibitor of the MBL VIM-2. The determined IC50 was in the low-micromolar range and hence needs to be further optimized. However, compound 1 has already been reported as an inhibitor of the MBLs ImiS and CcrA, and therefore is an interesting starting point for the development of broad-spectrum MBL inhibitors (Yang et al., 2015 ▸). Furthermore, the crystal structure of compound 1 in complex with VIM-2 is presented. To our knowledge, this is the first structure of a triazolylthioacetamide inhibitor bound to an MBL. The detailed information about the interactions involved in the binding of the inhibitor and the induced conformational changes of the conserved residue Trp87 will help to further optimize triazolylthioacetamide inhibitors and improve rational drug design targeting MBLs.
Supplementary Material
PDB reference: VIM-2 in complex with a triazolylthioacetamide inhibitor, 5lsc
Acknowledgments
We acknowledge Ørjan Samuelsen for supplying the P. aeruginosa isolate with VIM-2 and Trine Josefine O. Carlsen for the cloning and purification of VIM-2. The provision of beam time at BL14.1, BESSY II, Berlin, Germany is highly valued. This work was supported by the Tromsø Research Foundation and the Research Council of Norway (project Nos. 218539, SYNKNOYT 2011, and 213808, FRIMEDBIO 2011) and by grants 21272186, 21572179 and 81361138018 (to K-WY) from the National Natural Science Foundation of China.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Aitha, M., Marts, A. R., Bergstrom, A., Møller, A. J., Moritz, L., Turner, L., Nix, J. C., Bonomo, R. A., Page, R. C., Tierney, D. L. & Crowder, M. W. (2014). Biochemistry, 53, 7321–7331. [DOI] [PMC free article] [PubMed]
- Brem, J., van Berkel, S. S., Aik, W., Rydzik, A. M., Avison, M. B., Pettinati, I., Umland, K.-D., Kawamura, A., Spencer, J., Claridge, T. D. W., McDonough, M. A. & Schofield, C. J. (2014). Nature Chem. 6, 1084–1090. [DOI] [PubMed]
- Brem, J., van Berkel, S. S., Zollman, D., Lee, S. Y., Gileadi, O., McHugh, P. J., Walsh, T. R., McDonough, M. A. & Schofield, C. J. (2016). Antimicrob. Agents Chemother. 60, 142–150. [DOI] [PMC free article] [PubMed]
- Christopeit, T., Carlsen, T. J., Helland, R. & Leiros, H. K. (2015). J. Med. Chem. 58, 8671–8682. [DOI] [PubMed]
- Docquier, J.-D., Lamotte-Brasseur, J., Galleni, M., Amicosante, G., Frère, J.-M. & Rossolini, G. M. (2003). J. Antimicrob. Chemother. 51, 257–266. [DOI] [PubMed]
- Drawz, S. M. & Bonomo, R. A. (2010). Clin. Microbiol. Rev. 23, 160–201. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Evans, P. R. (2011). Acta Cryst. D67, 282–292. [DOI] [PMC free article] [PubMed]
- Evans, P. R. & Murshudov, G. N. (2013). Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed]
- Fast, W. & Sutton, L. D. (2013). Biochim. Biophys. Acta, 1834, 1648–1659. [DOI] [PubMed]
- Garau, G., García-Sáez, I., Bebrone, C., Anne, C., Mercuri, P., Galleni, M., Frère, J.-M. & Dideberg, O. (2004). Antimicrob. Agents Chemother. 48, 2347–2349. [DOI] [PMC free article] [PubMed]
- Garcia-Saez, I., Docquier, J.-D., Rossolini, G. M. & Dideberg, O. (2008). J. Mol. Biol. 375, 604–611. [DOI] [PubMed]
- Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D. & Bairoch, A. (2005). The Proteomics Protocols Handbook, edited by J. M. Walker, pp. 571–607. Totowa: Humana Press.
- Gelin, M., Delfosse, V., Allemand, F., Hoh, F., Sallaz-Damaz, Y., Pirocchi, M., Bourguet, W., Ferrer, J.-L., Labesse, G. & Guichou, J.-F. (2015). Acta Cryst. D71, 1777–1787. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- King, D. T., Worrall, L. J., Gruninger, R. & Strynadka, N. C. J. (2012). J. Am. Chem. Soc. 134, 11362–11365. [DOI] [PubMed]
- Lucasti, C., Vasile, L., Sandesc, D., Venskutonis, D., McLeroth, P., Lala, M., Rizk, M. L., Brown, M. L., Losada, M. C., Pedley, A., Kartsonis, N. A. & Paschke, A. (2016). Antimicrob Agents Chemother. 60, 6234–6243. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Mueller, U., Darowski, N., Fuchs, M. R., Förster, R., Hellmig, M., Paithankar, K. S., Pühringer, S., Steffien, M., Zocher, G. & Weiss, M. S. (2012). J. Synchrotron Rad. 19, 442–449. [DOI] [PMC free article] [PubMed]
- Poirel, L., Naas, T., Nicolas, D., Collet, L., Bellais, S., Cavallo, J. D. & Nordmann, P. (2000). Antimicrob. Agents Chemother. 44, 891–897. [DOI] [PMC free article] [PubMed]
- Wallace, A. C., Laskowski, R. A. & Thornton, J. M. (1995). Protein Eng. 8, 127–134. [DOI] [PubMed]
- World Health Organization (2014). Antimicrobial Resistance: Global Report on Surveillance. Geneva: World Health Organization. http://www.who.int/drugresistance/documents/surveillancereport/en/.
- Yamaguchi, Y., Jin, W., Matsunaga, K., Ikemizu, S., Yamagata, Y., Wachino, J., Shibata, N., Arakawa, Y. & Kurosaki, H. (2007). J. Med. Chem. 50, 6647–6653. [DOI] [PubMed]
- Yang, S.-K., Kang, J. S., Oelschlaeger, P. & Yang, K.-W. (2015). ACS Med. Chem. Lett. 6, 455–460. [DOI] [PMC free article] [PubMed]
- Zhai, L., Zhang, Y.-L., Kang, J. S., Oelschlaeger, P., Xiao, L., Nie, S.-S. & Yang, K.-W. (2016). ACS Med. Chem. Lett. 7, 413–417. [DOI] [PMC free article] [PubMed]
- Zhang, Y.-L., Yang, K.-W., Zhou, Y.-J., LaCuran, A. E., Oelschlaeger, P. & Crowder, M. W. (2014). ChemMedChem, 9, 2445–2448. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: VIM-2 in complex with a triazolylthioacetamide inhibitor, 5lsc